
Transcriptome Analysis of Post-Mortem Brain Tissue Reveals Up-Regulation of the Complement Cascade in a Subgroup of Schizophrenia Patients

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. RNA Extraction and Sequencing
2.3. Transcriptome Analysis
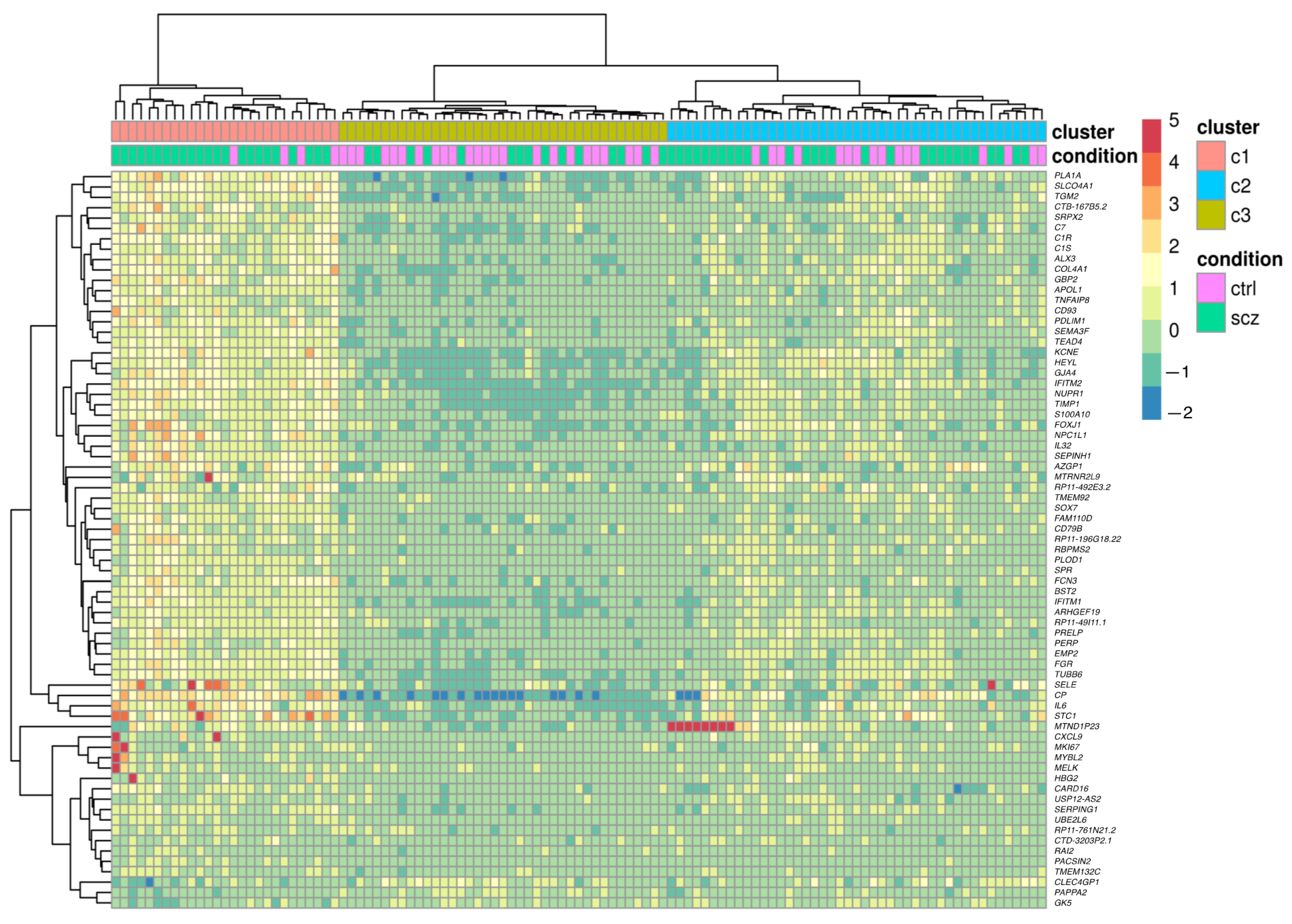
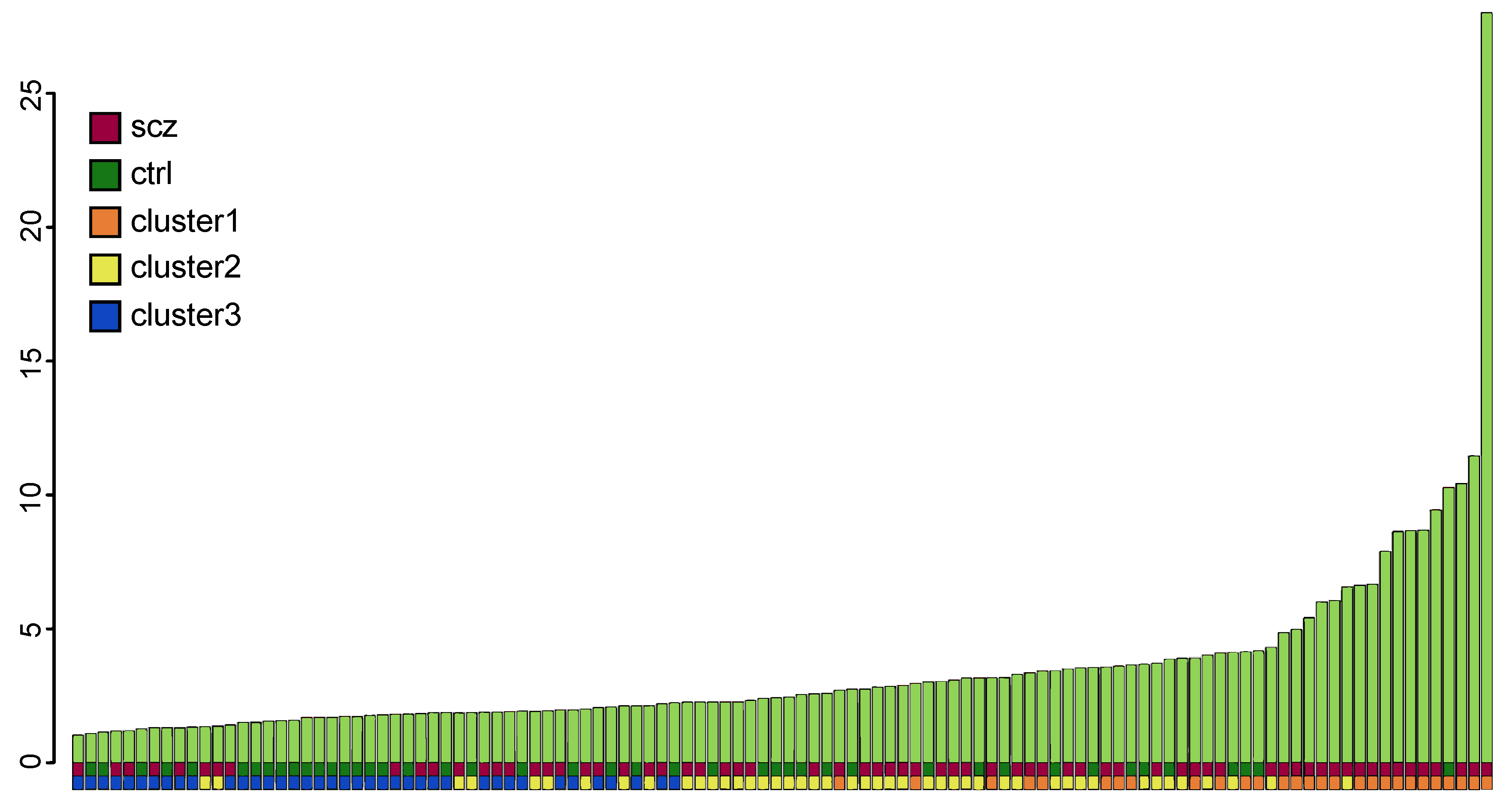
2.4. Cluster Analysis
2.5. Validation
2.6. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.7. Network Analysis of the High Expression Cluster
2.8. Module Preservation Analysis
2.9. Analysis of Overlaps with Previously Identified SCZ Genes
3. Results
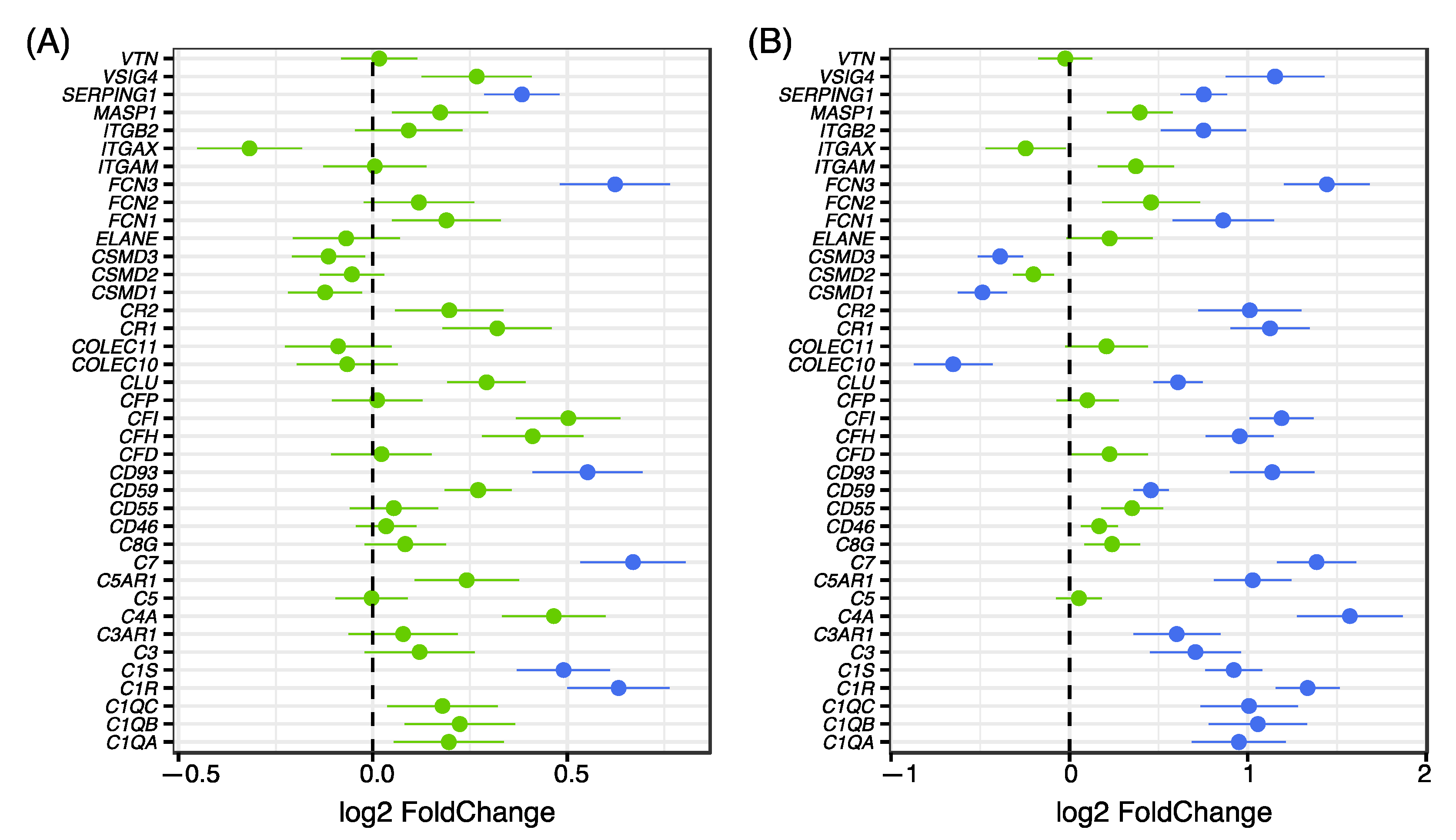
3.1. Investigation of Genes in the Complement System
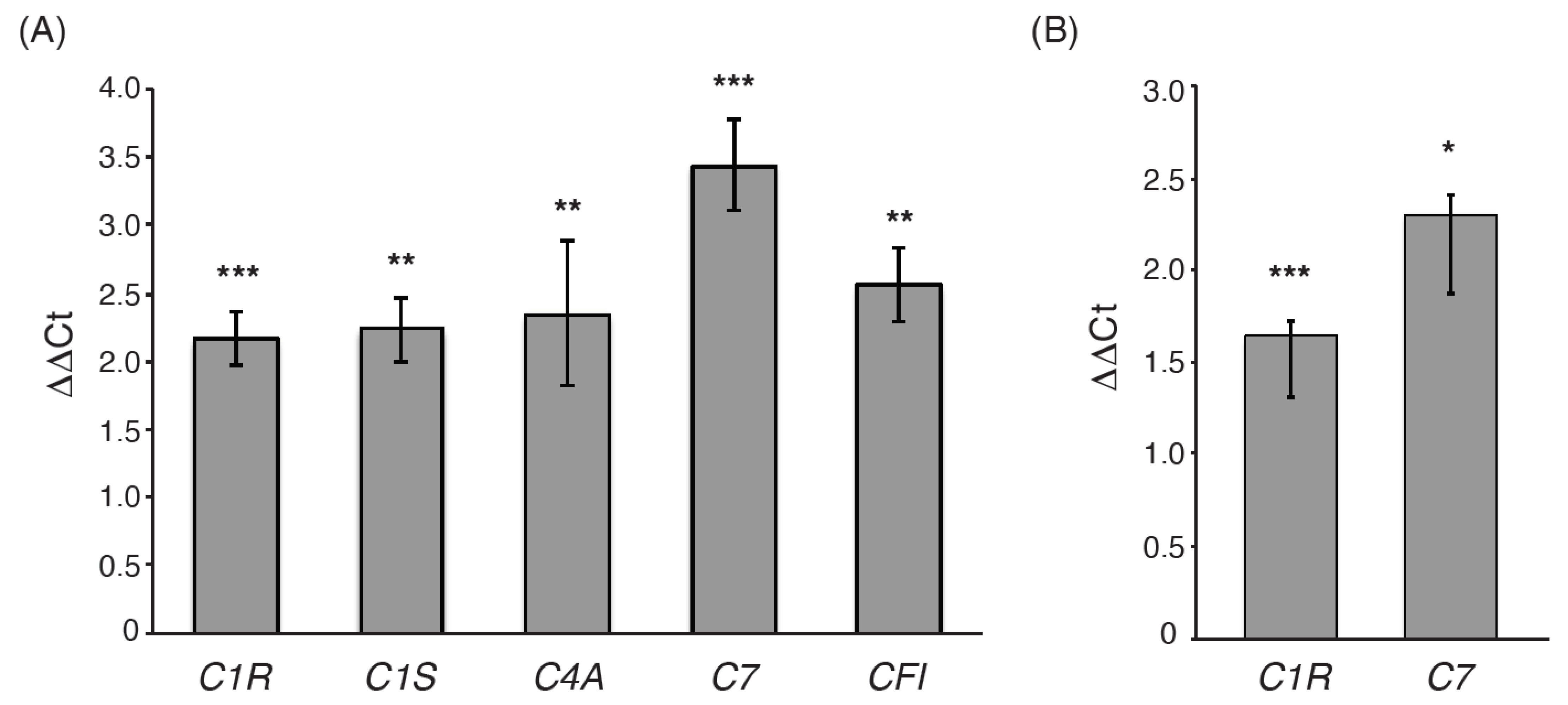
3.2. Validation of Differentially Expressed Genes
3.3. Network Analysis
3.4. Module Preservation Analysis
3.5. Gene Set Enrichment Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cardno, A.G.; Marshall, E.J.; Coid, B.; Macdonald, A.M.; Ribchester, T.R.; Davies, N.J. Heritability estimates for psychotic disorders: The Maudsley twin psychosis series. Arch. Gen. Psychiatry 1999, 56, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Ripke, S.; Neale, B.M.; Corvin, A.; Walters, J.T.; Farh, K.H.; Holmans, P.A.; Milanova, V. Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar]
- Malhotra, D.; Sebat, J. CNVs: Harbingers of a Rare Variant Revolution in Psychiatric Genetics. Cell 2012, 148, 1223–1241. [Google Scholar] [CrossRef] [Green Version]
