
Analysis of RNA Modifications by Second- and Third-Generation Deep Sequencing: 2020 Update



1
Université de Lorraine, CNRS, IMoPA (UMR7365), F54000 Nancy, France
2
Université de Lorraine, CNRS, INSERM, IBSLor (UMS2008/US40), Epitranscriptomics and RNA Sequencing Core Facility, F54000 Nancy, France
*
Authors to whom correspondence should be addressed.
Genes 2021, 12(2), 278; https://doi.org/10.3390/genes12020278
Submission received: 15 December 2020
/
Revised: 11 February 2021
/
Accepted: 12 February 2021
/
Published: 16 February 2021
(This article belongs to the Special Issue Functions and Dynamics of RNA Modifications)
{kind=link}
{kind=link}
{kind=link}
Abstract
:The precise mapping and quantification of the numerous RNA modifications that are present in tRNAs, rRNAs, ncRNAs/miRNAs, and mRNAs remain a major challenge and a top priority of the epitranscriptomics field. After the keystone discoveries of massive m6A methylation in mRNAs, dozens of deep sequencing-based methods and protocols were proposed for the analysis of various RNA modifications, allowing us to considerably extend the list of detectable modified residues. Many of the currently used methods rely on the particular reverse transcription signatures left by RNA modifications in cDNA; these signatures may be naturally present or induced by an appropriate enzymatic or chemical treatment. The newest approaches also include labeling at RNA abasic sites that result from the selective removal of RNA modification or the enhanced cleavage of the RNA ribose-phosphate chain (perhaps also protection from cleavage), followed by specific adapter ligation. Classical affinity/immunoprecipitation-based protocols use either antibodies against modified RNA bases or proteins/enzymes, recognizing RNA modifications. In this survey, we review the most recent achievements in this highly dynamic field, including promising attempts to map RNA modifications by the direct single-molecule sequencing of RNA by nanopores.
1. Introduction
Post-transcriptional RNA modifications (also called “Epitranscriptomics”) can be detected in RNA while using various methods and approaches exploiting the chemical and physico-chemical properties of these non-canonical RNA nucleotides. In addition to classical RNA techniques, such as 5’/3’ and specific internal labeling as well as nucleoside/RNA oligonucleotide analysis by Liquid Chromatography coupled to Mass Spectrometry (LC-MS) or tandem Mass Spectrometry (LC-MS/MS), methods that are based on second- (abbreviated as NGS for Next Generation Sequencing) and third- (NNGS, for Next-Next Generation Sequencing) generation sequencing become increasingly popular. These approaches aim to provide single-nucleotide resolution for the identification of the modified RNA position, but they may be less accurate in the exact nature of the modified residue due to a rather generic treatment used during the library preparation step. The most popular and reliable methods using NGS analysis rely on various specific chemical treatments that are applied to specifically alter RNA-modified residues to make them detectable either as RT-stop or as a mis-incorporation of nucleotides into cDNA. Actually, NNGS approaches mostly use ion-current profiles through the nanopore or kinetics of deoxynucleotide triphosphates (dNTP) incorporation in PacBio chips to deduce the presence of unusually modified nucleotides.
However, in both NGS (cluster sequencing) and NNGS (single-molecule sequencing) the nature of the signal may be only indirectly related to the chemical nature of the RNA modification, thus mis-identifications are not only possible, but actually rather frequent. In addition, if the analysis is performed at the whole-transcriptome scale (→106–107 nucleotides), even methods with an extremely good False Discovery Rate (FDR )<0.001 will still provide thousands of false positive hits. Thus, extreme care should be taken in the interpretation of large transcriptome-wide datasets claiming the presence of hundreds, or even thousands, of detected RNA modified nucleotides (as discussed in [1]).
The mapping of RNA modifications by NGS approaches is mainly based on: (1) altered base pairing during a reverse transcriptase (RT)-driven primer extension step, (2) altered chemical reactivity of the base due to a specific reagent (3) associated cleavage of the ribose-phosphate chain, and (4) a differential recognition of modified RNA nucleotides by specific antibodies (Ab) or proteins. In some instances, an enzymatic treatment or in vivo metabolic labeling can be used to exacerbate the chemical reactivity of a given modified nucleotide. Altered base-pairing is typically exploited for RNA modifications bearing extra chemical groups at the Watson–Crick (WC) edge of the base (so-called direct RT-signature) or for ‘RT-silent’ modifications after chemical derivatization affecting their WC edge. Methods that are based on the specific cleavage of the phosphodiester bond either rely on altered recognition by a specific enzyme or on the formation of an RNA abasic site, followed by a specific (and highly selective) ligation step. Antibody (protein enrichment) protocols exploit differential noncovalent or covalent binding to the modified site using UltraViolet light (UV) or chemical cross-linking steps.
Current approaches using NNGS (single-molecule sequencing) are mostly based on the use of direct nanopore RNA sequencing. Indeed, the profile of ion current registered for modified nucleotides passing through the nanopore is substantially altered when compared to the unmodified counterparts. While the experimental setup is generally rather straightforward and the RNA treatment is not different from classical RNA-Seq analysis, the extraction and analysis of raw nanopore sequencing data require complex and time-consuming bioinformatics treatment. Because of these limitations, NNGS methods are only at the emerging stage and cannot be considered to be an alternative to replace established NGS protocols. This will certainly evolve in the nearest future, and experience that accumulated in now-routine NGS analysis will be extremely helpful in the development of NNGS single-molecule analysis.
The analysis of RNA modifications by NGS is still a very recent topic (the first publications in the field are from 2012); however, a number of excellent review articles have already covered previous achievements [2,3,4,5,6,7]. In this comprehensive review, we will focus on the most recent and emerging approaches that have appeared during the last 2–3 years, and thye are not yet fully included in the relevant review literature.
2. Analysis of RNA Modifications by NGS
Different principles are currently employed for the detection of RNA modifications in the epitranscriptome using NGS and NNGS. We classify them in: (1) an analysis of RNA signatures that are visible in sequencing profiles (natural/enhanced or chemically induced), (2) the treatment-induced cleavage of the RNA phosphodiester chain followed by a selective ligation of sequencing adapters, and 3) affinity-based enrichment protocols exploiting the specificity of polyclonal or monoclonal antibodies and specific enzymes installing modifications in RNA. In many instances, the developed protocols use a combination of different principles (such as Ab-driven enrichment, followed by specific chemical treatment).
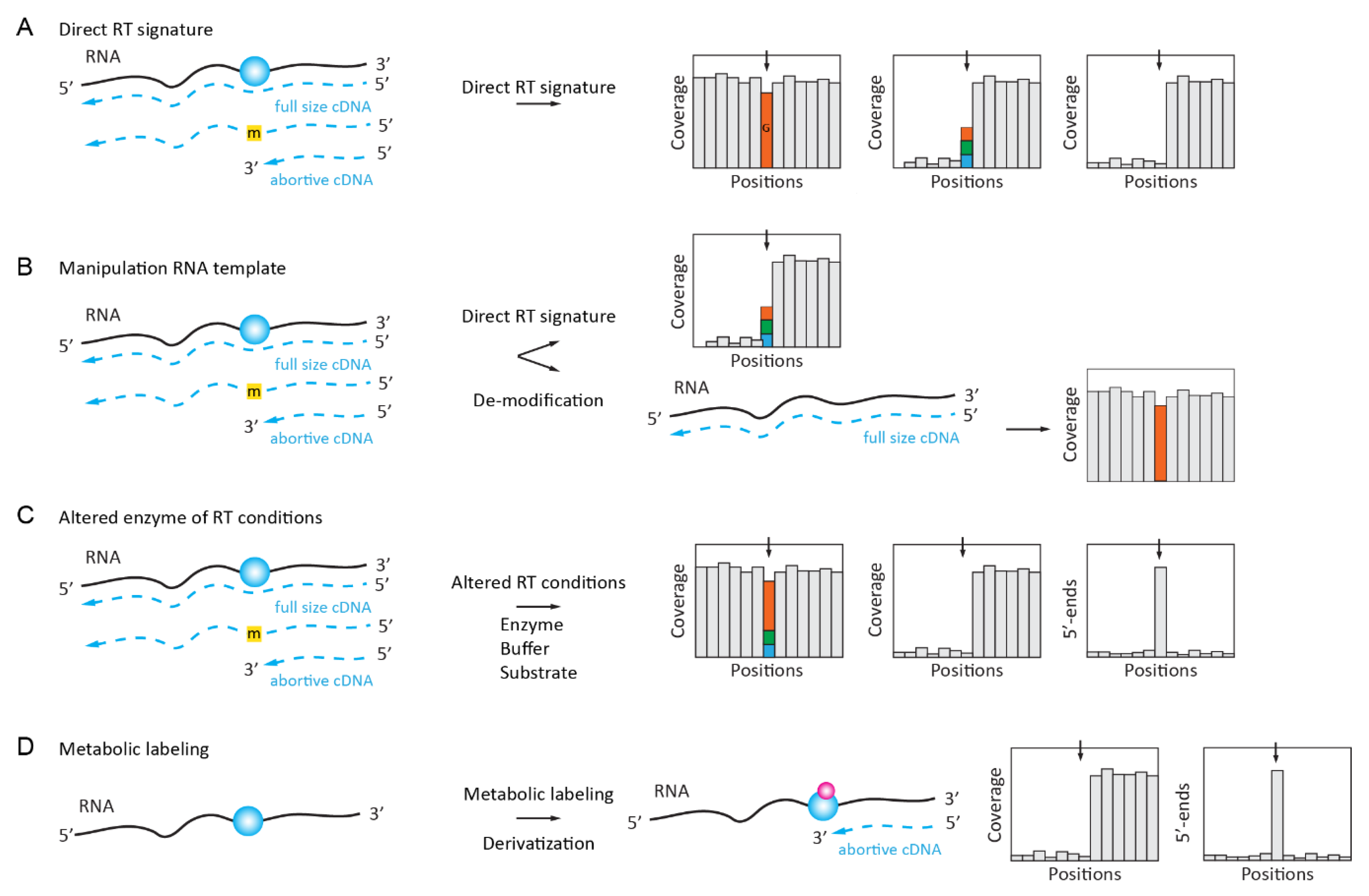
2.1. Naturally Existing RT Signatures of Modified Nucleotides
Natural RT signatures consist of the altered reading of the modified nucleotide during primer extension by an RNA-dependent RNA polymerase (reverse transcriptase, RT). Depending on the nature of the nucleotide, such a signature may represent a “mutation” as compared to the expected reference sequence or constitute a more complex profile composed of mis-incorporations at different proportions in addition to the abortive RT products that end at the modified nucleotide. Such RT signatures can be manipulated by the pre-treatment of the RNA template (e.g., by the de-modification/removal of modified residues), by the choice of a particular condition of the primer extension, or by the use of non-natural dNTP substrate(s) and mutants of the RT active site. The in vivo metabolic labeling described later in this review also allows for the incorporation of reactive chemical groups at positions of certain modifications (methyl groups) and, thus, also helps to alter the RT signature of the modified RNA template (Figure 1D).
2.1.1. Inosine “Mutation” RT Signature
The adenosine-to-inosine (A-to-I) deamination editing events in RNAs are catalyzed by enzymes from the ADAR/ADAT family. Because inosine base pairs with C and not with U (T) anymore, such events show up as A to G substitutions when the sequences of the genomic DNA and the cDNA are compared (Figure 1A). Such “mutational” RT signatures are the most straightforward approach for the detection of inosine residues in RNA. However, since the A-to-I conversion may only be partial, these inosine-generated “mutated” sequences are sometimes difficult to distinguish from sequencing errors or real SNPs in genomic DNA. Despite this limitation and the relatively high false detection rate, such a direct detection of the inosine residues is frequently used as an experimental design for the global analysis of the A-to-I editome [8,9].
2.1.2. Complex RT Signatures for m1A (m3U, m3C, m22G, etc.)
Unfortunately, only inosine shows a clear “mutational” RT signature out of →150 known RNA-modified nucleotides; all of the others are either totally silent during RT primer extension or generate more complex profiles due to their altered base pairing at the WC base edge. Such RT signatures comprise both the mis-incorporation of different bases at the position of modification and a strong RT-stop at the modified position. The proportion of both events (mis-incorporation and stop) depends on the nature of the RNA modification as well as that of the surrounding sequence [10,11] (Figure 1A). In line with these considerations, the HAMR method (High-throughput Annotation of Modified Ribonucleotides) [12,13] was initially used to map some potential m3C, m1A, m1I, m22G, and m1G sites in human tRNAs, of which a selection of predicted m3C sites was experimentally validated. Later, the application of a specific RT-signature was developed for m1A mapping in low-complexity RNA (such as rRNA and tRNAs) [10,11] and, more recently, the analysis of m1A-generated RT-signatures was extended to the analysis of the human transcriptome [14].
Even if a simple RT signature approach is now considered to be moderately reliable, the concept of “RNA-modification fingerprints” is still under further developments using ultra deep sequencing datasets allowing for the characterization of more complex events, such as deletions and truncations that are generated at the position of RNA modification [15]. tRNA molecules that contain a large proportion of RNA modifications affecting cDNA synthesis by RT seem to be good candidates for such analysis, as was demonstrated by a comprehensive analysis of Escherichia coli tRNA modifications using an optimized pipeline for library preparation and analysis [16].
