Pathogenicity Reclassification of RPE65 Missense Variants Related to Leber Congenital Amaurosis and Early-Onset Retinal Dystrophy

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
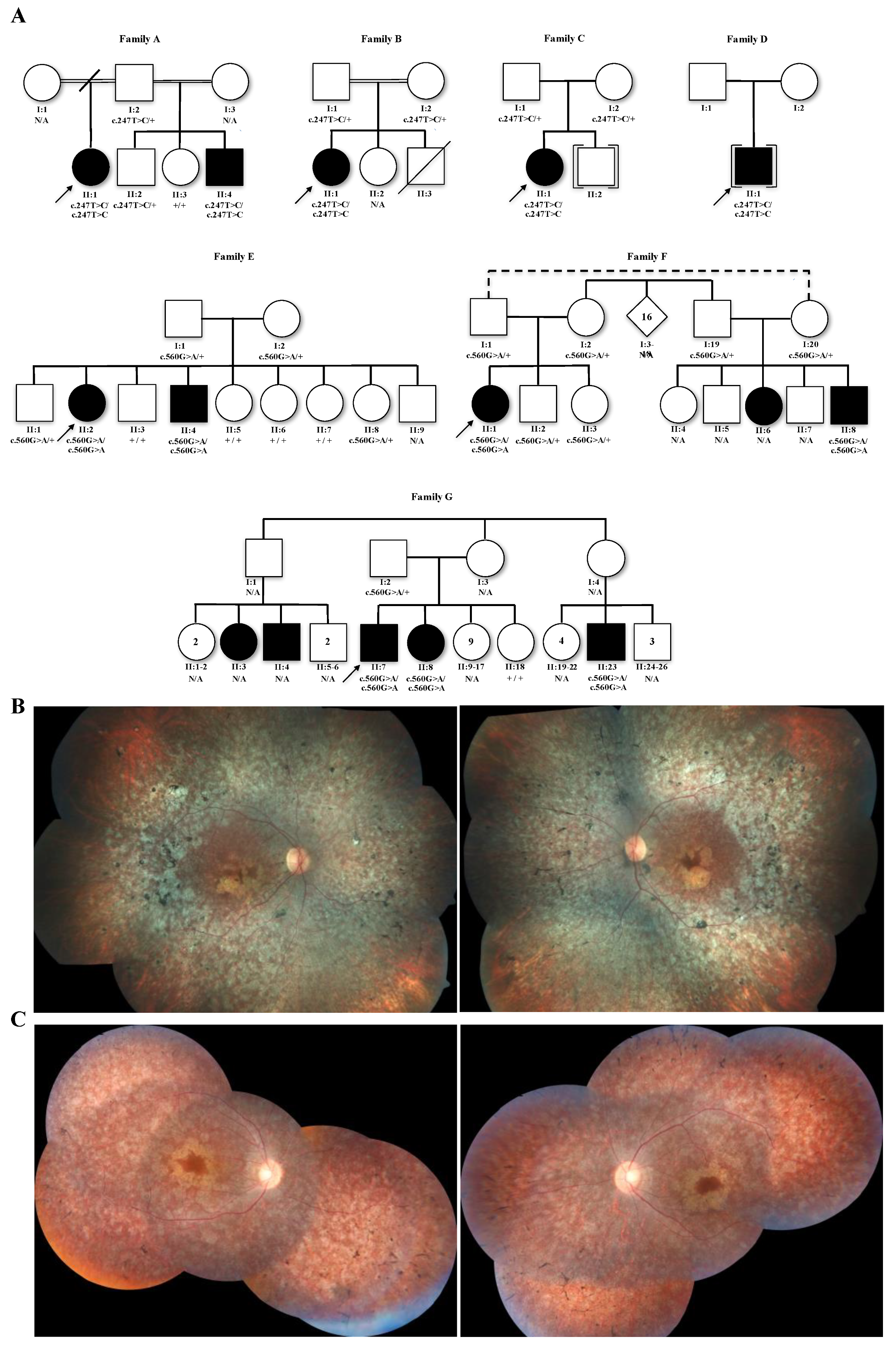
2.1. Subjects
2.2. Family Segregation
2.3. Variant Analysis
3. Results and Discussion
3.1. Pathogenicity Classification of RPE65 Variants c.247T>C (p.Phe83Leu) and c.560G>A (p.Gly187Glu) According to the Current Literature and Databases
3.2. Evaluation of the RPE65 Variants p.Phe83Leu and p.Gly187Glu in Brazilian Families with Inherited Retinal Dystrophies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Berger, W.; Kloeckener-Gruissem, B.; Neidhardt, J. The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 2010, 29, 335–375. [Google Scholar] [CrossRef] [Green Version]
- Motta, F.L.; Martin, R.P.; Filippelli-Silva, R.; Salles, M.V.; Sallum, J.M.F. Relative frequency of inherited retinal dystrophies in Brazil. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Daiger, S.P.; Sullivan, L.S.; Bowne, S.J.; Birch, D.G.; Heckenlively, J.R.; Pierce, E.A.; Weinstock, G.M. Targeted high-throughput DNA sequencing for gene discovery in retinitis pigmentosa. Adv. Exp. Med. Biol. 2010, 664, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Audo, I.; Bujakowska, K.M.; Léveillard, T.; Mohand-Saïd, S.; Lancelot, M.-E.; Germain, A.; Antonio, A.; Michiels, C.; Saraiva, J.-P.; Letexier, M.; et al. Development and application of a next-generation-sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet J. Rare Dis. 2012, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, J.P.; Trzupek, K. The current status of molecular diagnosis of inherited retinal dystrophies. Curr. Opin. Ophthalmol. 2015, 26, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Ellingford, J.M.; Barton, S.; Bhaskar, S.; Williams, S.G.; Sergouniotis, P.I.; O’Sullivan, J.; Lamb, J.A.; Perveen, R.; Hall, G.; Newman, W.G.; et al. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology 2016, 123, 1143–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Foxman, S.G.; Heckenlively, J.R.; Bateman, J.B.; Wirtschafter, J.D. Classification of Congenital and Early Onset Retinitis Pigmentosa. Arch. Ophthalmol. 1985, 103, 1502–1506. [Google Scholar] [CrossRef] [PubMed]
- Weleber, R.G.; Francis, P.J.; Trzupek, K.M.; Beattie, C. Leber Congenital Amaurosis. In GeneReviews®; Adam, M., Ardinger, H., Pagon, R.E., Wallace, S., Bean, L., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2004. [Google Scholar]
- Marlhens, F.; Griffoin, J.M.; Bareil, C.; Arnaud, B.; Claustres, M.; Hamel, C.P. Autosomal recessive retinal dystrophy associated with two novel mutations in the RPE65 gene. Eur. J. Hum. Genet. 1998, 6, 527–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, B.; Poliakov, E.; Schambeck, M.; Friedburg, C.; Preising, M.N.; Redmond, T.M. A Comprehensive Clinical and Biochemical Functional Study of a Novel RPE65 Hypomorphic Mutation. Investig. Opthalmol. Vis. Sci. 2008, 49, 5235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Izumi, T.; Hu, J.; Jin, H.H.; Siddiqui, A.