Identification of Two Independent COL5A1 Variants in Dogs with Ehlers–Danlos Syndrome

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics
2.2. Histopathology
2.2.1. Case 1
2.2.2. Cases 2 and 3
2.3. Transmission Electron Microscopy (TEM)
2.4. Biochemical Analysis of Dermis
2.5. DNA Extraction and Whole-Genome Sequencing
3. Results
3.1. Clinical Assessment
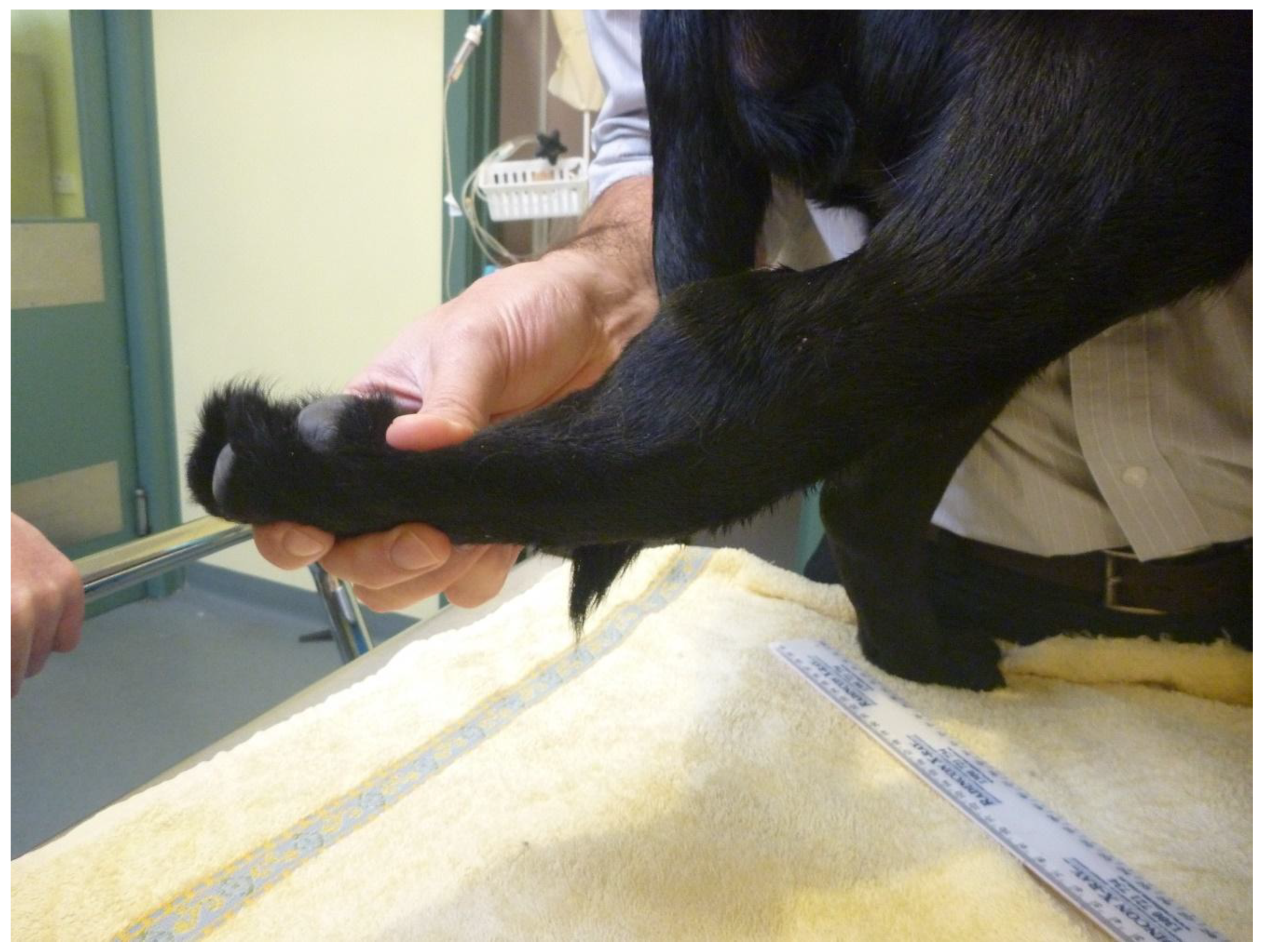
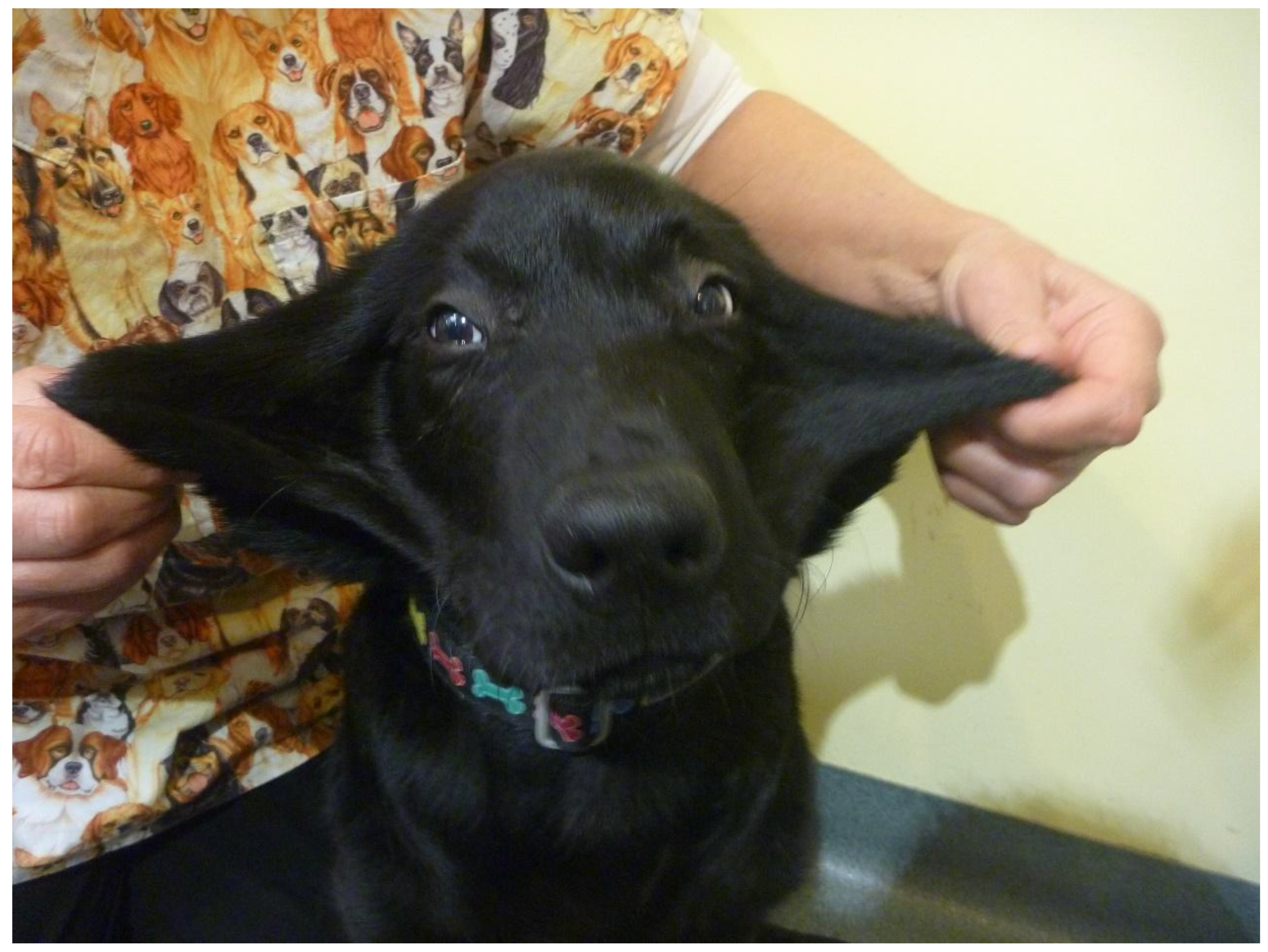
3.1.1. Case 1
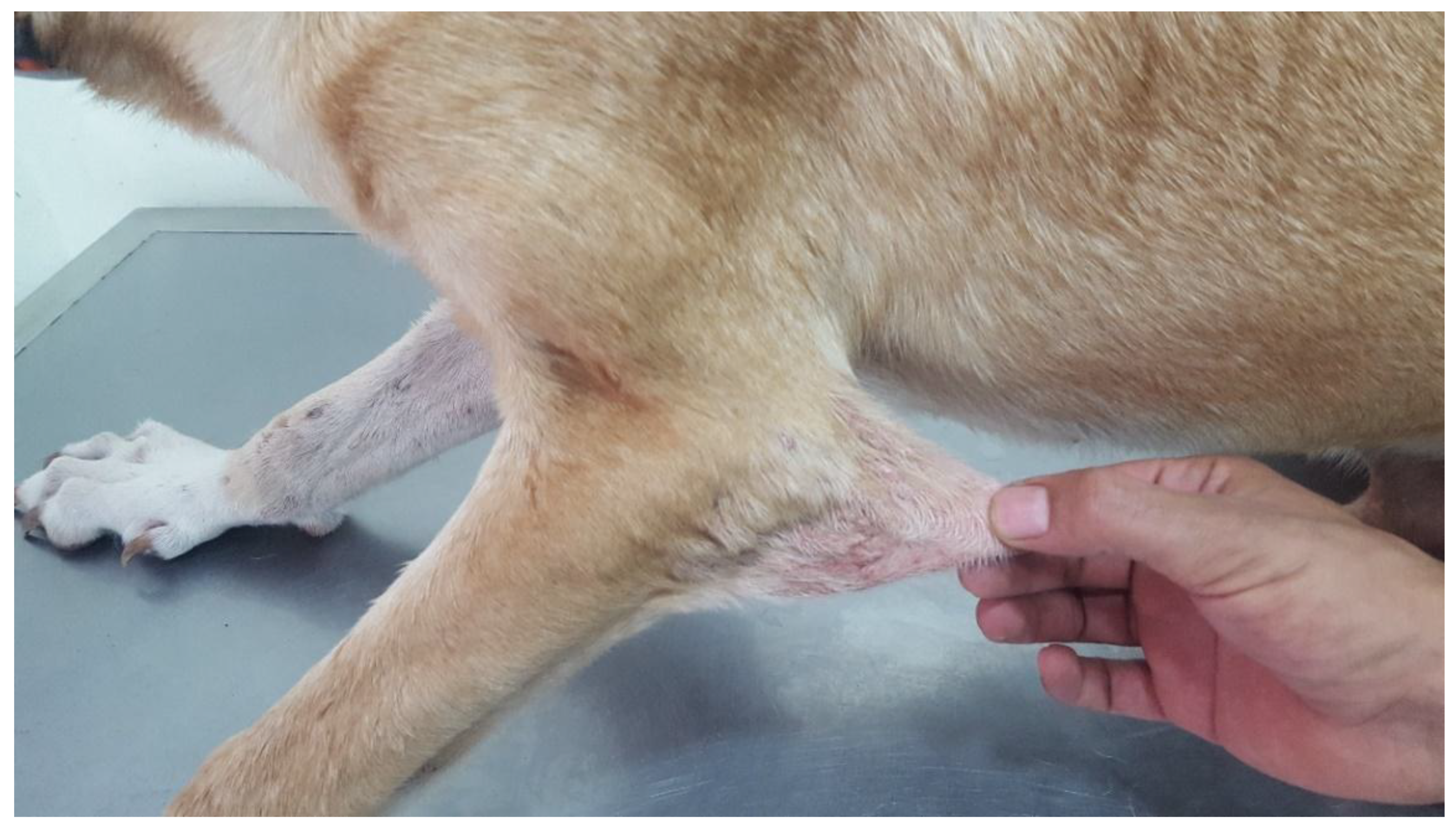
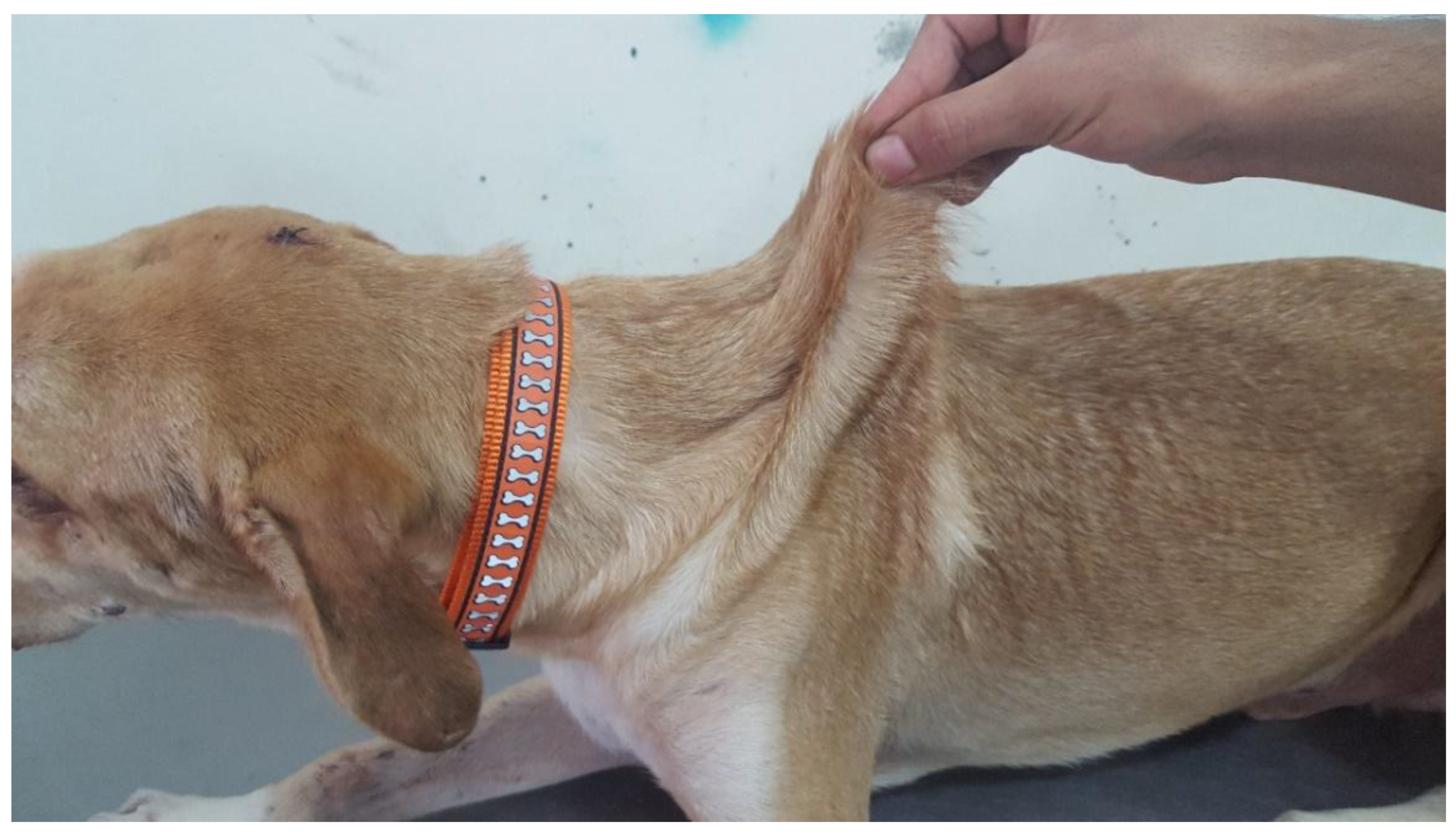
3.1.2. Cases 2 and 3
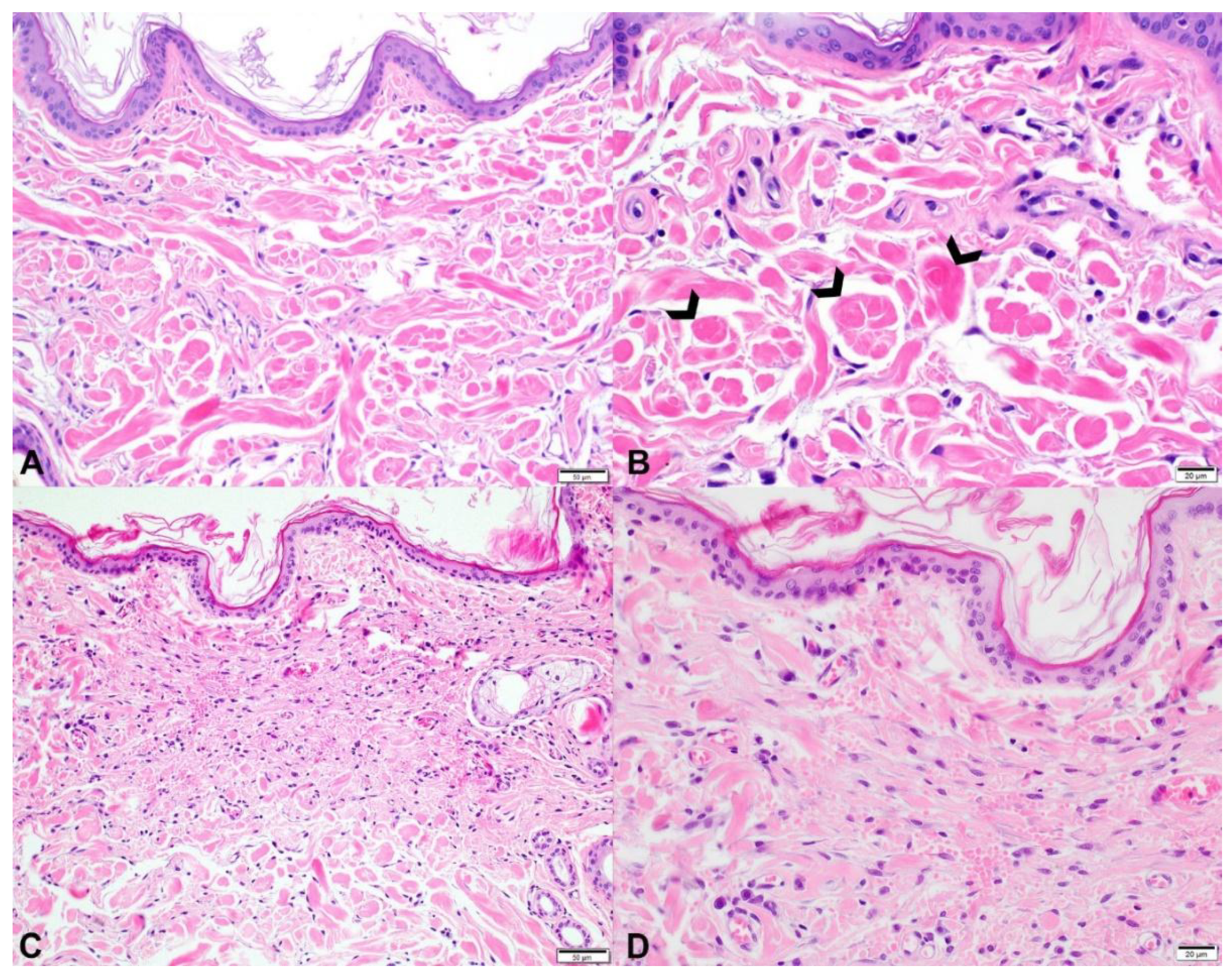
3.2. Histopathology
3.2.1. Case 1
3.2.2. Cases 2 and 3
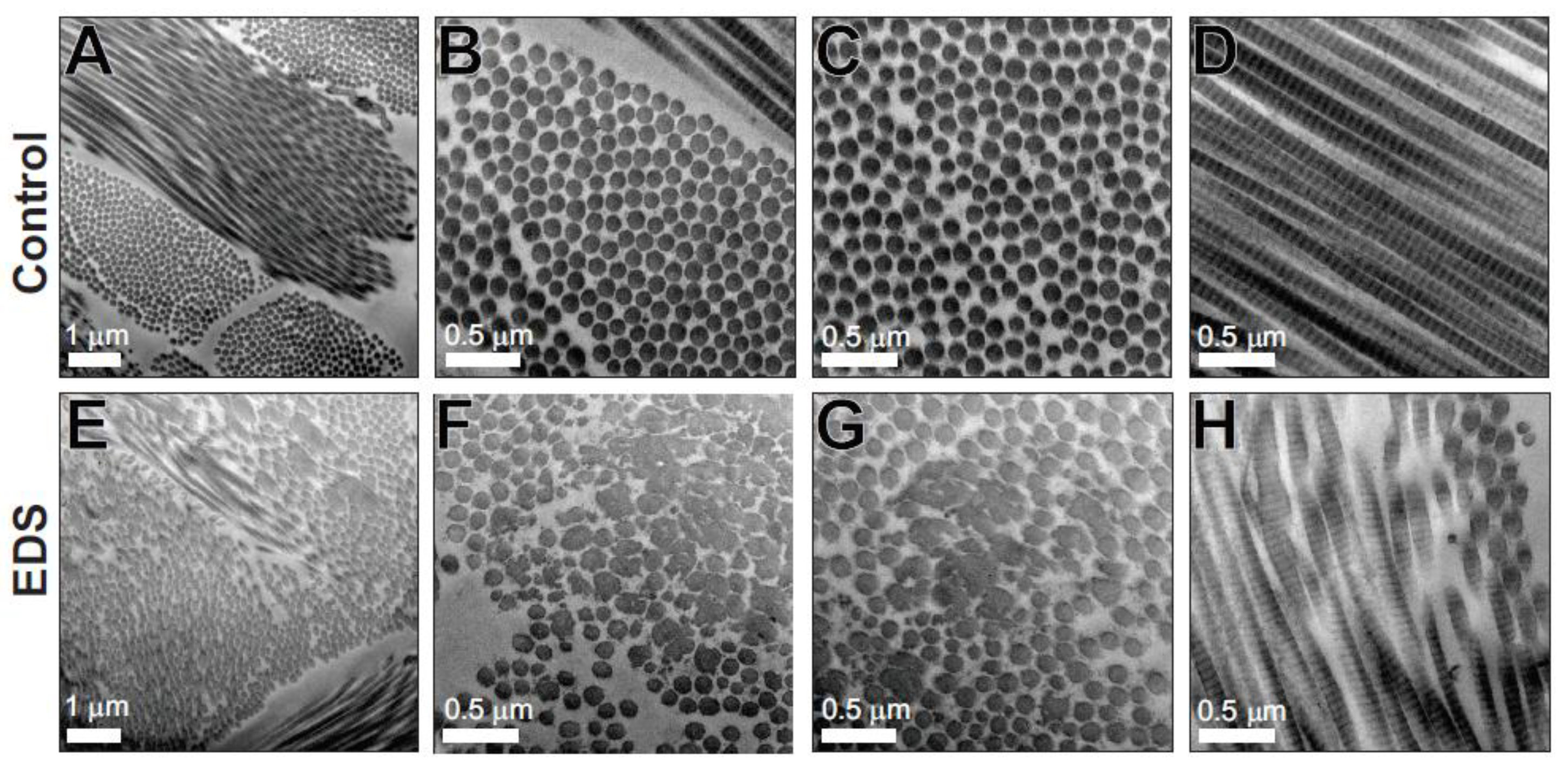
3.3. Transmission Electron Microscopy
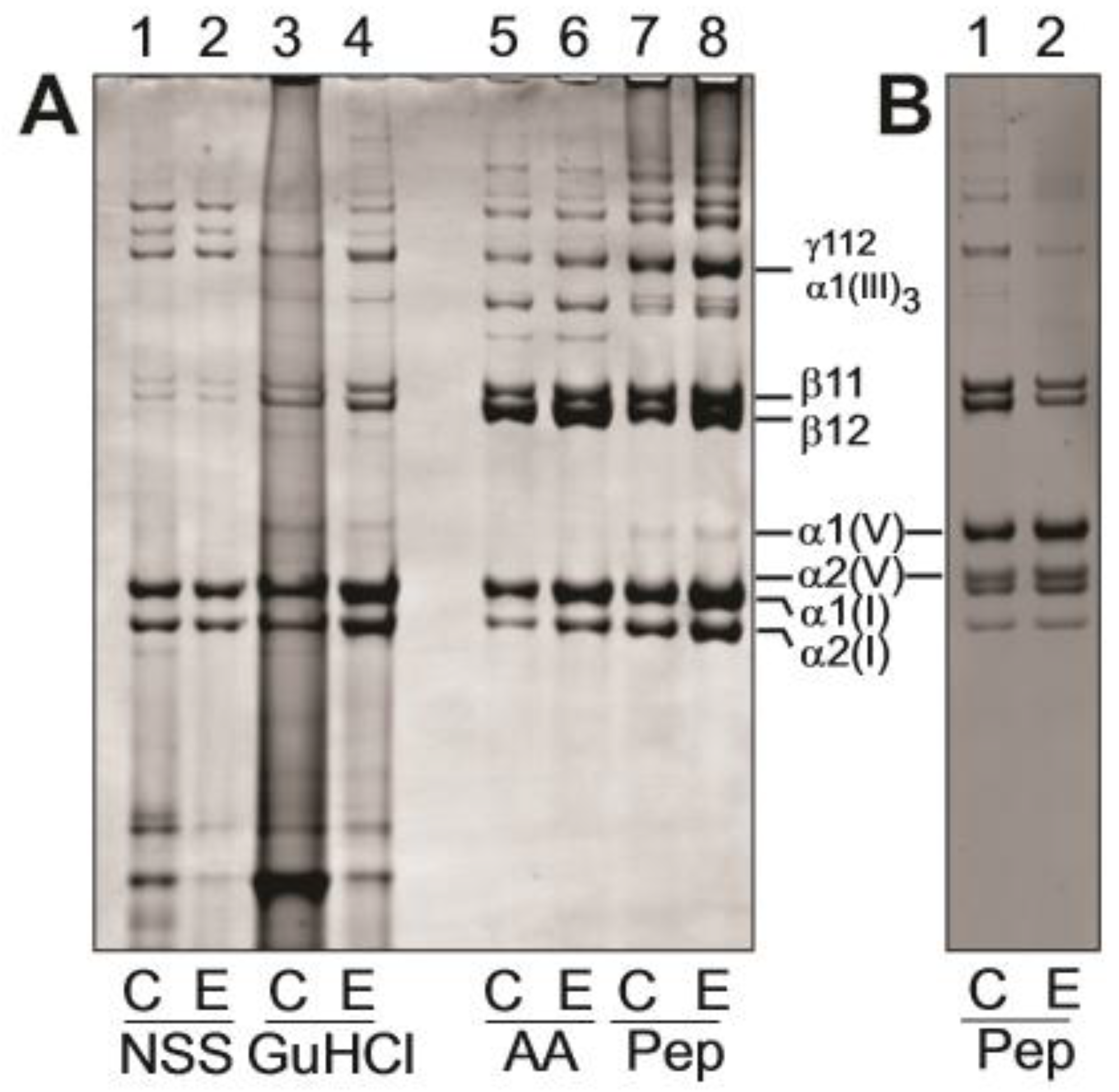
3.4. Biochemical Analysis of Dermis
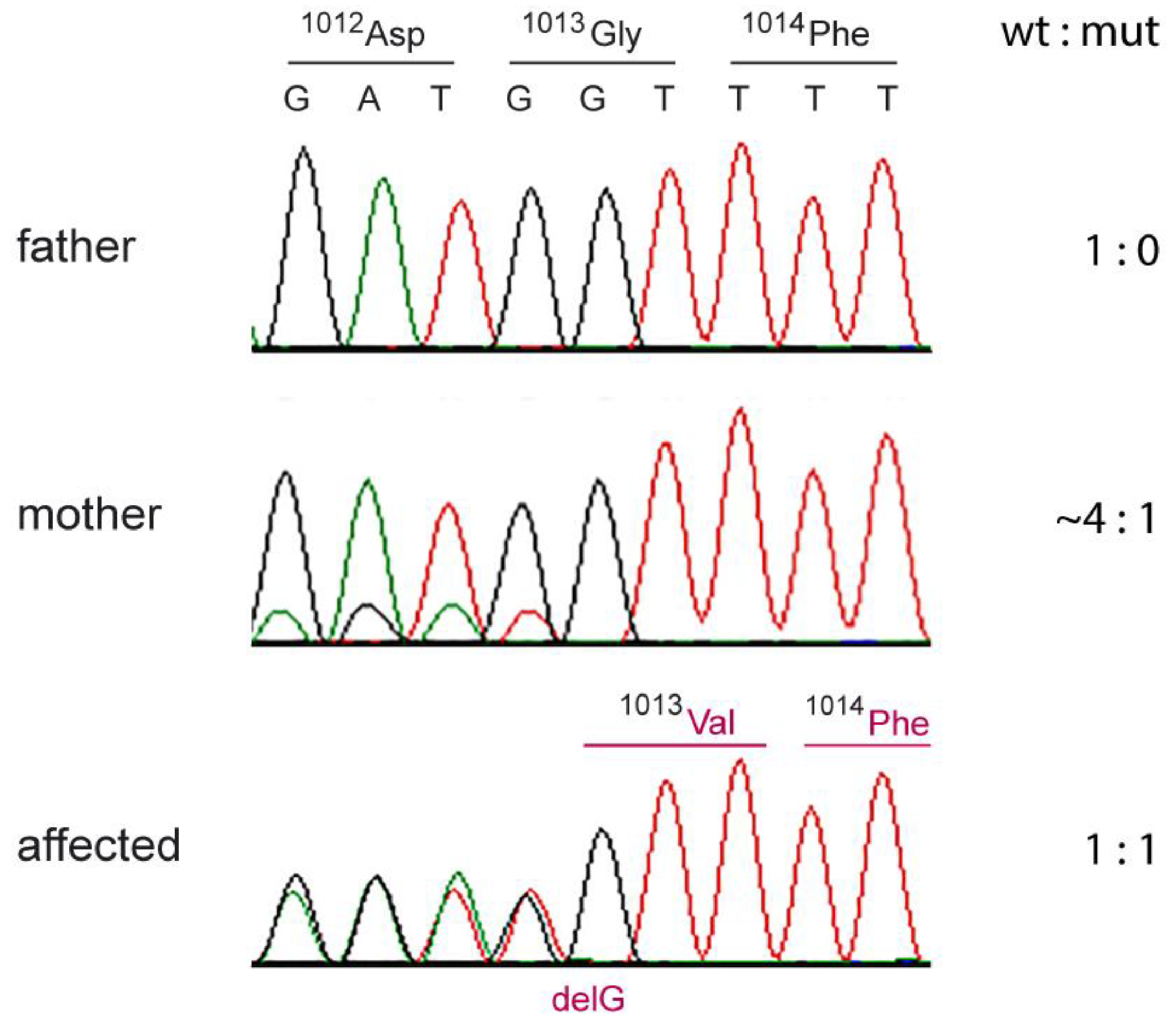
3.5. Genetic Analyses
3.5.1. Case 1
3.5.2. Cases 2 and 3
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Solomons, B. Equine cutis hyperelastica. Equine Vet. J. 1984, 16, 541–542. [Google Scholar] [CrossRef] [PubMed]
- Hegreberg, G.A. Animal model: Ehlers-Danlos syndrome in dogs and mink, canine cutaneous asthenia. Am. J. Pathol. 1975, 79, 383–386. [Google Scholar] [PubMed]
- Harvey, R.G.; Brown, P.J.; Young, R.D.; Whitbread, T.J. A connective tissue defect in two rabbits similar to the Ehlers-Danlos syndrome. Vet. Rec. 1990, 126, 130–132. [Google Scholar] [PubMed]
- Minor, R.R.; Lein, D.H.; Patterson, D.F.; Krook, L.; Porter, T.G.; Kane, A.C. Defects in collagen fibrillogenesis casuing hyperextensible, fragile skin in dogs. J. Am. Vet. Med. Assoc. 1983, 182, 142–148. [Google Scholar] [PubMed]
