Micronuclei Formation upon Radioiodine Therapy for Well-Differentiated Thyroid Cancer: The Influence of DNA Repair Genes Variants

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Genotype Analysis
2.3. Cytogenetic Analysis
2.4. Statistical Analysis
3. Results
3.1. Characteristics of the Study Population
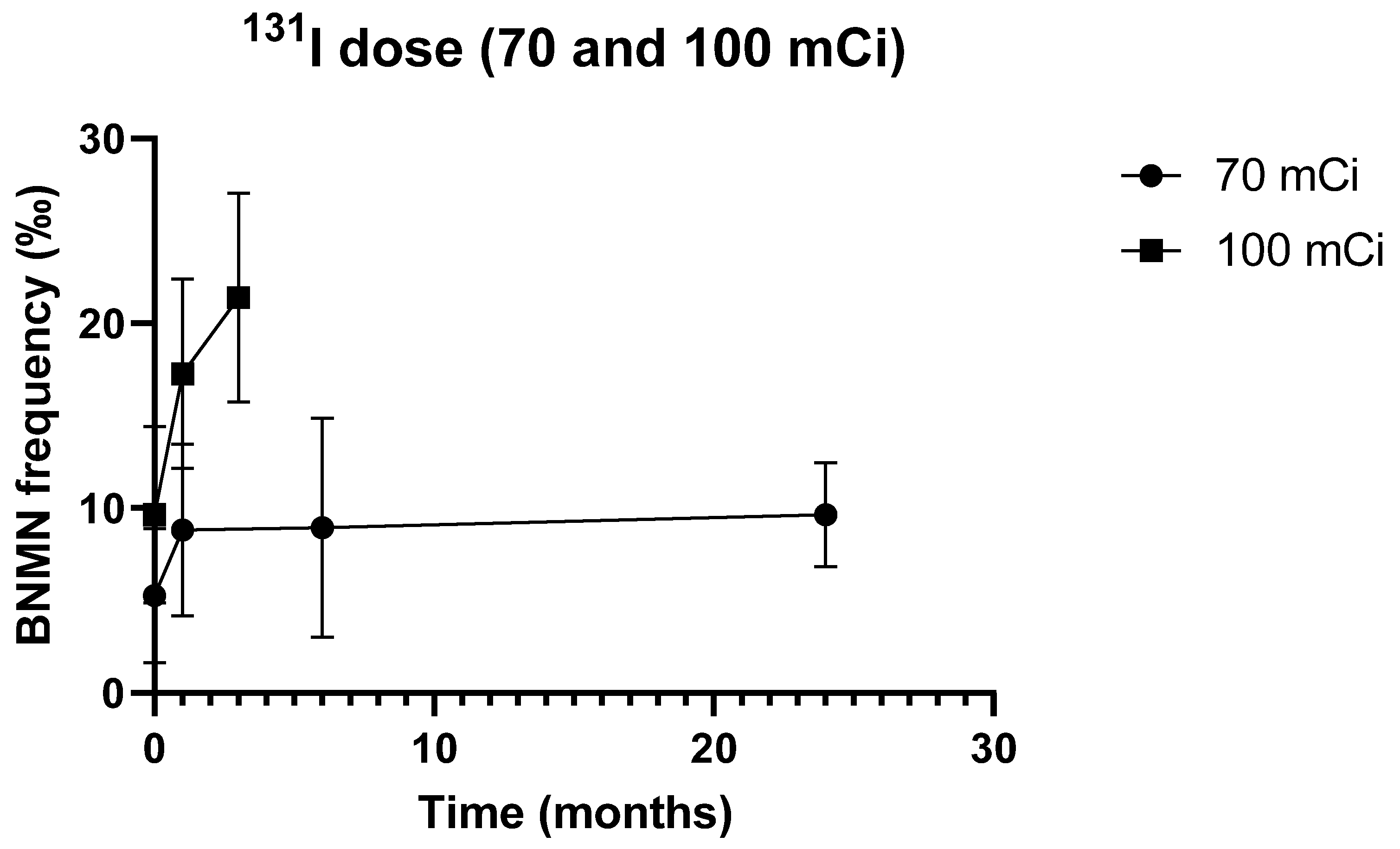
3.2. Cytogenetic Data
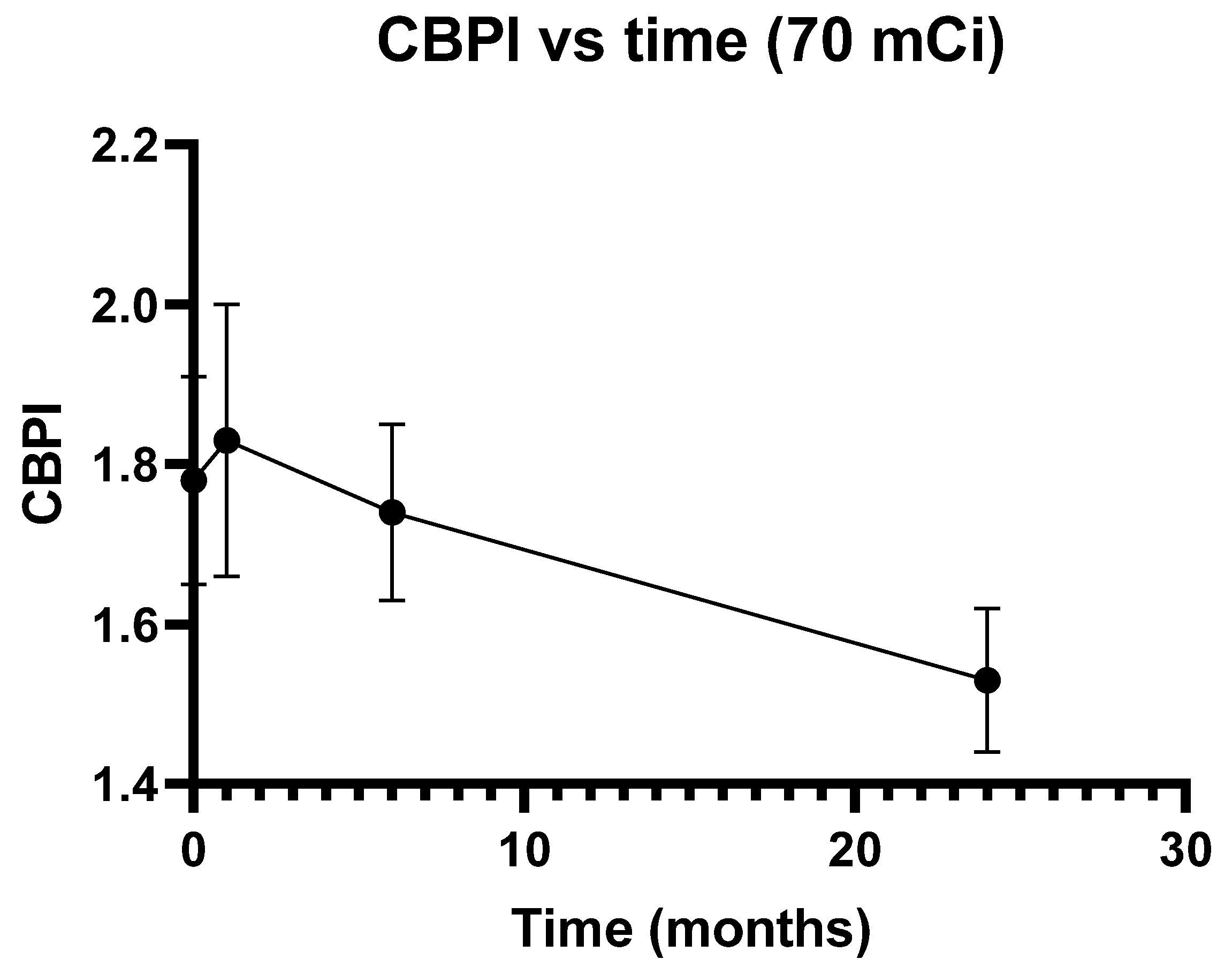
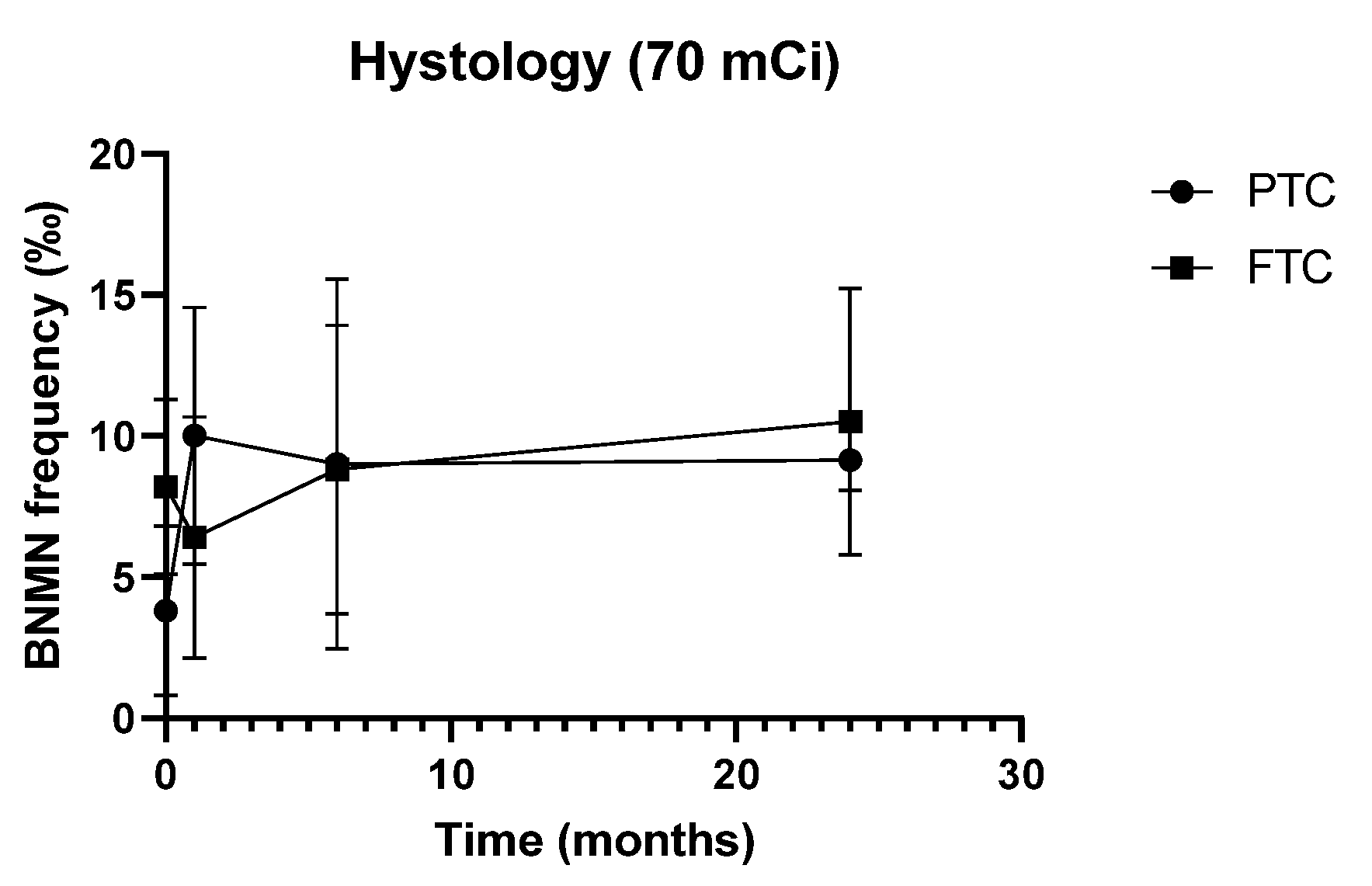
3.3. Characteristics of the Study Population and Cytogenetic Data
3.4. Distribution of DNA Repair SNPs in the Study Population
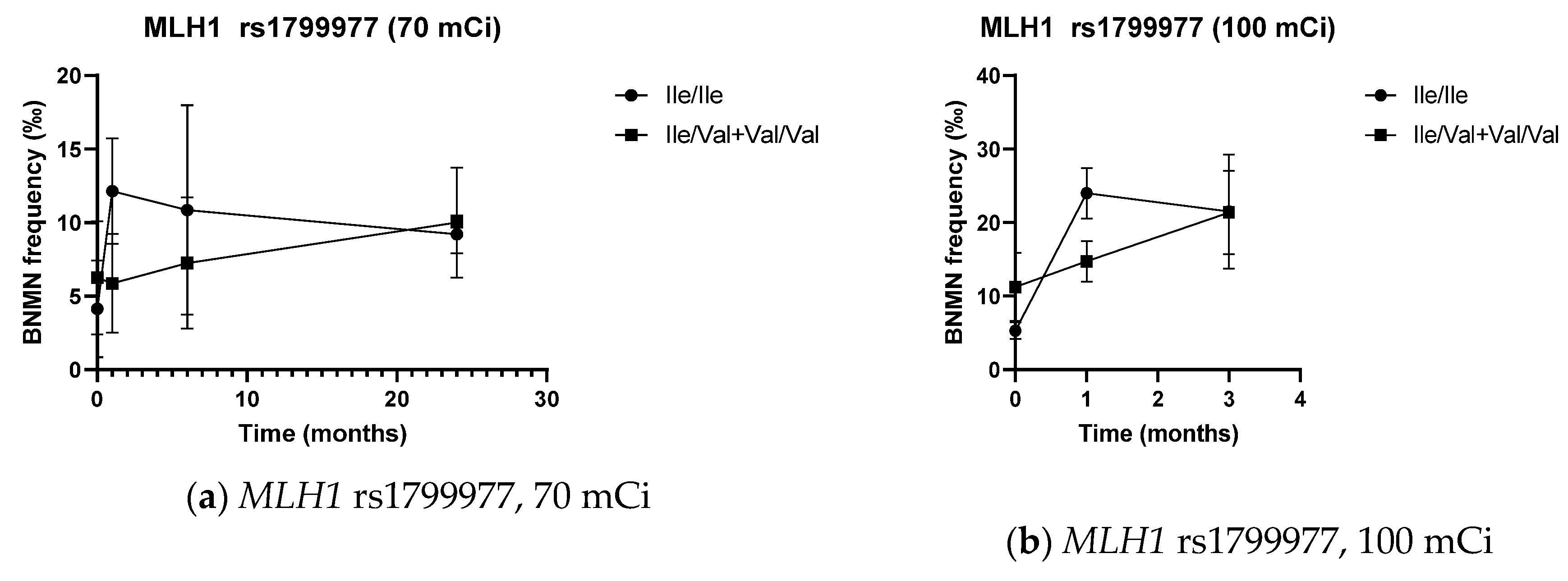
3.5. DNA Repair SNPs and Cytogenetic Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today. Available online: https://gco.iarc.fr/today (accessed on 28 May 2019).
- Kitahara, C.M.; Sosa, J.A. The changing incidence of thyroid cancer. Nat. Rev. Endocrinol. 2016, 12, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Lebastchi, A.H.; Callender, G.G. Thyroid cancer. Curr. Probl. Cancer 2014, 38, 48–74. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, M.H.; Kouhi, A.; Saeedi, M.; Bagherihagh, A.; Amirzade-Iranaq, M.H. Thyroid Cancers: Considerations, Classifications, and Managements. In Diagnosis and Management of Head and Neck Cancer; Akarslan, Z., Ed.; IntechOpen: London, UK, 2017; pp. 57–82. [Google Scholar] [CrossRef] [Green Version]
- Wild, C.; Weiderpass, E.; Stewart, B. (Eds.) World Cancer Report: Cancer Research for Cancer Prevention; International Agency for Research on Cancer: Lyon, France, 2020. [Google Scholar]
- Mayson, S.E.; Yoo, D.C.; Gopalakrishnan, G. The evolving use of radioiodine therapy in differentiated thyroid cancer. Oncology 2015, 88, 247–256. [Google Scholar] [CrossRef]
- Carballo, M.; Quiros, R.M. To treat or not to treat: The role of adjuvant radioiodine therapy in thyroid cancer patients. J. Oncol. 2012, 2012, 707156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid Off. J. Am. Thyroid Assoc. 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haugen, B.R. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: What is new and what has changed? Cancer 2017, 123, 372–381. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagenesis 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Collins, S.P.; Dritschilo, A. The mismatch repair and base excision repair pathways: An opportunity for individualized (personalized) sensitization of cancer therapy. Cancer Biol. Ther. 2009, 8, 1164–1166. [Google Scholar] [CrossRef] [Green Version]
- Doai, M.; Watanabe, N.; Takahashi, T.; Taniguchi, M.; Tonami, H.; Iwabuchi, K.; Kayano, D.; Fukuoka, M.; Kinuya, S. Sensitive immunodetection of radiotoxicity after iodine-131 therapy for thyroid cancer using gamma-H2AX foci of DNA damage in lymphocytes. Ann. Nucl. Med. 2013, 27, 233–238. [Google Scholar] [CrossRef]
- Eberlein, U.; Scherthan, H.; Bluemel, C.; Peper, M.; Lapa, C.; Buck, A.K.; Port, M.; Lassmann, M. DNA Damage in Peripheral Blood Lymphocytes of Thyroid Cancer Patients After Radioiodine Therapy. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2016, 57, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Simonelli, V.; Mazzei, F.; D’Errico, M.; Dogliotti, E. Gene susceptibility to oxidative damage: From single nucleotide polymorphisms to function. Mutat. Res. 2012, 731, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sameer, A.S.; Nissar, S. XPD-The Lynchpin of NER: Molecule, Gene, Polymorphisms, and Role in Colorectal Carcinogenesis. Front. Mol. Biosci. 2018, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Adjadj, E.; Schlumberger, M.; de Vathaire, F. Germ-line DNA polymorphisms and susceptibility to differentiated thyroid cancer. Lancet Oncol. 2009, 10, 181–190. [Google Scholar] [CrossRef]
- Gatzidou, E.; Michailidi, C.; Tseleni-Balafouta, S.; Theocharis, S. An epitome of DNA repair related genes and mechanisms in thyroid carcinoma. Cancer Lett. 2010, 290, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.S.; Gomes, B.C.; Bastos, H.N.; Gil, O.M.; Azevedo, A.P.; Ferreira, T.C.; Limbert, E.; Silva, S.N.; Rueff, J. Thyroid Cancer: The Quest for Genetic Susceptibility Involving DNA Repair Genes. Genes 2019, 10, 586. [Google Scholar] [CrossRef] [Green Version]
- Bastos, H.N.; Antao, M.R.; Silva, S.N.; Azevedo, A.P.; Manita, I.; Teixeira, V.; Pina, J.E.; Gil, O.M.; Ferreira, T.C.; Limbert, E.; et al. Association of polymorphisms in genes of the homologous recombination DNA repair pathway and thyroid cancer risk. Thyroid Off. J. Am. Thyroid Assoc. 2009, 19, 1067–1075. [Google Scholar] [CrossRef]
- Gomes, B.C.; Silva, S.N.; Azevedo, A.P.; Manita, I.; Gil, O.M.; Ferreira, T.C.; Limbert, E.; Rueff, J.; Gaspar, J.F. The role of common variants of non-homologous end-joining repair genes XRCC4, LIG4 and Ku80 in thyroid cancer risk. Oncol. Rep. 2010, 24, 1079–1085. [Google Scholar]
- Santos, L.S.; Silva, S.N.; Gil, O.M.; Ferreira, T.C.; Limbert, E.; Rueff, J. Mismatch repair single nucleotide polymorphisms and thyroid cancer susceptibility. Oncol. Lett. 2018, 15, 6715–6726. [Google Scholar] [CrossRef] [Green Version]
- Gil, O.M.; Oliveira, N.G.; Rodrigues, A.S.; Laires, A.; Ferreira, T.C.; Limbert, E.; Leonard, A.; Gerber, G.; Rueff, J. Cytogenetic alterations and oxidative stress in thyroid cancer patients after iodine-131 therapy. Mutagenesis 2000, 15, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Monteiro Gil, O.; Oliveira, N.G.; Rodrigues, A.S.; Laires, A.; Ferreira, T.C.; Limbert, E.; Rueff, J. Possible transient adaptive response to mitomycin C in peripheral lymphocytes from thyroid cancer patients after iodine-131 therapy. Int. J. Cancer 2002, 102, 556–561. [Google Scholar] [CrossRef]
