A Genome-Wide Association Study Revealed Key SNPs/Genes Associated With Salinity Stress Tolerance In Upland Cotton


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Germplasm Collection
2.2. Optimization of Salt Stress Concentration
2.3. Screening for Salt Tolerance
2.4. Statistical Analysis
2.5. Factor Analysis
2.6. GWAS and SNPs Annotation
2.7. q-RT PCR of Candidate Genes
3. Results
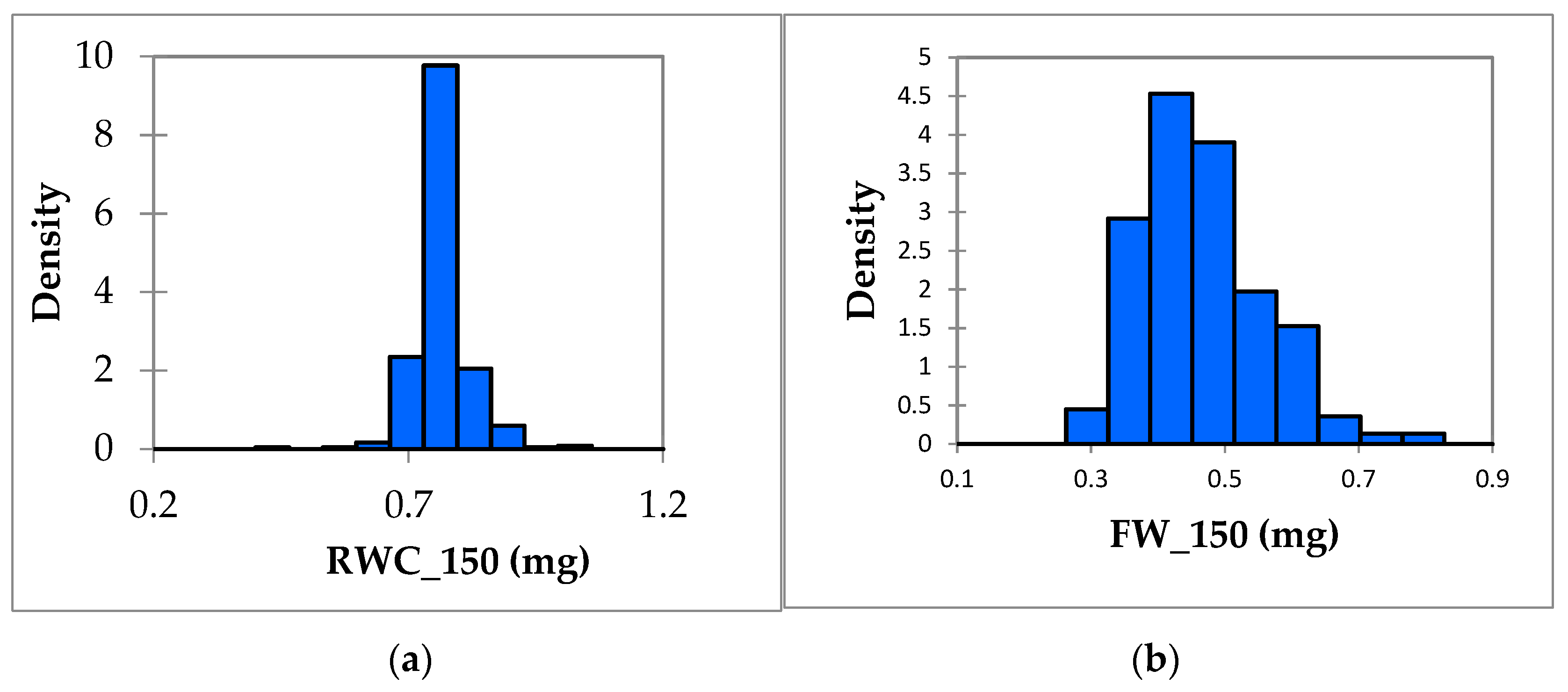
3.1. Phenotypic Variation in Salt Tolerance Traits
3.2. Evaluation and Stratification of Salt Tolerance
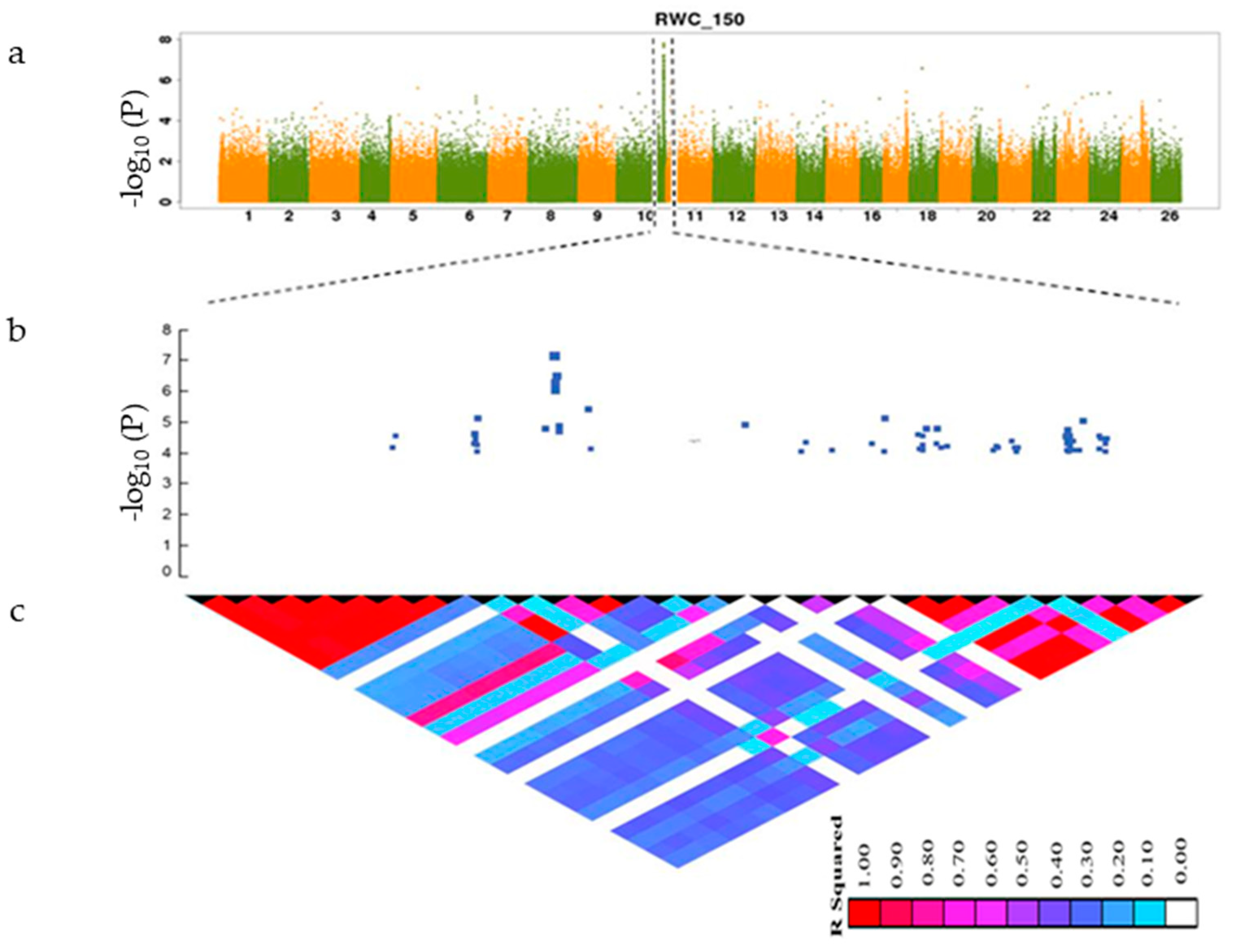
3.3. GWAS Analysis of Salt Tolerance-Related Traits
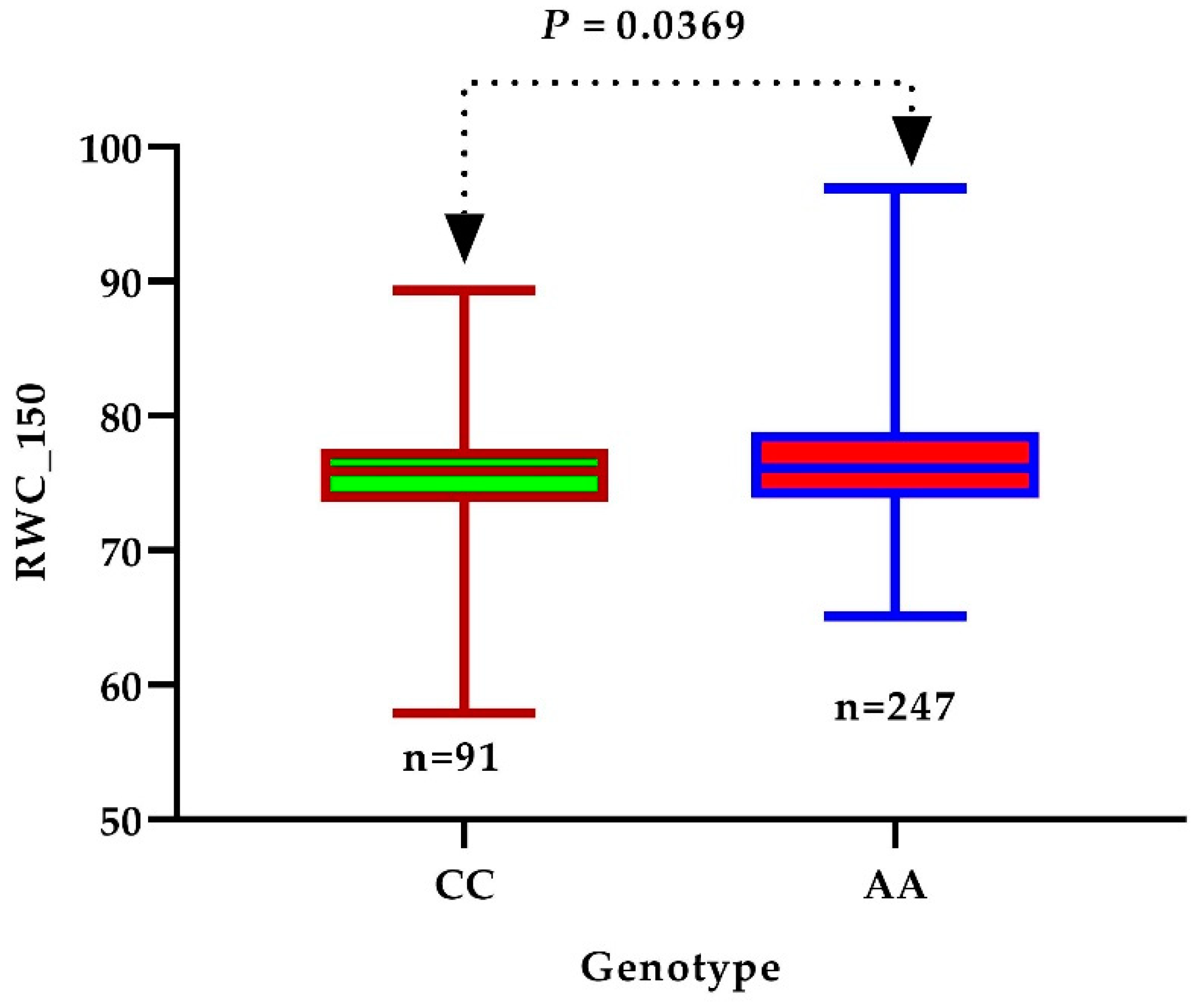
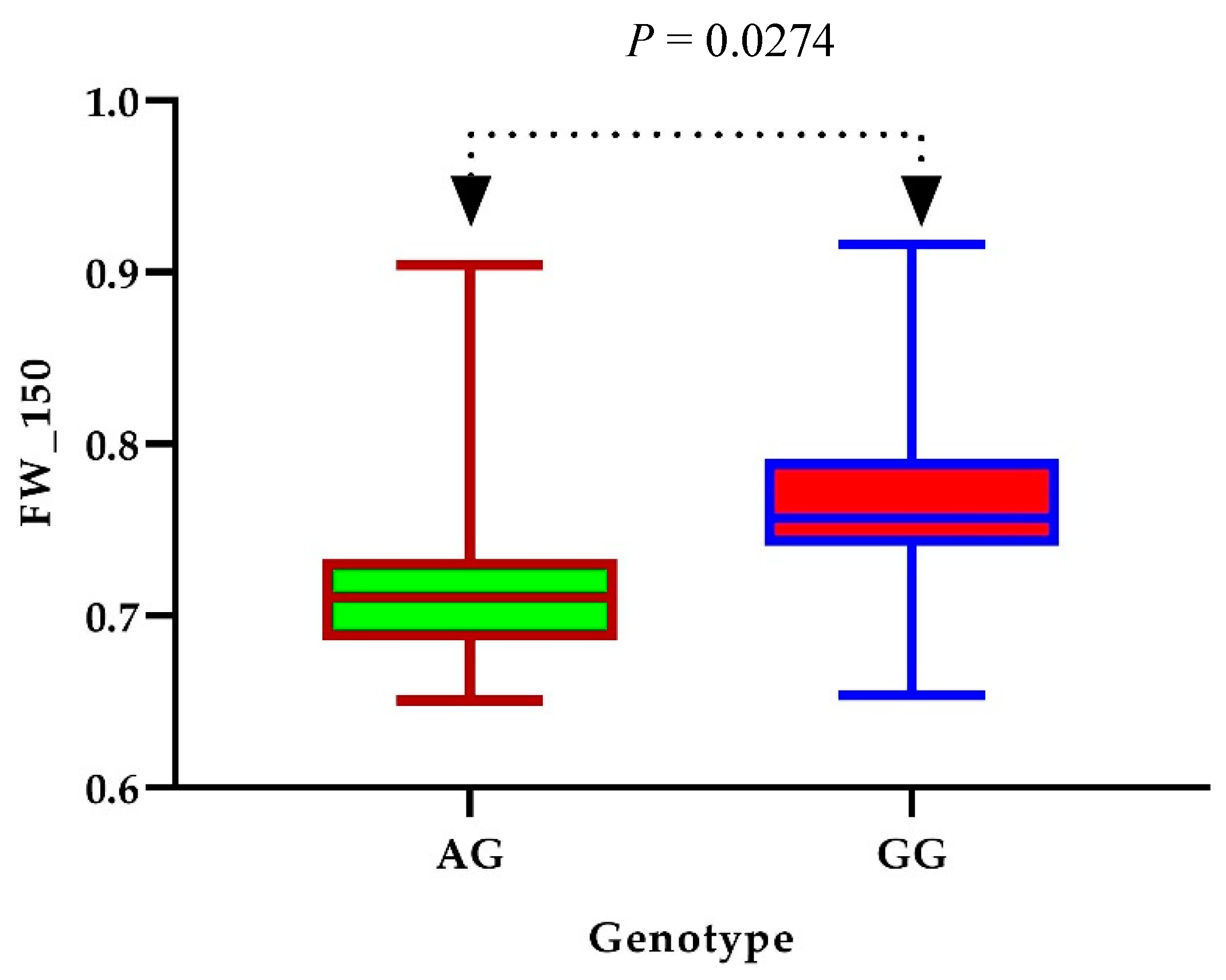
3.4. Identification of Candidate Genes
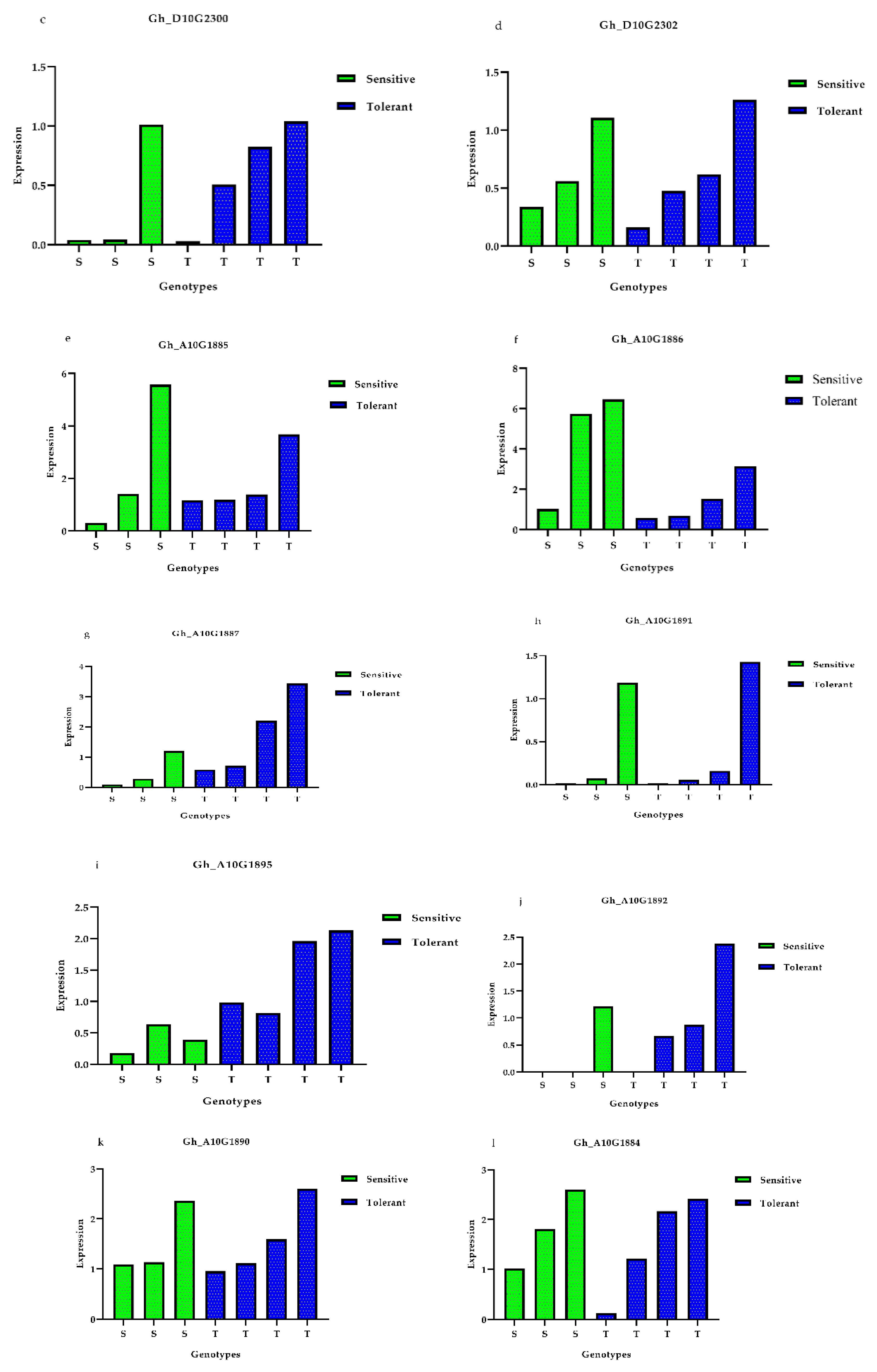
3.5. Expression Profiles of Presumed Candidate Genes via qRT-PCR Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, H.; Han, B.; Wang, T.; Chen, S.; Li, H.; Zhang, Y.; Dai, S. Mechanisms of plant salt response: Insights from proteomics. J. Proteome Res. 2011, 11, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.M.; Oosterhuis, D.; Heitholt, J.J.; Mauney, J.R. (Eds.) Cotton Physiology; The Cotton Foundation: Memphis, TN, USA, 1986. [Google Scholar]
- Wang, J.; Huang, X.; Zhong, T.; Chen, Z. Review on Sustainable Utilization of Salt-affected Land. Acta Geogr. Sin. 2011, 66, 673–684. [Google Scholar] [CrossRef]
- Zhu, J.-K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [PubMed]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef]
- Goossens, R.; Van Ranst, E. The use of remote sensing to map gypsiferous soils in the Ismailia Province (Egypt). Geoderma 1998, 87, 47–56. [Google Scholar] [CrossRef]
- Gilland, B.J. World population and food supply: Can food production keep pace with population growth in the next half-century? Food Policy 2002, 27, 47–63. [Google Scholar] [CrossRef]
- Brachi, B.; Morris, G.P.; Borevitz, J.O. Genome-wide association studies in plants: The missing heritability is in the field. Genome Biol. 2011, 12, 232. [Google Scholar] [CrossRef]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef]