- Persechino, A.G.; Roperto, F.; de Capraris, D.; Troncone, A. Rilievi clinici ed istopatologici in una collagenopatia del cane riportabile alla sindrome di Ehlers-Danlos dell’uomo [Collagenopathy in a dog resembling Ehlers-Danlos syndrome of man. Clinical and histopathological features]. Acta Med. Vet. 1983, 29, 67–79. [Google Scholar]
- Papparella, S.R.; Troncone, A. Sindrome di Elhers-Dan-los nel cane. Rilievi ultrastrutturali della cute [Elhers-Danlos syndrome in a dog. Ultrastructural findings of the skin]. Acta Med. Vet. 1988, 34, 207–213. [Google Scholar]
- Rodriguez, F.; Herráez, P.; Monteros, A.E.D.L.; Calabuig, P.; Rodríguez, J.L. Collagen dysplasia in a litter of Garafiano shepherd dogs. Zent. Fur Vet. Reihe A 1996, 43, 509–512. [Google Scholar]
- Barnett, K.C.; Cottrell, B.D. Ehlers-Danlos syndrome in a dog: Ocular, cutaneous and articular abnormalities. J. Small Anim. Pract. 1987, 28, 941–946. [Google Scholar] [CrossRef]
- Paciello, O.; Lamagna, F.; Lamagna, B.; Papparella, S. Ehlers-Danlos-like syndrome in 2 dogs: Clinical, histologic, and ultrastructural findings. Vet. Clin. Pathol. 2003, 32, 13–18. [Google Scholar] [CrossRef]
- Barrera, R.; Mane, C.; Duran, E.; Vives, M.A.; Zaragoza, C. Ehlers-Danlos syndrome in a dog. Can. Vet. J. 2004, 45, 355–356. [Google Scholar]
- Bellini, M.H.; Caldini, E.T.E.G.; Scapinelli, M.P.; Simões, M.J.; Machado, D.B.; Nürmberg, R.; Simões, M. Increased elastic microfibrils and thickening of fibroblastic nuclear lamina in canine cutaneous asthenia. Vet. Dermatol. 2009, 20, 139–143. [Google Scholar] [CrossRef]
