
Deleterious Mutations in the TPO Gene Associated with Familial Thyroid Follicular Cell Carcinoma in Dutch German Longhaired Pointers

 , and
, and
Abstract
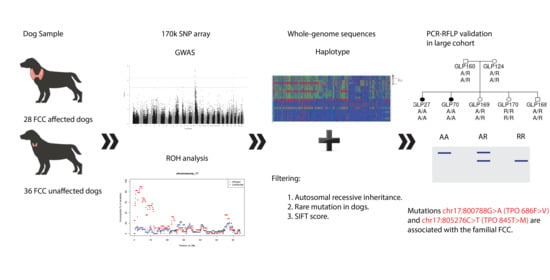
:
1. Introduction
2. Materials and Methods
2.1. Animal and Diagnosis
2.2. Genotyping
2.3. Whole Genome Sequencing (WGS)
2.4. RNA-Sequencing and Data Processing
2.5. GWAS
2.6. Runs of Homozygosity
2.7. Candidate SNPs PCR-RFLP Genotyping
2.8. Criteria for Candidate Variants
2.9. Amino Acid Conservation between Species
3. Results
3.1. Study Population
3.2. Genomic Region Associated with FCC
3.3. Deleterious Mutations in the TPO Gene
3.4. Deleterious Mutation in the SNTG2 Gene
3.5. Variants in the EIPR1 Gene
3.6. Validation by PCR-RFLP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cao, J.; Zhang, M.; Zhang, L.; Lou, J.; Zhou, F.; Fang, M. Non-coding RNA in thyroid cancer—Functions and mechanisms. Cancer Lett. 2021, 496, 117–126. [Google Scholar] [CrossRef]
- Bartsch, R.; Brinkmann, B.; Jahnke, G.; Laube, B.; Lohmann, R.; Michaelsen, S.; Neumann, I.; Greim, H. Human relevance of follicular thyroid tumors in rodents caused by non-genotoxic substances. Regul. Toxicol. Pharmacol. 2018, 98, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Campos, M.; Kool, M.M.; Daminet, S.; Ducatelle, R.; Rutteman, G.; Kooistra, H.S.; Galac, S.; Mol, J.A. Upregulation of the PI3K/Akt pathway in the tumorigenesis of canine thyroid carcinoma. J. Vet. Intern. Med. 2014, 28, 1814–1823. [Google Scholar] [CrossRef] [Green Version]
- Czene, K.; Lichtenstein, P.; Hemminki, K. Environmental and heritable causes of cancer among 9.6 million individuals in the swedish family-cancer database. Int. J. Cancer 2002, 99, 260–266. [Google Scholar] [CrossRef]
- Hińcza, K.; Kowalik, A.; Kowalska, A. Current knowledge of germline genetic risk factors for the development of non-medullary thyroid cancer. Genes 2019, 10, 482. [Google Scholar] [CrossRef] [Green Version]
- Yokote, K.; Chanprasert, S.; Lee, L.; Eirich, K.; Takemoto, M.; Watanabe, A.; Koizumi, N.; Lessel, D.; Mori, T.; Hisama, F.M.; et al. Wrn mutation update: Mutation spectrum, patient registries, and translational prospects. Hum. Mutat. 2017, 38, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, J.S.; da Silva, J.G.; Tomaz, R.A.; Pinto, A.E.; Bugalho, M.J.; Leite, V.; Cavaco, B.M. Identification of a novel germline foxe1 variant in patients with familial non-medullary thyroid carcinoma (fnmtc). Endocrine 2015, 49, 204–214. [Google Scholar] [CrossRef]
- He, H.; Bronisz, A.; Liyanarachchi, S.; Nagy, R.; Li, W.; Huang, Y.; Akagi, K.; Saji, M.; Kula, D.; Wojcicka, A.; et al. Srgap1 is a candidate gene for papillary thyroid carcinoma susceptibility. J. Clin. Endocrinol. Metab. 2013, 98, E973–E980. [Google Scholar] [CrossRef] [Green Version]
- Gara, S.K.; Jia, L.; Merino, M.J.; Agarwal, S.K.; Zhang, L.; Cam, M.; Patel, D.; Kebebew, E. Germline habp2 mutation causing familial nonmedullary thyroid cancer. N. Engl. J. Med. 2015, 373, 448–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wójcicka, A.; Czetwertyńska, M.; Świerniak, M.; Długosińska, J.; Maciąg, M.; Czajka, A.; Dymecka, K.; Kubiak, A.; Kot, A.; Płoski, R.; et al. Variants in the atm-chek2-brca1 axis determine genetic predisposition and clinical presentation of papillary thyroid carcinoma. Genes Chromosomes Cancer 2014, 53, 516–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siołek, M.; Cybulski, C.; Gąsior-Perczak, D.; Kowalik, A.; Kozak-Klonowska, B.; Kowalska, A.; Chłopek, M.; Kluźniak, W.; Wokołorczyk, D.; Pałyga, I.; et al. Chek2 mutations and the risk of papillary thyroid cancer. Int. J. Cancer 2015, 137, 548–552. [Google Scholar] [CrossRef]
- Gu, Y.; Yu, Y.; Ai, L.; Shi, J.; Liu, X.; Sun, H.; Liu, Y. Association of the atm gene polymorphisms with papillary thyroid cancer. Endocrine 2014, 45, 454–461. [Google Scholar] [CrossRef]