- Borevitz, J.O.; Nordborg, M. The impact of genomics on the study of natural variation in Arabidopsis. Plant Physiol. 2003, 132, 718–725. [Google Scholar] [CrossRef]
- Ogura, T.; Busch, W. From phenotypes to causal sequences: Using genome wide association studies to dissect the sequence basis for variation of plant development. Curr. Opin. Plant Biol. 2015, 23, 98–108. [Google Scholar] [CrossRef]
- Pantaliao, G.F.; Narciso, M.; Guimares, C.; Castro, A.; Colombari, J.M.; Breseghello, F.; Rodrigues, L.; Vianello, R.P.; Borba, T.O.; Brondani, C. Genome wide association study (GWAS) for grain yield in rice cultivated under water deficit. Genetica 2016, 144, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Dossa, K.; Zhang, Y.; Wei, X.; Wang, L.; Zhang, Y.; Liu, A.; Zhou, R.; Zhang, X. GWAS Uncovers Differential Genetic Bases for Drought and Salt Tolerances in Sesame at the Germination Stage. Genes 2018, 9, 87. [Google Scholar] [CrossRef] [PubMed]
- Chao, D.-Y.; Silva, A.; Baxter, I.; Huang, Y.S.; Nordborg, M.; Danku, J.; Lahner, B.; Yakubova, E.; Salt, D.E. Genome-wide association studies identify heavy metal ATPase3 as the primary determinant of natural variation in leaf cadmium in Arabidopsis thaliana. PLoS Genet. 2012, 8, e1002923. [Google Scholar] [CrossRef]
- Fang, L.; Wang, Q.; Hu, Y.; Jia, Y.; Chen, J.; Liu, B.; Zhang, Z.; Guan, X.; Chen, S.; Zhou, B.; et al. Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat. Genet. 2017, 49, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Ma, X.; Li, N.; Zhou, L.; Liu, Z.; Han, H.; Gui, Y.; Bao, Y.; Chen, J.; Dai, X. Genome-wide association study discovered candidate genes of Verticillium wilt resistance in upland cotton (Gossypium hirsutum L.). Plant Biotechnol. J. 2017, 15, 1520–1532. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, X.; Liu, Z.; Gu, Q.; Zhang, Y.; Li, Z.; Ke, H.; Yang, J.; Wu, J.; Wu, L.; et al. Genome-wide association study discovered genetic variation and candidate genes of fibre quality traits in Gossypium hirsutum L. Plant Biotechnol. J. 2017, 15, 982–996. [Google Scholar] [CrossRef]
