
A Homozygous AKNA Frameshift Variant Is Associated with Microcephaly in a Pakistani Family

 ,
,  , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
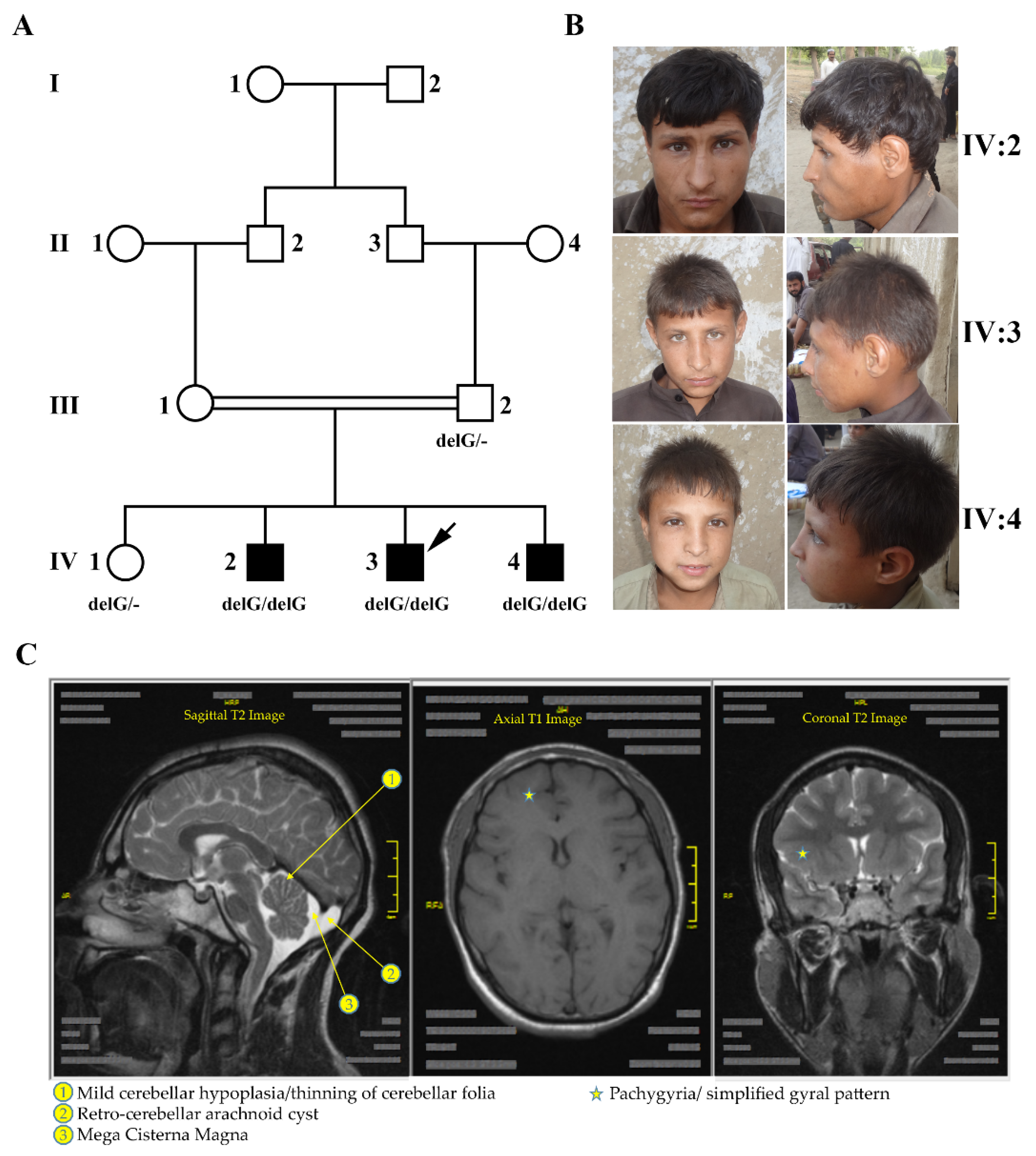
2.1. Clinical Manifestations
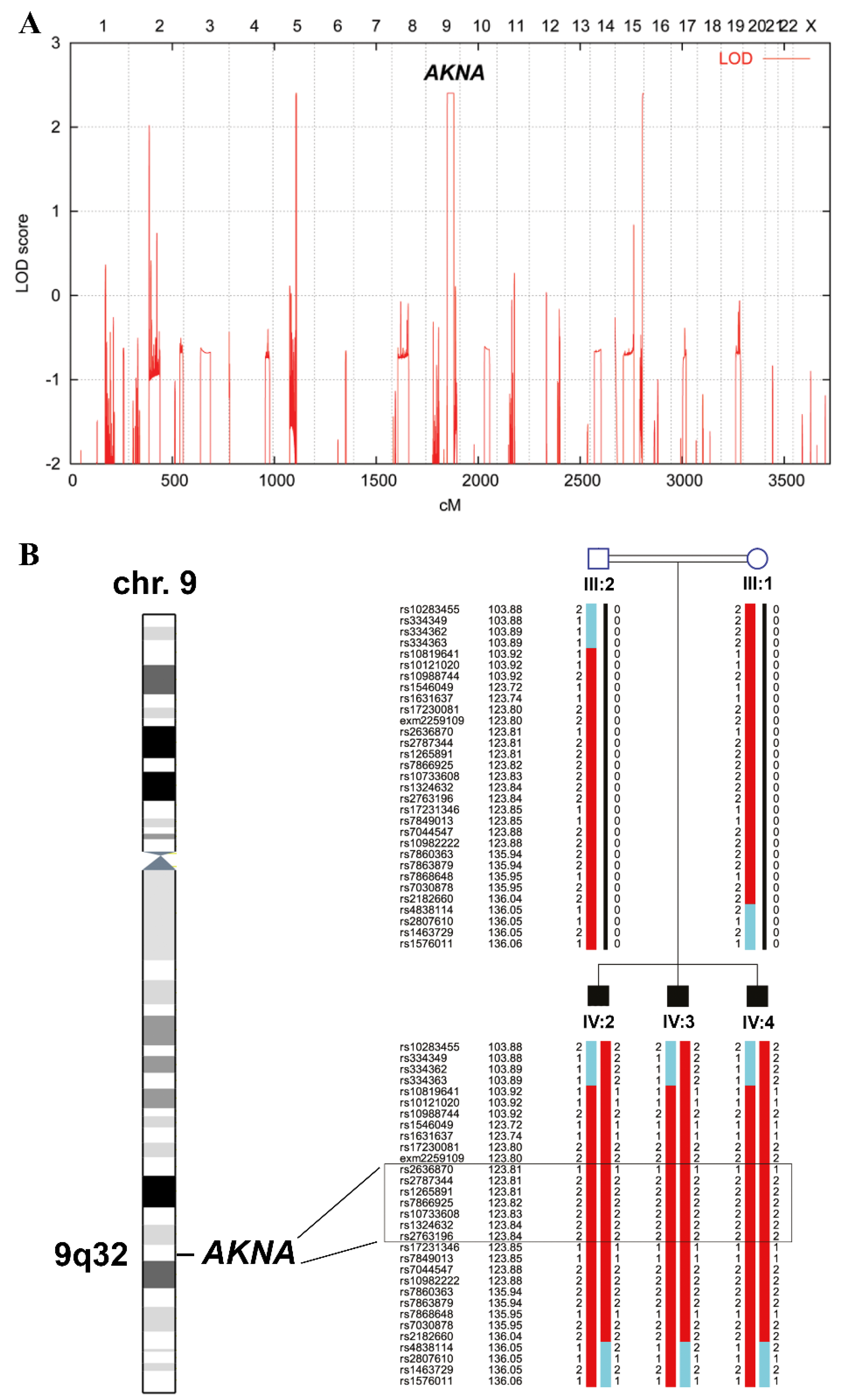
2.2. Linkage Analysis
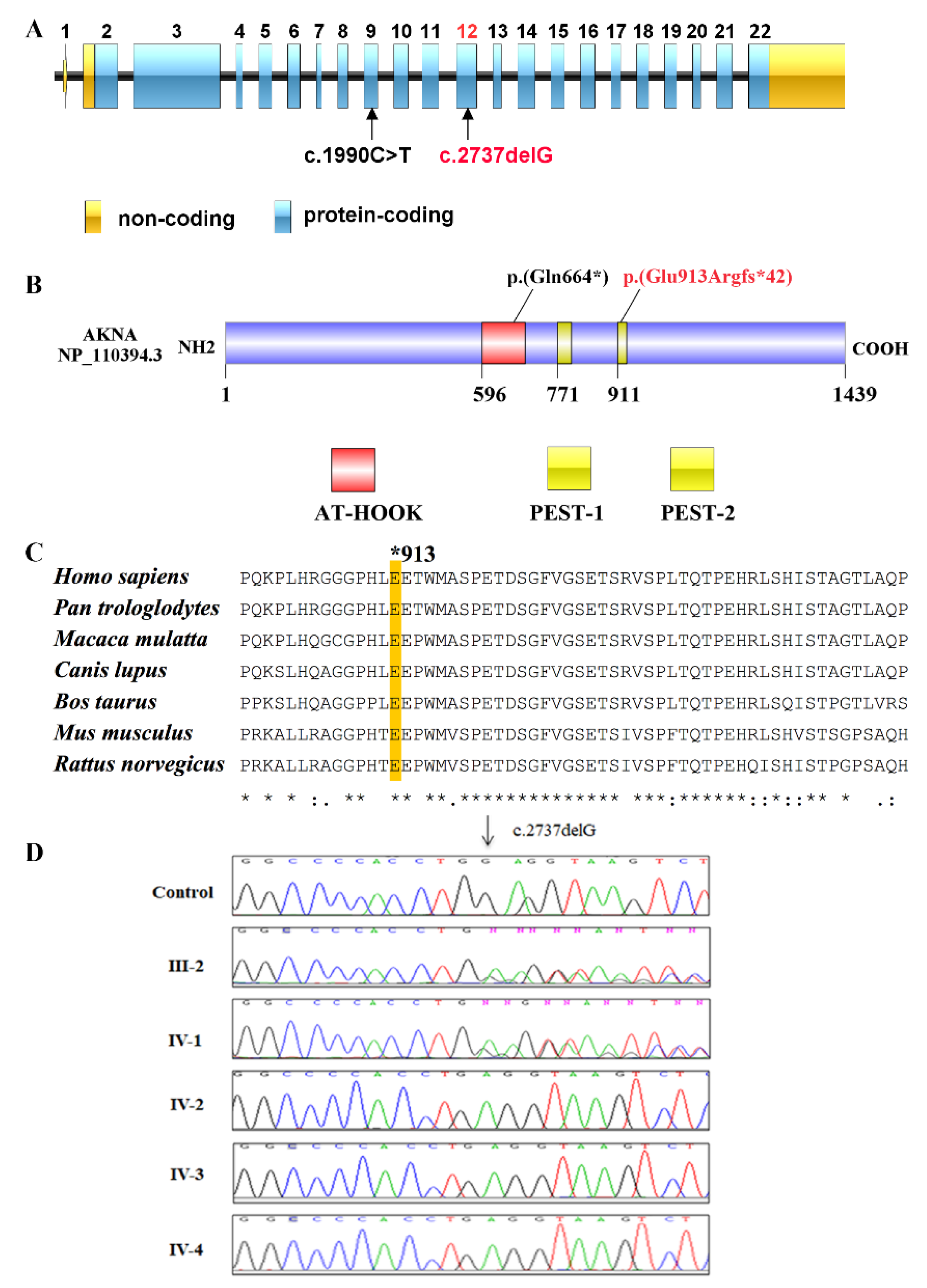
2.3. Next Generation Sequencing
2.4. In Silico Analyses of Identified Variants
2.5. Sanger Sequencing
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shaheen, R.; Maddirevula, S.; Ewida, N.; Alsahli, S.; Abdel-Salam, G.; Zaki, M.S.; Al Tala, S.; Alhashem, A.; Softah, A.; Al-Owain, M.; et al. Genomic and phenotypic delineation of congenital microcephaly. Genet. Med. 2018, 21, 545–552. [Google Scholar] [CrossRef]
- Cox, J.; Jackson, A.; Bond, J.; Woods, C.G. What primary microcephaly can tell us about brain growth. Trends Mol. Med. 2006, 12, 358–366. [Google Scholar] [CrossRef]
- Kaindl, A.M.; Passemard, S.; Kumar, P.; Kraemer, N.; Issa, L.; Zwirner, A.; Gerard, B.; Verloes, A.; Mani, S.; Gressens, P. Many roads lead to primary autosomal recessive microcephaly. Prog. Neurobiol. 2010, 90, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Thornton, G.K.; Woods, C.G. Primary microcephaly: Do all roads lead to Rome? Trends Genet. 2009, 25, 501–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, J. Physicians. With physician employment on the rise, new compensation models emerge. Hosp. Health Netw. 2010, 84, 10. [Google Scholar] [PubMed]
- Woods, C.G.; Bond, J.; Enard, W. Autosomal Recessive Primary Microcephaly (MCPH): A Review of Clinical, Molecular, and Evolutionary Findings. Am. J. Hum. Genet. 2005, 76, 717–728. [Google Scholar] [CrossRef] [Green Version]
- Zaqout, S.; Morris-Rosendahl, D.; Kaindl, A.M. Autosomal Recessive Primary Microcephaly (MCPH): An Update. Neuropediatrics 2017, 48, 135–142. [Google Scholar] [CrossRef]