- Gil, O.M.; Oliveira, N.G.; Rodrigues, A.S.; Laires, A.; Ferreira, T.C.; Limbert, E.; Rueff, J. No evidence of increased chromosomal aberrations and micronuclei in lymphocytes from nonfamilial thyroid cancer patients prior to radiotherapy. Cancer Genet. Cytogenet. 2000, 123, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Sole, X.; Guino, E.; Valls, J.; Iniesta, R.; Moreno, V. SNPStats: A web tool for the analysis of association studies. Bioinformatics 2006, 22, 1928–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- OECD. Test No. 487: In Vitro Mammalian Cell Micronucleus Test; OECD: Paris, France, 2016. [Google Scholar] [CrossRef]
- Hernández, A.; Xamena, N.; Gutiérrez, S.; Velázquez, A.; Creus, A.; Surrallés, J.; Galofré, P.; Marcos, R. Basal and induced micronucleus frequencies in human lymphocytes with different GST and NAT2 genetic backgrounds. Mutat. Res. 2006, 606, 12–20. [Google Scholar] [CrossRef]
- Gutiérrez, S.; Carbonell, E.; Galofré, P.; Creus, A.; Marcos, R. Cytogenetic damage after 131-iodine treatment for hyperthyroidism and thyroid cancer. A study using the micronucleus test. Eur. J. Nucl. Med. 1999, 26, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Livingston, G.K.; Foster, A.E.; Elson, H.R. Effect of in vivo exposure to iodine-131 on the frequency and persistence of micronuclei in human lymphocytes. J. Toxicol. Environ. Health 1993, 40, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, M.J.; Puerto, S.; Galofré, P.; Parry, E.M.; Parry, J.M.; Creus, A.; Marcos, R.; Surrallés, J. Multicolour FISH detection of radioactive iodine-induced 17cen-p53 chromosomal breakage in buccal cells from therapeutically exposed patients. Carcinogenesis 2000, 21, 1581–1586. [Google Scholar]
- Ramírez, M.J.; Surrallés, J.; Galofré, P.; Creus, A.; Marcos, R. Radioactive iodine induces clastogenic and age-dependent aneugenic effects in lymphocytes of thyroid cancer patients as revealed by interphase FISH. Mutagenesis 1997, 12, 449–455. [Google Scholar] [CrossRef]
- Monzen, S.; Mariya, Y.; Wojcik, A.; Kawamura, C.; Nakamura, A.; Chiba, M.; Hosoda, M.; Takai, Y. Predictive factors of cytotoxic damage in radioactive iodine treatment of differentiated thyroid cancer patients. Mol. Clin. Oncol. 2015, 3, 692–698. [Google Scholar] [CrossRef] [Green Version]
- Shakeri, M.; Zakeri, F.; Changizi, V.; Rajabpour, M.R.; Farshidpour, M.R. Cytogenetic effects of radiation and genetic polymorphisms of the XRCC1 and XRCC3 repair genes in industrial radiographers. Radiat. Environ. Biophys. 2019, 58, 247–255. [Google Scholar] [CrossRef]
- Müller, W.U.; Nüsse, M.; Miller, B.M.; Slavotinek, A.; Viaggi, S.; Streffer, C. Micronuclei: A biological indicator of radiation damage. Mutat. Res. 1996, 366, 163–169. [Google Scholar] [CrossRef]
- Sinitsky, M.Y.; Minina, V.I.; Asanov, M.A.; Yuzhalin, A.E.; Ponasenko, A.V.; Druzhinin, V.G. Association of DNA repair gene polymorphisms with genotoxic stress in underground coal miners. Mutagenesis 2017, 32, 501–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, N.; Yokoyama, K.; Kinuya, S.; Shuke, N.; Shimizu, M.; Futatsuya, R.; Michigishi, T.; Tonami, N.; Seto, H.; Goodwin, D.A. Radiotoxicity after iodine-131 therapy for thyroid cancer using the micronucleus assay. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 1998, 39, 436–440. [Google Scholar]
- Ballardin, M.; Gemignani, F.; Bodei, L.; Mariani, G.; Ferdeghini, M.; Rossi, A.M.; Migliore, L.; Barale, R. Formation of micronuclei and of clastogenic factor(s) in patients receiving therapeutic doses of iodine-131. Mutat. Res. 2002, 514, 77–85. [Google Scholar] [CrossRef]
- Livingston, G.K.; Khvostunov, I.K. Cytogenetic effects of radioiodine therapy: A 20-year follow-up study. Radiat. Environ. Biophys. 2016, 55, 203–213. [Google Scholar] [CrossRef]
- Puerto, S.; Marcos, R.; Ramírez, M.J.; Galofré, P.; Creus, A.; Surrallés, J. Equal induction and persistence of chromosome aberrations involving chromosomes 1, 4 and 10 in thyroid cancer patients treated with radioactive iodine. Mutat. Res. 2000, 469, 147–158. [Google Scholar] [CrossRef]
- Fenech, M.; Denham, J.; Francis, W.; Morley, A. Micronuclei in cytokinesis-blocked lymphocytes of cancer patients following fractionated partial-body radiotherapy. Int. J. Radiat. Biol. 1990, 57, 373–383. [Google Scholar] [CrossRef]
- M’Kacher, R.; Légal, J.D.; Schlumberger, M.; Aubert, B.; Beron-Gaillard, N.; Gaussen, A.; Parmentier, C. Sequential biological dosimetry after a single treatment with iodine-131 for differentiated thyroid carcinoma. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 1997, 38, 377–380. [Google Scholar]
- Livingston, G.K.; Escalona, M.; Foster, A.; Balajee, A.S. Persistent in vivo cytogenetic effects of radioiodine therapy: A 21-year follow-up study using multicolor FISH. J. Radiat. Res. 2018, 59, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Livingston, G.K.; Ryan, T.L.; Smith, T.L.; Escalona, M.B.; Foster, A.E.; Balajee, A.S. Detection of Simple, Complex, and Clonal Chromosome Translocations Induced by Internal Radioiodine Exposure: A Cytogenetic Follow-Up Case Study after 25 Years. Cytogenet. Genome Res. 2019, 159, 169–181. [Google Scholar] [CrossRef]
- Lindholm, C.; Acheva, A.; Salomaa, S. Clastogenic plasma factors: A short overview. Radiat. Environ. Biophys. 2010, 49, 133–138. [Google Scholar] [CrossRef]