- Ngeow, J.; Ni, Y.; Tohme, R.; Song Chen, F.; Bebek, G.; Eng, C. Germline alterations in rasal1 in cowden syndrome patients presenting with follicular thyroid cancer and in individuals with apparently sporadic epithelial thyroid cancer. J. Clin. Endocrinol. Metab. 2014, 99, E1316–E1321. [Google Scholar] [CrossRef] [Green Version]
- Tomsic, J.; He, H.; Akagi, K.; Liyanarachchi, S.; Pan, Q.; Bertani, B.; Nagy, R.; Symer, D.E.; Blencowe, B.J.; de la Chapelle, A. A germline mutation in srrm2, a splicing factor gene, is implicated in papillary thyroid carcinoma predisposition. Sci. Rep. 2015, 5, 10566. [Google Scholar] [CrossRef] [PubMed]
- Ryu, R.A.; Tae, K.; Min, H.J.; Jeong, J.H.; Cho, S.H.; Lee, S.H.; Ahn, Y.H. Xrcc1 polymorphisms and risk of papillary thyroid carcinoma in a korean sample. J. Korean Med. Sci. 2011, 26, 991–995. [Google Scholar] [CrossRef] [Green Version]
- Jendrzejewski, J.; He, H.; Radomska, H.S.; Li, W.; Tomsic, J.; Liyanarachchi, S.; Davuluri, R.V.; Nagy, R.; de la Chapelle, A. The polymorphism rs944289 predisposes to papillary thyroid carcinoma through a large intergenic noncoding RNA gene of tumor suppressor type. Proc. Natl. Acad. Sci. USA 2012, 109, 8646–8651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prevalence, clinicopathologic features, and somatic genetic mutation profile in familial versus sporadic nonmedullary thyroid cancer. Thyroid 2011, 21, 367–371. [CrossRef] [Green Version]
- Di Cristofaro, J.; Marcy, M.; Vasko, V.; Sebag, F.; Fakhry, N.; Wynford-Thomas, D.; de Micco, C. Molecular genetic study comparing follicular variant versus classic papillary thyroid carcinomas: Association of n-ras mutation in codon 61 with follicular variant. Hum. Pathol 2006, 37, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E. Molecular analysis of thyroid tumors. Mod. Pathol. 2011, 24, S34–S43. [Google Scholar] [CrossRef] [PubMed]
- Montero-Conde, C.; Leandro-Garcia, L.J.; Chen, X.; Oler, G.; Ruiz-Llorente, S.; Ryder, M.; Landa, I.; Sanchez-Vega, F.; La, K.; Ghossein, R.A.; et al. Transposon mutagenesis identifies chromatin modifiers cooperating with Ras in thyroid tumorigenesis and detects ATXN7 as a cancer gene. Proc. Natl. Acad. Sci. USA 2017, 114, E4951. [Google Scholar] [CrossRef] [Green Version]
- Miguel, A.Z.; Adrián, A.-R.; Marta, M.; Piero, C.; Pilar, S. Regulators of the ras-erk pathway as therapeutic targets in thyroid cancer. Endocr. Relat. Cancer 2019, 26, R319–R344. [Google Scholar]
- Liu, X.; Bishop, J.; Shan, Y.; Pai, S.; Liu, D.; Murugan, A.K.; Sun, H.; El-Naggar, A.K.; Xing, M. Highly prevalent tert promoter mutations in aggressive thyroid cancers. Endocr. Relat. Cancer 2013, 20, 603–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Wang, N.; Cao, J.; Sofiadis, A.; Dinets, A.; Zedenius, J.; Larsson, C.; Xu, D. The age- and shorter telomere-dependent tert promoter mutation in follicular thyroid cell-derived carcinomas. Oncogene 2014, 33, 4978–4984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elisei, R.; Tacito, A.; Ramone, T.; Ciampi, R.; Bottici, V.; Cappagli, V.; Viola, D.; Matrone, A.; Lorusso, L.; Valerio, L.; et al. Twenty-five years experience on ret genetic screening on hereditary mtc: An update on the prevalence of germline ret mutations. Genes 2019, 10, 698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.J.; Larsson, C.; Lui, W.O.; Höög, A.; von Euler, H. A dog pedigree with familial medullary thyroid cancer. Int. J. Oncol. 2006, 29, 1173–1182. [Google Scholar] [CrossRef] [Green Version]
- Sturgeon, C.; Clark, O.H. Familial nonmedullary thyroid cancer. Thyroid 2005, 15, 588–593. [Google Scholar] [CrossRef]
- Yu, Y.; Krupa, A.; Keesler, R.; Grinwis, G.C.M.; Ruijsscher, M.; Groenen, M.A.M.; Crooijmans, R.P.M.A. Familial thyroid follicular cell carcinomas in a large number of dutch german longhaired pointers. bioRxiv 2021. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. Plink: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. Fastqc: A Quality Control Tool for High Throughput Sequence Data [online]. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 June 2020).