- Jean, F.; Stuart, A.; Tarailo-Graovac, M. Dissecting the Genetic and Etiological Causes of Primary Microcephaly. Front. Neurol. 2020, 11, 570830. [Google Scholar] [CrossRef]
- Farooq, M.; Lindbæk, L.; Krogh, N.; Doganli, C.; Keller, C.; Mönnich, M.; Gonçalves, A.B.; Sakthivel, S.; Mang, Y.; Fatima, A.; et al. RRP7A links primary microcephaly to dysfunction of ribosome biogenesis, resorption of primary cilia, and neurogenesis. Nat. Commun. 2020, 11, 5816. [Google Scholar] [CrossRef] [PubMed]
- Parry, D.A.; Martin, C.A.; Greene, P.; Marsh, J.A.; Blyth, M.; Cox, H.; Donnelly, D.; Greenhalgh, L.; Greville-Heygate, S.; Harrison, V.; et al. Heterozygous lamin B1 and lamin B2 variants cause primary microcephaly and define a novel laminopathy. Genet. Med. 2021, 23, 408–414. [Google Scholar] [CrossRef]
- Wang, Y.J.; Zhou, X.K.; Xu, D. Update on autosomal recessive primary microcephaly (MCPH)-associated proteins. Yi Chuan 2019, 41, 905–918. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, D.; Bae, B.-I.; Walsh, C.A. The Genetics of Primary Microcephaly. Annu. Rev. Genom. Hum. Genet. 2018, 19, 177–200. [Google Scholar] [CrossRef] [Green Version]
- Siddiqa, A.; Sims-Mourtada, J.C.; Guzman-Rojas, L.; Rangel, R.; Guret, C.; Madrid-Marina, V.; Sun, Y.; Martinez-Valdez, H. Regulation of CD40 and CD40 ligand by the AT-hook transcription factor AKNA. Nature 2001, 410, 383–387. [Google Scholar] [CrossRef]
- Ma, W.; Ortiz-Quintero, B.; Rangel, R.; McKeller, M.R.; Herrera-Rodriguez, S.; Castillo, E.F.; Schluns, K.S.; Hall, M.; Zhang, H.; Suh, W.K.; et al. Coordinate activation of inflammatory gene networks, alveolar destruction and neonatal death in AKNA deficient mice. Cell Res. 2011, 21, 1564–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hug, P.; Anderegg, L.; Kehl, A.; Jagannathan, V.; Leeb, T. AKNA Frameshift Variant in Three Dogs with Recurrent Inflammatory Pulmonary Disease. Genes 2019, 10, 567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamseldin, H.E.; Al Mogarri, I.; Alqwaiee, M.M.; Alharbi, A.S.; Baqais, K.; AlSaadi, M.; AlAnzi, T.; Alhashem, A.; Saghier, A.; Ameen, W.; et al. An exome-first approach to aid in the diagnosis of primary ciliary dyskinesia. Hum. Genet. 