- Morgan, W.F. Is there a common mechanism underlying genomic instability, bystander effects and other nontargeted effects of exposure to ionizing radiation? Oncogene 2003, 22, 7094–7099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavragani, I.V.; Laskaratou, D.A.; Frey, B. Key mechanisms involved in ionizing radiation-induced systemic effects. A current review. Toxicol. Res. 2016, 5, 12–33. [Google Scholar] [CrossRef] [PubMed]
- Lorimore, S.A.; McIlrath, J.M.; Coates, P.J.; Wright, E.G. Chromosomal instability in unirradiated hemopoietic cells resulting from a delayed in vivo bystander effect of gamma radiation. Cancer Res. 2005, 65, 5668–5673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenech, M.; Bonassi, S. The effect of age, gender, diet and lifestyle on DNA damage measured using micronucleus frequency in human peripheral blood lymphocytes. Mutagenesis 2011, 26, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Fenech, M.; Holland, N.; Zeiger, E.; Chang, W.P.; Burgaz, S.; Thomas, P.; Bolognesi, C.; Knasmueller, S.; Kirsch-Volders, M.; Bonassi, S. The HUMN and HUMNxL international collaboration projects on human micronucleus assays in lymphocytes and buccal cells--past, present and future. Mutagenesis 2011, 26, 239–245. [Google Scholar] [CrossRef] [Green Version]
- Battershill, J.M.; Burnett, K.; Bull, S. Factors affecting the incidence of genotoxicity biomarkers in peripheral blood lymphocytes: Impact on design of biomonitoring studies. Mutagenesis 2008, 23, 423–437. [Google Scholar] [CrossRef] [Green Version]
- Bonassi, S.; Fenech, M.; Lando, C.; Lin, Y.P.; Ceppi, M.; Chang, W.P.; Holland, N.; Kirsch-Volders, M.; Zeiger, E.; Ban, S.; et al. HUman MicroNucleus project: International database comparison for results with the cytokinesis-block micronucleus assay in human lymphocytes: I. Effect of laboratory protocol, scoring criteria, and host factors on the frequency of micronuclei. Environ. Mol. Mutagenesis 2001, 37, 31–45. [Google Scholar] [CrossRef]
- Caria, P.; Vanni, R. Cytogenetic and molecular events in adenoma and well-differentiated thyroid follicular-cell neoplasia. Cancer Genet. Cytogenet. 2010, 203, 21–29. [Google Scholar] [CrossRef]
- Genutis, L.K.; Tomsic, J.; Bundschuh, R.A.; Brock, P.L.; Williams, M.D.; Roychowdhury, S.; Reeser, J.W.; Frankel, W.L.; Alsomali, M.; Routbort, M.J.; et al. Microsatellite Instability Occurs in a Subset of Follicular Thyroid Cancers. Thyroid Off. J. Am. Thyroid Assoc. 2019, 29, 523–529. [Google Scholar] [CrossRef]
- Lazzereschi, D.; Palmirotta, R.; Ranieri, A.; Ottini, L.; Veri, M.C.; Cama, A.; Cetta, F.; Nardi, F.; Colletta, G.; Mariani-Costantini, R. Microsatellite instability in thyroid tumours and tumour-like lesions. Br. J. Cancer 1999, 79, 340–345. [Google Scholar] [CrossRef] [Green Version]
- Migdalska-Sek, M.; Czarnecka, K.H.; Kusinski, M.; Pastuszak-Lewandoska, D.; Nawrot, E.; Kuzdak, K.; Brzezianska-Lasota, E. Clinicopathological Significance of Overall Frequency of Allelic Loss (OFAL) in Lesions Derived from Thyroid Follicular Cell. Mol. Diagn. Ther. 2019, 23, 369–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, L.S.; Brenta, G.; Medvedovic, M.; Fagin, J.A. Studies of allelic loss in thyroid tumors reveal major differences in chromosomal instability between papillary and follicular carcinomas. J. Clin. Endocrinol. Metab. 1998, 83, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, J.W.; Nasir, A.; Kaiser, H.E. Loss of heterozygosity in papillary and follicular thyroid carcinoma: A mini review. VIVO (AthensGreece) 2000, 14, 139–140. [Google Scholar]
- Xing, M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Rev. Cancer 2013, 13, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Sobrinho-Simoes, M.; Eloy, C.; Magalhaes, J.; Lobo, C.; Amaro, T. Follicular thyroid carcinoma. Mod. Pathol. 2011, 24, S10–S18. [Google Scholar] [CrossRef]
- Miller, A.C.; Gafner, J.; Clark, E.P.; Samid, D. Differences in radiation-induced micronuclei yields of human cells: Influence of ras gene expression and protein localization. Int. J. Radiat. Biol. 1993, 64, 547–554. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, D.S.; Luan, Z.S.; Zhang, F.; Liu, X.H.; Zhou, W.; Zhong, S.F.; Lai, H. Efficacy of radioiodine therapy for treating 20 patients with pulmonary metastases from differentiated thyroid cancer and a meta-analysis of the current literature. Clin. Transl. Oncol. 2018, 20, 928–935. [Google Scholar] [CrossRef] [Green Version]
- Eccles, L.J.; O’Neill, P.; Lomax, M.E. Delayed repair of radiation induced clustered DNA damage: Friend or foe? Mutat. Res. 2011, 711, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Sage, E.; Shikazono, N. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free Radic. Biol. Med. 2017, 107, 125–135. [Google Scholar] [CrossRef]
- Martin, L.M.; Marples, B.; Coffey, M.; Lawler, M.; Lynch, T.H.; Hollywood, D.; Marignol, L. DNA mismatch repair and the DNA damage response to ionizing radiation: Making sense of apparently conflicting data. Cancer Treat. Rev. 2010, 36, 518–527. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Sharma, N.; Taylor, L. Clustered DNA Double-Strand Breaks: Biological Effects and Relevance to Cancer Radiotherapy. Genes 2020, 11, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Xiong, Y.; Chen, J. DNA-protein cross-link repair: What do we know now? Cell Biosci. 2020, 10, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, T.; Xu, X.; Salem, A.M.H.; Shoulkamy, M.I.; Ide, H. Radiation-induced DNA-protein cross-links: Mechanisms and biological significance. Free Radic. Biol. Med. 2017, 107, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cheng, J.; Gao, J.; Wang, J.; Liu, X.; Xiong, L. Association between the NBS1 Glu185Gln polymorphism and lung cancer risk: A systemic review and meta-analysis. Mol. Biol. Rep. 2013, 40, 2711–2715. [Google Scholar] [CrossRef]