2.2. Enzymatically Enhanced Natural Signatures
The use of the natural RT signatures of RNA-modified nucleotides is straightforward and it does not require additional treatment steps, which might introduce uncontrolled biases. However, the real application of such methods is relatively limited, since additional controls are required to guarantee that the observed signal indeed corresponds to RNA modification and it is not an artefact due to the RNA 2D structure, local sequence context, or the presence of co-expressed tRNA isoforms only differing at a few positions. Thus, more advanced approaches exploiting RT signatures now include additional treatment steps that ensure that the observed signal is indeed RNA modification-dependent (Figure 1B). Such modulation of the RT signature can be achieved by the approaches that are described below.
2.2.1. Manipulation (De-Modification) of the RNA Template
The enzymatic removal of methyl groups from some RNA-modified nucleotides can be achieved by demethylase enzymes of the AlkB type. This was demonstrated for the m1A, m3C, and m1G RNA modifications, which are commonly found in tRNAs. The profiles obtained for native and de-methylated templates are compared and de-methylated residues are detected by the disappearance of the RT signature after enzymatic treatment (Figure 1B). The application of such an approach to tRNAs leads to AlkB-facilitated RNA methylation sequencing (ARM-Seq) [17] and demethylase tRNA sequencing (DM-tRNA-Seq) [18]. More recently, another AlkB variant (D135S/L118V) protein was shown to efficiently and selectively de-methylate m22G in tRNAs, thus improving tRNA deep sequencing and allowing for a more specific detection [19]. While both ARM-Seq and DM-tRNA-Seq share a similar de-modification concept, the experimental approach used is slightly different. ARM-Seq uses WT AlkB, while the demethylation enzymes used in DM-tRNA-Seq are optimized to broaden its specificity (including to m1G); moreover, the ligation-based library preparation protocol that is used in ARM-Seq only captures mutational signatures. The use of a thermophilic reverse transcriptase (TGIRT) and a template switch in DM-tRNA-Seq allows for the analysis of both RT arrest and mis-incorporation and, thus, may be preferred for analysis. De-modification is an attractive option, but the spectrum of enzymatically de-modified RNA modifications is limited to only few known substrates of alpha-ketoglutarate-dependent dioxygenases from the AlkB group. The successful application of DM-tRNA-Seq was described for the analysis of human microbiome tRNAome. De-methylation was found to improve the representativity of tRNA species and improve reliability in the assignment of Watson–Crick face base modifications [20].
An alternative approach for the manipulation of the RNA template was proposed in the fat-mass and obesity-associated (FTO)-assisted m6A selective chemical labeling method (termed m6A-SEAL) protocol [21]. Here, RT-silent m6A residues are first enzymatically oxidized to hm6A, followed by the chemical conversion of these unstable intermediates to N6-dithiolsitolmethyladenosine (dm6A) by DDT-mediated thiol addition. The resulting SH-containing modified bases can be labeled by biotin, pulled down by streptavidin beads, and they have their RNA fragments converted to a library and sequenced. The analysis is conducted similarly to the classical RNA immunoprecipitation (RIP) protocol.
The particular sensitivity of RNA exonucleases to the presence of modified RNA nucleotides was exploited in m6Am-Exo-Seq [22]. In this protocol, mRNA is uniformly fragmented and treated with a 5’ exonuclease to eliminate uncapped fragments, which results in a pool of capped mRNA 5’-end fragments enriched for m6Am and depleted for internal m6A. This selective enrichment is followed by de-capping and anti-m6A-immunoprecipitation (m6A-RIP). The resulting RNA fragments are analyzed by high-throughput sequencing.
2.2.2. Manipulation of the Conditions for Enzymatic Reaction or the Nature of dNTP Substrate
The parameters of an enzymatic reaction (temperature, pH, concentration of monovalent/divalent metal ions, and dNTP substrates) can affect the properties of an RT enzyme upon the reading of some “RT-silent” RNA modifications [23,24] (Figure 1C). For example, it was shown that ribose 2’-O-methylations can be detected as stops (or strong pauses) when the avian myeloblastosis virus (AMV) RT enzyme was used at highly reduced dNTP concentrations (1–5 µM final) during primer extension. This method was extensively used in the past for the mapping of 2’-O-Me groups in various rRNAs [25] and it was recently coupled to NGS for the mapping of 2’-O-Me residues [26], termed the 2OMe-seq protocol.
The modulation of divalent metal ions [Mg2+] can also affect the properties of RTs. This was explored for the detection of m1A, m22G, m1G, and m3C by replacing Mg2+ with Mn2+ in the reaction buffer of four different RTs. The arrest rate and mis-incorporation profiles are both strongly modulated under these altered conditions, with an increase in nucleotide skipping (deletions). Every RT polymerase shows an individual sensitivity to increased [Mn2+] and it depends on the nature of the RNA modification tested, allowing for a more reliable detection [27].
Finally, the detection of RT-silent RNA modifications can be achieved by the use of synthetic base-modified dNTP. Selenium substituted thymidine (4Se-dTTP) was found to induce RT stops at m6A due to the restricted base pairing and, thus, was used to generate a specific m6A signature in RNA [28].
2.2.3. Manipulation of the RT Enzyme Properties
Native DNA polymerases with RT activity (from Thermus thermophilus in the original study) show a relative selectivity during the incorporation of nucleotides opposite m6A in the RNA template [29]. This observation was used for developing both low-throughput (single-base elongation- and ligation-based qPCR amplification method, termed SELECT protocol) [30] and high-throughput (Locus-specific Extension of Annealed DNA probes targeting m6A and sequencing, LEAD-m6A-seq) approaches [31]. In both of the methods, cDNA extension that is mediated by the Bst DNA polymerase allows for the distinction of unmodified and m6A-modified residues at selected loci.
The properties of RT enzyme can also be modulated by point mutations introduced at the enzyme active site (Figure 1C). This was explored with an engineered version of a thermostable KlenTaq DNA polymerase, which has a significant intrinsic RT activity. Generated mutant KlenTaq DNA polymerase displayed sensitivity to 2’-O-Me RNA residues, even at normal dNTP concentrations, and it can be used for the RTL-P-like [32] detection of modifications [33]. Another KlenTaq variant displayed sensitivity to m6A and exhibits increased mis-incorporation at respective sites of m6A in the template [34]. These engineered enzymes have not yet been coupled to the NGS detection of these modifications.
A similar approach was also applied for the selection of HIV-1 RT variants showing a particular sensitivity towards N1-methyladenosine (m1A). The selected mutant carrying six amino acid replacements shows both a robust RT primer extension activity and a well-defined RT signature at the modified position, suitable for the mapping and quantitative assessment of m1A modification [35].
2.2.4. Manipulation of RNA Template by in Vivo Metabolic Labeling
A proof of concept for metabolic RNA labelling while using the S-adenosyl-L-methionine (SAM) analog was first demonstrated by the enzyme-catalyzed transfer of alkynyl moiety onto RNA [36]. However, the efficiency of transfer was quite low and further improvements were required for in vivo application.
Such an application was developed for m6A mapping in cellular RNAs by the combination of SAM analog bearing the biorthogonal propargyl group and the “click-chemistry”-driven biotin labeling of the modified RNA molecules. For in vivo labeling, the SAM analog was replaced by propargyl-L-selenohomocysteine (Figure 1D). This enables the detection of METTL3 target sites by NGS as RT stops [37].
Another variant of SAM-analog metabolic labeling was also proposed to detect m6A at the transcriptome-wide level with single-nucleotide resolution and called ‘m6A-label-seq’. The living cells were fed with Se-allyl-L-selenohomocysteine, which substitutes the methyl group on the enzyme cofactor SAM with the allyl group. Allyl-modified a6A undergoes cyclization upon iodine treatment and Cyc-A is detected by NGS as RT mis-incorporation sites [38].
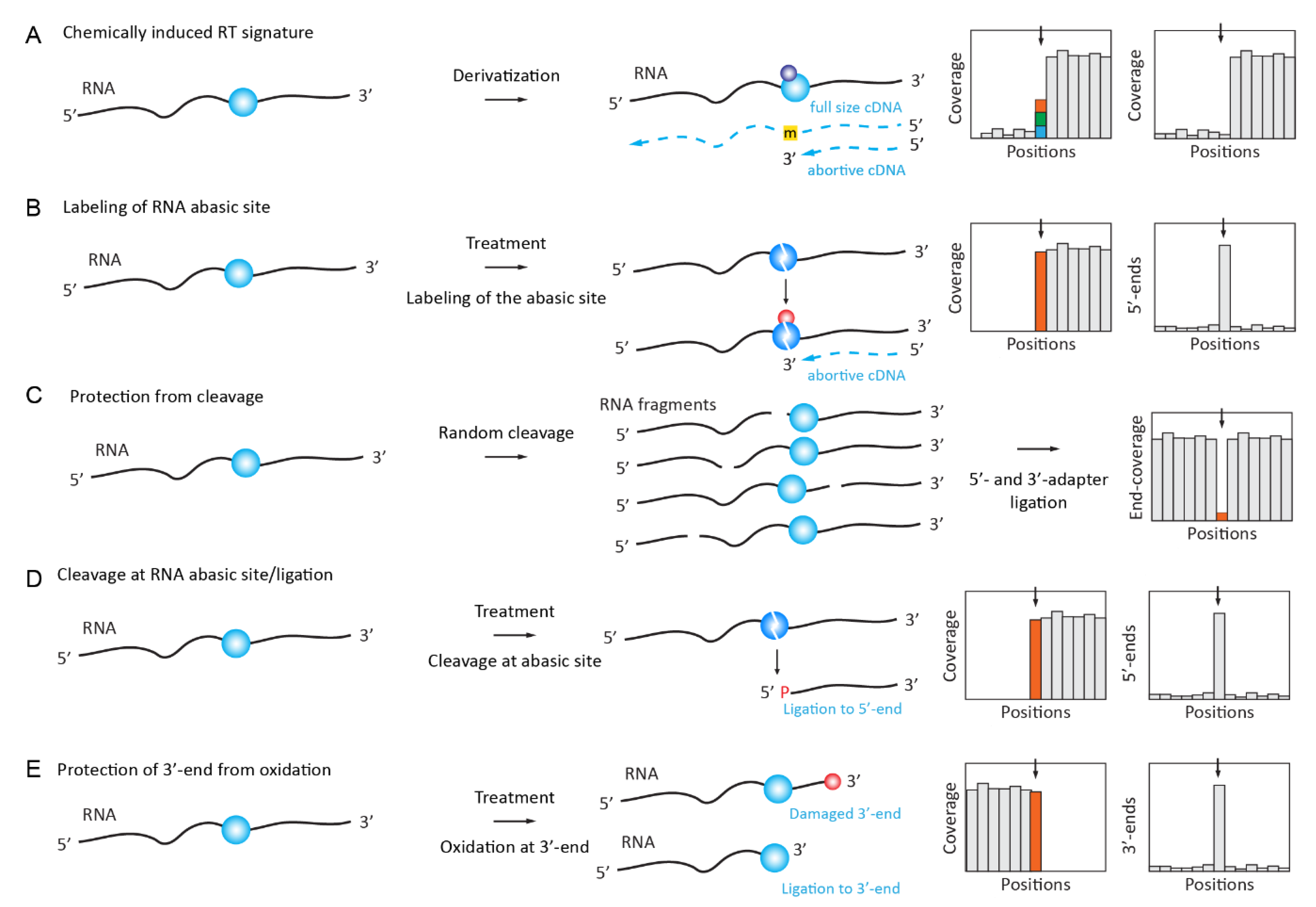
2.3. Chemically Induced RT Signatures or RT Stops
2.3.1. N1-Alkylation of Inosine (I)
The detection of inosine can be achieved on the basis of its A→G RT signature in the NGS data, as described above. However, multiple false-positive hits due to sequencing errors and SNPs contaminate such datasets. Treatment with acrylonitrile was shown to chemically modify inosines by alkylation, leading to RT arrest at inosine sites (Figure 2A) and ultimately allowing for the identification of inosines in a transcriptome-wide search with increased confidence (ICE-Seq [42]).
Another inosine labeling approach uses bulky acrylamidofluorescein for selective N1-inosine derivatization. Such labeling does not require subsequent reactions for affinity capture with specific antibodies and it can be used for the comprehensive transcriptome-wide analysis of A-to-I editing [43].
2.3.2. S4-Alkylation of 4-Thiouridine (s4U)
Thio-substitued uridine at position 4 is a natural modified nucleotide that is found at position 8 in bacterial tRNAs, but it is rather known as an artificially introduced random RNA modification widely used in Photoactivatable Ribonucleoside-Enhanced Crosslinking and Immunoprecipitation (PAR-CLIP) experiments [44]. It was well known that the reverse transcription profile of s4U displays a certain percentage of mis-incorporation events, resulting in a U→C transition in the cDNA. The alkylation of s4U with iodoacetamide (ICH2-CONH2) that is used in SLAM-seq [45] alters the hydrogen binding pattern of s4U, which causes s4U to be retrotranscribed as a C, increasing the apparent transition rate to near-quantitative. While this method was mostly applied to trace the incorporation of s4U nucleoside into a nascent RNA in metabolic labeling experiments, it is certainly suited to trace posttranscriptional s4U modifications in bacterial tRNAs.