- Sun, H.; Meng, M.; Yan, Z.; Lin, Z.; Nie, X.; Yang, X. Genome-wide association mapping of stress-tolerance traits in cotton. Crop J. 2019, 7, 77–88. [Google Scholar] [CrossRef]
- Cai, C.; Zhu, G.; Zhang, T.; Guo, W. High-density 80 K SNP array is a powerful tool for genotyping G. hirsutum accessions and genome analysis. BMC Genom. 2017, 18, 654. [Google Scholar] [CrossRef]
- Diouf, L.; Pan, Z.; He, S.-P.; Gong, W.-F.; Jia, Y.H.; Magwanga, R.O.; Romy, K.R.E.; Rashid, H.O.; Kirungu, J.N.; Du, X. High-Density Linkage Map Construction and Mapping of Salt-Tolerant QTLs at Seedling Stage in Upland Cotton Using Genotyping by Sequencing (GBS). Int. J. Mol. Sci. 2017, 18, 2622. [Google Scholar] [CrossRef]
- Sun, Z.; Li, H.; Zhang, Y.; Li, Z.; Ke, H.; Wu, L.; Zhang, G.; Wang, X.; Ma, Z. Identification of SNPs and Candidate Genes Associated With Salt Tolerance at the Seedling Stage in Cotton (Gossypium hirsutum L.). Front. Plant Sci. 2018, 9, 1011. [Google Scholar] [CrossRef]
- Lacape, J.-M.; Nguyen, T.-B.; Courtois, B.; Belot, J.-L.; Giband, M.; Gourlot, J.-P.; Gawryziak, G.; Roques, S.; Hau, B. QTL analysis of cotton fiber quality using multiple Gossypium hirsutum × Gossypium barbadense backcross generations. Crop Sci. 2005, 45, 123–140. [Google Scholar] [CrossRef]
- Chartzoulakis, K.; Klapaki, G. Response of two greenhouse pepper hybrids to NaCl salinity during different growth stages. Sci. Hortic. 2000, 86, 247–260. [Google Scholar] [CrossRef]
- Sexton, P.; Gerard, C. Emergence Force of Cotton Seedlings as Influenced by Salinity 1. Agron. J. 1982, 74, 699–702. [Google Scholar] [CrossRef]
- Ma, Z.; He, S.; Wang, X.; Sun, J.; Zhang, Y.; Zhang, G.; Wu, L.; Li, Z.; Liu, Z.; Sun, G.; et al. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 2018, 50, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Maqbool, M.A.; Aslam, M.; Ali, H. Breeding for improved drought tolerance in Chickpea (Cicer arietinum L.). Plant Breed. 2017, 136, 300–318. [Google Scholar] [CrossRef]