- Rasch, S.N. Surgical and medical treatment of ocular disease in a dog with Ehlers–Danlos syndrome. Clin. Case Rep. 2017, 5, 880–886. [Google Scholar] [CrossRef]
- Uri, M.; Verin, R.; Ressel, L.; Buckley, L.; McEwan, N. Ehlers–Danlos Syndrome Associated with Fatal Spontaneous Vascular Rupture in a Dog. J. Comp. Pathol. 2015, 152, 211–216. [Google Scholar] [CrossRef]
- Ueda, K.; Kawai, T.; Senoo, H.; Shimizu, A.; Ishiko, A.; Nagata, M. Histopathological and electron microscopic study in dogs with patellar luxation and skin hyperextensibility. J. Vet. Med. Sci. 2018, 80, 1309–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Counts, D.F.; Byers Holbrook, K.A.; Hegreberg, G.A. Dermatosparaxis in a Himalyan cat. J. Investig. Dermatol. 1980, 74, 96–99. [Google Scholar] [CrossRef]
- Sequeira, J.L.; Bandarra, E.P.; Figueiredo, L.M.A.; Eugenio, F.R.; Rocha, N.S. Collagen Dysplasia (Cutaneous Asthenia) in a Cat. Vet. Pathol. 1999, 36, 603–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holbrook, K.A.; Byers, P.H.; Counts, D.F.; Hegreberg, G.A. Dermatosparaxis in a Himalayan cat. II. Ultrastructural studies of dermal collagen. J. Investig. Dermatol. 1980, 74, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.W.; Miller, W.H.; Griffin, C.E. Congenital and Hereditary Diseases. In Muller and Kirk’s Small Animal Dermatology, 6th ed.; Scott, D.W., Miller, W.H., Griffin, C.E., Eds.; WB Saunders: Philadelphia, PA, USA, 2001; pp. 913–1003. [Google Scholar]