-A.A.; Jacobson, S.G.; Bok, D.; Jin, M. Rescue of enzymatic function for disease-associated RPE65 proteins containing various missense mutations in non-active sites. J. Biol. Chem. 2014, 289, 18943–18956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Merico, D.; Yuen, R.K.C.; Hua, Y.; Gueroussov, S.; Najafabadi, H.S.; Hughes, T.R.; et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.-F.; Huang, F.; Wu, K.-C.; Wu, J.; Chen, J.; Pang, C.-P.; Lu, F.; Qu, J.; Jin, Z.-B. Genotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet. Med. 2015, 17, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.; Li, S.; Hu, J.; Jin, H.H.; Jacobson, S.G.; Bok, D. Functional Rescue of Retinal Degeneration-Associated Mutant RPE65 Proteins. Adv. Exp. Med. Biol. 2016, 854, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Simonelli, F.; Ziviello, C.; Testa, F.; Rossi, S.; Fazzi, E.; Bianchi, P.E.; Fossarello, M.; Signorini, S.; Bertone, C.; Galantuomo, S.; et al. Clinical and molecular genetics of Leber’s congenital amaurosis: A multicenter study of Italian patients. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4284–4290. [Google Scholar] [CrossRef]
- Thompson, D.A.; Gyürüs, P.; Fleischer, L.L.; Bingham, E.L.; McHenry, C.L.; Apfelstedt-Sylla, E.; Zrenner, E.; Lorenz, B.; Richards, J.E.; Jacobson, S.G.; et al. Genetics and phenotypes of RPE65 mutations in inherited retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2000, 41, 4293–4299. [Google Scholar]
- Philp, A.R.; Jin, M.; Li, S.; Schindler, E.I.; Iannaccone, A.; Lam, B.L.; Weleber, R.G.; Fishman, G.A.; Jacobson, S.G.; Mullins, R.F.; et al. Predicting the pathogenicity of RPE65 mutations. Hum. Mutat. 2009, 30, 1183–1188. [Google Scholar] [CrossRef] [Green Version]
- Chung, D.C.; Bertelsen, M.; Lorenz, B.; Pennesi, M.E.; Leroy, B.P.; Hamel, C.P.; Pierce, E.; Sallum, J.; Larsen, M.; Stieger, K.; et al. The Natural History of Inherited Retinal Dystrophy Due to Biallelic Mutations in the RPE65 Gene. Am. J. Ophthalmol. 2019, 199, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Morimura, H.; Fishman, G.A.; Grover, S.A.; Fulton, A.B.; Berson, E.L.; Dryja, T.P. Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc. Natl. Acad. Sci. USA 1998, 95, 3088–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maguire, A.M.; High, K.A.; Auricchio, A.; Wright, J.F.; Pierce, E.A.; Testa, F.; Mingozzi, F.; Bennicelli, J.L.; Ying, G.; Rossi, S.; et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: A phase 1 dose-escalation trial. Lancet 2009, 374, 1597–1605. [Google Scholar] [CrossRef] [Green Version]
- Redmond, T.M.; Poliakov, E.; Yu, S.; Tsai, J.-Y.; Lu, Z.; Gentleman, S. Mutation of key residues of RPE65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proc. Natl. Acad. Sci. USA 2005, 102, 13658–13663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019, 531210. [Google Scholar] [CrossRef] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA. Available online: http://evs.gs.washington.edu/EVS/ (accessed on 05 November 2019).
