
Metal-Coordinated Dynamics and Viscoelastic Properties of Double-Network Hydrogels
by
 , , ,
, , ,
Shilei Zhu
1,†,
Yan Wang
2,†,
Zhe Wang
2,
Lin Chen
2,
Fengbo Zhu
2,3,
Yanan Ye
2,3,*,
Yong Zheng
4,*,
Wenwen Yu
2,3 and
Qiang Zheng
2,5 1
College of Physics, Taiyuan University of Technology, Taiyuan 030024, China
2
College of Materials Science & Engineering, Taiyuan University of Technology, Taiyuan 030024, China
3
Shanxi-Zheda Institute of Advanced Materials and Chemical Engineering, Taiyuan 030024, China
4
Institute for Chemical Reaction Design and Discovery, Hokkaido University, Sapporo 001-0021, Japan
5
Ministry of Education Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Gels 2023, 9(2), 145; https://doi.org/10.3390/gels9020145
Submission received: 15 January 2023
/
Revised: 31 January 2023
/
Accepted: 4 February 2023
/
Published: 9 February 2023
(This article belongs to the Special Issue Properties and Structure of Hydrogel-Related Materials)
Abstract
:Biological soft tissues are intrinsically viscoelastic materials which play a significant role in affecting the activity of cells. As potential artificial alternatives, double-network (DN) gels, however, are pure elastic and mechanically time independent. The viscoelasticization of DN gels is an urgent challenge in enabling DN gels to be used for advanced development of biomaterial applications. Herein, we demonstrate a simple approach to regulate the viscoelasticity of tough double-network (DN) hydrogels by forming sulfonate–metal coordination. Owing to the dynamic nature of the coordination bonds, the resultant hydrogels possess highly viscoelastic, mechanical time-dependent, and self-recovery properties. Rheological measurements are performed to investigate the linear dynamic mechanical behavior at small strains. The tensile tests and cyclic tensile tests are also systematically performed to evaluate the rate-dependent large deformation mechanical behaviors and energy dissipation behaviors of various ion-loaded DN hydrogels. It has been revealed based on the systematic analysis that robust strong sulfonate–Zr4+ coordination interactions not only serve as dynamic crosslinks imparting viscoelastic rate-dependent mechanical performances, but also strongly affect the relative strength of the first PAMPS network, thereby increasing the yielding stress σy and the fracture stress at break σb and reducing the stretch ratio at break λb. It is envisioned that the viscoelasticization of DN gels enables versatile applications in the biomedical and engineering fields.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Hydrogels have recently emerged as promising candidates for use in artificial cartilage engineering because they possess cartilage tissue-like features such as ease of fabrication, toughness, surface smoothness, biocompatibility, and appropriate biochemical properties. However, conventional hydrogels are usually too mechanically weak and brittle to sustain external loads, owing to the inhomogeneous and amorphous structure of the polymer network; however, the natural cartilage is mechanically strong and tough, and can even sustain compressive stress as great as tens of MPa [1,2]. These huge gaps in the mechanical properties between conventional synthetic weak hydrogels and strong load-bearing biological tissues significantly impede the advanced functional applications of these hydrogels as biomaterials.
To achieve better mechanical properties for conventionally weak hydrogels, in the last few decades, many great efforts have been made in fabricating strong and tough hydrogels through chemical modification methods [3,4,5,6,7] (e.g., by introducing sacrificial bonds [8,9,10,11] and making a homogeneous polymer network structure [4,6]) and by incorporating reinforcing nanofillers [12,13,14,15,16,17]. Among these great efforts, the double-network (DN) approach stands out as an extraordinarily effective method for developing strong and tough materials, and the “sacrificial bonds” mechanism has been proved to efficiently increase energy dissipation for improving toughness and strength synchronously [5,10]. A hydrogel with DN structure showing extremely enhanced mechanical strength and toughness is designed to be composed of two types of interpenetrating polymer network with contrasting natures, i.e., a densely crosslinked stiff and brittle polyelectrolyte first network, and a loosely crosslinked soft and ductile neutral second network [10]. Upon deformation, the brittle first network can break into fragments and dissipate a large amount of energy, while the stretchable second network can keep its integrity without causing catastrophic failure of the whole hydrogel [10].