In another sequencing approach (named TUC-Seq), s4U was first introduced by metabolic labeling followed by conversion to cytidine residues by osmium (OsO4) treatment, thus abolishing the RT signature and allowing for a direct assessment of s4U-labeled RNA [46].
2.3.3. Deamination and Oxidation of 5-methylcytosine (m5C)
The conversion of C-to-U residues in DNA and RNA by bisulfite is probably the most used chemical reaction in nucleic acid chemistry. Bisulfite RNA sequencing, which was adapted from 5mC detection in DNA, was described for m5C mapping and quantification by Schaefer and Lyko [47]. The method is generally considered to be relatively robust for abundant RNAs, such as tRNA and rRNA [48,49,50,51], but the reliability of the data obtained on less abundant RNAs, including mRNAs and lncRNAs [52,53,54,55], is still being debated [56,57,58].
Optimized and presumably more reliable bisulfite conversion protocols have been proposed with improved C-to-U conversion rates in the presence of formamide [58] and with a preferential amplification of only C→U converted RNA using a random ACT-containing DNA primer for the RT step [57].
As a further extension of RNA bisulfite deamination chemistry, approaches for the detection of f5C in RNA were suggested based on DNA-related protocols [59,60]. This nucleotide is not distinguishable from C by bisulfite treatment, but it can be reduced to hm5C by NaBH4, and hm5C in RNA is detected by its resistance to deamination, similarly to m5C. Alternatively, f5C is first protected by O-ethylhydroxylamine (fCAB-Seq for RNA), and this adduct becomes resistant to subsequent deamination [61].
A bisulfite-free single-base-resolution mapping protocol for m5C/hm5C in RNA was also suggested. It uses the selective oxidation of hm5C to trihydroxylated-thymine (thT) that is mediated by peroxotungstate. The thT residue base pairs with A during the RT step and, thus, becomes T in sequencing. If combined with the TET-mediated oxidation of m5C to hm5C, this method is also suitable for base-resolution m5C mapping [62].
Of note, in DNA 5hmC can be oxidized to f5C by potassium oxoruthenates (K2RuO4), followed by an aldol-type addition-elimination-cyclization sequence [63], and the ensuing alteration of base-pairing properties was exploited for sequencing. However, this technique still awaits potential adaptation to hm5C detection in RNA.
Recently, the global deamination of all RNA bases by nitrous acid treatment was used for m6A mapping and quantification. The protocol, termed NOSeq [64], exploits the resistance of m6A to chemical deamination under conditions where all other RNA nucleotides are efficiently converted. The protocol allows for targeted sequencing for the confirmation of m6A’s presence and the relative quantification of the modified residue.
2.3.4. Derivatization of Pseudouridine (Ψ) by Soluble Carbodiimide
Classical approaches for RT-silent pseudouridine (Ψ) detection are based on the use of soluble carbodiimide (N-Cyclohexyl-N’-(2-morpholinoethyl)carbodiimide metho-p-toluenesulfonate, usually abbreviated as CMCT) [65] and they were developed in the 1990s for low-throughput Ψ mapping. Several groups successfully coupled this protocol to NGS almost simultaneously and independently [66,67,68,69,70,71,72]. A promising variant of the method used a “clickable” CMCT derivative for the subsequent enrichment of derivatized, pseudouridine-containing RNA [73,74]. Open questions include the moderate overlap of pseudouridine sites from different studies [75], potentially a consequence of variable sequencing depth (discussed in [76]). Carbodiimide-modified pseudouridine (CMC-Ψ) is generally detected as a strong RT stop, but sequence-dependent mutational signatures can also be detected and used for a more reliable detection of the modification [77].
Alternative chemistry for Ψ detection in RNA was proposed in RBS-Seq, the protocol that uses RNA bisulfite deamination under optimized conditions. In the presence of Mg2+ ions, the Ψ-monobisulfite adduct undergoes heat-induced ribose ring opening and Mg2+-assisted reorientation, causing base skipping during cDNA synthesis. These 1–2 base deletions in the read sequence can be detected and used for precise Ψ mapping by NGS [58].
2.3.5. Dimroth Rearrangement of 1-Methyladenosine (m1A) to m6A under Alkaline Conditions
Under alkaline conditions, m1A, which causes mis-incorporation and abortive cDNA synthesis [10], undergoes Dimroth rearrangement to RT-silent m6A. This reaction was essentially used for validation by observing the disappearance of m1A RT-signatures in RNA incubated under alkaline conditions at high temperatures [14,78].
2.3.6. Sodium Borohydride Reduction of 4-Acetylcytidine (ac4C)
For the 4-acetylcytidine (ac4C), which is present in tRNAs and rRNAs, a chemical reduction with sodium borohydride (NaBH4) leading to the saturation of the 5,6 double-bond in acetylated cytidines was employed. Reduced ac4C, in turn, produced ~20–30% of mis-incorporation signals in cDNA, as determined by Sanger sequencing [79,80]. This protocol was further improved by using sodium cyanoborohydride (NaCNBH3) under acidic conditions. This new mapping approach did not confirm previously reported mRNA ac4C data, but revealed a temperature-dependent modulation of rRNA/ncRNA and mRNA acetylation from hyperthermophilic archaea [81].
2.3.7. Sodium Borohydride Reduction of 7-Methylguanosine (m7G)
A selective chemical reaction for m7G reduction by NaBH4, followed by aniline cleavage, was popularized in the 1970s [23,82,83,84]. However, this protocol was only recently applied to m7G detection using NGS.
Samples were treated with NaBH4 to generate a visible RT signature from this otherwise RT-silent RNA modification, but subsequent aniline cleavage was omitted to generate RNA abasic sites with mutational signatures during RT primer extension. The method, called m7G Mutational Profiling sequencing (m7G-MaP-seq), allows for the high-throughput detection of m7G modifications at a single-nucleotide resolution [85].
The same NaBH4 reduction of m7G was used for its profiling in human miRNAs, but the resulting RNA abasic site was used for Schiff base formation with Biotin-ARP (Aldehyde Reactive Probe) reagent containing the reactive NH2 group for the selective enrichment of the modified sites (Borohydride Reduction sequencing, BoRed-seq) [86] (Figure 2B). The presence of m7G in miRNA is still under debate [87,88].
2.4. Chemically Induced Cleavage of the Ribose-Phosphate Backbone and Selective Ligation
2.4.1. Detection and Quantification of Nm Residues by RiboMethSeq
Several variants of the so-called RiboMethSeq approach, which were first published by the Nielsen lab and further independently developed by two other groups [89,90,91,92], are based on a relative protection of the phosphodiester bond in RNA when the 5’-neighboring ribose is 2’-O-methylated (Figure 2C). These protocols are now widely used for the analysis of rRNA and tRNA 2’-O-methylations in different biological systems, including various pathologies [93,94,95,96,97,98,99].
2.4.2. Detection of 7-methylguanosine (m7G) by TRAC-Seq and AlkAnilineSeq
The reduction of m7G with NaBH4, followed by aniline-induced cleavage (see above), led to the scission of the tRNA chain at the modified nucleotide and the position of the cleavage can be followed as RT-stop in RNA-Seq data in a combination termed TRAC-seq [100,101]. Demethylation by AlkB was used to remove major tRNA modifications, and the method was also coupled to m7G-RIP to enrich the modified targets.
Another recently established technique for m7G detection, named AlkAnilineSeq [102], relies on an alternative approach for the selective fragment enrichment of abasic sites, which were created by alkaline hydrolysis at positions containing m7G (as well as by m3C, D, and ho5C). Aniline cleavage results in downstream RNA fragments with 5’-phosphate, and a selective 5’-primer ligation ultimately led to a positive selection of fragments and, thus, to an exquisite sensitivity (Figure 2D).
2.4.3. Mapping and Quantification of Pseudouridine (Ψ) by HydraPsiSeq
Like the CMCT-based approaches that are described above, the recent HydraPsiSeq protocol exploits the resistance of pseudouridine residues to hydrazine cleavage while unmodified Us are readily cleaved under these conditions [83,103]. Conceptually, this method is a mid-point between AlkAniline-Seq (for the ligation of 5’-phosphates in RNA) and RiboMethSeq, since it uses a similar concept of ”negative” detection by protection against cleavage. The obtained signal directly reflects the pseudouridine level and it can be used for precise quantification. Normalization to natural unmodified RNA and/or to synthetic in vitro transcripts allows for the absolute measurement of modification levels. HydraPsiSeq requires minute amounts of RNA (as low as 10–50 ng), which makes it compatible with the high-throughput profiling of diverse biological and clinical samples.
2.4.4. Profiling of m3C Using Hydrazine Cleavage
The same chemical reagent, hydrazine, was also proposed for the specific mapping of m3C residues in RNAs [104]. The technique of HAC-Seq uses preferential hydrazine-driven cleavage at m3C sites, followed by the decomposition of RNA abasic site by aniline, as in the AlkAniline-Seq and HydraPsiSeq protocols. Because other nucleotides (mostly U, see above) are also sensitive to hydrazine, a comparison between native and AlkB-treated (to remove m3C residues) samples is mandatory.
2.4.5. Detection of Nm RNA Residues by Their Resistance to IO4 Oxidation (RibOxi-Seq/Nm-Seq)
Two conceptually similar methods, Nm-Seq [105] and RibOxi-Seq [106], exploit the stability of 2’-O-Me ribose against periodate (NaIO4) oxidation. RNA is first randomly fragmented either chemically (Zn2+) through cleavage at 95 °C (Nm-Seq) or enzymatically by benzonase (RibOxi-Seq), (de-phosphorylated if necessary) and subjected to 3’-terminal ribose oxidations by NaIO4. 3’-terminal Nm residues resist cleavage and remain competent for the ligation of 3’-adapter, while unmodified riboses are converted to a non-ligatable dialdehyde structure (Figure 2E). Because only a few (if any) 3’-Nm are directly exposed after a random cleavage, the removal of at least one (RibOxi-Seq) or multiple (Nm-Seq) terminal nucleotide(s) by successive NaIO4 oxidation/aniline treatment/de-phosphorylation is strictly required. Better enrichment is achieved when multiple cycles (up to eight cycles used in Nm-Seq protocol) of 3’-terminal nucleotide removal are performed. In comparison, Nm-Seq provides a better sensitivity in Nm detection, but it requires substantial amounts of input RNA, due to the inevitable loss occurring during NaIO4-oxidation/aniline treatment/de-phosphorylation steps.
2.4.6. Selective Protection of m6A Methylated Motifs against MazF Cleavage (MAZTER-Seq)
The systematic quantitative profiling of m6A at a single-nucleotide resolution was achieved by use of MazF endonuclease, which is sensitive to the m6A methylation status of ACA motifs, overlapping with the classical DRACH (where D = A, G or U; R = A or G; H = A, C or U) motif for m6A modification by METTL3 [107]. RNA is totally fragmented by MazF, fragments are end-repaired, and the 3’-adapter is ligated. After a RT step, the second adapter is ligated to the cDNA 3’-end and the fragments are sequenced. Because of m6A-related protection, some ACA sites are absent from the library and this proportion can be quantified.