- Kinsella, T.J. Coordination of DNA mismatch repair and base excision repair processing of chemotherapy and radiation damage for targeting resistant cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 1853–1859. [Google Scholar] [CrossRef] [Green Version]
- Edelbrock, M.A.; Kaliyaperumal, S.; Williams, K.J. Structural, molecular and cellular functions of MSH2 and MSH6 during DNA mismatch repair, damage signaling and other noncanonical activities. Mutat. Res. 2013, 743, 53–66. [Google Scholar] [CrossRef] [Green Version]
- Iyama, T.; Wilson, D.M., 3rd. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Iarmarcovai, G.; Bonassi, S.; Botta, A.; Baan, R.A.; Orsière, T. Genetic polymorphisms and micronucleus formation: A review of the literature. Mutat. Res. 2008, 658, 215–233. [Google Scholar] [CrossRef]
- Plotz, G.; Raedle, J.; Spina, A.; Welsch, C.; Stallmach, A.; Zeuzem, S.; Schmidt, C. Evaluation of the MLH1 I219V alteration in DNA mismatch repair activity and ulcerative colitis. Inflamm. Bowel Dis. 2008, 14, 605–611. [Google Scholar] [CrossRef]
- Milanizadeh, S.; Khanyaghma, M.; Haghighi, M.M.; Mohebbi, S.; Damavand, B.; Almasi, S.; Azimzadeh, P.; Zali, M. Molecular analysis of imperative polymorphisms of MLH1 gene in sporadic colorectal cancer. Cancer Biomark. Sect. A Dis. Markers 2013, 13, 427–432. [Google Scholar] [CrossRef]
- Kim, J.C.; Roh, S.A.; Koo, K.H.; Ka, I.H.; Kim, H.C.; Yu, C.S.; Lee, K.H.; Kim, J.S.; Lee, H.I.; Bodmer, W.F. Genotyping possible polymorphic variants of human mismatch repair genes in healthy Korean individuals and sporadic colorectal cancer patients. Fam. Cancer 2004, 3, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Rasi, S.; Di Rocco, A.; Fabbri, A.; Forconi, F.; Gloghini, A.; Bruscaggin, A.; Franceschetti, S.; Fangazio, M.; De Paoli, L.; et al. The host genetic background of DNA repair mechanisms is an independent predictor of survival in diffuse large B-cell lymphoma. Blood 2011, 117, 2405–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.Q.; Gong, W.D.; Wang, S.Z.; Zhang, Z.D.; Rui, X.P.; Wu, G.Z.; Ren, F. Polymorphisms of mismatch repair gene hMLH1 and hMSH2 and risk of gastric cancer in a Chinese population. Oncol. Lett. 2012, 3, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Dreussi, E.; Cecchin, E.; Polesel, J.; Canzonieri, V.; Agostini, M.; Boso, C.; Belluco, C.; Buonadonna, A.; Lonardi, S.; Bergamo, F.; et al. Pharmacogenetics Biomarkers and Their Specific Role in Neoadjuvant Chemoradiotherapy Treatments: An Exploratory Study on Rectal Cancer Patients. Int. J. Mol. Sci. 2016, 17, 1482. [Google Scholar] [CrossRef] [Green Version]
- Damaraju, S.; Murray, D.; Dufour, J.; Carandang, D.; Myrehaug, S.; Fallone, G.; Field, C.; Greiner, R.; Hanson, J.; Cass, C.E.; et al. Association of DNA repair and steroid metabolism gene polymorphisms with clinical late toxicity in patients treated with conformal radiotherapy for prostate cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 2545–2554. [Google Scholar] [CrossRef] [Green Version]
- Morales, F.; Vásquez, M.; Santamaría, C.; Cuenca, P.; Corrales, E.; Monckton, D.G. A polymorphism in the MSH3 mismatch repair gene is associated with the levels of somatic instability of the expanded CTG repeat in the blood DNA of myotonic dystrophy type 1 patients. DNA Repair 2016, 40, 57–66. [Google Scholar] [CrossRef]
- Nogueira, G.A.; Lourenço, G.J.; Oliveira, C.B.; Marson, F.A.; Lopes-Aguiar, L.; Costa, E.F.; Lima, T.R.; Liutti, V.T.; Leal, F.; Santos, V.C.; et al. Association between genetic polymorphisms in DNA mismatch repair-related genes with risk and prognosis of head and neck squamous cell carcinoma. Int. J. Cancer 2015, 137, 810–818. [Google Scholar] [CrossRef]
- Vogelsang, M.; Wang, Y.; Veber, N.; Mwapagha, L.M.; Parker, M.I. The cumulative effects of polymorphisms in the DNA mismatch repair genes and tobacco smoking in oesophageal cancer risk. PLoS ONE 2012, 7, e36962. [Google Scholar] [CrossRef] [Green Version]
- Mangoni, M.; Bisanzi, S.; Carozzi, F.; Sani, C.; Biti, G.; Livi, L.; Barletta, E.; Costantini, A.S.; Gorini, G. Association between genetic polymorphisms in the XRCC1, XRCC3, XPD, GSTM1, GSTT1, MSH2, MLH1, MSH3, and MGMT genes and radiosensitivity in breast cancer patients. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 52–58. [Google Scholar] [CrossRef]
- Xu, X.L.; Yao, Y.L.; Xu, W.Z.; Feng, J.G.; Mao, W.M. Correlation of MSH3 polymorphisms with response and survival in advanced non-small cell lung cancer patients treated with first-line platinum-based chemotherapy. Genet. Mol. Res. Gmr 2015, 14, 3525–3533. [Google Scholar] [CrossRef]