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Naslavsky, M.S.; Yamamoto, G.L.; de Almeida, T.F.; Ezquina, S.A.M.; Sunaga, D.Y.; Pho, N.; Bozoklian, D.; Sandberg, T.O.M.; Brito, L.A.; Lazar, M.; et al. Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum. Mutat. 2017, 38, 751–763. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [Green Version]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions using Hidden Markov Models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef]
- Shihab, H.A.; Rogers, M.F.; Gough, J.; Mort, M.; Cooper, D.N.; Day, I.N.M.; Gaunt, T.R.; Campbell, C. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 2015, 31, 1536–1543. [Google Scholar] [CrossRef] [Green Version]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Jhong, J.-H.; Lee, J.; Koo, J.-Y. Meta-analytic support vector machine for integrating multiple omics data. BioData Min. 2017, 10, 2. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; NISC Comparative Sequencing Program; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porto, F.; Jones, E.; Branch, J.; Soens, Z.; Maia, I.; Sena, I.; Sampaio, S.; Simões, R.; Chen, R. Molecular Screening of 43 Brazilian Families Diagnosed with Leber Congenital Amaurosis or Early-Onset Severe Retinal Dystrophy. Genes 2017, 8, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The Human Genomic Variant Search Engine. bioRxiv 2018. [Google Scholar] [CrossRef] [PubMed]
- Flannery, J.G. Transgenic Animal Models for the Study of Inherited Retinal Dystrophies. ILAR J. 1999, 40, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veleri, S.; Lazar, C.H.; Chang, B.; Sieving, P.A.; Banin, E.; Swaroop, A. Biology and therapy of inherited retinal degenerative disease: Insights from mouse models. Dis. Model. Mech. Mech. 2015, 8, 109–129. [Google Scholar] [CrossRef] [Green Version]
- Petersen-Jones, S.M.; Komáromy, A.M. Dog Models for Blinding Inherited Retinal Dystrophies. Hum. Gene Ther. Clin. Dev. 2015, 26, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Slijkerman, R.W.N.; Song, F.; Astuti, G.D.N.; Huynen, M.A.; van Wijk, E.; Stieger, K.; Collin, R.W.J. The pros and cons of vertebrate animal models for functional and therapeutic research on inherited retinal dystrophies. Prog. Retin. Eye Res. 2015, 48, 137–159. [Google Scholar] [CrossRef]
- Katz, M.L.; Redmond, T.M. Effect of Rpe65 knockout on accumulation of lipofuscin fluorophores in the retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2001, 42, 3023–3030. [Google Scholar]