- Wenstrup, R.J.; Florer, J.B.; Davidson, J.M.; Phillips, C.L.; Pfeiffer, B.J.; Menezes, D.W.; Chervoneva, I.; Birk, D.E. Murine Model of the Ehlers-Danlos Syndrome COL5A1 haploinsufficiency disrupts collagen fibril assembly at multiple stages. J. Biol. Chem. 2006, 281, 12888–12895. [Google Scholar] [CrossRef]
- Bouma, P. COL5A1 Exon 14 Splice Acceptor Mutation Causes a Functional Null Allele, Haploinsufficiency of alpha 1(V) and Abnormal Heterotypic Interstitial Fibrils in Ehlers-Danlos Syndrome II. J. Biol. Chem. 2001, 276, 13356–13364. [Google Scholar] [CrossRef]
- Symoens, S.; Syx, D.; Malfait, F.; Callewaert, B.; De Backer, J.; Vanakker, O.; Coucke, P.; De Paepe, A. Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Hum. Mutat. 2012, 33, 1485–1493. [Google Scholar] [CrossRef]
- Wenstrup, R.J.; Langland, G.T.; Willing, M.C.; D’Souza, V.N.; Cole, W.G. A splice-junction mutation in the region of COL5A1 that codes for the carboxyl propeptide of pro alpha 1(V) chains results in the gravis form of the Ehlers-Danlos syndrome (type I). Hum. Mol. Genet. 1996, 5, 1733–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Paepe, A.; Nuytinck, L.; Hausser, I.; Anton-Lamprecht, I.; Naeyaert, J.M. Mutations in the COL5A1 gene are causal in the Ehlers-Danlos syndromes I and II. Am. J. Hum. Genet. 1997, 60, 547–554. [Google Scholar] [PubMed]