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From fastq data to high confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.11–11.10.33. [Google Scholar]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Li, H. A statistical framework for snp calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buels, R.; Yao, E.; Diesh, C.M.; Hayes, R.D.; Munoz-Torres, M.; Helt, G.; Goodstein, D.M.; Elsik, C.G.; Lewis, S.E.; Stein, L.; et al. Jbrowse: A dynamic web platform for genome visualization and analysis. Genome Biol. 2016, 17, 66. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with hisat2 and hisat-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. Featurecounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C.; Henikoff, S. Sift: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chan, A.P. Provean web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Thomas, P.D. Panther-psep: Predicting disease-causing genetic variants using position-specific evolutionary preservation. Bioinformatics 2016, 32, 2230–2232. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plassais, J.; Kim, J.; Davis, B.W.; Karyadi, D.M.; Hogan, A.N.; Harris, A.C.; Decker, B.; Parker, H.G.; Ostrander, E.A. Whole genome sequencing of canids reveals genomic regions under selection and variants influencing morphology. Nat. Commun. 2019, 10, 1489. [Google Scholar] [CrossRef] [PubMed]
- Ruf, J.; Carayon, P. Structural and functional aspects of thyroid peroxidase. Arch. Biochem. Biophys. 2006, 445, 269–277. [Google Scholar] [CrossRef]
- Williams, D.E.; Le, S.N.; Hoke, D.E.; Chandler, P.G.; Gora, M.; Godlewska, M.; Banga, J.P.; Buckle, A.M. Structural studies of thyroid peroxidase show the monomer interacting with autoantibodies in thyroid autoimmune disease. Endocrinology 2020, 161, bqaa016. [Google Scholar] [CrossRef]
- Gershlick, D.C.; Schindler, C.; Chen, Y.; Bonifacino, J.S. Tssc1 is novel component of the endosomal retrieval machinery. Mol. Biol Cell 2016, 27, 2867–2878. [Google Scholar] [CrossRef] [Green Version]
- Leroy, G. Genetic diversity, inbreeding and breeding practices in dogs: Results from pedigree analyses. Vet. J. 2011, 189, 177–182. [Google Scholar] [CrossRef]
- Ujvari, B.; Klaassen, M.; Raven, N.; Russell, T.; Vittecoq, M.; Hamede, R.; Thomas, F.; Madsen, T. Genetic diversity, inbreeding and cancer. Proc. R. Soc. B Biol. Sci. 2018, 285, 20172589. [Google Scholar] [CrossRef] [Green Version]
- Piluso, G.; Mirabella, M.; Ricci, E.; Belsito, A.; Abbondanza, C.; Servidei, S.; Puca, A.A.; Tonali, P.; Puca, G.A.; Nigro, V. Gamma1- and gamma2-syntrophins, two novel dystrophin-binding proteins localized in neuronal cells. J. Biol. Chem. 2000, 275, 15851–15860. [Google Scholar] [CrossRef] [Green Version]
- Neto, L.H.J.; Wicik, Z.; Torres, G.H.F.; Takayama, L.; Caparbo, V.F.; Lopes, N.H.M.; Pereira, A.C.; Pereira, R.M.R. Overexpression of sntg2, traf3ip2, and itga6 transcripts is associated with osteoporotic vertebral fracture in elderly women from community. Mol. Genet. Genom. Med. 2020, 8, e1391. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, J.A.; Ballif, B.C.; Torchia, B.S.; Sahoo, T.; Ravnan, J.B.; Schultz, R.; Lamb, A.; Bejjani, B.A.; Shaffer, L.G. Copy number variations associated with autism spectrum disorders contribute to a spectrum of neurodevelopmental disorders. Genet. Med. 2010, 12, 694–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cipollini, M.; Pastor, S.; Gemignani, F.; Castell, J.; Garritano, S.; Bonotti, A.; Biarnés, J.; Figlioli, G.; Romei, C.; Marcos, R.; et al. Tpo genetic variants and risk of differentiated thyroid carcinoma in two european populations. Int. J. Cancer 2013, 133, 2843–2851. [Google Scholar]