- Rees, E.; Walters, J.; Chambert, K.D.; O’Dushlaine, C.; Szatkiewicz, J.; Richards, A.L.; Georgieva, L.; Mahoney-Davies, G.; Legge, S.; Moran, J.; et al. CNV analysis in a large schizophrenia sample implicates deletions at 16p12.1 and SLC1A1 and duplications at 1p36.33 and CGNL1. Hum. Mol. Genet. 2014, 23, 1669–1676. [Google Scholar] [CrossRef] [PubMed]
- Szatkiewicz, J.P.; O’Dushlaine, C.; Chen, G.; Chambert, K.; Moran, J.L.; Neale, B.M.; Fromer, M.; Ruderfer, U.; Akterin, S.; Bergen, S.E.; et al. Copy number variation in schizophrenia in Sweden. Mol. Psychiatry 2014, 19, 762–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fromer, M.; Pocklington, A.; Kavanagh, D.H.; Williams, H.; Dwyer, S.; Gormley, P.; Georgieva, L.; Rees, E.; Palta, P.; Ruderfer, D.; et al. De novo mutations in schizophrenia implicate synaptic networks. Nat. Cell Biol. 2014, 506, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Network, T. Network, Pathway Analysis Subgroup of Psychiatric Genomics C. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat. Neurosci. 2015, 18, 199–209. [Google Scholar]
- Barnes, M.; Huxley-Jones, J.; Maycox, P.R.; Lennon, M.; Thornber, A.; Kelly, F.; Bates, S.; Taylor, A.; Reid, J.; Jones, N.; et al. Transcription and pathway analysis of the superior temporal cortex and anterior prefrontal cortex in schizophrenia. J. Neurosci. Res. 2011, 89, 1218–1227. [Google Scholar] [CrossRef]
- Fillman, S.G.; Cloonan, N.; Catts, V.; Miller, L.C.; Wong, J.; McCrossin, T.; Cairns, M.; Weickert, C.S. Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol. Psychiatry 2012, 18, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Hwang, Y.; Kim, J.; Shin, J.-Y.; Seo, J.