- Veske, A.; Nilsson, S.E.G.; Narfström, K.; Gal, A. Retinal Dystrophy of Swedish Briard/Briard–Beagle Dogs Is Due to a 4-bp Deletion inRPE65. Genomics 1999, 57, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Znoiko, S.L.; Rohrer, B.; Lu, K.; Lohr, H.R.; Crouch, R.K.; Ma, J.-X. Downregulation of cone-specific gene expression and degeneration of cone photoreceptors in the Rpe65-/- mouse at early ages. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1473–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samardzija, M.; von Lintig, J.; Tanimoto, N.; Oberhauser, V.; Thiersch, M.; Remé, C.E.; Seeliger, M.; Grimm, C.; Wenzel, A. R91W mutation in Rpe65 leads to milder early-onset retinal dystrophy due to the generation of low levels of 11-cis-retinal. Hum. Mol. Genet. 2008, 17, 281–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Yu, S.; Duncan, T.; Li, Y.; Liu, P.; Gene, E.; Cortes-Pena, Y.; Qian, H.; Dong, L.; Redmond, T.M. Mouse model of human RPE65 P25L hypomorph resembles wild type under normal light rearing but is fully resistant to acute light damage. Hum. Mol. Genet. 2015, 24, 4417–4428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, E.H.; Suh, S.; Sander, C.L.; Hernandez, C.J.O.; Bulman, E.R.; Khadka, N.; Dong, Z.; Shi, W.; Palczewski, K.; Kiser, P.D. Insights into the pathogenesis of dominant retinitis pigmentosa associated with a D477G mutation in RPE65. Hum. Mol. Genet. 2018, 27, 2225–2243. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, A.; Reme, C.E.; Williams, T.P.; Hafezi, F.; Grimm, C. The Rpe65 Leu450Met variation increases retinal resistance against light-induced degeneration by slowing rhodopsin regeneration. J. Neurosci. 2001, 21, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, A.; Grimm, C.; Samardzija, M.; Remé, C.E. The genetic modifier Rpe65Leu(450): Effect on light damage susceptibility in c-Fos-deficient mice. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2798–2802. [Google Scholar] [CrossRef] [Green Version]
- Silva, E.; Dharmaraj, S.; Li, Y.Y.; Pina, A.L.; Carter, R.C.; Loyer, M.; Traboulsi, E.; Theodossiadis, G.; Koenekoop, R.; Sundin, O.; et al. A missense mutation in GUCY2D acts as a genetic modifier in RPE65-related Leber Congenital Amaurosis. Ophthalmic Genet. 2004, 25, 205–217. [Google Scholar] [CrossRef]
- Samardzija, M.; Wenzel, A.; Naash, M.; Remé, C.E.; Grimm, C. Rpe65 as a modifier gene for inherited retinal degeneration. Eur. J. Neurosci. 2006, 23, 1028–1034. [Google Scholar] [CrossRef] [Green Version]
- Narfström, K.; Vaegan; Katz, M.; Bragadottir, R.; Rakoczy, E.P.; Seeliger, M. Assessment of structure and function over a 3-year period after gene transfer in RPE65-/- dogs. Doc. Ophthalmol. 2005, 111, 39–48. [Google Scholar] [CrossRef]
- Nusinowitz, S.; Ridder, W.H.; Pang, J.J.; Chang, B.; Noorwez, S.M.; Kaushal, S.; Hauswirth, W.W.; Heckenlively, J.R. Cortical visual function in the rd12 mouse model of Leber Congenital Amarousis (LCA) after gene replacement therapy to restore retinal function. Vis. Res. 2006, 46, 3926–3934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, G.K.; Komáromy, A.M.; Cideciyan, A.V.; Brainard, D.H.; Aleman, T.S.; Roman, A.J.; Avants, B.B.; Gee, J.C.; Korczykowski, M.; Hauswirth, W.W.; et al. Canine and human visual cortex intact and responsive despite early retinal blindness from RPE65 mutation. PLoS Med. 2007, 4, e230. [Google Scholar] [CrossRef] [PubMed]