- Kaashyap, M.; Ford, R.; Kudapa, H.; Jain, M.; Edwards, D.; Varshney, R.; Mantri, N. Differential regulation of genes involved in root morphogenesis and cell wall modification is associated with salinity tolerance in chickpea. Sci. Rep. 2018, 8, 4855. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Com—Putting. R Core Team: Vienna, Austria, 2017. [Google Scholar]
- Wei, T.; Simko, V. R Package “Corrplot”: Visualization of a Correlation Matrix. github. 2017. [Google Scholar]
- Cerny, B.A.; Kaiser, H.F. A Study Of A Measure Of Sampling Adequacy for Factor-Analytic Correlation. Multivar. Behav. Res. 1977, 12, 43–47. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 2012, 44, 1098–1103. [Google Scholar] [CrossRef]
- Wang, M.; Tu, L.; Lin, M.; Lin, Z.; Wang, P.; Yang, Q.; Ye, Z.; Shen, C.; Li, J.; Zhang, L. Asymmetric subgenome selection and cis-regulatory divergence during cotton domestication. Nat. Genet. 2017, 49, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Said, J.I.; Lin, Z.; Zhang, X.; Song, M.; Zhang, J. A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genom. 2013, 14, 776. [Google Scholar] [CrossRef]
- Yano, K.; Yamamoto, E.; Aya, K.; Takeuchi, H.; Lo, P.C.; Hu, L.; Yamasaki, M.; Yoshida, S.; Kitano, H.; Hirano, K.; et al. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat Genet 2016, 48, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Flowers, T.J. Improving crop salt tolerance. J. Exp. Bot. 2004, 55, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Mackay, T.F.; Stone, E.A.; Ayroles, J.F. The genetics of quantitative traits: Challenges and prospects. Nat. Rev. Genet. 2009, 10, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Turner, M.K.; Chao, S.; Kolmer, J.; Anderson, J.A. Genome Wide Association Study of Seedling and Adult Plant Leaf Rust Resistance in Elite Spring Wheat Breeding Lines. PLoS ONE 2016, 11, e0148671. [Google Scholar] [CrossRef] [PubMed]