2020, 139, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Camargo Ortega, G.; Falk, S.; Johansson, P.A.; Peyre, E.; Broix, L.; Sahu, S.K.; Hirst, W.; Schlichthaerle, T.; De Juan Romero, C.; Draganova, K.; et al. The centrosome protein AKNA regulates neurogenesis via microtubule organization. Nature 2019, 567, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Rüschendorf, F.; Nürnberg, P. ALOHOMORA: A tool for linkage analysis using 10K SNP array data. Bioinformatics 2005, 21, 2123–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abecasis, G.R.; Cherny, S.S.; Cookson, W.O.C.; Cardon, L.R. GRR: Graphical representation of relationship errors. Bioinformatics 2001, 17, 742–743. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, J.R.; Weeks, D.E. PedCheck: A program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 1998, 63, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Abecasis, G.R.; Cherny, S.; Cookson, W.O.; Cardon, L.R. Merlin—Rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2001, 30, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Thiele, H.; Nürnberg, P. HaploPainter: A tool for drawing pedigrees with complex haplotypes. Bioinformatics 2005, 21, 1730–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, I.; Baig, S.M.; Abdulkareem, A.R.; Hussain, M.S.; Sur, I.; Toliat, M.R.; Nürnberg, G.; Dalibor, N.; Moawia, A.; Waseem, S.S.; et al. Genetic heterogeneity in Pakistani microcephaly families revisited. Clin. Genet. 2017, 92, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Rasool, S.; Baig, J.M.; Moawia, A.; Ahmad, I.; Iqbal, M.; Waseem, S.S.; Asif, M.; Abdullah, U.; Makhdoom, E.U.H.; Kaygusuz, E.; et al. An update of pathogenic variants in ASPM, WDR62, CDK5RAP2, STIL, CENPJ, and CEP135 underlying autosomal recessive primary microcephaly in 32 consanguineous families from Pakistan. Mol. Genet. Genom. Med. 2020, 8, e1408. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.S.; Baig, S.M.; Neumann, S.; Nürnberg, G.; Farooq, M.; Ahmad, I.; Alef, T.; Hennies, H.C.; Technau, M.; Altmüller, J.; et al. A Truncating Mutation of CEP135 Causes Primary Microcephaly and Disturbed Centrosomal Function. Am. J. Hum. Genet. 2012, 90, 871–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basto, R.; Lau, J.; Vinogradova, T.; Gardiol, A.; Woods, C.G.; Khodjakov, A.; Raff, J.W. Flies without Centrioles. Cell 2006, 125, 1375–1386. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, E.; Wason, A.; Ramani, A.; Gooi, L.M.; Keller, P.; Pozniakovsky, A.; Poser, I.; Noack, F.; Telugu, N.S.; Calegari, F.; et al. CPAP promotes timely cilium disassembly to maintain neural progenitor pool. EMBO J. 2016, 35, 803–819. [Google Scholar] [CrossRef]
- Thawani, A.; Kadzik, R.S.; Petry, S. XMAP215 is a microtubule nucleation factor that functions synergistically with the γ-tubulin ring complex. Nat. Cell Biol. 2018, 20, 575–585. [Google Scholar] [CrossRef]
- Fernandez, V.; Llinares-Benadero, C.; Borrell, V. Cerebral cortex expansion and folding: What have we learned? EMBO J. 2016, 35, 1021–1044. [Google Scholar] [CrossRef]
- Sims-Mourtada, J.C.; Bruce, S.; McKeller, M.R.; Rangel, R.; Guzman-Rojas, L.; Cain, K.; Lopez, C.; Zimonjic, D.B.; Popescu, N.C.; Gordon, J.; et al. The human AKNA gene expresses multiple transcripts and protein isoforms as a result of alternative promoter usage, splicing, and polyadenylation. DNA Cell Biol. 2005, 24, 325–338. [Google Scholar] [CrossRef]
- Zarudnaya, M.I.; Kolomiets, I.M.; Potyahaylo, A.L.; Hovorun, D.M. Downstream elements of mammalian pre-mRNA polyadenylation signals: Primary, secondary and higher-order structures. Nucleic Acids Res. 2003, 31, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Mirra, V.; Werner, C.; Santamaria, F. Primary Ciliary Dyskinesia: An Update on Clinical Aspects, Genetics, Diagnosis, and Future Treatment Strategies. Front. Pediatr. 2017, 5, 135. [Google Scholar] [CrossRef] [PubMed]