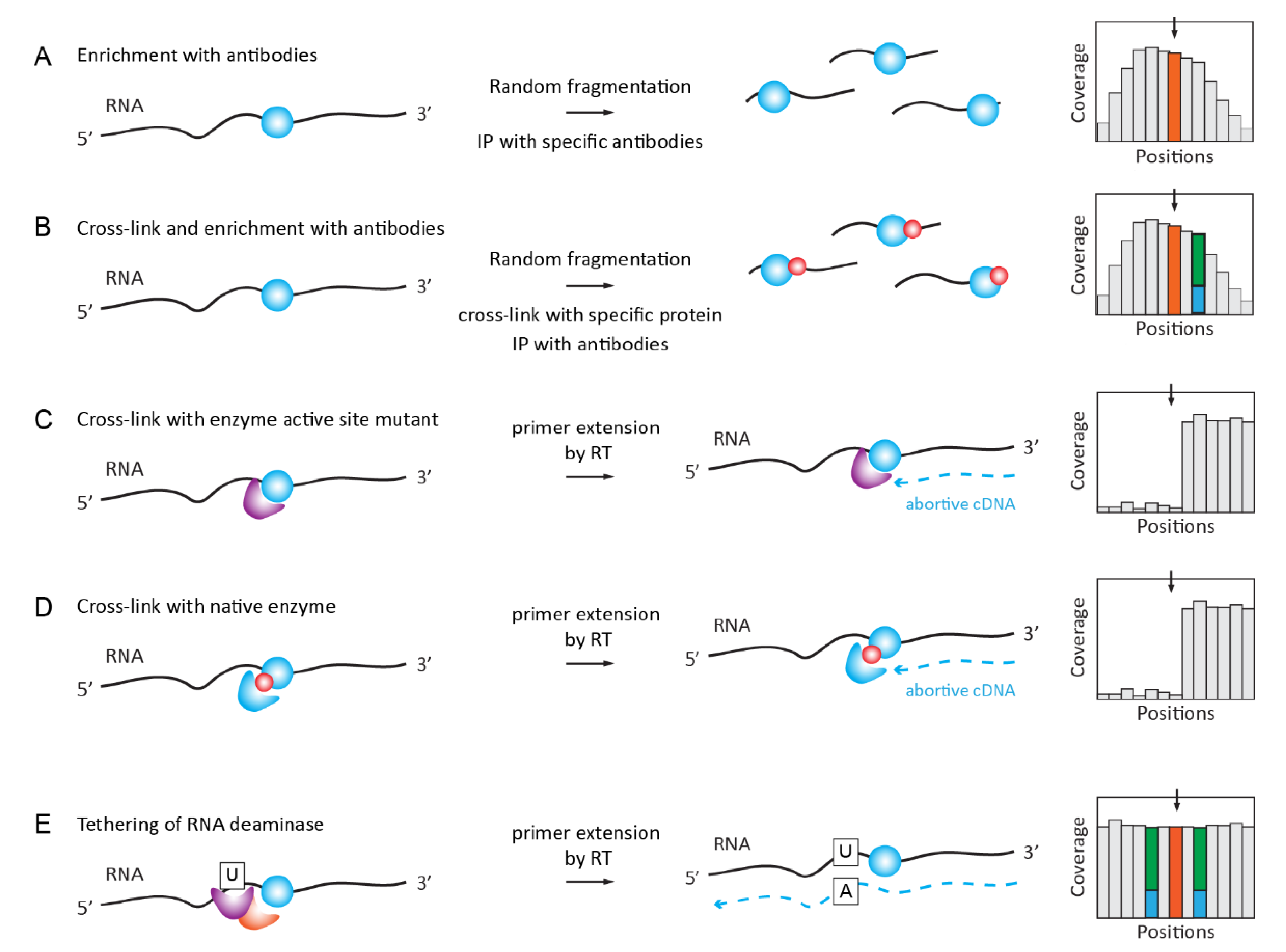
2.5. Antibody-Based Enrichment Methods (MeRIP-Seq, i/miCLIP)
The use of specific antibodies for the detection of RNA modifications was proposed and successfully implemented in the late 1970s [108,109,110], and numerous polyclonal and monoclonal antibodies were used for analysis of DNA- and RNA-modified nucleotides (as reviewed in [111]). This development is still ongoing and highly specific antibodies can be obtained in this manner (see [112]). However, the majority of antibodies against modified nucleotides/nucleosides have a poor affinity and specificity [113] and, therefore, enrichment factors for modified RNA are only very modest [114]. Taking these considerations into account, it is not surprising that multiple artifacts in RNA modification mapping result from antibody cross-reactivity and uncertain specificity (as discussed in [115,116,117]). Despite these limitations, RNA modification-specific antibodies are widely used in the RNA Immunoprecipitation (RIP) and cross-linking and immunoprecipitation (CLIP) protocols that are applied in RNA modification mapping.
2.5.1. RNA ImmunoPrecipitation (RIP) for m6A, m1A, hm5C, ac4C
MeRIP-Seq is the most straightforward application of specific antibodies for RNA modification analysis. RNA is fragmented into pieces (100–150 nt in length), immunoprecipitated with RNA modification-specific antibody, and the enriched fragments are converted to libraries and sequenced (Figure 3A). A comparison with the profile that was obtained in the input fraction is mandatory and only broad enrichment peaks can be called, precluding a single-nucleotide resolution. The consensus sequence and the exact modification site inside the fragment are generally deduced by the conservation of the significantly enriched motifs.
The applications of MeRIP-Seq for RNA modification detection are rather numerous, starting with cornerstone publications on m6A detection [118,119]. A similar approach was also applied for m1A mapping [78], as reviewed in [120]. More recently, MeRIP-Seq was also used for the mapping of hm5C [121], ac4C [122], and m7G [123].
2.5.2. iCLIP/PAR-CLIP with Antibodies and Specific Proteins/Enzymes
Further improvements in MeRIP-Seq specificity and resolution were brought about by the inclusion of a UV-crosslinking step in addition to Ab-driven immunoprecipitation (Figure 3B). Specificity is improved by the use of more stringent wash conditions that are allowed by the covalent link between the RNA ligand and the Ab, and a single-nucleotide resolution is obtained due to the C→U mutational signature present in close proximity to the modification site. Successful applications of the iCLIP/miCLIP approaches include the mapping of m6A and m6Am [124,125], as reviewed in [126]. For specific m6Am detection by miCLIP, differential maps were generated using PCIF1 KO [127]. PAR-CLIP methods are based on the use of a photoactivatable nucleotide (generally s4U) and cross-linking by mild UV-light irradiation at 365 nm [128].
2.5.3. Covalent Cross-Linking with RNA Target Mediated by RNA Modification Enzyme or Specific Reader Protein
The idea to use an RNA modification enzyme (or its catalytic mutant) for covalent attachment to the target sequence in RNA is based on a two-step mechanism of m5C-MTases, including two distinct steps: the methylation of the target base and the recycling of the covalent intermediate to release the enzyme from the covalent complex with RNA [129,130,131].
A high-throughput implementation was developed for human NSUN2 catalytic mutant (C271A) and it was called individual-nucleotide-resolution crosslinking and immunoprecipitation (iCLIP) protocol (Figure 3C). This allowed for the identification of numerous NSUN2 targets in coding and non-coding RNAs [132]. Recently, the same protocol was updated for mapping the RNA substrates of the E. coli radical SAM m8A-MTase RlmN by miCLIP-MaPseq [133]; for a review of catalytic crosslinking-based methods, see [134].
An alternative cross-linking protocol, called Aza-IP (AzaIP-Seq), uses 5-azacytidine-mediated RNA immunoprecipitation to form a covalent bond between an RNA substrate and native m5C-MTase in vivo [135]. Crosslinked targets are subsequently identified by high-throughput sequencing [136]. The use of thermostable group II intron reverse transcriptase (TGIRT) allows for the direct detection of the crosslinked nucleotides by mismatches [137].
The formation of covalent adducts with RNA substrate was also known for m5U54-MTase RUMT [138], and the incorporation of the 5-fluorouracil into the RNA substrate allows to trap covalent reaction intermediates [139]. Thus, the method Fluorouracil-Induced-Catalytic-Crosslinking-Sequencing (FICC-Seq) was used for a genome-wide single-nucleotide resolution mapping of human TRMT2A modification sites [140] (Figure 3D).
Finally, the fusion of a m6A-reader YTH domain with a cytidine deaminase APOBEC1 allows for the specific binding of YHT to m6A-modified RNA and the site-specific APOBEC1-catalyzed deamination of adjacent C residues (Figure 3D). This Deamination Adjacent to RNA modification Targets (DART-seq) protocol has identified thousands of m6A sites in cells and it can detect m6A accumulation over time [141].
2.6. Data Analysis and Interpretation
An essential step in the application of all high-throughput procedures is the error-free base calling of the candidate modified sites and the application of appropriate statistical tests to evaluate the probability of false-positive/false-negative identifications [1,142]. Only recently have these aspects come to be seriously considered when many highly optimistic RNA modification datasets were released. There are a number of publications that provide such analyses and discuss the reasons for the poor accuracy and specificity of high-throughput applications; they also provide strategies that minimize false positives and other pitfalls that are associated with mapping and measuring epitranscriptomic modifications [142,143]. Another analysis points out a surprising number of experimental and bioinformatics artifacts, which can ultimately lead to substantially inflated estimates of the abundance of diverse modifications [144]. Such discrepancies being observed in the analysis of even identical experimental raw data led to an extensive discussion of the real number of m1A sites in the human transcriptome, showing that mis-annotations, mis-mapping, SNPs, and sequencing errors may greatly contribute to the over- or under-estimation of the real number of modifications [145,146].
3. Analysis of RNA Modifications by NNGS (Single-Molecule Sequencing)
The use of single-molecule sequencing approaches (NNGS or third-generation deep sequencing) is an attractive alternative to classical cluster sequencing protocols. Indeed, cluster sequencing involves an amplification step, providing only an average picture of modifications in a population of RNA molecules. Single-molecule analysis should be performed to obtain information regarding the exact combinations of modifications in a given RNA chain (individual modification pattern) [153].
The proof of principle for the analysis of RNA modifications (namely, m6A) by single-molecule sequencing was established seven years ago by using the PacBio single-molecule, real-time (SMRT) technology. HIV-1 and AMV RT were loaded to a zero-mode waveguide (ZMW) chip and the extension of DNA primer on the RNA template was monitored [154]. Even if the precision of the RNA sequencing remains limited, the analysis of the RT kinetics can be used to identify the RNA base modifications. This work was not pursued further, most probably because HIV-1 RT containing ZMW chips for PacBio machines is not commercially available.
More recent examples of single-molecule RNA sequencing for the detection of RNA modifications concern direct RNA sequencing by nanopores (Oxford Nanopores). Using direct RNA sequencing, it was demonstrated that m6A RNA modifications can be detected with a high accuracy in the form of systematic errors and decreased base-calling qualities [155]. With appropriate training datasets containing m6A-modified and -unmodified synthetic sequences, the prediction of m6A RNA modifications can be achieved with ~90% accuracy. The analysis of ion current profiles for the direct MinION nanopore sequencing of full-length 16S rRNA revealed conserved and aminoglycoside antibiotic resistance-related 7-methylguanosines (m7G) as well as pseudouridine modifications [156].
It is clear that the major challenge in the field of direct RNA sequencing and RNA modification mapping by nanopores consists of the use of appropriate data analysis software and algorithms. Analysis can be either conducted by standard base calling and the identification of “sequencing signatures” or by the extremely laborious, but direct, analysis of ion current traces. The first solution is implemented in the software MINES (m6A Identification using Nanopore Sequencing), which assigns m6A methylation status to more than 13,000 previously unannotated DRACH (D = A/G/U, R = A/G, H = A/C/U) sites in endogenous HEK293T transcripts and identifies more than 40,000 sites with isoform-level resolution in a human mammary epithelial cell line [157].
The direct analysis of nanopore ion current profiles is extremely computationally heavy, but it certainly provides more valuable information. The bioinformatic tool, called Epitranscriptional Landscape Inferring from Glitches of ONT signals (ELIGOS), was trained on various types of synthetic modified RNA and applied to rRNA and mRNA sequencing. ELIGOS is able to accurately predict known classes of RNA methylation sites (AUC > 0.93) in rRNAs from E. coli, yeast, and human cells [158]. Model-based base calling from ionic current signal levels is certainly required for reliable analysis [159]. Another intermediate solution, a workflow for the analysis of direct RNA sequencing reads, termed MasterOfPores, converts raw current intensities into multiple types of processed data, including FASTQ and BAM, providing metrics of the quality of the run, quality filtering, demultiplexing, base calling, and mapping. In a second step, the pipeline performs downstream analyses of the mapped reads, including the prediction of RNA modifications and the estimation of polyA tail lengths [160].
In the context of the COVID-19 pandemic, the direct RNA nanopore sequencing of full-length coronavirus genomic RNA allowed for us to predict multiple sites of m5C modification in SARS-Cov-2 [161]. However, the existence of these modifications in SARS-Cov-2 is still controversial, since another study utilizing nanopore sequencing with more rigorous controls did not confirm their presence [162].
In conclusion, direct RNA modification analysis by nanopore sequencing is rapidly developing and improving in reliability, but it has still not reached maturity for routine application in RNA epitranscriptomics. Thus, classical approaches are still widely used in the routine analysis of RNA modifications, and the use of nanopores is only envisaged as an alternative validation technique.