- Atwell, S.; Huang, Y.S.; Vilhjalmsson, B.J.; Willems, G.; Horton, M.; Li, Y.; Meng, D.; Platt, A.; Tarone, A.M.; Hu, T.T.; et al. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 2010, 465, 627–631. [Google Scholar] [CrossRef]
- Su, J.; Fan, S.; Li, L.; Wei, H.; Wang, C.; Wang, H.; Song, M.; Zhang, C.; Gu, L.; Zhao, S.; et al. Detection of Favorable QTL Alleles and Candidate Genes for Lint Percentage by GWAS in Chinese Upland Cotton. Front. Plant Sci. 2016, 7, 1576. [Google Scholar] [CrossRef]
- Jia, Y.-H.; Sun, J.-L.; Wang, X.-W.; Zhou, Z.-L.; Pan, Z.-E.; He, S.-P.; Pang, B.-Y.; Wang, L.-R.; Du, X.-M. Molecular Diversity and Association Analysis of Drought and Salt Tolerance in Gossypium hirsutum L. Germplasm. J. Integr. Agric. 2014, 13, 1845–1853. [Google Scholar] [CrossRef]
- Garcia-Hernandez, M.; Berardini, T.; Chen, G.; Crist, D.; Doyle, A.; Huala, E.; Knee, E.; Lambrecht, M.; Miller, N.; Mueller, L.A.; et al. TAIR: A resource for integrated Arabidopsis data. Funct. Integr. Genom. 2002, 2, 239–253. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X.-M.; Zhou, L.; He, Y.; Wang, D.; Qi, Y.-H.; Jiang, D.-A. Rubisco Activase Is Also a Multiple Responder to Abiotic Stresses in Rice. PLoS ONE 2015, 10, e0140934. [Google Scholar] [CrossRef]
- Bi, H.; Liu, P.; Jiang, Z.; Ai, X. Overexpression of the rubisco activase gene improves growth and low temperature and weak light tolerance in Cucumis sativus L. Physiol. Plant. 2017, 161, 224–234. [Google Scholar] [CrossRef]
- Robin, A.H.K.; Matthew, C.; Uddin, M.J.; Bayazid, K.N. Salinity-induced reduction in root surface area and changes in major root and shoot traits at the phytomer level in wheat. J. Exp. Bot. 2016, 67, 3719–3729. [Google Scholar] [CrossRef] [PubMed]
- Cao, J. Analysis of the Prefoldin Gene Family in 14 Plant Species. Front. Plant Sci. 2016, 7, 317. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Milla, M.A.; Salinas, J. Prefoldins 3 and 5 Play an Essential Role in Arabidopsis Tolerance to Salt Stress. Mol. Plant 2009, 2, 526–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Hao, L.; Pan, J.; Zheng, X.; Jiang, G.; Jin, Y.; Gu, Z.; Qian, Q.; Zhai, W.; Ma, B. SPL5, a cell death and defense-related gene, encodes a putative splicing factor 3b subunit 3 (SF3b3) in rice. Mol. Breed. 2012, 30, 939–949. [Google Scholar] [CrossRef]