- Petersen-Jones, S.M.; Annear, M.J.; Bartoe, J.T.; Mowat, F.M.; Barker, S.E.; Smith, A.J.; Bainbridge, J.W.; Ali, R.R. Gene Augmentation Trials Using the Rpe65-Deficient Dog: Contributions Towards Development and Refinement of Human Clinical Trials. In Retinal Degenerative Diseases. Advances in Experimental Medicine and Biology; LaVail, M., Ash, J., Anderson, R., Hollyfield, J., Grimm, C., Eds.; Springer: Boston, MA, USA, 2012; Volume 723, pp. 177–182. [Google Scholar]
- Zheng, Q.; Ren, Y.; Tzekov, R.; Zhang, Y.; Chen, B.; Hou, J.; Zhao, C.; Zhu, J.; Zhang, Y.; Dai, X.; et al. Differential proteomics and functional research following gene therapy in a mouse model of Leber congenital amaurosis. PLoS ONE 2012, 7, e44855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, D.H.; Song, D.W.; Cho, C.S.; Kim, U.G.; Lee, K.J.; Lee, K.; Park, S.W.; Kim, D.; Kim, J.H.; Kim, J.-S.; et al. CRISPR-Cas9-mediated therapeutic editing of Rpe65 ameliorates the disease phenotypes in a mouse model of Leber congenital amaurosis. Sci. Adv. 2019, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Moiseyev, G.; Ma, J. Identification of key residues determining isomerohydrolase activity of human RPE65. J. Biol. Chem. 2014, 289, 26743–26751. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Gao, G.; Wang, P.; Song, X.; Xu, P.; Xie, B.; Zhou, T.; Pan, G.; Peng, F.; Zhang, Q.; et al. Generation and Characterization of Induced Pluripotent Stem Cells and Retinal Organoids From a Leber’s Congenital Amaurosis Patient With Novel RPE65 Mutations. Front. Mol. Neurosci. 2019, 12, 212. [Google Scholar] [CrossRef] [Green Version]
- Yang, U.; Gentleman, S.; Gai, X.; Gorin, M.B.; Borchert, M.S.; Lee, T.C.; Villanueva, A.; Koenekoop, R.; Maguire, A.M.; Bennett, J.; et al. Utility of In Vitro Mutagenesis of RPE65 Protein for Verification of Mutational Pathogenicity Before Gene Therapy. JAMA Ophthalmol. 2019, 1. [Google Scholar] [CrossRef]


{kind=link}
| c.247T>C; p.Phe83Leu | c.560G>A; p.Gly187Glu | |
|---|---|---|
| Population Genetics Databases | ||
| gnomAD total | No data | 2 in 251,106 alleles |
| ExAC total | No data | 1 in 120,880 alleles |
| ESP | No data | No data |
| 1000 Genomes | No data | No data |
| ABraOM | No data | No data |
| Human Variations and Phenotypes Databases | ||
| ClinVar | Not reported | Not reported |
| UniProt | Not reported | Not reported |
| HGMD | Disease-Causing Mutation [22] | Disease-Causing Mutation [46] |
| Prediction or Score | ||
|---|---|---|
| In Silico Prediction Tool | c.247T>C; p.Phe83Leu | c.560G>A; p.Gly187Glu |
| PolyPhen2 | Possibly damaging | Probably damaging |
| SIFT | Damaging | Damaging |
| DANN (max. damaging score 1) | 0.9955 | 0.9979 |
| PROVEAN | Damaging | Damaging |
| LRT | Deleterious | Deleterious |
| MutationTaster | Disease-causing | Disease-causing |
| MutationAssessor | Medium impact | Medium impact |
| FATHMM [38] | Damaging | Damaging |