Previous studies have shown that a conventional DN gel composed of poly(2-acrylamido-2-methyl propanesulfonic acid) (PAMPS) as the brittle first network and poly(N,N-dimethylacrylamide) (PDMA) or polyacrylamide (PAAm) as the ductile second network is a promising biomaterial for biological tissues (e.g., artificial cartilages) [18,19]. It has been demonstrated that DN gel has excellent biocompatibility and a low degree of toxicity in vivo, high compressive strength, extremely low friction, and low wear production in vitro and in ex vivo friction tests [20,21,22,23,24]. On the other hand, natural biological tissues are intrinsically viscoelastic materials [25,26], and recent studies have shown that the viscoelasticity, especially the time-dependent stiffness, plays a significant role in affecting the activity of cells [27,28,29,30,31]. Unlike natural biological tissues, traditional DN hydrogels are rate independent and elastic. Thus, achieving their viscoelastic transformation is a pressing issue in advancing conventional DN gel for biomaterial applications, and can help deepen our understanding of the regulated viscoelasticity and activity of biological tissue cells. Introducing dynamic bonds is a common strategy to impart viscoelasticity to materials [9,32,33,34]. The dynamic bonds include many types of physical interactions, such as van der Waals interactions [35,36], π−π stacking interactions [37,38], hydrogen-bonding interactions [33,39], ionic interactions [9,40], metal−ligand interactions [32,41,42], and so on.
Taking advantage of sulfonate groups in the PAMPS first network, herein, we demonstrate a simple approach to regulate the viscoelasticity of traditional DN hydrogels. The traditionally rate-independent elastic PAMPS/PAAm DN hydrogels were tailored to exhibit rate-dependent viscoelastic mechanical responses by forming sulfonate–Mn+ coordination complexes that act as dynamic crosslinks to confer viscoelasticity [41]. The PAMPS first network, with plenty of sulfonate (–SO3−) functional groups, provides functional sites for forming metal–coordination complexes [41,42]. Here, the Zr4+ was chosen as the model ion, and Fe3+ and Na+ ions were chosen for comparisons. We first performed rheological measurements to investigate the linear dynamic mechanical behavior at small strain. Further tensile tests and cyclic tensile tests were then systematically conducted to evaluate the rate-dependent large deformation mechanical behaviors and energy dissipation behaviors for various ion-loaded DN hydrogels. The effect of robust sulfonate–Zr4+ coordination interactions on the rate-dependent mechanical performances is carefully detailed in the discussion section. This work offers a simple strategy for tuning the viscoelasticity of conventional elastic DN hydrogels, which could expand the application of DN hydrogels as biological artificial tissues.