- Yang, X.; Liang, Z.; Wen, X.; Lu, C. Genetic engineering of the biosynthesis of glycinebetaine leads to increased tolerance of photosynthesis to salt stress in transgenic tobacco plants. Plant Mol. Biol. 2008, 66, 73. [Google Scholar] [CrossRef]
- Tahara, M.; Carver, B.F.; Johnson, R.C.; Smith, E.L. Relationship between relative water content during reproductive development and winter wheat grain yield. Euphytica 1990, 49, 255–262. [Google Scholar]
- Xu, J.; Xing, X.-J.; Tian, Y.-S.; Peng, R.-H.; Xue, Y.; Zhao, W.; Yao, Q.-H. Transgenic Arabidopsis Plants Expressing Tomato Glutathione S-Transferase Showed Enhanced Resistance to Salt and Drought Stress. PLoS ONE 2015, 10, e0136960. [Google Scholar] [CrossRef]
- Kumar, S.; Asif, M.H.; Chakrabarty, D.; Tripathi, R.D.; Dubey, R.S.; Trivedi, P.K. Expression of a rice Lambda class of glutathione S-transferase, OsGSTL2, in Arabidopsis provides tolerance to heavy metal and other abiotic stresses. J. Hazard. Mater. 2013, 248, 228–237. [Google Scholar] [CrossRef]
- Chan, K.X.; Wirtz, M.; Phua, S.Y.; Estavillo, G.M.; Pogson, B.J. Balancing metabolites in drought: The sulfur assimilation conundrum. Trends Plant Sci. 2013, 18, 18–29. [Google Scholar] [CrossRef]
- Ernst, L.; Goodger, J.Q.; Alvarez, S.; Marsh, E.L.; Berla, B.; Lockhart, E.; Jung, J.; Li, P.; Bohnert, H.J.; Schachtman, D.P. Sulphate as a xylem-borne chemical signal precedes the expression of ABA biosynthetic genes in maize roots. J. Exp. Bot. 2010, 61, 3395–3405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallardo, K.; Courty, P.-E.; Le Signor, C.; Wipf, D.; Vernoud, V. Sulfate transporters in the plant’s response to drought and salinity: Regulation and possible functions. Front. Plant Sci. 2014, 5, 580. [Google Scholar] [CrossRef] [PubMed]