Author Contributions
Y.M. and V.M., writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Grand Est Region, France, FRCR grants EpiARN and ViroMOD to Yuri Motorin.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wiener, D.; Schwartz, S. The Epitranscriptome beyond m6A. Nat. Rev. Genet. 2020, 1–13. [Google Scholar] [CrossRef]
- Helm, M.; Motorin, Y. Detecting RNA Modifications in the Epitranscriptome: Predict and Validate. Nat. Rev. Genet. 2017, 18, 275–291. [Google Scholar] [CrossRef]
- Motorin, Y.; Helm, M. Methods for RNA Modification Mapping Using Deep Sequencing: Established and New Emerging Technologies. Genes 2019, 10, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krogh, N.; Nielsen, H. Sequencing-Based Methods for Detection and Quantitation of Ribose Methylations in RNA. Methods 2019, 156, 5–15. [Google Scholar] [CrossRef]
- Hartstock, K.; Rentmeister, A. MappingN6-Methyladenosine (m6A) in RNA: Established Methods, Remaining Challenges, and Emerging Approaches. Chem. A Eur. J. 2019, 25, 3455–3464. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-Y.; Song, J.; Liu, Y.; Song, C.-X.; Yi, C. Mapping the Epigenetic Modifications of DNA and RNA. Protein Cell 2020, 11, 792–808. [Google Scholar] [CrossRef]
- Linder, B.; Jaffrey, S.R. Discovering and Mapping the Modified Nucleotides That Comprise the Epitranscriptome of mRNA. Cold Spring Harb. Perspect. Biol. 2019, 11, a032201. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, Z.; Ren, C.; Liu, F.; An, G.; Bo, X.; Shu, W. The Landscape of the A-to-I RNA Editome from 462 Human Genomes. Sci. Rep. 2018, 8, 12069. [Google Scholar] [CrossRef] [PubMed]
- Oakes, E.; Vadlamani, P.; Hundley, H.A. Methods for the Detection of Adenosine-to-Inosine Editing Events in Cellular RNA. In Advanced Structural Safety Studies; Springer International Publishing: Berlin, Germany, 2017; Volume 1648, pp. 103–127. [Google Scholar]
- Hauenschild, R.; Tserovski, L.; Schmid, K.; Thuring, K.L.; Winz, M.-L.; Sharma, S.; Entian, K.-D.; Wacheul, L.; Lafontaine, D.L.J.; Anderson, J.; et al. The reverse Transcription Signature ofN-1-Methyladenosine in RNA-Seq is Sequence Dependent. Nucleic Acids Res. 2015, 43, 9950–9964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tserovski, L.; Marchand, V.; Hauenschild, R.; Blanloeil-Oillo, F.; Helm, M.; Motorin, Y. High-Throughput Sequencing for 1-Methyladenosine (m1a) Mapping in RNA. Methods 2016, 107, 110–121. [Google Scholar] [CrossRef]
- Ryvkin, P.; Leung, Y.Y.; Silverman, I.M.; Childress, M.; Valladares, O.; Dragomir, I.; Gregory, B.D.; Wang, L.-S. HAMR: High-Throughput Annotation of Modified Ribonucleotides. RNA 2013, 19, 1684–1692. [Google Scholar] [CrossRef] [Green Version]
- Kuksa, P.P.; Leung, Y.Y.; Vandivier, L.E.; Anderson, Z.; Gregory, B.D.; Wang, L.-S. In Silico Identification of RNA Modifications from High-Throughput Sequencing Data Using HAMR. Methods Mol. Biol. 2017, 1562, 211–229. [Google Scholar] [CrossRef]
- Safra, M.; Sas-Chen, A.; Nir, R.; Winkler, R.; Nachshon, A.; Bar-Yaacov, D.; Erlacher, M.; Rossmanith, W.; Stern-Ginossar, N.; Schwartz, S. The m1A Landscape on Cytosolic and Mitochondrial mRNA at Single-Base Resolution. Nat. Cell Biol. 2017, 551, 251–255. [Google Scholar] [CrossRef]
- Kietrys, A.M.; Velema, W.A.; Kool, E.T. Fingerprints of Modified RNA Bases from Deep Sequencing Profiles. J. Am. Chem. Soc. 2017, 139, 17074–17081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Toffano-Nioche, C.; Lorieux, F.; Gautheret, D.; Lehmann, J. Accurate Characterization of Escherichia Coli Trna Modifications with a Simple Method of Deep-Sequencing Library Preparation. RNA Biol. 2021, 18, 33–46. [Google Scholar] [CrossRef]
- Cozen, A.E.; Quartley, E.; Holmes, A.D.; Hrabeta-Robinson, E.; Phizicky, E.M.; Lowe, T.M. ARM-Seq: AlkB-Facilitated RNA Methylation Sequencing Reveals a Complex Landscape of Modified tRNA Fragments. Nat. Methods 2015, 12, 879–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, G.; Qin, Y.; Clark, W.C.; Dai, Q.; Yi, C.; He, C.; Lambowitz, A.M.; Pan, T. Efficient and Quantitative High-Throughput rRNA Sequencing. Nat. Methods 2015, 12, 835–837. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Zheng, G.; Schwartz, M.H.; Clark, W.C.; Pan, T. Selective Enzymatic Demethylation of N 2, N 2-Dimethylguanosine in RNA and Its Application in High-Throughput tRNA Sequencing. Angew. Chem. Int. Ed. 2017, 56, 5017–5020. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, M.H.; Wang, H.; Pan, J.N.; Clark, W.C.; Cui, S.; Eckwahl, M.J.; Pan, D.W.; Parisien, M.; Owens, S.M.; Cheng, B.L.; et al. Microbiome Characterization by High-Throughput Transfer RNA Sequencing and Modification Analysis. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, Y.; Dong, S.; Yu, Q.; Jia, G. Antibody-Free Enzyme-Assisted Chemical Approach for Detection of N6-Methyladenosine. Nat. Chem. Biol. 2020, 16, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Sendinc, E.; Valle-Garcia, D.; Dhall, A.; Chen, H.; Henriques, T.; Navarrete-Perea, J.; Sheng, W.; Gygi, S.P.; Adelman, K.; Shi, Y. PCIF1 Catalyzes m6Am mRNA Methylation to Regulate Gene Expression. Mol. Cell 2019, 75, 620–630.e9. [Google Scholar] [CrossRef]
- Behm-Ansmant, I.; Helm, M.; Motorin, Y. Use of Specific Chemical Reagents for Detection of Modified Nucleotides in RNA. J. Nucleic Acids 2011, 2011, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Motorin, Y.; Muller, S.; Behm-Ansmant, I.; Branlant, C. Identification of Modified Residues in RNAs by Reverse Transcription-Based Methods. Methods Enzymol. 2007, 425, 21–53. [Google Scholar] [CrossRef]
- Maden, B.H. Mapping 2′-O-Methyl Groups in Ribosomal RNA. Methods 2001, 25, 374–382. [Google Scholar] [CrossRef]
- Incarnato, D.; Anselmi, F.; Morandi, E.; Neri, F.; Maldotti, M.; Rapelli, S.; Parlato, C.; Basile, G.; Oliviero, S. High-Throughput Single-Base Resolution Mapping of RNA 2’-O-Methylated Residues. Nucleic Acids Res. 2017, 45, 1433–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristen, M.; Plehn, J.; Marchand, V.; Friedland, K.; Motorin, Y.; Helm, M.; Werner, S. Manganese Ions Individually Alter the Reverse Transcription Signature of Modified Ribonucleosides. Genes 2020, 11, 950. [Google Scholar] [CrossRef]
- Hong, T.; Yuan, Y.; Chen, Z.; Xi, K.; Wang, T.; Xie, Y.; He, Z.; Su, H.; Zhou, Y.; Tan, Z.-J.; et al. Precise Antibody-Independent m6A Identification via 4SedTTP-Involved and FTO-Assisted Strategy at Single-Nucleotide Resolution. J. Am. Chem. Soc. 2018, 140, 5886–5889. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, E.M.; Ehrenschwender, T.; Batista, P.J.; Chang, H.Y.; Kool, E.T. Identification of a Selective Polymerase Enables Detection of N6-Methyladenosine in RNA. J. Am. Chem. Soc. 2013, 135, 19079–19082. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Wang, Y.; Tang, Q.; Wei, L.; Zhang, X.; Jia, G. An Elongation-and Ligation-Based qPCR Amplification Method for the Radiolabeling-Free Detection of Locus-Specific N 6 -Methyladenosine Modification. Angew. Chem. Int. Ed. 2018, 57, 15995–16000. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Z.; Sepich-Poore, C.; Zhang, L.; Xiao, Y.; He, C. LEAD-m 6 A-seq for Locus-Specific Detection of N 6-Methyladenosine and Quantification of Differential Methylation. Angew. Chem. Int. Ed. 2021, 60, 873–880. [Google Scholar] [CrossRef]
- Dong, Z.-W.; Shao, P.; Diao, L.-T.; Zhou, H.; Yu, C.-H.; Qu, L.-H. RTL-P: A Sensitive Approach for Detecting Sites of 2′-O-Methylation in RNA Molecules. Nucleic Acids Res. 2012, 40, e157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aschenbrenner, J.; Marx, A. Direct and Site-Specific Quantification of RNA 2′-O-Methylation by PCR with an Engineered DNA Polymerase. Nucleic Acids Res. 2016, 44, 3495–3502. [Google Scholar] [CrossRef]
- Aschenbrenner, J.; Werner, S.; Marchand, V.; Adam, M.; Motorin, Y.; Helm, M.; Marx, A. Engineering of a DNA Polymerase for Direct m6A Sequencing. Angew. Chem. Int. Ed. 2018, 57, 417–421. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Rauch, S.; Dai, Q.; Cui, X.; Zhang, Z.; Nachtergaele, S.; Sepich, C.; He, C.; Dickinson, B.C. Evolution of a Reverse Transcriptase to Map N1-Methyladenosine in Human Messenger RNA. Nat. Methods 2019, 16, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Motorin, Y.; Burhenne, J.; Teimer, R.; Koynov, K.; Willnow, S.; Weinhold, E.; Helm, M. Expanding the Chemical Scope of RNA: Methyltransferases to Site-Specific Alkynylation of RNA for Click Labeling. Nucleic Acids Res. 2010, 39, 1943–1952. [Google Scholar] [CrossRef] [PubMed]
- Hartstock, K.; Nilges, B.S.; Ovcharenko, A.; Cornelissen, N.V.; Puellen, N.; Lawrence-Dörner, A.-M.; Leidel, S.A.; Rentmeister, A. Enzymatic or In Vivo Installation of Propargyl Groups in Combination with Click Chemistry for the Enrichment and Detection of Methyltransferase Target Sites in RNA. Angew. Chem. Int. Ed. 2018, 57, 6342–6346. [Google Scholar] [CrossRef]
- Shu, X.; Cao, J.; Cheng, M.; Xiang, S.; Gao, M.; Li, T.; Ying, X.; Wang, F.; Yue, Y.; Lu, Z.; et al. A Metabolic Labeling Method Detects m6A Transcriptome-Wide at Single Base Resolution. Nat. Chem. Biol. 2020, 16, 887–895. [Google Scholar] [CrossRef]
- Holstein, J.M.; Rentmeister, A. Current Covalent Modification Methods for Detecting RNA in Fixed and Living Cells. Methods 2016, 98, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Ovcharenko, A.; Rentmeister, A. Emerging Approaches for Detection of Methylation Sites in RNA. Open Biol. 2018, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Muthmann, N.; Hartstock, K.; Rentmeister, A. Chemo-Enzymatic Treatment of RNA to Facilitate Analyses. Wiley Interdiscip. Rev. RNA 2020, 11, e1561. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Ueda, H.; Okada, S.; Sakurai, M. Transcriptome-Wide Identification of Adenosine-to-Inosine Editing Using the ICE-Seq Method. Nat. Protoc. 2015, 10, 715–732. [Google Scholar] [CrossRef]
- Knutson, S.D.; Ayele, T.M.; Heemstra, J.M. Chemical Labeling and Affinity Capture of Inosine-Containing RNAs Using Acrylamidofluorescein. Bioconjugate Chem. 2018, 29, 2899–2903. [Google Scholar] [CrossRef]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M.; Jungkamp, A.-C.; Munschauer, M.; et al. Transcriptome-Wide Identification of RNA-Binding Protein and MicroRNA Target Sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzog, V.A.; Reichholf, B.; Neumann, T.; Rescheneder, P.; Bhat, P.; Burkard, T.R.; Wlotzka, W.; Von Haeseler, A.; Zuber, J.; Ameres, S.L. Thiol-Linked Alkylation of RNA to Assess Expression Dynamics. Nat. Methods 2017, 14, 1198–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riml, C.; Amort, T.; Rieder, D.; Gasser, C.; Lusser, A.; Micura, R. Osmium-Mediated Transformation of 4-Thiouridine to Cytidine as Key to Study RNA Dynamics by Sequencing. Angew. Chem. Int. Ed. 2017, 56, 13479–13483. [Google Scholar] [CrossRef]
- Schaefer, M.; Pollex, T.; Hanna, K.; Lyko, F. RNA Cytosine Methylation Analysis by Bisulfite Sequencing. Nucleic Acids Res. 2008, 37, e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, M. RNA 5-Methylcytosine Analysis by Bisulfite Sequencing. Methods Enzymol. 2015, 560, 297–329. [Google Scholar] [CrossRef]
- Bourgeois, G.; Ney, M.; Gaspar, I.; Aigueperse, C.; Schaefer, M.; Kellner, S.; Helm, M.; Motorin, Y. Eukaryotic rRNA Modification by Yeast 5-Methylcytosine-Methyltransferases and Human Proliferation-Associated Antigen p120. PLoS ONE 2015, 10, e0133321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, M.; Hartmann, M.; Schuster, I.; Bender, S.; Thüring, K.L.; Helm, M.; Katze, J.R.; Nellen, W.; Lyko, F.; Ehrenhofer-Murray, A.E. Dynamic Modulation of Dnmt2-Dependent tRNA Methylation by the Micronutrient Queuine. Nucleic Acids Res. 2015, 43, 10952–10962. [Google Scholar] [CrossRef] [Green Version]