- Nicholls, A.C.; E Oliver, J.; McCarron, S.; Harrison, J.B.; Greenspan, D.S.; Pope, F.M. An exon skipping mutation of a type V collagen gene (COL5A1) in Ehlers-Danlos syndrome. J. Med. Genet. 1996, 33, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.J.; Martin, S.; Nicholls, A.C.; Harrison, J.B.; Pope, F.M.; Burrows, N.P. A single base mutation in COL5A2 causes Ehlers-Danlos syndrome type II. J. Med. Genet. 1998, 35, 846–848. [Google Scholar] [CrossRef] [PubMed]
- Toriello, H.V.; Glover, T.W.; Takahara, K.; Byers, P.H.; Miller, D.E.; Higgins, J.V.; Greenspan, D.S. A translocation interrupts the COL5A1 gene in a patient with Ehlers–Danlos syndrome and hypomelanosis of Ito. Nat. Genet. 1996, 13, 361–365. [Google Scholar] [CrossRef]
- Birk, D.E. Type V collagen: Heterotypic type I/V collagen interactions in the regulation of fibril assembly. Micron (Oxf. Engl. 1993) 2001, 32, 223–237. [Google Scholar] [CrossRef]
- Hausser, I.; Anton-Lamprecht, I. Differential ultrastructural aberrations of collagen fibrils in Ehlers-Danlos syndrome types I–IV as a means of diagnostics and classification. Qual. Life Res. 1994, 93, 394–407. [Google Scholar] [CrossRef]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [Green Version]
- Bateman, J.F.; Golub, S.B. Deposition and selective degradation of structurally-abnormal type I collagen in a collagen matrix produced by osteogenesis imperfecta fibroblasts in vitro. Matrix Biol. 1994, 14, 251–262. [Google Scholar] [CrossRef]