- Hyung, D.; Lee, C.; Kim, J.-H.; Yoo, D.; Seo, Y.-S.; Jeong, S.-C.; Lee, J.-H.; Chung, Y.; Jung, K.-H.; Cook, D.R. Cross-family translational genomics of abiotic stress-responsive genes between Arabidopsis and Medicago truncatula. PLoS ONE 2014, 9, e91721. [Google Scholar] [CrossRef] [PubMed]
- Schutzendubel, A.; Polle, A. Plant responses to abiotic stresses: Heavy metal-induced oxidative stress and protection by mycorrhization. J. Exp. Bot. 2002, 53, 1351–1365. [Google Scholar] [CrossRef]
- Flora, S.J. Structural, chemical and biological aspects of antioxidants for strategies against metal and metalloid exposure. Oxidative Med. Cell. Longev. 2009, 2, 191–206. [Google Scholar] [CrossRef]
- Gustin, J.L.; Zanis, M.J.; Salt, D.E. Structure and evolution of the plant cation diffusion facilitator family of ion transporters. BMC Evol. Biol. 2011, 11, 76. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Minimum | Maximum | Mean | Std. Deviation | CV% | Skewness (Pearson) | Kurtosis (Pearson) |
|---|---|---|---|---|---|---|---|
| GR_0 | 22 | 100 | 80.676 | 15.48 | 19.187 | –1.746 | 4.404 |
| REC_0 | 0.002 | 0.87 | 0.476 | 0.102 | 21.428 | –0.509 | 4.814 |
| MDA_0 | 0.001 | 0.093 | 0.033 | 0.012 | 36.363 | 0.913 | 3.028 |
| FW_0 | 0.369 | 1.07 | 0.577 | 0.087 | 15.077 | 0.765 | 2.245 |
| SL_0 | 4.84 | 12.667 | 7.559 | 1.068 | 14.128 | 0.417 | 1.018 |
| RWC_0 | 0.561 | 1.06 | 0.91 | 0.048 | 5.2747 | –1.913 | 13.762 |
| CHL_0 | 26.492 | 87.053 | 50.665 | 4.851 | 9.5746 | 0.535 | 9.043 |
| Variable | Minimum | Maximum | Mean | Std. Deviation | CV% | Skewness (Pearson) | Kurtosis (Pearson) |
|---|---|---|---|---|---|---|---|
| GR_150 | 13.33 | 98 | 74.209 | 16.152 | 21.765 | −1.262 | 1.083 |
| REC_150 | 0.426 | 3.057 | 1.083 | 0.319 | 29.455 | 2.291 | 9.09 |
| MDA_150 | 0.001 | 0.111 | 0.027 | 0.014 | 51.851 | 1.258 | 3.633 |
| FW_150 | 0.28 | 0.818 | 0.464 | 0.092 | 19.827 | 0.773 | 0.567 |
| SL_150 | 2.487 | 8.91 | 5.801 | 1.134 | 19.548 | 0.211 | −0.43 |
| RWC_150 | 0.434 | 1.054 | 0.766 | 0.055 | 7.180 | 0.51 | 7.185 |
| CHL_150 | 25.74 | 60.933 | 48.348 | 5.571 | 11.522 | −0.572 | 0.724 |