-S.; Webster, M.J.; Lee, D.; Kim, S. Gene expression profiling by mRNA sequencing reveals increased expression of immune/inflammation-related genes in the hippocampus of individuals with schizophrenia. Transl. Psychiatry 2013, 3, e321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fromer, M.; Roussos, P.; Sieberts, S.K.; Johnson, J.S.; Kavanagh, D.H.; Perumal, T.M. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 2016, 19, 1442–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandal, M.J.; Zhang, P.; Hadjimichael, E.; Walker, R.L.; Chen, C.; Liu, S.; Won, H.; Van Bakel, H.; Varghese, M.; Wang, Y.; et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018, 362, eaat8127. [Google Scholar] [CrossRef] [Green Version]
- Sekar, A.; Adolfsson, R.; Bialas, A.R.; De Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephan, A.H.; Barres, B.A.; Stevens, B. The Complement System: An Unexpected Role in Synaptic Pruning during Development and Disease. Annu. Rev. Neurosci. 2012, 35, 369–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.; Mehalow, A.; Huberman, A.D.; Stafford, B.; et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, I. Schizophrenia: Caused by a fault in programmed synaptic elimination during adolescence? J. Psychiatr. Res. 1982, 17, 319–334. [Google Scholar] [CrossRef]
- Zhu, H.; Urban, D.J.; Blashka, J.; McPheeters, M.T.; Kroeze, W.K.; Mieczkowski, P.; Overholser, J.C.; Jurjus, G.J.; Dieter, L.; Mahajan, G.J.; et al. Quantitative analysis of focused A-to-I RNA editing sites by Ultra-High-Throughput Sequencing in psychiatric disorders. PLoS ONE 2012, 7, e43227. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Kirov, G.; Rees, E.; Walters, J.T.; Escott-Price, V.; Georgieva, L.; Richards, A.L.; Chambert, K.D.; Davies, G.; Legge, S.E.; Moran, J.L.; et al. The Penetrance of Copy Number Variations for Schizophrenia and Developmental Delay. Biol. Psychiatry 2014, 75, 378–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girard, S.; Gauthier, J.; Noreau, A.; Xiong, L.; Zhou, S.; Jouan, L.; Dionne-Laporte, A.; Spiegelman, D.; Henrion, E.; Diallo, O.; et al. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat. Genet. 2011, 43, 860–863. [Google Scholar] [CrossRef]
- Xu, B.; Ionita-Laza, I.; Roos, J.; Boone, B.; Woodrick, S.; Sun, Y.; Levy, S.; A Gogos, J.; Karayiorgou, M. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat. Genet. 2012, 44, 1365–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulsuner, S.; Walsh, T.; Watts, A.C.; Lee, M.K.; Thornton, A.M.; Casadei, S.; Rippey, C.; Shahin, H.; Nimgaonkar, V.L.; Go, R.C.; et al. Spatial and Temporal Mapping of De Novo Mutations in Schizophrenia to a Fetal Prefrontal Cortical Network. Cell 2013, 154, 518–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, S.E.; Gillis, J.; Kramer, M.R.; Lihm, J.; Yoon, S.; Berstein, Y.; Mistry, M.; Pavlidis, P.; Solomon, R.; Ghiban, E.; et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol. Psychiatry 2014, 19, 652–658. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Gaujoux, R.; Seoighe, C. CellMix: A comprehensive toolbox for gene expression deconvolution. Bioinformatics 2013, 29, 2211–2212. [Google Scholar] [CrossRef]