- Chu, Y.L.; Wu, X.; Xu, Y.; Her, C. MutS homologue hMSH4: Interaction with eIF3f and a role in NHEJ-mediated DSB repair. Mol. Cancer 2013, 12, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.L.; Hu, L.M.; Huang, M.D.; Zhao, W.; Yin, Y.M.; Hu, Z.B.; Ma, H.X.; Shen, H.B.; Shu, Y.Q. Genetic variants of NBS1 predict clinical outcome of platinum-based chemotherapy in advanced non-small cell lung cancer in Chinese. Asian Pac. J. Cancer Prev. Apjcp 2012, 13, 851–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, T.R.; Liu-Mares, W.; Van Emburgh, B.O.; Levine, E.A.; Allen, G.O.; Hill, J.W.; Reis, I.M.; Kresty, L.A.; Pegram, M.D.; Miller, M.S.; et al. Genetic polymorphisms of multiple DNA repair pathways impact age at diagnosis and TP53 mutations in breast cancer. Carcinogenesis 2011, 32, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Qiu, F.; Zhang, L.; Deng, J.; Zhang, H.; Yang, L.; Zhou, Y.; Lu, J. The functional polymorphism of NBS1 p.Glu185Gln is associated with an increased risk of lung cancer in Chinese populations: Case-control and a meta-analysis. Mutat. Res. 2014, 770, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Lu, J.; Yang, X.; Yang, M.; Tan, H.; Yun, B.; Shi, L. Association between the NBS1 E185Q polymorphism and cancer risk: A meta-analysis. BMC Cancer 2009, 9, 124. [Google Scholar] [CrossRef] [Green Version]
- Goricar, K.; Erculj, N.; Zadel, M.; Dolzan, V. Genetic polymorphisms in homologous recombination repair genes in healthy Slovenian population and their influence on DNA damage. Radiol. Oncol. 2012, 46, 46–53. [Google Scholar] [CrossRef]
- Gdowicz-Klosok, A.; Widel, M.; Rzeszowska-Wolny, J. The influence of XPD, APE1, XRCC1, and NBS1 polymorphic variants on DNA repair in cells exposed to X-rays. Mutat. Res. 2013, 755, 42–48. [Google Scholar] [CrossRef]
- Mumbrekar, K.D.; Goutham, H.V.; Vadhiraja, B.M.; Bola Sadashiva, S.R. Polymorphisms in double strand break repair related genes influence radiosensitivity phenotype in lymphocytes from healthy individuals. Dna Repair 2016, 40, 27–34. [Google Scholar] [CrossRef]
- Yin, M.; Liao, Z.; Huang, Y.J.; Liu, Z.; Yuan, X.; Gomez, D.; Wang, L.E.; Wei, Q. Polymorphisms of homologous recombination genes and clinical outcomes of non-small cell lung cancer patients treated with definitive radiotherapy. PLoS ONE 2011, 6, e20055. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, G.H.; Manjunath, V.B.; Mumbrekar, K.D.; Negi, H.; Fernandes, D.J.; Sharan, K.; Banerjee, S.; Bola Sadashiva, S.R. Polymorphisms in radio-responsive genes and its association with acute toxicity among head and neck cancer patients. PLoS ONE 2014, 9, e89079. [Google Scholar] [CrossRef]
- Chang-Claude, J.; Ambrosone, C.B.; Lilla, C.; Kropp, S.; Helmbold, I.; von Fournier, D.; Haase, W.; Sautter-Bihl, M.L.; Wenz, F.; Schmezer, P.; et al. Genetic polymorphisms in DNA repair and damage response genes and late normal tissue complications of radiotherapy for breast cancer. Br. J. Cancer 2009, 100, 1680–1686. [Google Scholar] [CrossRef] [PubMed]
- Kerns, S.L.; Stock, R.G.; Stone, N.N.; Blacksburg, S.R.; Rath, L.; Vega, A.; Fachal, L.; Gómez-Caamaño, A.; De Ruysscher, D.; Lammering, G.; et al. Genome-wide association study identifies a region on chromosome 11q14.3 associated with late rectal bleeding following radiation therapy for prostate cancer. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2013, 107, 372–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, C.; Zhang, H.; Chen, K.; Zhao, C.; Gao, J. Genetic variability of DNA repair mechanisms influences treatment outcome of gastric cancer. Oncol. Lett. 2015, 10, 1997–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erčulj, N.; Kovač, V.; Hmeljak, J.; Franko, A.; Dodič-Fikfak, M.; Dolžan, V. DNA repair polymorphisms and treatment outcomes of patients with malignant mesothelioma treated with gemcitabine-platinum combination chemotherapy. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2012, 7, 1609–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, K.; Rachakonda, P.S.; Panzram, B.; Keller, G.; Lordick, F.; Becker, K.; Langer, R.; Buechler, M.; Hemminki, K.; Kumar, R. DNA repair gene and MTHFR gene polymorphisms as prognostic markers in locally advanced adenocarcinoma of the esophagus or stomach treated with cisplatin and 5-fluorouracil-based neoadjuvant chemotherapy. Ann. Surg. Oncol. 2011, 18, 2688–2698. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, Z.Y.; Li, C.B.; Gao, S.; Ding, L.H.; Wu, X.L.; Wang, Z.Y. Genetic polymorphisms of DNA repair pathways influence the response to chemotherapy and overall survival of gastric cancer. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 3017–3023. [Google Scholar] [CrossRef]
- Jiang, Y.H.; Xu, X.L.; Ruan, H.H.; Xu, W.Z.; Li, D.; Feng, J.G.; Han, Q.B.; Mao, W.M. The impact of functional LIG4 polymorphism on platinum-based chemotherapy response and survival in non-small cell lung cancer. Med. Oncol. 2014, 31, 959. [Google Scholar] [CrossRef]
- Sinitsky, M.Y.; Larionov, A.V.; Asanov, M.A.; Druzhinin, V.G. Associations of DNA-repair gene polymorphisms with a genetic susceptibility to ionizing radiation in residents of areas with high radon (222Rn) concentration. Int. J. Radiat. Biol. 2015, 91, 486–494. [Google Scholar] [CrossRef]
- Senghore, T.; Wang, W.C.; Chien, H.T. Polymorphisms of Mismatch Repair Pathway Genes Predict Clinical Outcomes in Oral Squamous Cell Carcinoma Patients Receiving Adjuvant Concurrent Chemoradiotherapy. Cancers 2019, 11, 598. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Valentin, M.; Drost, M.; Therkildsen, C.; Rambech, E.; Ehrencrona, H.; Angleys, M.; Lau Hansen, T.; de Wind, N.; Nilbert, M.; Juel Rasmussen, L. Functional implications of the p.Cys680Arg mutation in the MLH1 mismatch repair protein. Mol. Genet. Genom. Med. 2014, 2, 352–355. [Google Scholar] [CrossRef]