- Niyibizi, C.; Fietzek, P.P.; van der Rest, M. Human placenta type V collagens. Evidence for the existence of an alpha 1(V) alpha 2(V) alpha 3(V) collagen molecule. J. Biol. Chem. 1984, 259, 14170–14174. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, B.; Zhao, W.M.; Tang, B.X.; Wang, Y.Q.; Wang, L.; Zhang, Z.; Yang, H.C.; Liu, Y.H.; Zhu, J.W.; Irwin, D.M.; et al. DoGSD: The dog and wolf genome SNP database. Nucleic Acids Res. 2015, 43, D777–D783. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Patterson, D.F.; Minor, R.R. Hereditary fragility and hyper-extensibility of the skin of cats. Lab. Investig. 1977, 37, 170. [Google Scholar] [PubMed]
- Freeman, L.J.; Hegreberg, G.A.; Robinette, J.D. Ehlers-Danlos syndrome in dogs and cats. Semin. Vet. Med. Surg. Small Anim. 1987, 2, 221–227. [Google Scholar] [PubMed]
- Joseph, A.W.; Joseph, S.S.; Francomano, C.A.; Kontis, T.C. Characteristics, Diagnosis, and Management of Ehlers-Danlos Syndromes: A Review. JAMA Facial Plast. Surg. 2018, 20, 70–75. [Google Scholar] [CrossRef]
- Mauldin, E.; Peters-Kennedy, J. Integumentary System. In Jubb, Kennedy, and Palmer’s Pathology of Domestic Animals, 6th ed.; Grant Maxie, M., St Louis, M.O., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2016; pp. 542–546. [Google Scholar]
- Fang, Y.; Bateman, J.F.; Mercer, J.F.; Lamande, S.R. Nonsense-mediated mRNA decay of collagen-emerging complexity in RNA surveillance mechanisms. J. Cell Sci. 2013, 126, 2551–2560. [Google Scholar] [CrossRef]
- DeNigris, J.; Yao, Q.; Birk, E.K.; Birk, D.E. Altered dermal fibroblast behavior in a collagen V haploinsufficient murine model of classic Ehlers-Danlos syndrome. Connect Tissue Res. 2016, 57, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bateman, J.F.; Boot-Handford, R.P.; Lamandé, S.R. Genetic diseases of connective tissues: Cellular and extracellular effects of ECM mutations. Nat. Rev. Genet. 