- Costa, V.; Aprile, M.; Esposito, R.; Ciccodicola, A. RNA-Seq and human complex diseases: Recent accomplishments and future perspectives. Eur. J. Hum. Genet. 2013, 21, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Busse, S.; Busse, M.; Schiltz, K.; Bielau, H.; Gos, T.; Brisch, R.; Mawrin, C.; Schmitt, A.; Jordan, W.; Müller, U.J.; et al. Different distribution patterns of lymphocytes and microglia in the hippocampus of patients with residual versus paranoid schizophrenia: Further evidence for disease course-related immune alterations? Brain Behav. Immun. 2012, 26, 1273–1279. [Google Scholar] [CrossRef]
- Schlaaff, K.; Dobrowolny, H.; Frodl, T.; Mawrin, C.; Gos, T.; Steiner, J.; Bogerts, B. Increased densities of T and B lymphocytes indicate neuroinflammation in subgroups of schizophrenia and mood disorder patients. Brain Behav. Immun. 2020, 88, 497–506. [Google Scholar] [CrossRef]
- Dean, B.; Tawadros, N.; Scarr, E.; Gibbons, A.S. Regionally-specific changes in levels of tumour necrosis factor in the dorsolateral prefrontal cortex obtained postmortem from subjects with major depressive disorder. J. Affect. Disord. 2010, 120, 245–248. [Google Scholar] [CrossRef]
- van Kesteren, C.F.; Gremmels, H.; de Witte, L.D.; Hol, E.M.; Van Gool, A.R.; Falkai, P.G. Immune involvement in the pathogenesis of schizophrenia: A meta-analysis on postmortem brain studies. Transl. Psychiatry 2017, 7, e1075. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Hancock, A.M.; Bradner, J.; Chung, K.A.; Quinn, J.F.; Peskind, E.R. Complement 3 and factor h in human cerebrospinal fluid in Parkinson’s disease, Alzheimer’s disease, and multiple-system atrophy. Am. J. Pathol. 2011, 178, 1509–1516. [Google Scholar] [CrossRef]