- Dominguez-Valentin, M.; Wernhoff, P.; Cajal, A.R.; Kalfayan, P.G.; Piñero, T.A.; Gonzalez, M.L.; Ferro, A.; Sammartino, I.; Causada Calo, N.S.; Vaccaro, C.A. MLH1 Ile219Val Polymorphism in Argentinean Families with Suspected Lynch Syndrome. Front. Oncol. 2016, 6, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasi, M.F.; Ventura, I.; Aquilina, G.; Degan, P.; Bertario, L.; Bassi, C.; Radice, P.; Bignami, M. A human cell-based assay to evaluate the effects of alterations in the MLH1 mismatch repair gene. Cancer Res. 2006, 66, 9036–9044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, P.T.; Curtin, K.; Ulrich, C.M.; Samowitz, W.S.; Bigler, J.; Velicer, C.M.; Caan, B.; Potter, J.D.; Slattery, M.L. Mismatch repair polymorphisms and risk of colon cancer, tumour microsatellite instability and interactions with lifestyle factors. Gut 2009, 58, 661–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentin, M.D.; Da Silva, F.C.; Santos, E.M.; Da Silva, S.D.; De Oliveira Ferreira, F.; Aguiar Junior, S.; Gomy, I.; Vaccaro, C.; Redal, M.A.; Della Valle, A.; et al. Evaluation of MLH1 I219V polymorphism in unrelated South American individuals suspected of having Lynch syndrome. Anticancer Res. 2012, 32, 4347–4351. [Google Scholar]
- Nejda, N.; Iglesias, D.; Moreno Azcoita, M.; Medina Arana, V.; González-Aguilera, J.J.; Fernández-Peralta, A.M. A MLH1 polymorphism that increases cancer risk is associated with better outcome in sporadic colorectal cancer. Cancer Genet. Cytogenet. 2009, 193, 71–77. [Google Scholar] [CrossRef]
- Li, S.; Zheng, Y.; Tian, T.; Wang, M.; Liu, X.; Liu, K.; Zhai, Y.; Dai, C.; Deng, Y.; Li, S.; et al. Pooling-analysis on hMLH1 polymorphisms and cancer risk: Evidence based on 31,484 cancer cases and 45,494 cancer-free controls. Oncotarget 2017, 8, 93063–93078. [Google Scholar] [CrossRef] [Green Version]
- Zare, M.; Jafari-Nedooshan, J. Relevance of hMLH1 -93G>A, 655A>G and 1151T>A polymorphisms with colorectal cancer susceptibility: A meta-analysis based on 38 case-control studies. Rev. Assoc. Med. Bras. (1992) 2018, 64, 942–951. [Google Scholar] [CrossRef]
- Zhang, Y.; Rohde, L.H.; Emami, K.; Hammond, D.; Casey, R.; Mehta, S.K.; Jeevarajan, A.S.; Pierson, D.L.; Wu, H. Suppressed expression of non-DSB repair genes inhibits gamma-radiation-induced cytogenetic repair and cell cycle arrest. DNA Repair 2008, 7, 1835–1845. [Google Scholar] [CrossRef]
- Bakhtiari, E.; Monfared, A.S.; Niaki, H.A.; Borzoueisileh, S.; Niksirat, F.; Fattahi, S.; Monfared, M.K.; Gorji, K.E. The expression of MLH1 and MSH2 genes among inhabitants of high background radiation area of Ramsar, Iran. J. Environ. Radioact. 2019, 208–209, 106012. [Google Scholar] [CrossRef]
- Yang, J.; Huang, Y.; Feng, Y.; Li, H.; Feng, T.; Chen, J.; Yin, L.; Wang, W.; Wang, S.; Liu, Y.; et al. Associations of Genetic Variations in Mismatch Repair Genes MSH3 and PMS1 with Acute Adverse Events and Survival in Patients with Rectal Cancer Receiving Postoperative Chemoradiotherapy. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2019, 51, 1198–1206. [Google Scholar] [CrossRef] [Green Version]
- Miao, H.K.; Chen, L.P.; Cai, D.P.; Kong, W.J.; Xiao, L.; Lin, J. MSH3 rs26279 polymorphism increases cancer risk: A meta-analysis. Int. J. Clin. Exp. Pathol. 2015, 8, 11060–11067. [Google Scholar] [PubMed]
- Ma, X.; Zhang, B.; Zheng, W. Genetic variants associated with colorectal cancer risk: Comprehensive research synopsis, meta-analysis, and epidemiological evidence. Gut 2014, 63, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Beeghly-Fadiel, A.; Long, J.; Zheng, W. Genetic variants associated with breast-cancer risk: Comprehensive research synopsis, meta-analysis, and epidemiological evidence. Lancet Oncol. 2011, 12, 477–488. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Pearlman, A.H.; Hsieh, P. DNA mismatch repair and the DNA damage response. DNA Repair 2016, 38, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Crouse, G.F. Non-canonical actions of mismatch repair. DNA Repair 2016, 38, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Conde, J.; Silva, S.N.; Azevedo, A.P.; Teixeira, V.; Pina, J.E.; Rueff, J.; Gaspar, J.F. Association of common variants in mismatch repair genes and breast cancer susceptibility: A multigene study. BMC Cancer 2009, 9, 344. [Google Scholar] [CrossRef] [Green Version]
- Doherty, J.A.; Sakoda, L.C.; Loomis, M.M.; Barnett, M.J.; Julianto, L.; Thornquist, M.D.; Neuhouser, M.L.; Weiss, N.S.; Goodman, G.E.; Chen, C. DNA repair genotype and lung cancer risk in the beta-carotene and retinol efficacy trial. Int. J. Mol. Epidemiol. Genet. 2013, 4, 11–34. [Google Scholar]
- Kappil, M.; Terry, M.B.; Delgado-Cruzata, L.; Liao, Y.; Santella, R.M. Mismatch Repair Polymorphisms as Markers of Breast Cancer Prevalence in the Breast Cancer Family Registry. Anticancer Res. 2016, 36, 4437–4441. [Google Scholar] [CrossRef] [Green Version]
- Clark, N.; Wu, X.; Her, C. MutS Homologues hMSH4 and hMSH5: Genetic Variations, Functions, and Implications in Human Diseases. Curr. Genom. 2013, 14, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.L.; Wu, X.; Xu, J.; Watts, J.L.; Her, C. DNA damage induced MutS homologue hMSH4 acetylation. Int. J. Mol. Sci. 2013, 14, 20966–20982. [Google Scholar] [CrossRef] [Green Version]