| FATHMM-MKL | Damaging | Damaging |
| M-CAP | Damaging | Damaging |
| CADD (>20 more likely the SNV is damaging) | 24.9 | 32 |
| MetaSVM | Damaging | Damaging |
| MetaLR | Damaging | Damaging |
| PhyloP 20way (max. conservation score 1.199000) | 1.199000 | 0.807000 |
| PhyloP 100way (max. conservation score 10.003000) | 8.735000 | 7.461000 |
| GERP RS (max. conservation score 6.17) | 5.03 | 5.54 |
| Family | Patient | Diagnosis | Age of Onset | Current Age | Visual Acuity (OD;OE) | Other Findings |
|---|---|---|---|---|---|---|
| A | II:1 | LCA | since birth | 27 years | 20/1600; 20/800 | Nystagmus, Light-seeking behavior |
| II:4 | LCA | since birth | 20 years | 20/200; 20/200 | Nystagmus, Light-seeking behavior | |
| B | II:1 | LCA | before 1 year | 37 years | 20/800; 20/1600 | Nystagmus |
| C | II:1 | LCA | before 1 year | 49 years | Light Perception | Nystagmus, Optic nerve drusen |
| D | II:1 | LCA | before 1 year | 13 years | N/A | Nystagmus |
| E | II:2 | EORD | N/A | 39 years | 20/400; 20/400 | Nystagmus |
| II:4 | EORD | N/A | 34 years | N/A | N/A | |
| F | II:1 | LCA | since birth | 35 years | 20/60; 20/60 | No nystagmus |
| II:8 | LCA | since birth | 37 years | 20/400; 20/200 | No nystagmus | |
| G | II:7 | EORD | N/A | 54 years | 20/800; 20/500 | N/A |
| II:8 | EORD | 7 years | 51 years | Hand Movement | N/A | |
| II:23 | EORD | N/A | 71 years | 20/500; 20/500 | N/A |
| Nucleotide Change * | Consequence | Brazilian IRD Patients | |||
|---|---|---|---|---|---|
| Allele Count | Allele Number | Allele Frequency | Homozygotes | ||
| c.247T>C | p.Phe83Leu | 8 | 1026 | 0.0078 | 4 |
| c.560G>A | p.Gly187Glu | 6 | 1026 | 0.0058 | 3 |
| Genotype | LCA/EORD (n = 93) | Other IRD (n = 420) | OR * (97.5%CI) † |
|---|---|---|---|
| c.247T>C;p.Phe83Leu Homozygous | 4 | 0 | 42.285 (1.482–206.268) |
| c.247T>C;p.Phe83Leu Non-Homozygous | 89 | 420 | |
| c.560G>A;p.Gly187Glu Homozygous | 3 | 0 | 32.525 (1.088–972.642) |
| c.560G>A;p.Gly187Glu Non-Homozygous | 90 | 420 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motta, F.L.; Martin, R.P.; Porto, F.B.O.; Wohler, E.S.; Resende, R.G.; Gomes, C.P.; Pesquero, J.B.; Sallum, J.M.F. Pathogenicity Reclassification of RPE65 Missense Variants Related to Leber Congenital Amaurosis and Early-Onset Retinal Dystrophy. Genes 2020, 11, 24. https://doi.org/10.3390/genes11010024
Motta FL, Martin RP, Porto FBO, Wohler ES, Resende RG, Gomes CP, Pesquero JB, Sallum JMF. Pathogenicity Reclassification of RPE65 Missense Variants Related to Leber Congenital Amaurosis and Early-Onset Retinal Dystrophy. Genes. 2020; 11(1):24. https://doi.org/10.3390/genes11010024
Chicago/Turabian StyleMotta, Fabiana L., Renan P. Martin, Fernanda B. O. Porto, Elizabeth S. Wohler, Rosane G. Resende, Caio P. Gomes, João B. Pesquero, and Juliana M. F. Sallum. 2020. "Pathogenicity Reclassification of RPE65 Missense Variants Related to Leber Congenital Amaurosis and Early-Onset Retinal Dystrophy" Genes 11, no. 1: 24. https://doi.org/10.3390/genes11010024