2. Results and Discussion
2.1. Hydrogel Fabrication Process
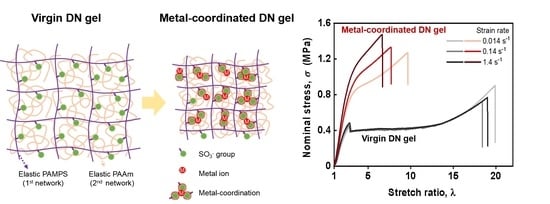
The conventional PAMPS/PAAm DN hydrogels [43] were selected as model systems in this work due to the presence of plenty of sulfonate (–SO3−) functional groups in the PAMPS of the first network providing functional sites for the formation of metal coordination, and the extensive research on their use as artificial tissue materials in previous works [18,19,20,21,22,23,24]. Here, the multivalent Zr4+ and Fe3+ were chosen as metal ions for their coordination abilities with sulfonates, as reported in previous studies [41,42]; conversely, the monovalent Na+ ion, which lacks coordination ability with sulfonates, was chosen as a reference. Figure 1 illustrates the fabrication procedure of ion-coordinated viscoelastic DN hydrogels from virgin elastic DN hydrogels through immersion in metal–ion solutions and subsequent dialysis in water. For simplification, the DN hydrogels equilibrated in metal–ion solution (in the as-prepared state) and the hydrogels subsequently equilibrated in water are denoted as “DN−Mn+−Cm−AP” and “DN−Mn+−Cm−E” hydrogels, respectively, where Mn+ represents Zr4+, Fe3+ and Na+ ions in this work, and Cm (M) represents the molar concentration of metal–ion in solution. In the process of transformation from DN−Mn+−Cm−AP gel to DN−Mn+−Cm−E gel, counter ions were dialyzed out from the network, and more dynamic coordination bonds were formed, giving the latter a stronger viscoelasticity. By forming robust sulfonate–Zr4+ or sulfonate–Fe3+ coordination complexes, the purely elastic PAMPS network will be transformed into a viscoelastic one, imparting rate-dependency and viscoelasticity to the DN hydrogels. For the Zr4+ and Fe3+ ions-loaded DN hydrogels, the concentration of metal ion solution Cm is fixed at a relatively small concentration of 0.1 M, while for the Na+ ion-loaded DN hydrogels, the concentration of metal ion solution Cm is fixed at a high concentration of 1.0 M to demonstrate the ineffective effect of Na+ ions. Note that the monomer concentration of AMPS monomer for the PAMPS network in the final DN hydrogels can be estimated as 0.05 M, which means that the effective concentration of sulfonate (–SO3−) functional groups is ~0.05 M.
2.2. Linear Dynamic Mechanical Behavior at Small Strain
Since the dynamic metal coordination bonds are sensitive to temperature, we chose frequency sweep rheological measurements at different temperatures to verify our proposed approach. Here, Zr4+−coordinated DN gels were selected as a typical example. Figure 2 depicts the frequency (ω) sweep of G′, G″, and tanδ from 0.1 to 100 rad/s at various temperatures ranging from 8 °C to 88 °C for the DN−Zr4+−0.1 M−AP and DN−Zr4+−0.1 M−E hydrogels. For the virgin DN gel, the G′ is frequency independent in the full frequency range. Furthermore, with an increase in temperature, the G′ shows negligible change, indicating that the virgin DN gel is pure elastic, and time–temperature superposition fails for such kinds of material. Different from the virgin DN gel, the G′ for the DN−Zr4+−0.1 M−AP and DN−Zr4+−0.1 M−E hydrogels show strong frequency-dependent and temperature-dependent behaviors, indicating that the dynamic metal coordination bonds were successfully introduced into the DN gels. Besides, it is interestingly found that compared to the DN−Zr4+−0.1 M−AP hydrogel, the DN−Zr4+−0.1 M−E hydrogel shows a much higher G′ at the same frequency and temperature. The lower G′ for the DN−Zr4+−0.1 M−AP hydrogel could be attributed to the excess of Zr4+ ions in DN−Zr4+−0.1 M−AP hydrogels, which weakens the sulfonate–Zr4+ coordination bonds in the first PAMPS network; by moving the gels to DI water, the excessive free Zr4+ ions will be dialyzed out from the gel to achieve a new equilibrium, helping to form stronger sulfonate–Zr4+ coordination in the gels. To confirm the formation of metal coordination, we further studied the rheological behaviors of DN−Na+−1.0 M−AP and DN−Na+−1.0 M−E hydrogels, where the Na+ lacks the coordination ability with sulfonate. As shown in Figure S1, both gels show frequency and temperature independence similar to that of virgin gels. The above comparison demonstrated that only the metal ions with coordination ability with sulfonate can be used to achieve the viscoelasticization of DN gels.