- He, Y.Z.; Chi, X.S.; Zhang, Y.C.; Deng, X.B.; Wang, J.R.; Lv, W.Y.; Zhou, Y.H.; Wang, Z.Q. NBS1 Glu185Gln polymorphism and cancer risk: Update on current evidence. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Ma, N.; Li, M.; Tian, Q.B.; Liu, D.W. Functional variants in NBS1 and cancer risk: Evidence from a meta-analysis of 60 publications with 111 individual studies. Mutagenesis 2013, 28, 683–697. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Fang, Y.; Chen, B.; Jin, F.; Wang, S. Association between the NBS1 Glu185Gln polymorphism and breast cancer risk: A meta-analysis. Tumour Biol. J. Int. Soc. Oncodeve. Biol. Med. 2013, 34, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Stern, M.C.; Lin, J.; Figueroa, J.D.; Kelsey, K.T.; Kiltie, A.E.; Yuan, J.M.; Matullo, G.; Fletcher, T.; Benhamou, S.; Taylor, J.A.; et al. Polymorphisms in DNA repair genes, smoking, and bladder cancer risk: Findings from the international consortium of bladder cancer. Cancer Res. 2009, 69, 6857–6864. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, Q.; Yuan, S.; Xie, W.; Liu, Y.; Xiang, Y.; Wu, N.; Wu, L.; Ma, X.; Cai, T.; et al. Genetic predisposition to lung cancer: Comprehensive literature integration, meta-analysis, and multiple evidence assessment of candidate-gene association studies. Sci. Rep. 2017, 7, 8371. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, Z.; Xu, Y. Carriage of NBN polymorphisms and acute leukemia risk. Int. J. Clin. Exp. Med. 2015, 8, 3769–3776. [Google Scholar]
- Zhang, Y.; Huang, Y.S.; Lin, W.Q.; Zhang, S.D.; Li, Q.W.; Hu, Y.Z.; Zheng, R.L.; Tang, T.; Li, X.Z.; Zheng, X.H. NBS1 Glu185Gln polymorphism and susceptibility to urinary system cancer: A meta-analysis. Tumour Biol. J. Int. Soc. Oncodeve. Biol. Med. 2014, 35, 10723–10729. [Google Scholar] [CrossRef]
- Vineis, P.; Manuguerra, M.; Kavvoura, F.K.; Guarrera, S.; Allione, A.; Rosa, F.; Di Gregorio, A.; Polidoro, S.; Saletta, F.; Ioannidis, J.P.; et al. A field synopsis on low-penetrance variants in DNA repair genes and cancer susceptibility. J. Natl. Cancer Inst. 2009, 101, 24–36. [Google Scholar] [CrossRef]
- Sud, A.; Hemminki, K.; Houlston, R.S. Candidate gene association studies and risk of Hodgkin lymphoma: A systematic review and meta-analysis. Hematol. Oncol. 2017, 35, 34–50. [Google Scholar] [CrossRef]
- Mehdinejad, M.; Sobhan, M.R.; Mazaheri, M.; Zare Shehneh, M.; Neamatzadeh, H.; Kalantar, S.M. Genetic Association between ERCC2, NBN, RAD51 Gene Variants and Osteosarcoma Risk: A Systematic Review and Meta-Analysis. Asian Pac. J. Cancer Prev. Apjcp 2017, 18, 1315–1321. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Location | DB SNP Cluster ID (RS NO.) | Base Change | Amino Acid Change | MAF (%) a | AB Assay ID |
|---|---|---|---|---|---|---|
| MLH1 | 3p22.2 | rs1799977 | A → G | Ile219Val | 23.3 | C___1219076_20 |
| MSH3 | 5q14.1 | rs26279 | A → G | Thr1045Ala | 27.1 | C____800002_1_ |
| MSH4 | 1p31.1 | rs5745325 | G → A | Ala97Thr | 26.0 | C___3286081_10 |
| PMS1 | 2q32.2 | rs5742933 | G → C | -- b | 23.4 | C__29329633_10 |
| MSH6 | 2p16.3 | rs1042821 | C → T | Gly39Glu | 18.2 | C___8760558_10 |
| RAD51 | 15q15.1 | rs1801321 | G → T | -- b | 33.2 | C___7482700_10 |
| NBN | 8q21.3 | rs1805794 | G → C | Glu185Gln | 34.7 | C__26470398_30 |
| XRCC3 | 14q32.33 | rs861539 | C → T | Thr241Met | 29.0 | -- d |
| XRCC5 | 2q35 | rs2440 | C → T | -- c | 36.3 | C___3231046_10 |
| Characteristics | Study Population n (%) | 70 mCi n (%) | 100 mCi n (%) | p Value c |
|---|---|---|---|---|
| Gender | ||||
| Male | 3 (11.5) | 1 (6.7) | 2 (18.2) | 0.556 |
| Female | 23 (88.5) | 14 (93.3) | 9 (81.8) | |
| Age a | 52.54 ± 11.62 b | 52.07 ± 10.26 b | 53.18 ± 13.76 b | 0.815 |
| ≤54 | 14 (53.8) | 8 (53.3) | 6 (54.5) | 1.000 |
| >54 | 12 (46.2) | 7 (46.7) | 5 (45.5) | |
| Smoking habits | ||||
| Non-smokers | 22 (84.6) | 13 (86.7) | 9 (81.8) | 1.000 |
| Smokers | 4 (15.4) | 2 (13.3) | 2 (18.2) | |
| Histology | ||||
| Papillary | 18 (69.2) | 10 (66.7) | 8 (72.7) | 1.000 |
| Follicular | 8 (30.8) | 5 (33.3) | 3 (27.3) | |
| Genotype | 70 mCi (n = 15) | 100 mCi (n = 11) | TOTAL (n = 26) | |||
|---|---|---|---|---|---|---|
| MAF | Genotype Frequency n (%) | MAF | Genotype Frequency n (%) | MAF | Genotype Frequency n (%) | |
| MLH1 rs1799977 | ||||||
| Ile/Ile | G: 0.30 | 7 (46.7) | G: 0.45 | 3 (27.3) | G: 0.37 | 10 (38.5) |
| Ile/Val | 7 (46.7) | 6 (54.5) | 13 (50.0) | |||
| Val/Val | 1 (6.7) | 2 (18.2) | 3 (11.5) | |||
| Ile/Val+Val/Val | 8 (53.3) | 8 (72.7) | 16 (61.5) | |||
| MSH3 rs26279 | ||||||
| Thr/Thr | G: 0.23 | 10 (66.7) | G: 0.14 | 8 (72.7) | G: 0.19 | 18 (69.2) |
| Thr/Ala | 3 (20.0) | 3 (27.3) | 6 (23.1) | |||
| Ala/Ala | 2 (13.3) | 0 (0.0) | 2 (7.7) | |||
| Thr/Ala+Ala/Ala | 5 (33.3) | 3 (27.3) | 8 (30.8) | |||
| MSH4 rs5745325 | ||||||
| Ala/Ala | A: 0.13 | 11 (73.3) | A: 0.32 | 4 (36.4) | A: 0.21 | 15 (57.7) |
| Ala/Thr | 4 (26.7) | 7 (63.6) | 11 (42.3) | |||
| Thr/Thr | 0 (0.0) | 0 (0.0) | 0 (0.0) | |||
| Ala/Thr+Thr/Thr | 4 (26.7) | 7 (63.6) | 11 (42.3) | |||
| PMS1 rs5742933 | ||||||
| G/G | C: 0.18 | 10 (71.4) | C: 0.14 | 9 (81.8) | C: 0.16 | 19 (76.0) |
| G/C | 3 (21.4) | 1 (9.1) | 4 (16.0) | |||
| C/C | 1 (7.1) | 1 (9.1) | 2 (8.0) | |||
| G/C+C/C | 4 (28.6) | 2 (18.2) | 6 (24.0) | |||
| MSH6 rs1042821 | ||||||
| Gly/Gly | T: 0.17 | 10 (66.7) | T: 0.09 | 9 (81.8) | T: 0.13 | 19 (73.1) |
| Gly/Glu | 5 (33.3) | 2 (18.2) | 7 (26.9) | |||
| Glu/Glu | 0 (0.0) | 0 (0.0) | 0 (0.0) | |||
| Gly/Glu+Glu/Glu | 5 (33.3) | 2 (18.2) | 7 (26.9) | |||
| RAD51 rs1801321 | ||||||
| T/T | G: 0.50 | 4 (26.7) | G: 0.45 | 4 (36.4) | G: 0.48 | 8 (30.8) |
| T/G | 7 (46.7) | 4 (36.4) | 11 (42.3) | |||
| G/G | 4 (26.7) | 3 (27.3) | 7 (26.9) | |||
| T/G+G/G | 11 (73.3) | 7 (63.6) | 18 (69.2) | |||
| NBN rs1805794 | ||||||
| Glu/Glu | C: 0.30 | 7 (46.7) | C: 0.14 | 8 (72.7) | C: 0.23 | 15 (57.7) |
| Glu/Gln | 7 (46.7) | 3 (27.3) | 10 (38.5) | |||
| Gln/Gln | 1 (6.7) | 0 (0.0) | 1 (3.8) | |||
| Glu/Gln+Gln/Gln | 8 (53.3) | 3 (27.3) | 11 (42.3) | |||