- Horani, A.; Ferkol, T.W.; Dutcher, S.K.; Brody, S.L. Genetics and biology of primary ciliary dyskinesia. Paediatr. Respir. Rev. 2015, 18, 18–24. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, C.W.; Preston, C.C.; Finn, R.; Eyster, K.M.; Faustino, R.S.; Lee, L. Strain-specific differences in brain gene expression in a hydrocephalic mouse model with motile cilia dysfunction. Sci. Rep. 2018, 8, 13370. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.; Mader, M.T.; Irmler, M.; Gentilini, M.; Santoni, F.; Drechsel, D.; Blum, R.; Stahl, R.; Bulfone, A.; Malatesta, P.; et al. Prospective isolation of functionally distinct radial glial subtypes—Lineage and transcriptome analysis. Mol. Cell Neurosci. 2008, 38, 15–42. [Google Scholar] [CrossRef] [PubMed]
- Betts, J.C.; Nabel, G.J. Differential regulation of NF-kappaB2(p100) processing and control by amino-terminal sequences. Mol. Cell Biol. 1996, 16, 6363–6371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stennicke, H.R.; Salvesen, G.S. Caspases-controlling intracellular signals by protease zymogen activation. Biochim. Biophys. Acta 2000, 1477, 299–306. [Google Scholar] [CrossRef]
- Salmerón, A.; Janzen, J.; Soneji, Y.; Bump, N.; Kamens, J.; Allen, H.; Ley, S.C. Direct Phosphorylation of NF-κB1 p105 by the IκB Kinase Complex on Serine 927 Is Essential for Signal-induced p105 Proteolysis. J. Biol. Chem. 2001, 276, 22215–22222. [Google Scholar] [CrossRef] [Green Version]
- Rogers, S.; Wells, R.; Rechsteiner, M. Amino acid sequences common to rapidly degraded proteins: The PEST hypothesis. Science 1986, 234, 364–368. [Google Scholar] [CrossRef]