| Trait | Downstream | Exonic | Intergenic | Intronic | Splicing | Upstream | Upstream; Downstream | Total |
|---|---|---|---|---|---|---|---|---|
| CHL_0 | 18 | 34 | 115 | 39 | 37 | 9 | 252 | |
| CHL_150 | 13 | 11 | 228 | 9 | 21 | 4 | 286 | |
| CHL_150_0 | 8 | 4 | 124 | 6 | 2 | 3 | 147 | |
| FW_0 | 534 | 221 | 5179 | 1607 | 1 | 507 | 117 | 8166 |
| FW_150 | 15 | 17 | 140 | 28 | 53 | 14 | 267 | |
| FW_150_0 | 8 | 5 | 184 | 13 | 11 | 4 | 225 | |
| GR_0 | 10 | 18 | 253 | 13 | 9 | 7 | 310 | |
| GR_150 | 7 | 125 | 1 | 3 | 1 | 137 | ||
| GR_150_0 | 2 | 1 | 74 | 6 | 8 | 1 | 92 | |
| MDA_0 | 23 | 4 | 199 | 17 | 16 | 8 | 267 | |
| MDA_150 | 14 | 11 | 210 | 12 | 31 | 5 | 283 | |
| MDA_150_0 | 11 | 5 | 122 | 6 | 12 | 4 | 160 | |
| REC_0 | 31 | 23 | 332 | 41 | 30 | 17 | 474 | |
| REC_150 | 50 | 41 | 259 | 32 | 45 | 18 | 445 | |
| REC_150_0 | 268 | 140 | 2294 | 319 | 1 | 343 | 78 | 3443 |
| RWC_0 | 33 | 9 | 568 | 33 | 39 | 8 | 690 | |
| RWC_150 | 30 | 8 | 217 | 30 | 24 | 12 | 321 | |
| RWC_150_0 | 17 | 6 | 368 | 11 | 15 | 10 | 427 | |
| SL_0 | 10 | 4 | 125 | 13 | 1 | 8 | 1 | 162 |
| SL_150 | 11 | 10 | 103 | 15 | 21 | 14 | 174 | |
| SL_150_0 | 28 | 16 | 412 | 20 | 48 | 12 | 536 | |
| Grand Total | 1141 | 588 | 11,631 | 2271 | 3 | 1283 | 347 | 17,264 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasir, M.; He, S.; Sun, G.; Geng, X.; Pan, Z.; Gong, W.; Jia, Y.; Du, X. A Genome-Wide Association Study Revealed Key SNPs/Genes Associated With Salinity Stress Tolerance In Upland Cotton. Genes 2019, 10, 829. https://doi.org/10.3390/genes10100829
Yasir M, He S, Sun G, Geng X, Pan Z, Gong W, Jia Y, Du X. A Genome-Wide Association Study Revealed Key SNPs/Genes Associated With Salinity Stress Tolerance In Upland Cotton. Genes. 2019; 10(10):829. https://doi.org/10.3390/genes10100829
Chicago/Turabian StyleYasir, Muhammad, Shoupu He, Gaofei Sun, Xiaoli Geng, Zhaoe Pan, Wenfang Gong, Yinhua Jia, and Xiongming Du. 2019. "A Genome-Wide Association Study Revealed Key SNPs/Genes Associated With Salinity Stress Tolerance In Upland Cotton" Genes 10, no. 10: 829. https://doi.org/10.3390/genes10100829