- Tuorto, F.; Liebers, R.; Musch, T.; Schaefer, M.; Hofmann, S.; Kellner, S.; Frye, M.; Helm, M.; Stoecklin, G.; Lyko, F. RNA Cytosine Methylation by Dnmt2 and NSun2 Promotes tRNA Stability and Protein Synthesis. Nat. Struct. Mol. Biol. 2012, 19, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Amort, T.; Rieder, D.; Wille, A.; Khokhlova-Cubberley, D.; Riml, C.; Trixl, L.; Jia, X.-Y.; Micura, R.; Lusser, A. Distinct 5-Methylcytosine Profiles in Poly(A) RNA from Mouse Embryonic Stem Cells and Brain. Genome Biol. 2017, 18, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, R.; Burgess, A.; Parker, B.; Li, J.; Pulsford, K.; Sibbritt, T.; Preiss, T.; Searle, I.R. Transcriptome-Wide Mapping of RNA 5-Methylcytosine in Arabidopsis mRNAs and Noncoding RNAs. Plant Cell 2017, 29, 445–460. [Google Scholar] [CrossRef] [Green Version]
- Edelheit, S.; Schwartz, S.; Mumbach, M.R.; Wurtzel, O.; Sorek, R. Transcriptome-Wide Mapping of 5-Methylcytidine RNA Modifications in Bacteria, Archaea, and Yeast Reveals m5C within Archaeal mRNAs. PLoS Genet. 2013, 9, e1003602. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Panneerdoss, S.; Timilsina, S.; Zhu, J.; Mohammad, T.A.; Lu, Z.-L.; De Magalhães, J.P.; Chen, Y.; Rong, R.; Huang, Y.; et al. Topological Characterization of Human and Mouse m5C Epitranscriptome Revealed by Bisulfite Sequencing. Int. J. Genom. 2018, 2018, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Legrand, C.; Tuorto, F.; Hartmann, M.; Liebers, R.; Jacob, D.; Helm, M.; Lyko, F. Statistically Robust Methylation Calling for Whole-Transcriptome Bisulfite Sequencing Reveals Distinct Methylation Patterns for Mouse RNAs. Genome Res. 2017, 27, 1589–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-S.; Ma, H.-L.; Yang, Y.; Lai, W.-Y.; Sun, B.-F.; Yang, Y.-G. 5-Methylcytosine Analysis by RNA-BisSeq. In Advanced Structural Safety Studies; Springer International Publishing: Berlin, Germany, 2018; Volume 1870, pp. 237–248. [Google Scholar]
- Khoddami, V.; Yerra, A.; Mosbruger, T.L.; Fleming, A.M.; Burrows, C.J.; Cairns, B.R. Transcriptome-Wide Profiling of Multiple RNA Modifications Simultaneously at Single-Base Resolution. Proc. Natl. Acad. Sci. USA 2019, 116, 6784–6789. [Google Scholar] [CrossRef] [Green Version]
- Song, C.-X.; Szulwach, K.E.; Dai, Q.; Fu, Y.; Mao, S.-Q.; Lin, L.; Street, C.; Li, Y.; Poidevin, M.; Wu, H.; et al. Genome-Wide Profiling of 5-Formylcytosine Reveals Its Roles in Epigenetic Priming. Cell 2013, 153, 678–691. [Google Scholar] [CrossRef] [Green Version]
- Booth, M.J.; Marsico, G.; Bachman, M.; Beraldi, D.; Balasubramanian, S. Quantitative Sequencing of 5-Formylcytosine in DNA at Single-Base Resolution. Nat. Chem. 2014, 6, 435–440. [Google Scholar] [CrossRef] [Green Version]
- Van Haute, L.; Dietmann, S.; Kremer, L.; Hussain, S.; Pearce, S.F.; Powell, C.A.; Rorbach, J.; Lantaff, R.; Blanco, S.; Sauer, S.; et al. Deficient Methylation and Formylation of mt-tRNAMet Wobble Cytosine in a Patient Carrying Mutations in NSUN3. Nat. Commun. 2016, 7, 12039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, F.; Bi, Y.; Siejka-Zielinska, P.; Zhou, Y.-L.; Zhang, X.-X.; Song, C.-X. Bisulfite-Free and Base-Resolution Analysis of 5-Methylcytidine and 5-Hydroxymethylcytidine in RNA with Peroxotungstate. Chem. Commun. 2019, 55, 2328–2331. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; He, B.; Xia, B.; Bai, D.; Lu, X.; Cai, J.; Chen, L.; Zhou, A.; Zhu, C.; Meng, H.; et al. Bisulfite-Free, Nanoscale Analysis of 5-Hydroxymethylcytosine at Single Base Resolution. J. Am. Chem. Soc. 2018, 140, 13190–13194. [Google Scholar] [CrossRef]
- Werner, S.; Galliot, A.; Pichot, F.; Kemmer, T.; Marchand, V.; Sednev, M.V.; Lence, T.; Roignant, J.-Y.; König, J.; Höbartner, C.; et al. NOseq: Amplicon Sequencing Evaluation Method for RNA m6A Sites after Chemical Deamination. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Bakin, A.; Ofengand, J. Four Newly Located Pseudouridylate Residues in Escherichia Coli 23S Ribosomal RNA Are All at the Peptidyltransferase Center: Analysis by the Application of a New Sequencing Technique. Biochemical 1993, 32, 9754–9762. [Google Scholar] [CrossRef]
- Carlile, T.M.; Rojas-Duran, M.F.; Zinshteyn, B.; Shin, H.; Bartoli, K.M.; Gilbert, W.V. Pseudouridine Profiling Reveals Regulated mRNA Pseudouridylation in Yeast and Human Cells. Nat. Cell Biol. 2014, 515, 143–146. [Google Scholar] [CrossRef] [Green Version]
- Carlile, T.M.; Rojas-Duran, M.F.; Gilbert, W.V. Transcriptome-Wide Identification of Pseudouridine Modifications Using Pseudo-Seq. Curr. Protoc. Mol. Biol. 2015, 112, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Carlile, T.M.; Rojas-Duran, M.F.; Gilbert, W.V. Pseudo-Seq. Methods Enzymol. 2015, 560, 219–245. [Google Scholar] [CrossRef]
- Lovejoy, A.F.; Riordan, D.P.; Brown, P.O. Transcriptome-Wide Mapping of Pseudouridines: Pseudouridine Synthases Modify Specific mRNAs in S. cerevisiae. PLOS ONE 2014, 9, e110799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamoto, M.A.; Lovejoy, A.F.; Cygan, A.M.; Boothroyd, J.C. mRNA Pseudouridylation Affects RNA Metabolism in the Parasite Toxoplasma gondii. RNA 2017, 23, 1834–1849. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, S.; Bernstein, D.A.; Mumbach, M.R.; Jovanovic, M.; Herbst, R.H.; León-Ricardo, B.X.; Engreitz, J.M.; Guttman, M.; Satija, R.; Lander, E.S.; et al. Transcriptome-Wide Mapping Reveals Widespread Dynamic-Regulated Pseudouridylation of ncRNA and mRNA. Cell 2014, 159, 148–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajan, K.S.; Doniger, T.; Cohen-Chalamish, S.; Chen, D.; Semo, O.; Aryal, S.; Saar, E.G.; Chikne, V.; Gerber, D.; Unger, R.; et al. Pseudouridines on Trypanosoma Brucei Spliceosomal Small Nuclear RNAs and Their Implication for RNA and Protein Interac-Tions. Nucleic Acids Res. 2019, 47, 7633–7647. [Google Scholar] [CrossRef]
- Li, X.; Zhu, P.; Ma, S.; Song, J.; Bai, J.; Sun, F.; Yi, C. Chemical Pulldown Reveals Dynamic Pseudouridylation of the Mammalian Transcriptome. Nat. Chem. Biol. 2015, 11, 592–597. [Google Scholar] [CrossRef]
- Li, X.; Ma, S.; Yi, C. Pseudouridine Chemical Labeling and Profiling. Methods Enzymol. 2015, 560, 247–272. [Google Scholar] [CrossRef]
- Zaringhalam, M.; Papavasiliou, F.N. Pseudouridylation Meets Next-Generation Sequencing. Methods 2016, 107, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safra, M.; Nir, R.; Farouq, D.; Slutskin, I.V.; Schwartz, S. TRUB1 Is the Predominant Pseudouridine Synthase Acting on Mammalian mRNA via a Predictable and Conserved Code. Genome Res. 2017, 27, 393–406. [Google Scholar] [CrossRef] [Green Version]
- Zhou, K.I.; Clark, W.C.; Pan, D.W.; Eckwahl, M.J.; Dai, Q.; Pan, T. Pseudouridines Have Context-Dependent Mutation and Stop Rates in High-Throughput Sequencing. RNA Biol. 2018, 15, 892–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominissini, D.; Nachtergaele, S.; Moshitch-Moshkovitz, S.; Peer, E.; Kol, N.; Ben-Haim, M.S.; Dai, Q.; Di Segni, A.; Salmon-Divon, M.; Clark, W.C.; et al. The Dynamic N1-Methyladenosine Methylome in Eukaryotic Messenger RNA. Nat. Cell Biol. 2016, 530, 441–446. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.M.; Briney, C.A.; Nance, K.D.; Lopez, J.E.; Thorpe, A.L.; Fox, S.D.; Bortolin-Cavaille, M.-L.; Sas-Chen, A.; Arango, D.; Oberdoerffer, S.; et al. A Chemical Signature for Cytidine Acetylation in RNA. J. Am. Chem. Soc. 2018, 140, 12667–12670. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.M.; Bryson, K.M.; Meier, J.L. Nucleotide Resolution Sequencing of N4-Acetylcytidine in RNA. Methods Enzymol. 2019, 621, 31–51. [Google Scholar] [CrossRef] [PubMed]
- Sas-Chen, A.; Thomas, J.M.; Matzov, D.; Taoka, M.; Nance, K.D.; Nir, R.; Bryson, K.M.; Shachar, R.; Liman, G.L.S.; Burkhart, B.W.; et al. Dynamic RNA Acetylation Revealed by Quantitative Cross-Evolutionary Mapping. Nat. Cell Biol. 2020, 583, 638–643. [Google Scholar] [CrossRef]
- Wintermeyer, W.; Zachau, H.G. Tertiary Structure Interactions of 7-Methylguanosine in Yeast TRNA Phe as Studied by Borohydride Reduction. FEBS Lett. 1975, 58, 306–309. [Google Scholar] [CrossRef] [Green Version]
- Peattie, D.A. Direct Chemical Method for Sequencing RNA. Proc. Natl. Acad. Sci. USA 1979, 76, 1760–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zueva, V.S.; Mankin, A.S.; Bogdanov, A.A.; Baratova, L.A. Specific Fragmentation of tRNA and rRNA at a 7-Methylguanine Residue in the Presence of Methylated Carrier RNA. JBIC J. Biol. Inorg. Chem. 1985, 146, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Enroth, C.; Poulsen, L.D.; Iversen, S.; Kirpekar, F.; Albrechtsen, A.; Vinther, J. Detection of Internal N7-Methylguanosine (m7G) RNA Modifications by Mutational Profiling Sequencing. Nucleic Acids Res. 2019, 47, e126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandolfini, L.; Barbieri, I.; Bannister, A.J.; Hendrick, A.; Andrews, B.; Webster, N.; Murat, P.; Mach, P.; Brandi, R.; Robson, S.C.; et al. METTL1 Promotes Let-7 MicroRNA Processing via m7G Methylation. Mol. Cell 2019, 74, 1278–1290.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinther, J. No Evidence for N7-Methylation of Guanosine (m7G) in Human Let-7e. Mol. Cell 2020, 79, 199–200. [Google Scholar] [CrossRef]
- Kouzarides, T.; Pandolfini, L.; Barbieri, I.; Bannister, A.J.; Andrews, B. Further Evidence Supporting N7-Methylation of Guanosine (m7G) in Human MicroRNAs. Mol. Cell 2020, 79, 201–202. [Google Scholar] [CrossRef]
- Birkedal, U.; Christensen-Dalsgaard, M.; Krogh, N.; Sabarinathan, R.; Gorodkin, J.; Nielsen, H. Profiling of Ribose Methylations in RNA by High-Throughput Sequencing. Angew. Chem. Int. Ed. 2014, 54, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Gumienny, R.; Jedlinski, D.J.; Schmidt, A.; Gypas, F.; Martin, G.; Vina-Vilaseca, A.; Zavolan, M. High-Throughput Identification of C/D box snoRNA Targets with CLIP and RiboMeth-seq. Nucleic Acids Res. 2016, 45, 2341–2353. [Google Scholar] [CrossRef] [Green Version]
- Marchand, V.; Blanloeil-Oillo, F.; Helm, M.; Motorin, Y. Illumina-Based RiboMethSeq Approach for Mapping of 2′-O-Me Residues in RNA. Nucleic Acids Res. 2016, 44, e135. [Google Scholar] [CrossRef] [Green Version]
- Marchand, V.; Ayadi, L.; El Hajj, A.; Blanloeil-Oillo, F.; Helm, M.; Motorin, Y. High-Throughput Mapping of 2′-O-Me Residues in RNA Using Next-Generation Sequencing (Illumina RiboMethSeq Protocol). Breast Cancer 2017, 1562, 171–187. [Google Scholar] [CrossRef]
- Ringeard, M.; Marchand, V.; Decroly, E.; Motorin, Y.; Bennasser, Y. FTSJ3 is an RNA 2′-O-Methyltransferase Recruited by HIV to Avoid Innate Immune Sensing. Nat. Cell Biol. 2019, 565, 500–504. [Google Scholar] [CrossRef]
- Erales, J.; Marchand, V.; Panthu, B.; Gillot, S.; Belin, S.; Ghayad, S.E.; Garcia, M.; Laforêts, F.; Marcel, V.; Baudin-Baillieu, A.; et al. Evidence for rRNA 2′-O-Methylation Plasticity: Control of Intrinsic Translational Capabilities of Human Ribosomes. Proc. Natl. Acad. Sci. USA 2017, 114, 12934–12939. [Google Scholar] [CrossRef] [Green Version]
- Hebras, J.; Krogh, N.; Marty, V.; Nielsen, H.; Cavaillé, J. Developmental Changes of rRNA Ribose Methylations in the Mouse. RNA Biol. 2020, 17, 150–164. [Google Scholar] [CrossRef] [PubMed]