- Waters, A.M.; Asfahani, R.; Carroll, P.; Bicknell, L.; Lescai, F.; Bright, A.; Chanudet, E.; Brooks, A.; Christou-Savina, S.; Osman, G.; et al. The kinetochore protein, CENPF, is mutated in human ciliopathy and microcephaly phenotypes. J. Med. Genet. 2015, 52, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Coppieters, F.; Lefever, S.; Leroy, B.P.; De Baere, E. CEP290, a gene with many faces: Mutation overview and presentation of CEP290base. Hum. Mutat. 2010, 31, 1097–1108. [Google Scholar] [CrossRef] [Green Version]
- Sha, Y.W.; Xu, X.; Mei, L.B.; Li, P.; Su, Z.Y.; He, X.Q.; Li, L. A homozygous CEP135 mutation is associated with multiple morphological abnormalities of the sperm flagella (MMAF). Gene 2017, 633, 48–53. [Google Scholar] [CrossRef]
- Bamborschke, D.; Daimagüler, H.S.; Hahn, A.; Hussain, M.S.; Nürnberg, P.; Cirak, S. Mutation in CEP135 causing primary microcephaly and subcortical heterotopia. Am. J. Med. Genet. A 2020, 182, 2450–2453. [Google Scholar] [CrossRef] [PubMed]
- Nerakh, G.; Mounika, K.; Geeta, K.; Nalluri, H.B. CEP135 associated primary Microcephaly-A rare presentation in early second trimester. Eur. J. Med. Genet. 2021, 64, 104233. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patient Characteristics | Patients with a Novel Homozygous Frameshift Variant in AKNA (c.2737delG) | |||
|---|---|---|---|---|
| Patient ID | IV-2 | IV-3 | IV-4 | |
| Origin | Pakistani | Pakistani | Pakistani | |
| Gender | Male | Male | Male | |
| Age | 15 years | 9 years | 6 years | |
| Pregnancy | Normal | Normal | Normal | |
| Head circumference (HC) | 43 cm (−7.5 SD) | 43 cm (−7.0 SD) | 42 cm (−7.5 SD) | |
| Hypotonia | No | No | No | |
| Delay of motor milestones | Not reported | Not reported | Not reported | |
| Seizures | No | No | No | |
| Cardiac problems | No | No | No | |
| Facial anomalies | No | No | No | |
| Ophthalmologic findings | No | No | No | |
| Hearing impairment | Yes (can hear only loud sounds) | Yes (can hear only loud sounds) | Yes (can hear only loud sounds) | |
| Gait abnormality | No | No | No | |