- Lindsberg, P.; Ohman, J.; Lehto, T.; Wuorimaa, T.; Meri, S.; Karjalainen-Lindsberg, M.-L.; Paetau, A.; Carpén, O.; Kaste, M. Complement activation in the central nervous system following blood-brain barrier damage in man. Ann. Neurol. 1996, 40, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Bajic, G.; E Degn, S.; Thiel, S.; Andersen, G.R. Complement activation, regulation, and molecular basis for complement-related diseases. EMBO J. 2015, 34, 2735–2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merle, N.S.; Noé, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef] [Green Version]
- Purves-Tyson, T.D.; Robinson, K.; Brown, A.M.; Boerrigter, D.; Cai, H.Q.; Weissleder, C.; Owens, S.J.; Rothmond, D.A.; Weickert, C.S. Increased Macrophages and C1qA, C3, C4 Transcripts in the Midbrain of People with Schizophrenia. Front. Immunol. 2020, 11, 2002. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Haney, J.R.; Zhang, P.; Hernandez, L.M.; Wang, L.K.; Perez-Cano, L. Brain gene co-expression networks link complement signaling with convergent synaptic pathology in schizophrenia. Nat. Neurosci. 2021, 24, 799–809. [Google Scholar] [CrossRef]
- Arion, D.; Unger, T.; Lewis, D.A.; Levitt, P.; Mirnics, K. Molecular evidence for increased expression of genes related to immune and chaperone function in the prefrontal cortex in schizophrenia. Biol. Psychiatry 2007, 62, 711–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, L.W.; Wayland, M.; Lan, M.; Ryan, M.; Giger, T.; Lockstone, H.; Wuethrich, I.; Mimmack, M.; Wang, L.; Kotter, M.; et