- Zhu, H.; Peng, Y.G.; Ma, S.G.; Liu, H. Tpo gene mutations associated with thyroid carcinoma: Case report and literature review. Cancer Biomark. 2015, 15, 909–913. [Google Scholar] [CrossRef]
- Fyfe, J.C.; Lynch, M.; Olsen, J.; Louёr, E. A thyroid peroxidase (tpo) mutation in dogs reveals a canid-specific gene structure. Mamm. Genome 2013, 24, 127–133. [Google Scholar] [CrossRef]
- Pastor, S.; Akdi, A.; González, E.R.; Castell, J.; Biarnés, J.; Marcos, R.; Velázquez, A. Common genetic variants in pituitary-thyroid axis genes and the risk of differentiated thyroid cancer. Endocr. Connect. 2012, 1, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Son, H.-Y.; Hwangbo, Y.; Yoo, S.-K.; Im, S.-W.; Yang, S.D.; Kwak, S.-J.; Park, M.S.; Kwak, S.H.; Cho, S.W.; Ryu, J.S.; et al. Genome-wide association and expression quantitative trait loci studies identify multiple susceptibility loci for thyroid cancer. Nat. Commun. 2017, 8, 15966. [Google Scholar] [CrossRef] [Green Version]
- Bann, D.V.; Jin, Q.; Sheldon, K.E.; Houser, K.R.; Nguyen, L.; Warrick, J.I.; Baker, M.J.; Broach, J.R.; Gerhard, G.S.; Goldenberg, D. Genetic variants implicate dual oxidase-2 in familial and sporadic nonmedullary thyroid cancer. Cancer Res. 2019, 79, 5490–5499. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, Y.; Ebrhim, R.S.; Abdullah, M.A.; Weiss, R.E. A novel missense mutation in the slc5a5 gene in a sudanese family with congenital hypothyroidism. Thyroid 2018, 28, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, L.S.; Qamri, Z.; Kari, S.; Ashtekar, A. Mouse models of thyroid cancer: A 2015 update. Mol. Cell Endocrinol. 2016, 421, 18–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Liu, M.; Sa, R.; Fu, H.; Cheng, L.; Chen, L. Mouse models of thyroid cancer: Bridging pathogenesis and novel therapeutics. Cancer Lett. 2020, 469, 35–53. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.S.; Jeong, Y.W.; Kim, J.J.; Park, S.W.; Ko, K.H.; Kang, M.; Kim, Y.K.; Jung, E.M.; Moon, C.; Hyun, S.H.; et al. A canine model of alzheimer’s disease generated by overexpressing a mutated human amyloid precursor protein. Int. J. Mol. Med. 2014, 33, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Specht, A.; Fiske, L.; Erger, K.; Cossette, T.; Verstegen, J.; Campbell-Thompson, M.; Struck, M.B.; Lee, Y.M.; Chou, J.Y.; Byrne, B.J.; et al. Glycogen storage disease type ia in canines: A model for human metabolic and genetic liver disease. J. Biomed. Biotechnol. 2011, 2011, 646257. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Number of Variants | 12,248,323 |
|---|---|
| Variants in the candidate region | 23,338 |
| Of which are homozygous | 6171 |
| Of which are private for cases | 2374 |
| Of which are exonic | 18 |
| Of which are missense | 7 |
| Of which are deleterious | 3 |
| Chromosome Coordination | SV-Type | Location | SV-Length |
|---|---|---|---|