- Freund, I.; Buhl, D.K.; Boutin, S.; Kotter, A.; Pichot, F.; Marchand, V.; Vierbuchen, T.; Heine, H.; Motorin, Y.; Helm, M.; et al. 2′-O-Methylation within Prokaryotic and Eukaryotic tRNA Inhibits Innate Immune Activation by Endosomal Toll-like Receptors but Does Not Affect Recognition of Whole Organisms. RNA 2019, 25, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Rajan, K.S.; Zhu, Y.; Adler, K.; Doniger, T.; Cohen-Chalamish, S.; Srivastava, A.; Shalev-Benami, M.; Matzov, D.; Unger, R.; Tschudi, C.; et al. The Large Repertoire of 2’-O-Methylation Guided by C/D snoRNAs on Trypanosoma Brucei rRNA. RNA Biol. 2020, 17, 1018–1039. [Google Scholar] [CrossRef]
- Ramachandran, S.; Krogh, N.; Jørgensen, T.E.; Johansen, S.D.; Nielsen, H.; Babiak, I. The Shift from Early to Late Types of Ribosomes in Zebrafish Development Involves Changes at a Subset of rRNA 2′-O-Me Sites. RNA 2020, 26, 1919–1934. [Google Scholar] [CrossRef] [PubMed]
- Angelova, M.T.; Dimitrova, D.G.; Da Silva, B.; Marchand, V.; Jacquier, C.; Achour, C.; Brazane, M.; Goyenvalle, C.; Bourguignon-Igel, V.; Shehzada, S.; et al. tRNA 2′-O-Methylation by a Duo of TRM7/FTSJ1 Proteins Modulates Small RNA Silencing in Drosophila. Nucleic Acids Res. 2020, 48, 2050–2072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Liu, Q.; Jiang, Y.-Z.; Gregory, R.I. Nucleotide Resolution Profiling of m7G tRNA Modification by TRAC-Seq. Nat. Protoc. 2019, 14, 3220–3242. [Google Scholar] [CrossRef]
- Lin, S.; Liu, Q.; Lelyveld, V.S.; Choe, J.; Szostak, J.W.; Gregory, R.I. Mettl1/Wdr4-Mediated m7G tRNA Methylome Is Required for Normal mRNA Translation and Embryonic Stem Cell Self-Renewal and Differentiation. Mol. Cell 2018, 71, 244–255.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchand, V.; Ayadi, L.; Ernst, F.G.M.; Hertler, J.; Bourguignon-Igel, V.; Galvanin, A.; Kotter, A.; Helm, M.; Lafontaine, D.L.J.; Motorin, Y. AlkAniline-Seq: Profiling of m 7 G and m 3 C RNA Modifications at Single Nucleotide Resolution. Angew. Chem. Int. Ed. 2018, 57, 16785–16790. [Google Scholar] [CrossRef]
- Marchand, V.; Pichot, F.; Neybecker, P.; Ayadi, L.; Bourguignon-Igel, V.; Wacheul, L.; Lafontaine, D.L.J.; Pinzano, A.; Helm, M.; Motorin, Y. HydraPsiSeq: A Method for Systematic and Quantitative Mapping of Pseudouridines in RNA. Nucleic Acids Res. 2020, 48, e110. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Liu, Q.; Sendinc, E.; Shi, Y.; I Gregory, R. Nucleotide Resolution Profiling of m3C RNA Modification by HAC-seq. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Dai, Q.; Moshitch-Moshkovitz, S.; Han, D.; Kol, N.; Amariglio, N.; Rechavi, G.; Dominissini, D.; He, C. Nm-seq Maps 2′-O-Methylation Sites in Human mRNA with Base Precision. Nat. Methods 2017, 14, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Pirnie, S.P.; Carmichael, G.G. High-Throughput and Site-Specific Identification of 2′-O-Methylation Sites Using Ribose Oxidation Sequencing (RibOxi-seq). RNA 2017, 23, 1303–1314. [Google Scholar] [CrossRef] [Green Version]
- Pandey, R.R.; Pillai, R.S. Counting the Cuts: MAZTER-Seq Quantifies m6A Levels Using a Methylation-Sensitive Ribonuclease. Cell 2019, 178, 515–517. [Google Scholar] [CrossRef]
- Milstone, D.S.; Vold, B.S.; Glitz, D.G.; Shutt, N. Antibodies to N6-(Δ2-isopentenyl) Adenosine and Its Nucleotide: Interaction with Purified tRNAs and with Bases, Nucleosides and Nucleotides of the Isopen-Tenyladenosine Family. Nucleic Acids Res. 1978, 5, 3439–3455. [Google Scholar] [CrossRef]
- Vold, B.S.; Nolen, H.W. A Unique Method Utilizing Antinucleotide Antibodies for Evaluating Changes in the Levels of Modified Nucleosides of tRNAs from Crude Extracts of Whole Cells. Nucleic Acids Res. 1979, 7, 971–980. [Google Scholar] [CrossRef] [Green Version]
- Woodsworth, M.L.; Latimer, L.J.; Janzer, J.J.; McLennan, B.D.; Lee, J.S. Characierization of Monoclonal Antibodies Specific for Isopentenyl Adenosine Derivatives Occurring in Transfer RNA. Biochem. Biophys. Res. Commun. 1983, 114, 791–796. [Google Scholar] [CrossRef]
- Feederle, R.; Schepers, A. Antibodies Specific for Nucleic Acid Modifications. RNA Biol. 2017, 14, 1089–1098. [Google Scholar] [CrossRef]
- Matsuzawa, S.; Wakata, Y.; Ebi, F.; Isobe, M.; Kurosawa, N. Development and Validation of Monoclonal Antibodies against N6-Methyladenosine for the Detection of RNA Modifications. PLoS ONE 2019, 14, e0223197. [Google Scholar] [CrossRef]
- Mishima, E.; Jinno, D.; Akiyama, Y.; Itoh, K.; Nankumo, S.; Shima, H.; Kikuchi, K.; Takeuchi, Y.; Elkordy, A.; Suzuki, T.; et al. Immuno-Northern Blotting: Detection of RNA Modifications by Using Antibodies against Modified Nucleosides. PLoS ONE 2015, 10, e0143756. [Google Scholar] [CrossRef] [Green Version]
- Slama, K.; Galliot, A.; Weichmann, F.; Hertler, J.; Feederle, R.; Meister, G.; Helm, M. Determination of Enrichment Factors for Modified RNA in MeRIP Experiments. Methods 2019, 156, 102–109. [Google Scholar] [CrossRef]
- Grozhik, A.V.; Olarerin-George, A.O.; Sindelar, M.; Li, X.; Gross, S.S.; Jaffrey, S.R. Antibody Cross-Reactivity Accounts for Widespread Appearance of m1A in 5’UTRs. Nat. Commun. 2019, 10, 5126. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, A.B.R.; Gokhale, N.S.; Cerchietti, L.; Jaffrey, S.R.; Horner, S.M.; Mason, C.E. Limits in the Detection of m6A Changes Using MeRIP/m6A-seq. Sci. Rep. 2020, 10, 6590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helm, M.; Lyko, F.; Motorin, Y. Limited Antibody Specificity Compromises Epitranscriptomic Analyses. Nat. Commun. 2019, 10, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Divon, S.M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the Human and Mouse m6A RNA Methylomes revealed by m6A-seq. Nat. Cell Biol. 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Salmon-Divon, M.; Amariglio, N.; Rechavi, G. Transcriptome-Wide Mapping of N6-Methyladenosine by m6A-Seq Based on Immunocapturing and Massively Parallel Sequencing. Nat. Protoc. 2013, 8, 176–189. [Google Scholar] [CrossRef]
- Vandivier, L.E.; Gregory, B.D. Reading the Epitranscriptome. Chick Chorioallantoic Membr. Model Precis. Cancer Ther. 2017, 41, 269–298. [Google Scholar] [CrossRef]
- Delatte, B.; Wang, F.; Ngoc, L.V.; Collignon, E.; Bonvin, E.; Deplus, R.; Calonne, E.; Hassabi, B.; Putmans, P.; Awe, S.; et al. Transcriptome-Wide Distribution and Function of RNA Hydroxymethylcytosine. Science 2016, 351, 282–285. [Google Scholar] [CrossRef] [Green Version]
- Arango, D.; Sturgill, D.; Alhusaini, N.; Dillman, A.A.; Sweet, T.J.; Hanson, G.; Hosogane, M.; Sinclair, W.R.; Nanan, K.K.; Mandler, M.D.; et al. Acetylation of Cytidine in mRNA Promotes Translation Efficiency. Cell 2018, 175, 1872–1886.e24. [Google Scholar] [CrossRef] [Green Version]
- Malbec, L.; Zhang, T.; Chen, Y.-S.; Sun, B.-F.; Shi, B.-Y.; Zhao, Y.-L.; Yang, Y.; Yang, Y.-G. Dynamic Methylome of Internal mRNA N7-Methylguanosine and Its Regulatory Role in Translation. Cell Res. 2019, 29, 927–941. [Google Scholar] [CrossRef]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-Nucleotide-resolution Mapping of m6A and m6Am throughout the Transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Hawley, B.R.; Jaffrey, S.R. Transcriptome-Wide Mapping of m6A and m6Am at Single-Nucleotide Resolution Using miCLIP. Curr. Protoc. Mol. Biol. 2019, 126, e88. [Google Scholar] [CrossRef] [PubMed]
- Grozhik, A.V.; Linder, B.; Olarerin-George, A.O.; Jaffrey, S.R. Mapping m6A at Individual-Nucleotide Resolution Using Crosslinking and Immunoprecipitation (miCLIP). Methods Mol. Biol. 2017, 1562, 55–78. [Google Scholar] [CrossRef] [Green Version]
- Boulias, K.; Toczydłowska-Socha, D.; Hawley, B.R.; Liberman, N.; Takashima, K.; Zaccara, S.; Guez, T.; Vasseur, J.-J.; Debart, F.; Aravind, L.; et al. Identification of the m6Am Methyltransferase PCIF1 Reveals the Location and Functions of m6Am in the Transcriptome. Mol. Cell 2019, 75, 631–643.e8. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N6-Methyladenosine-Dependent RNA Structural Switches Regulate RNA–Protein Interactions. Nat. Cell Biol. 2015, 518, 560–564. [Google Scholar] [CrossRef] [Green Version]
- King, M.Y.; Redman, K.L. RNA Methyltransferases Utilize Two Cysteine Residues in the Formation of 5-Methylcytosine. Biochemical 2002, 41, 11218–11225. [Google Scholar] [CrossRef]
- Redman, K.L. Assembly of Protein−RNA Complexes Using Natural RNA and Mutant Forms of an RNA Cytosine Methyltransferase. Biomacromolecules 2006, 7, 3321–3326. [Google Scholar] [CrossRef]
- Moon, H.J.; Redman, K.L. Trm4 and Nsun2 RNA:m5C Methyltransferases Form Metabolite-Dependent, Covalent Adducts with Previously Methylated RNA. Biochemical 2014, 53, 7132–7144. [Google Scholar] [CrossRef]
- Hussain, S.; Sajini, A.A.; Blanco, S.; Dietmann, S.; Lombard, P.; Sugimoto, Y.; Paramor, M.; Gleeson, J.G.; Odom, D.T.; Ule, J.; et al. NSun2-Mediated Cytosine-5 Methylation of Vault Noncoding RNA Determines Its Processing into Regulatory Small RNAs. Cell Rep. 2013, 4, 255–261. [Google Scholar] [CrossRef]
- Stojković, V.; Chu, T.; Therizols, G.; Weinberg, D.E.; Fujimori, D.G. miCLIP-MaPseq, a Substrate Identification Approach for Radical SAM RNA Methylating Enzymes. J. Am. Chem. Soc. 2018, 140, 7135–7143. [Google Scholar] [CrossRef]
- Hussain, S. Catalytic Crosslinking-Based Methods for Enzyme-Specified Profiling of RNA Ribonucleotide Modifications. Methods 2019, 156, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Khoddami, V.; Cairns, B.R. Identification of Direct Targets and Modified Bases of RNA Cytosine Methyltransferases. Nat. Biotechnol. 2013, 31, 458–464. [Google Scholar] [CrossRef] [Green Version]
- Khoddami, V.; Cairns, B.R. Transcriptome-Wide Target Profiling of RNA Cytosine Methyltransferases Using the Mechanism-Based Enrichment Procedure Aza-IP. Nat. Protoc. 2014, 9, 337–361. [Google Scholar] [CrossRef] [PubMed]
- Khoddami, V.; Yerra, A.; Cairns, B.R. Experimental Approaches for Target Profiling of RNA Cytosine Methyltransferases. Methods Enzymol. 2015, 560, 273–296. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Santi, D.V. Covalent Adducts between tRNA (m5U54)-Methyltransferase and RNA Substrates. Biochemical 1992, 31, 10295–10302. [Google Scholar] [CrossRef]
- Kealey, J.T.; Santi, D.V. Stereochemistry of tRNA(m5U54)-Methyltransferase Catalysis: 19F NMR Spectroscopy of an Enzyme-FUraRNA Covalent Complex. Biochemical 1995, 34, 2441–2446. [Google Scholar] [CrossRef]
- Carter, J.-M.; Emmett, W.; Mozos, I.R.; Kotter, A.; Helm, M.; Ule, J.; Hussain, S. FICC-Seq: A Method for Enzyme-Specified Profiling of Methyl-5-Uridine in Cellular RNA. Nucleic Acids Res. 2019, 47, e113. [Google Scholar] [CrossRef]
- Meyer, K.D. DART-seq: An Antibody-Free Method for Global m6a Detection. Nat. Methods 2019, 16, 1275–1280. [Google Scholar] [CrossRef] [PubMed]
- Grozhik, A.V.; Jaffrey, S.R. Distinguishing RNA Modifications from Noise in Epitranscriptome Maps. Nat. Chem. Biol. 2018, 14, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Grozhik, A.V.; Jaffrey, S.R. Shrinking Maps of RNA Modifications. Nat. Cell Biol. 2017, 551, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Sas-Chen, A.; Schwartz, S. Misincorporation Signatures for Detecting Modifications in mRNA: Not as Simple as It Sounds. Methods 2019, 156, 53–59. [Google Scholar] [CrossRef]