| Speech impairment | Yes (only few words, no sentences) | Yes (only few words, no sentences) | Yes (only few words, no sentences) | |
| Cognitive skills (problem solving, judgement power) | Not age appropriate | Not age appropriate | Not age appropriate | |
| Self-feeding | Yes | Yes | Yes | |
| Self-clothing | Yes | Yes | Yes | |
| Ataxia | No | No | No | |
| Learning disability | Yes (no schooling at all) | Yes (no schooling at all) | Yes (no schooling at all) | |
| Limb/trunk deformities | No | No | No | |
| Muscle function (muscle tone, strength, endurance) | Normal | Normal | Normal | |
| Nasal voice | Yes | Yes | Yes | |
| Recurrent fever | Yes | Yes | Yes | |
| MRI brain | Age at scan | NR | 15 years | NR |
| Simplified gyral pattern | NR | Yes | NR | |
| Microcephaly | NR | Yes | NR | |
| Thin filea | NR | Yes | NR | |
| Mega cisterna magna | NR | Yes | NR | |
| Thick nasal mucosa | NR | Yes | NR | |
| Brain stem | NR | Normal | NR | |
| Others | NR | Normal | NR | |
| Chest CT | Lung | NR | Normal | NR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waseem, S.S.; Moawia, A.; Budde, B.; Tariq, M.; Khan, A.; Ali, Z.; Khan, S.; Iqbal, M.; Malik, N.A.; Haque, S.u.; et al. A Homozygous AKNA Frameshift Variant Is Associated with Microcephaly in a Pakistani Family. Genes 2021, 12, 1494. https://doi.org/10.3390/genes12101494
Waseem SS, Moawia A, Budde B, Tariq M, Khan A, Ali Z, Khan S, Iqbal M, Malik NA, Haque Su, et al. A Homozygous AKNA Frameshift Variant Is Associated with Microcephaly in a Pakistani Family. Genes. 2021; 12(10):1494. https://doi.org/10.3390/genes12101494
Chicago/Turabian StyleWaseem, Syeda Seema, Abubakar Moawia, Birgit Budde, Muhammad Tariq, Ayaz Khan, Zafar Ali, Sheraz Khan, Maria Iqbal, Naveed Altaf Malik, Saif ul Haque, and et al. 2021. "A Homozygous AKNA Frameshift Variant Is Associated with Microcephaly in a Pakistani Family" Genes 12, no. 10: 1494. https://doi.org/10.3390/genes12101494