| XRCC3 rs861539 | ||||||
| Thr/Thr | C: 0.47 | 5 (33.3) | T: 0.36 | 5 (45.5) | T: 0.46 | 10 (38.5) |
| Thr/Met | 4 (26.7) | 4 (36.4) | 8 (30.8) | |||
| Met/Met | 6 (40.0) | 2 (18.2) | 8 (30.8) | |||
| Thr/Met+Met/Met | 10 (66.7) | 6 (54.5) | 16 (61.5) | |||
| XRCC5 rs2440 | ||||||
| T/T | C: 0.47 | 5 (33.3) | C: 0.50 | 2 (22.2) | C: 0.48 | 7 (29.2) |
| T/C | 6 (40.0) | 5 (55.6) | 11 (45.8) | |||
| C/C | 4 (26.7) | 2 (22.2) | 6 (25.0) | |||
| T/C+C/C | 10 (66.7) | 7 (77.8) | 17 (70.8) | |||
| Genotype | 70 mCi Group (n = 15), ‰BNMN (Mean ± SD) | 100 mCi Group (n = 11), ‰BNMN (Mean ± SD) | 70 + 100 mCi Groups (n = 26), ‰BNMN (Mean ± SD) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| t0 | t1 | t6 | t24 | t0 | t1 | t3 | t0 | t1 | |
| MLH1 rs1799977 | |||||||||
| Ile/Ile | 4.14 ± 3.29 | 12.14 ± 3.58 | 10.86 ± 7.11 | 9.20 ± 1.30 | 5.33 ± 1.16 | 24.00 ± 3.46 | 21.50 ± 7.78 | 4.50 ± 2.80 | 15.70 ± 6.63 |
| Ile/Val + Val/Val | 6.25 ± 3.85 | 5.88 ± 3.36 * | 7.25 ± 4.46 | 10.00 ± 3.74 | 11.25 ± 4.62 * | 14.75 ± 2.77 * | 21.38 ± 5.71 | 8.75 ± 4.85 * | 10.31 ± 5.46 * |
| MSH3 rs26279 | |||||||||
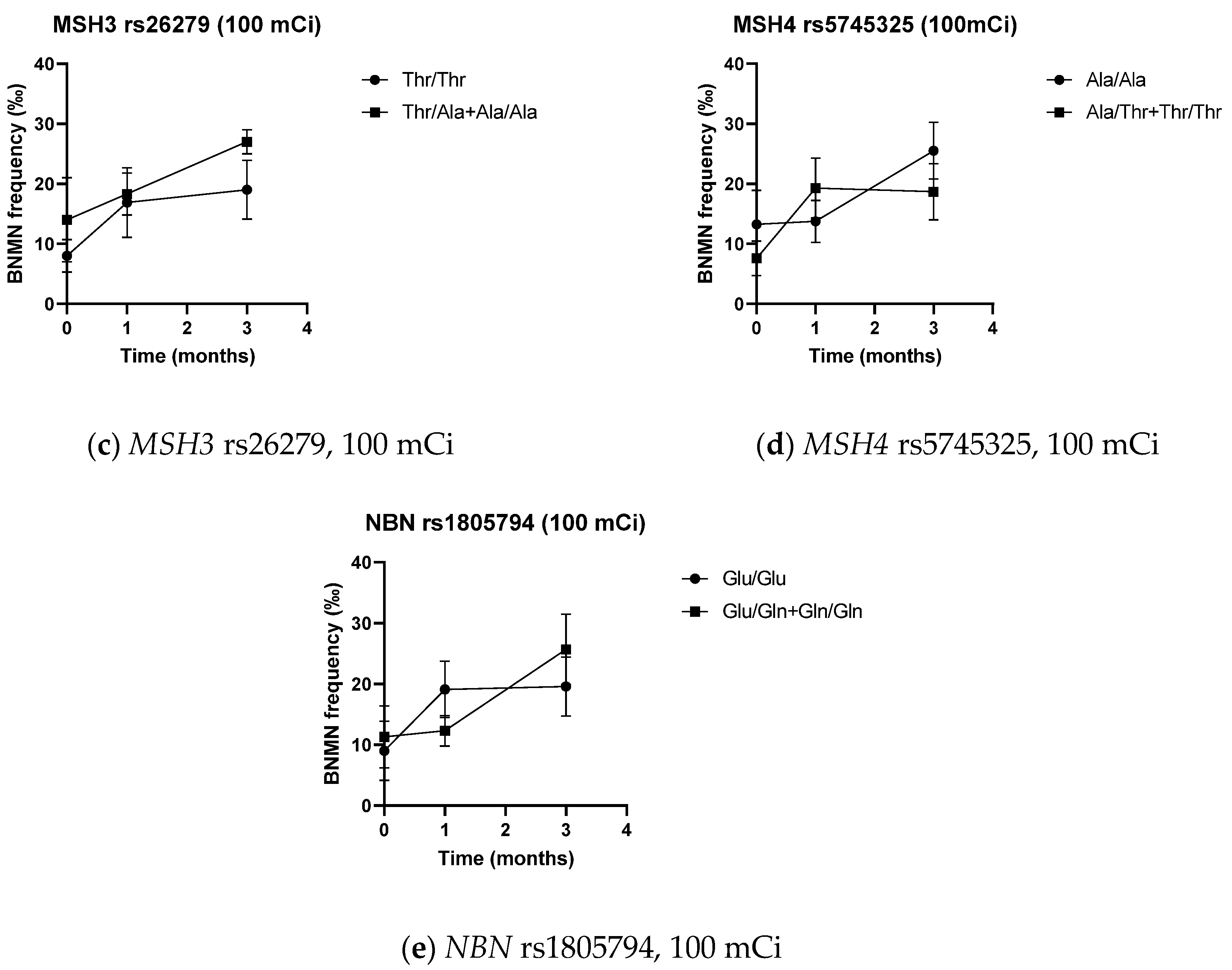
| Thr/Thr | 5.50 ± 3.63 | 8.90 ± 3.81 | 9.90 ± 7.09 | 10.13 ± 1.64 | 8.00 ± 2.73 | 16.88 ± 5.79 | 19.00 ± 4.93 | 6.61 ± 3.42 | 12.44 ± 6.18 |
| Thr/Ala + Ala/Ala | 4.80 ± 4.03 | 8.60 ± 6.54 | 7.00 ± 1.58 | 8.33 ± 5.13 | 14.00 ± 7.00 | 18.33 ± 3.51 | 27.00 ± 2.00 * | 8.25 ± 6.78 | 12.25 ± 7.31 |
| MSH4 rs5745325 | |||||||||
| Ala/Ala | 5.18 ± 3.79 | 8.91 ± 5.07 | 9.09 ± 6.64 | 9.63 ± 3.34 | 13.25 ± 5.68 | 13.75 ± 3.50 | 25.50 ± 4.73 | 7.33 ± 5.55 | 10.20 ± 5.09 |
| Ala/Thr + Thr/Thr | 5.50 ± 3.70 | 8.50 ± 3.87 | 8.50 ± 4.04 | 9.67 ± 0.58 | 7.57 ± 2.88 | 19.29 ± 4.99 | 18.67 ± 4.68 | 6.82 ± 3.19 | 15.36 ± 7.00 * |
| NBN rs1805794 | |||||||||
| Glu/Glu | 5.43 ± 4.61 | 10.00 ± 4.51 | 8.14 ± 4.56 | 9.86 ± 2.12 | 9.00 ± 4.84 | 19.13 ± 4.64 | 19.57 ± 4.89 | 7.33 ± 4.92 | 14.87 ± 6.46 |
| Glu/Gln + Gln/Gln | 5.13 ± 2.85 | 7.75 ± 4.80 | 9.63 ± 7.15 | 9.25 ± 4.11 | 11.33 ± 5.13 | 12.33 ± 2.52 * | 25.67 ± 5.77 | 6.82 ± 4.40 | 9.00 ± 4.69 * |
| Genotype | 70 mCi Group (n = 15), ‰BNMN (mean ± SD) | 100 mCi Group (n = 11), ‰BNMN (mean ± SD) | 70 + 100 mCi Groups (n = 26), ‰BNMN (mean ± SD) | |||
|---|---|---|---|---|---|---|
| Δt1 | Δt6 | Δt24 | Δt1 | Δt3 | Δt1 | |
| MLH1 rs1799977 | ||||||
| Ile/Ile | 8.00 ± 4.97 | 6.71 ± 6.85 | 5.00 ± 3.39 | 18.67 ± 3.06 | 16.50 ± 6.36 | 11.20 ± 6.71 |
| Ile/Val + Val/Val | −0.38 ± 3.70 * | 1.00 ± 4.90 | 3.50 ± 4.37 | 3.50 ± 4.57 * | 10.13 ± 5.28 | 1.56 ± 4.49 * |
| MSH4 rs5745325 | ||||||
| Ala/Ala | 3.73 ± 6.83 | 3.91 ± 7.05 | 4.13 ± 3.91 | 0.50 ± 3.11 | 12.25 ± 5.32 | 2.87 ± 6.13 |
| Ala/Thr + Thr/Thr | 3.00 ± 3.56 | 3.00 ± 4.90 | 4.33 ± 4.51 | 11.71 ± 7.27 * | 10.83 ± 6.49 | 8.55 ± 7.41 * |
| Gene | DB SNP Cluster ID (RS NO.) | Functional Impact | Clinical Association Studies (Radio and/or Chemosensitivity) |
|---|---|---|---|
| MLH1 | rs1799977 | Missense SNP located in a highly conserved N-terminal ATPase domain, vital for MLH1 function [73]; G allele associated with reduced expression [74,75,76,77]. | GG genotype associated with increased radiosensitivity in cancer patients, translating into increased efficacy [78] or toxicity [79] of radiotherapy (alone or combined with chemotherapy). |
| MSH3 | rs26279 | Missense SNP located in the ATPase domain, critical for protein activity [80]; altered expression has been suggested [81] but not confirmed [82]. | GG genotype associated with decreased incidence of radiation dermatitis in breast cancer patients receiving radiotherapy [83], decreased overall survival in head and neck squamous cell carcinoma patients submitted to radiochemotherapy [81] and decreased response to platinum-based chemotherapy in advanced non-small cell lung cancer patients [84]. |
| MSH4 | rs5745325 | Missense SNP located in the N-terminal domain, involved in the interaction with eIF3f [85]. | None to be reported. |
| NBN | rs1805794 | Missense SNP located in the BRCT domain, a region involved in the interaction with BRCA1 [86,87,88,89]; conflicting results from functional studies [88,90,91,92]. | No association detected in most studies focusing on response to radiotherapy [79,93,94,95,96] or chemotherapy [97,98,99]; conflicting results also reported as the C allele has been associated with either improved [86,100] or worse [68,101] prognosis upon platinum-based chemotherapy; increased frequency of binucleated lymphocytes with nucleoplasmic bridges in Glu/Gln children with high IR exposure, opposite to Gln/Gln children [102]. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
S. Santos, L.; M. Gil, O.; N. Silva, S.; C. Gomes, B.; C. Ferreira, T.; Limbert, E.; Rueff, J. Micronuclei Formation upon Radioiodine Therapy for Well-Differentiated Thyroid Cancer: The Influence of DNA Repair Genes Variants. Genes 2020, 11, 1083. https://doi.org/10.3390/genes11091083
S. Santos L, M. Gil O, N. Silva S, C. Gomes B, C. Ferreira T, Limbert E, Rueff J. Micronuclei Formation upon Radioiodine Therapy for Well-Differentiated Thyroid Cancer: The Influence of DNA Repair Genes Variants. Genes. 2020; 11(9):1083. https://doi.org/10.3390/genes11091083
Chicago/Turabian StyleS. Santos, Luís, Octávia M. Gil, Susana N. Silva, Bruno C. Gomes, Teresa C. Ferreira, Edward Limbert, and José Rueff. 2020. "Micronuclei Formation upon Radioiodine Therapy for Well-Differentiated Thyroid Cancer: The Influence of DNA Repair Genes Variants" Genes 11, no. 9: 1083. https://doi.org/10.3390/genes11091083