- Schwartz, S. m1A Within Cytoplasmic mRNAs at Single Nucleotide Resolution: A Reconciled Transcriptome-Wide Map. RNA 2018, 24, 1427–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.; Li, X.; Wang, K.; Yi, C. Perspectives on Topology of the Human m1A Methylome at Single Nucleotide Resolution. RNA 2018, 24, 1437–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diroma, M.A.; Ciaccia, L.; Pesole, G.; Picardi, E. Elucidating the Editome: Bioinformatics Approaches for RNA Editing Detection. Briefings Bioinform. 2019, 20, 436–447. [Google Scholar] [CrossRef]
- Parker, B.J. Statistical Methods for Transcriptome-Wide Analysis of RNA Methylation by Bisulfite Sequencing. In Advanced Structural Safety Studies; Springer International Publishing: Berlin, Germany, 2017; Volume 1562, pp. 155–167. [Google Scholar]
- Hauenschild, R.; Werner, S.; Tserovski, L.; Hildebrandt, A.; Motorin, Y.; Helm, M. CoverageAnalyzer (CAn): A Tool for Inspection of Modification Signatures in RNA Sequencing Profiles. Biomolecules 2016, 6, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piechotta, M.; Wyler, E.; Ohler, U.; Landthaler, M.; Dieterich, C. JACUSA: Site-Specific Identification of RNA Editing Events from Replicate Sequencing Data. BMC Bioinform. 2017, 18, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Busan, S.; Weeks, K.M. Accurate Detection of Chemical Modifications in RNA by Mutational Profiling (MaP) with ShapeMapper 2. RNA 2018, 24, 143–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bormann, F.; Tuorto, F.; Cirzi, C.; Lyko, F.; Legrand, C. BisAMP: A Web-Based Pipeline for Targeted RNA Cytosine-5 Methylation Analysis. Methods 2019, 156, 121–127. [Google Scholar] [CrossRef]
- Xu, L.; Seki, M. Recent Advances in the Detection of Base Modifications Using the Nanopore Sequencer. J. Hum. Genet. 2020, 65, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Vilfan, I.D.; Tsai, Y.-C.; A Clark, T.; Wegener, J.; Dai, Q.; Yi, C.; Pan, T.; Turner, S.W.; Korlach, J. Analysis of RNA Base Modification and Structural Rearrangement by Single-Molecule Real-Time Detection of Reverse Transcription. J. Nanobiotechnol. 2013, 11, 8. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Begik, O.; Lucas, M.C.; Ramirez, J.M.; Mason, C.E.; Wiener, D.; Schwartz, S.; Mattick, J.S.; Smith, M.A.; Novoa, E.M. Accurate Detection of m6A RNA Modifications in Native RNA Sequences. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.M.; Jain, M.; Mulroney, L.; Garalde, D.R.; Akeson, M. Reading Canonical and Modified Nucleobases in 16S Ribosomal RNA Using Nanopore Native RNA Sequencing. PLoS ONE 2019, 14, e0216709. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, D.A.; Sathe, S.; Einstein, J.M.; Yeo, G.W. Direct RNA Sequencing Enables m6A Detection in Endogenous Transcript Isoforms at Base-Specific Resolution. RNA 2020, 26, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Jenjaroenpun, P.; Wongsurawat, T.; Wadley, T.D.; Wassenaar, T.M.; Liu, J.; Dai, Q.; Wanchai, V.; Akel, N.S.; Jamshidi-Parsian, A.; Franco, A.T.; et al. Decoding the Epitranscriptional Landscape from Native RNA Sequences. Nucleic Acids Res. 2021, 49, e7. [Google Scholar] [CrossRef]
- Ding, H.; Bailey, A.D.; Jain, M.; Olsen, H.; Paten, B. Gaussian Mixture Model-Based Unsupervised Nucleotide Modification Number Detection Using Nanopore-Sequencing Readouts. Bioinformatics 2020, 36, 4928–4934. [Google Scholar] [CrossRef]
- Cozzuto, L.; Liu, H.; Pryszcz, L.P.; Pulido, T.H.; Delgado-Tejedor, A.; Ponomarenko, J.; Novoa, E.M. MasterOfPores: A Workflow for the Analysis of Oxford Nanopore Direct RNA Sequencing Datasets. Front. Genet. 2020, 11, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viehweger, A.; Krautwurst, S.; Lamkiewicz, K.; Madhugiri, R.; Ziebuhr, J.; Hölzer, M.; Marz, M. Direct RNA Nanopore Sequencing of Full-Length Coronavirus Genomes Provides Novel Insights into Structural Variants and Enables Modification Analysis. Genome Res. 2019, 29, 1545–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef]
Figure 1.
Direct detection of RNA modifications by natural or induced reverse-transcription (RT) signatures. (A): natural abortive/mutational RT signatures formed in cDNA. The signature may be seen as a pure mutation (different nucleotides are shown by color), a combination of abortive cDNA synthesis with mis-incorporation, or as a simple abortive cDNA synthesis. (B): reverse transcription signature is altered after the de-modification of RNA template. (C): RT signature is induced by specific RT conditions (buffer, substrate concentration), by altered deoxynucleotide triphosphates (dNTP), or by a mutation in the RT active site. (D): RT signature induced by metabolically labeled RNA-modified nucleotide (applied to RNA methylations with SAM analog).
Figure 1.
Direct detection of RNA modifications by natural or induced reverse-transcription (RT) signatures. (A): natural abortive/mutational RT signatures formed in cDNA. The signature may be seen as a pure mutation (different nucleotides are shown by color), a combination of abortive cDNA synthesis with mis-incorporation, or as a simple abortive cDNA synthesis. (B): reverse transcription signature is altered after the de-modification of RNA template. (C): RT signature is induced by specific RT conditions (buffer, substrate concentration), by altered deoxynucleotide triphosphates (dNTP), or by a mutation in the RT active site. (D): RT signature induced by metabolically labeled RNA-modified nucleotide (applied to RNA methylations with SAM analog).

Figure 2.
Detection of RNA modifications using chemically induced RT signatures or specific cleavage of the RNA ribose-phosphate backbone. (A): RNA treatment by a specific chemical reagent induces RT signature at the modified RNA residue in the form of a mis-incorporation profile (different nucleotides are shown by color) or abortive cDNA synthesis products. (B): chemical treatment selectively removes RNA-modified base by the cleavage of the N-glycosidic bond, releasing an RNA abasic site which can be labeled using an aldehyde-specific probe. (C): RNA modification induces the protection of the RNA backbone against cleavage (negative detection in RiboMethSeq and HydraPsiSeq protocols), thus excluding fragments starting and ending at the modified nucleotide. The signal is seen as a characteristic “gap” in 5’/3’-end coverage. (D): chemical treatment induces the cleavage of the RNA backbone and the release of a 5’-phosphate, which is a competent in 5’-adapter ligation. Panel (E): RNA modification (here, 3’-terminal Nm residues released by random RNA fragmentation) protects RNA from NaIO4 oxidation, thus maintaining the efficient ligation of the 3’ adapter.
Figure 2.
Detection of RNA modifications using chemically induced RT signatures or specific cleavage of the RNA ribose-phosphate backbone. (A): RNA treatment by a specific chemical reagent induces RT signature at the modified RNA residue in the form of a mis-incorporation profile (different nucleotides are shown by color) or abortive cDNA synthesis products. (B): chemical treatment selectively removes RNA-modified base by the cleavage of the N-glycosidic bond, releasing an RNA abasic site which can be labeled using an aldehyde-specific probe. (C): RNA modification induces the protection of the RNA backbone against cleavage (negative detection in RiboMethSeq and HydraPsiSeq protocols), thus excluding fragments starting and ending at the modified nucleotide. The signal is seen as a characteristic “gap” in 5’/3’-end coverage. (D): chemical treatment induces the cleavage of the RNA backbone and the release of a 5’-phosphate, which is a competent in 5’-adapter ligation. Panel (E): RNA modification (here, 3’-terminal Nm residues released by random RNA fragmentation) protects RNA from NaIO4 oxidation, thus maintaining the efficient ligation of the 3’ adapter.

Figure 3.
Detection of RNA modifications by immunoaffinity and other affinity-related methods. (A): A classical Me-RIP approach first applied to m6A and further extended also to non-methylated modified nucleotides. RNA is fragmented and the modification-containing fraction is enriched by immunoprecipitation. The signal of Me-RIP is somehow asymmetric, and a modified nucleotide may be displaced from the peak summit. Panel (B): immunoprecipitation-based individual-nucleotide resolution UV crosslinking and immunoprecipitation/Photoactivatable Ribonucleoside-Enhanced Crosslinking and Immunoprecipitation (iCLIP)/PAR-CLIP approaches using the covalent cross-linking of the specific antibody to RNA in the proximity of RNA modification. RT signature (shown in blue/green and resulting from RNA/protein cross-link may be visible in proximity to the modified nucleotide. Panel (C): A covalent cross-linking with the modified residue when the enzyme active site mutant is used. Described for m5C and m5U methylating enzymes, which use a two-step reaction mechanism. Panel (D): cross-linking with the native enzyme may be achieved by the incorporation of non-natural nucleotides in RNA in vitro or in vivo (metabolic labeling). Panel (E): Reader protein for RNA modification is fused with the RNA deaminase domain (APOBEC) and, thus, tethered to the modified RNA residue (shown in orange), inducing partial C→U deamination in close proximity to RNA modification. These partial deamination signatures are visible in the sequencing profiles (shown in green/blue).
Figure 3.
Detection of RNA modifications by immunoaffinity and other affinity-related methods. (A): A classical Me-RIP approach first applied to m6A and further extended also to non-methylated modified nucleotides. RNA is fragmented and the modification-containing fraction is enriched by immunoprecipitation. The signal of Me-RIP is somehow asymmetric, and a modified nucleotide may be displaced from the peak summit. Panel (B): immunoprecipitation-based individual-nucleotide resolution UV crosslinking and immunoprecipitation/Photoactivatable Ribonucleoside-Enhanced Crosslinking and Immunoprecipitation (iCLIP)/PAR-CLIP approaches using the covalent cross-linking of the specific antibody to RNA in the proximity of RNA modification. RT signature (shown in blue/green and resulting from RNA/protein cross-link may be visible in proximity to the modified nucleotide. Panel (C): A covalent cross-linking with the modified residue when the enzyme active site mutant is used. Described for m5C and m5U methylating enzymes, which use a two-step reaction mechanism. Panel (D): cross-linking with the native enzyme may be achieved by the incorporation of non-natural nucleotides in RNA in vitro or in vivo (metabolic labeling). Panel (E): Reader protein for RNA modification is fused with the RNA deaminase domain (APOBEC) and, thus, tethered to the modified RNA residue (shown in orange), inducing partial C→U deamination in close proximity to RNA modification. These partial deamination signatures are visible in the sequencing profiles (shown in green/blue).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Motorin, Y.; Marchand, V. Analysis of RNA Modifications by Second- and Third-Generation Deep Sequencing: 2020 Update. Genes 2021, 12, 278. https://doi.org/10.3390/genes12020278
AMA Style
Motorin Y, Marchand V. Analysis of RNA Modifications by Second- and Third-Generation Deep Sequencing: 2020 Update. Genes. 2021; 12(2):278. https://doi.org/10.3390/genes12020278
Chicago/Turabian StyleMotorin, Yuri, and Virginie Marchand. 2021. "Analysis of RNA Modifications by Second- and Third-Generation Deep Sequencing: 2020 Update" Genes 12, no. 2: 278. https://doi.org/10.3390/genes12020278
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.