2009, 10, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Spycher, M.; Bauer, A.; Jagannatha, V.; Frizzi, M.; De Lucia, M.; Leeb, T. A frameshift variant in the COL5A1 gene in a cat with Ehlers-Danlos syndrome. Anim. Genet. 2018, 49, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Jaffey, J.A.; Bullock, G.; Teplin, E.; Guo, J.; Villani, N.A.; Mhlanga-Mutangadura, T.; Schnabel, R.D.; Cohn, L.A.; Johnson, G.S. A homozygous ADAMTS2 nonsense mutation in a Doberman Pinscher dog with Ehlers Danlos syndrome and extreme skin fragility. Anim. Genet. 2019, 50, 543–545. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.; de Lucia, M.; Leuthard, F.; Jagannathan, V.; Leeb, T. Compound heterozygosity for TNXB genetic variants in a mixed-breed dog with Ehlers-Danlos syndrome. Anim. Genet. 2019, 50, 546–549. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Filtering Step | Number of Variants | |
|---|---|---|
| Case 1 | Case 2 | |
| homozygous variants in the whole-genome | 2,791,220 | 2,384,739 |
| homozygous protein-changing variants in the whole-genome | 12,121 | 11,065 |
| homozygous private protein-changing variants | 4 | 9 |
| homozygous private protein-changing variants in candidate genes | 0 | 0 |
| heterozygous variants in the whole-genome | 3,080,790 | 4,346,318 |
| heterozygous protein-changing variants in the whole-genome | 10,310 | 13,789 |
| heterozygous private protein-changing variants | 31 | 276 |