al. The Cerebral Microvasculature in Schizophrenia: A Laser Capture Microdissection Study. PLoS ONE 2008, 3, e3964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, D.; Whiteside, T.L.; Kasid, U. Identification of a novel tumor necrosis factor-α-inducible gene, SCC-S2, containing the consensus sequence of a death effector domain of fas-associated death domain-like interleukin- 1beta-converting enzyme-inhibitory protein. J. Biol. Chem. 2000, 275, 2973–2978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leeuwenberg, J.F.; Smeets, E.F.; Neefjes, J.; A Shaffer, M.; Cinek, T.; Jeunhomme, T.M.; Ahern, T.J.; A Buurman, W. E-selectin and intercellular adhesion molecule-1 are released by activated human endothelial cells in vitro. Immunology 1992, 77, 543–549. [Google Scholar]
- Mimmack, M.L.; Ryan, M.; Baba, H.; Navarro-Ruiz, J.; Iritani, S.; Faull, R.; McKenna, P.; Jones, P.; Arai, H.; Starkey, M.; et al. Gene expression analysis in schizophrenia: Reproducible up-regulation of several members of the apolipoprotein L family located in a high-susceptibility locus for schizophrenia on chromosome 22. Proc. Natl. Acad. Sci. USA 2002, 99, 4680–4685. [Google Scholar] [CrossRef] [Green Version]
- Saetre, P.; Emilsson, L.; Axelsson, E.; Kreuger, J.; Lindholm, E.; Jazin, E. Inflammation-related genes up-regulated in schizophrenia brains. BMC Psychiatry 2007, 7, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Xu, J.; Chen, J.; Kim, S.; Reimers, M.; Bacanu, S.-A.; Yu, H.; Liu, C.; Sun, J.; Wang, Q.; et al. Transcriptome sequencing and genome-wide association analyses reveal lysosomal function and actin cytoskeleton remodeling in schizophrenia and bipolar disorder. Mol. Psychiatry 2015, 20, 563–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