| chr17:370832-370908 | deletion | intergenic | 77 |
| chr17:379867-380195 | deletion | intergenic | 328 |
| chr17:491919-492126 | deletion | intergenic | 208 |
| chr17:503763-503968 | deletion | intergenic | 206 |
| chr17:533005-533231 | deletion | intergenic | 227 |
| chr17:551908-551961 | deletion | intergenic | 54 |
| chr17:626018 | insertion | intergenic | 95 |
| chr17:731556 | insertion | Intron-SNTG2 | 55 |
| chr17:885408-885477 | deletion | downstream | 70 |
| chr17:900381-900486 | deletion | intergenic | 106 |
| chr17:1633933 | insertion | intergenic | 57 |
| chr17:1859631 | insertion | intergenic | 214 |
| chr17:1938475-1938524 | deletion | Intron-EIPR1 | 50 |
| chr17:2136862-2137405 | deletion | upstream | 544 |
| Genomic Coordinates | Gene | Amino Acid Change | SIFT | PROVEAN SCORE (Cutoff = −0.25) | PANTHER | PolyPhen-2 | Genotype Counts 1 | ||
|---|---|---|---|---|---|---|---|---|---|
| Affected GLPs | Unaffected GLPs | 722 Dogs 2 | |||||||
| Chr17:800788G>A | TPO | 686F>V | Deleterious (0) | −6.775 | 0.89 | 0.999 | 9/1/1 | 0/9/2 | - |
| Chr17:805276C>T | TPO | 845T>M | Tolerated (0.06) | −4.042 | 0.89 | 1 | 9/1/1 | 0/9/2 | 6/15/637 |
| Chr17:743943T>C | SNTG2 | 360F>S | Deleterious (0) | −1.901 | 0.27 | 0.337 | 9/1/1 | 0/9/2 | 4/16/615 |
| Chromosome | Position | Gene | Element | Genotype Counts 1 | ||
|---|---|---|---|---|---|---|
| Case | Control | 722 Dogs 2 | ||||
| 17 | 1869833 | EIPR1 | Downstream gene | 11/0/0 | 0/10/1 | 38/93/536 |
| 17 | 1898881 | EIPR1 | 4th intron | 11/0/0 | 0/10/1 | 169/190/318 |
| 17 | 1898831 | EIPR1 | 4th intron | 11/0/0 | 0/10/1 | 37/117/528 |
| 17 | 1898919 | EIPR1 | 4th intron | 11/0/0 | 0/9/2 | 163/190/326 |
| 17 | 1901542 | EIPR1 | 4th intron | 11/0/0 | 0/10/1 | 112/170/376 |
| 17 | 1901551 | EIPR1 | 4th intron | 11/0/0 | 0/10/1 | 102/166/384 |
| 17 | 1901564 | EIPR1 | 4th intron | 11/0/0 | 0/10/1 | 104/172/373 |
| 17 | 1905133 | EIPR1 | 4th intron | 11/0/0 | 0/10/1 | 182/214/315 |
| 17 | 1933629 | EIPR1 | 3rd intron | 11/0/0 | 0/10/1 | 174/222/315 |
| 17 | 1948595 | EIPR1 | 3rd intron | 11/0/0 | 0/10/1 | 161/182/338 |
| SNP | AA | AR | RR | Relative Risk of AA | Relative Risk of AR |
|---|---|---|---|---|---|
| chr17:800788G>A | 45/9 | 11/56 | 3/58 | 16.94 (p-value 2.20 × 10−16) | 3.34 (p-value 0.07) |
| chr17:805276C>T | 45/10 | 11/59 | 3/58 | 16.64 (p-value 2.24 × 10−16) | 3.20 (p-value 0.09) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Y.; Bovenhuis, H.; Wu, Z.; Laport, K.; Groenen, M.A.M.; Crooijmans, R.P.M.A. Deleterious Mutations in the TPO Gene Associated with Familial Thyroid Follicular Cell Carcinoma in Dutch German Longhaired Pointers. Genes 2021, 12, 997. https://doi.org/10.3390/genes12070997
Yu Y, Bovenhuis H, Wu Z, Laport K, Groenen MAM, Crooijmans RPMA. Deleterious Mutations in the TPO Gene Associated with Familial Thyroid Follicular Cell Carcinoma in Dutch German Longhaired Pointers. Genes. 2021; 12(7):997. https://doi.org/10.3390/genes12070997
Chicago/Turabian StyleYu, Yun, Henk Bovenhuis, Zhou Wu, Kimberley Laport, Martien A. M. Groenen, and Richard P. M. A. Crooijmans. 2021. "Deleterious Mutations in the TPO Gene Associated with Familial Thyroid Follicular Cell Carcinoma in Dutch German Longhaired Pointers" Genes 12, no. 7: 997. https://doi.org/10.3390/genes12070997