| heterozygous private protein-changing variants in candidate genes | 1 | 1 |
| Major criteria | 1. Skin hyperextensibility and atrophic scarring 2. Generalized joint hypermobility |
| Minor criteria | 1. Easy bruising |
| 2. Soft, doughty skin | |
| 3. Skin fragility (or traumatic splitting) | |
| 4. Molluscoid pseudotumors | |
| 5. Subcutaneous spheroids | |
| 6. Hernia (history thereof) | |
| 7. Epicanthal folds | |
| 8. Complications of joint hypermobility (e.g., sprains, luxation/subluxation, pain, flexible flatfoot) | |
| 9. Family history of a first-degree relative who meets clinical criteria |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauer, A.; Bateman, J.F.; Lamandé, S.R.; Hanssen, E.; Kirejczyk, S.G.M.; Yee, M.; Ramiche, A.; Jagannathan, V.; Welle, M.; Leeb, T.; et al. Identification of Two Independent COL5A1 Variants in Dogs with Ehlers–Danlos Syndrome. Genes 2019, 10, 731. https://doi.org/10.3390/genes10100731
Bauer A, Bateman JF, Lamandé SR, Hanssen E, Kirejczyk SGM, Yee M, Ramiche A, Jagannathan V, Welle M, Leeb T, et al. Identification of Two Independent COL5A1 Variants in Dogs with Ehlers–Danlos Syndrome. Genes. 2019; 10(10):731. https://doi.org/10.3390/genes10100731
Chicago/Turabian StyleBauer, Anina, John F. Bateman, Shireen R. Lamandé, Eric Hanssen, Shannon G.M. Kirejczyk, Mark Yee, Ali Ramiche, Vidyha Jagannathan, Monika Welle, Tosso Leeb, and et al. 2019. "Identification of Two Independent COL5A1 Variants in Dogs with Ehlers–Danlos Syndrome" Genes 10, no. 10: 731. https://doi.org/10.3390/genes10100731