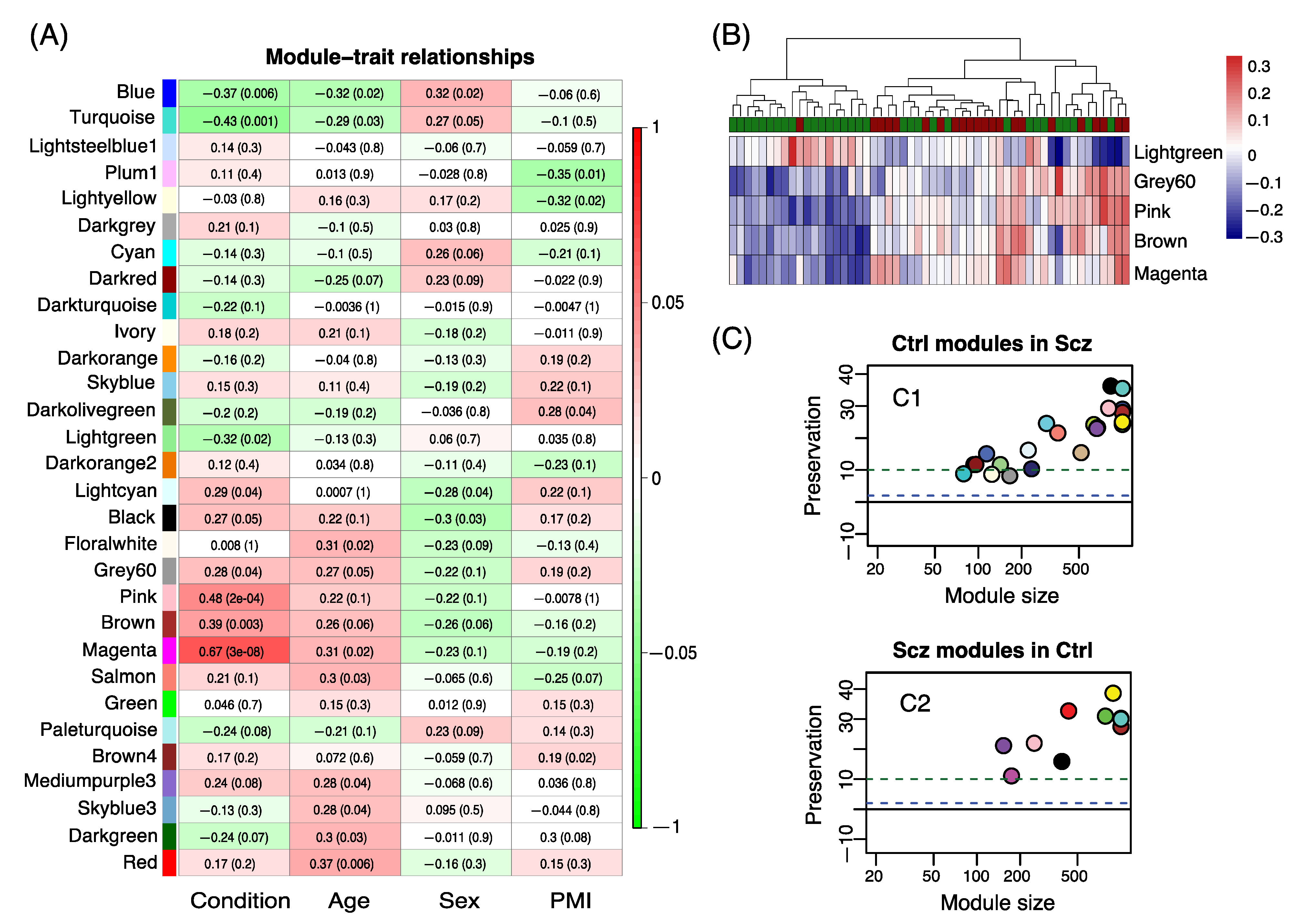{kind=link}
| Demographics | Schizophrenia (SCZ) | Control |
|---|---|---|
| Age | 52.5 ± 16.9 | 58.6 ± 15.3 |
| Sex | 32F:41M | 21F:31M |
| DLPFC:OFC | 67:6 | 40:12 |
| pH ± s.d | 6.5 ± 0.2 | 6.3 ± 0.1 |
| PMI (hours) ± s.d | 32 ± 21.9 | 38 ± 19.9 |
| Average RIN | 6.9 ± 1.1 | 6.7 ± 1.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lindholm Carlström, E.; Niazi, A.; Etemadikhah, M.; Halvardson, J.; Enroth, S.; Stockmeier, C.A.; Rajkowska, G.; Nilsson, B.; Feuk, L. Transcriptome Analysis of Post-Mortem Brain Tissue Reveals Up-Regulation of the Complement Cascade in a Subgroup of Schizophrenia Patients. Genes 2021, 12, 1242. https://doi.org/10.3390/genes12081242
Lindholm Carlström E, Niazi A, Etemadikhah M, Halvardson J, Enroth S, Stockmeier CA, Rajkowska G, Nilsson B, Feuk L. Transcriptome Analysis of Post-Mortem Brain Tissue Reveals Up-Regulation of the Complement Cascade in a Subgroup of Schizophrenia Patients. Genes. 2021; 12(8):1242. https://doi.org/10.3390/genes12081242
Chicago/Turabian StyleLindholm Carlström, Eva, Adnan Niazi, Mitra Etemadikhah, Jonatan Halvardson, Stefan Enroth, Craig A. Stockmeier, Grazyna Rajkowska, Bo Nilsson, and Lars Feuk. 2021. "Transcriptome Analysis of Post-Mortem Brain Tissue Reveals Up-Regulation of the Complement Cascade in a Subgroup of Schizophrenia Patients" Genes 12, no. 8: 1242. https://doi.org/10.3390/genes12081242