The dynamic mechanical study can be used to clarify the strength of the dynamic bonds within the hydrogel system [9]. Based on the above isothermal frequency sweep data collected over a wide range of temperatures, we constructed the master curve according to the time–temperature superposition (TTS) principle for DN−Zr4+−0.1 M−AP and DN−Zr4+−0.1 M−E hydrogels [44,45]. The reference temperature was set at 24 °C. For each gel, the frequency sweep curves were shifted horizontally with a temperature-dependent shift factor aT, and no vertical shift was performed. As shown in Figure 3a,b, both the DN−Zr4+−0.1 M−AP and DN−Zr4+−0.1 M−E hydrogels show well-superposed time–temperature master curves. The Arrhenius plot for the shift factor of the master curve, aT, is shown in Figure 3c for the DN−Zr4+−0.1 M−AP gels and Figure 3d for the DN−Zr4+−0.1 M−E hydrogels, from which the activation energy Ea can be extracted from the Arrhenius equation: aT = AeEa/RT, where R is the ideal gas constant and A is a constant. The aT-1/T plot gives an apparent activation energy (Ea) of ≈80.8 kJ mol−1 for the DN−Zr4+−0.1 M−AP gels. This relatively low apparent activation energy (Ea) is considered to be due to weakened metal coordinated bonds caused by the existence of excessive ions. Thus, over an ordinary time scale, these bonds have a relatively short relaxation time, which easily dissociates during deformation. Meanwhile, for the DN−Zr4+−0.1 M−E hydrogels, the plot of ln aT verse 1/T shows very different slopes at low and high temperature regimes (Figure 3d). Based on this plot, two apparent activation energy values of 48.6 kJ/mol and 160.9 kJ/mol were obtained, indicating two kinds of physical bond with different bonding strengths. The differentiation of bonding strengths in the DN−Zr4+−0.1 M−E hydrogels could be attributed to the inhomogeneous redistribution of coordinated chains during the dialysis of excess ions from gels, confirmed in charge-balanced polyampholytes gels in previous publications [46]. For DN−Zr4+−0.1 M−AP gels, the gels show a relatively homogeneous structure in the presence of excess ions. When immersing the gels in DI water, the rapid dialysis of ions out of the gels leads to the redistribution of polymer chains into the polymer’s dense and dilute phases. Due to the high density of the polymer chain in the dense phase, the multivalent metal ions are more tightly coordinated with sulfonate, resulting in a larger bond energy, which means that more energy is required to break this coordination bond (i.e., a larger Ea); conversely, in the dilute phase, the loose molecular chains make the metal coordination interaction weaker, leading to lower coordination strength (and a lower Ea).
2.3. Large Deformation Mechanical Behaviors and Energy Dissipation Behaviors
The above rheological results clarified the bond strength change during the gel fabrication. However, these rheological results can only be used to evaluate the small-strain dynamic mechanical behaviors of DN hydrogels. Next, we evaluate the large deformation mechanical behaviors of various DN hydrogels by performing tensile tests under different deformation rates ranging from 10 to 1000 mm/min, corresponding to a strain rate range of 0.014 to 1.4 s−1. Figure 4a–c shows the tensile curves for the virgin DN hydrogels as well as the Zr4+ ion-loaded DN−Zr4+−0.1 M−AP and DN−Zr4+−0.1 M−E hydrogels, respectively. With increasing strain rate over two orders from 0.014 to 1.4 s−1, the tensile curves of virgin DN hydrogels are perfectly overlapped, indicating the rate-independency at large deformation. This clearly shows that the virgin DN hydrogels consisting of two elastic polymer networks are purely elastic with negligible interactions, consistent with the small-strain rheological results shown in Figure 2a–c. Meanwhile, for the Zr4+ ion-loaded DN−Zr4+−0.1 M−AP and DN−Zr4+−0.1 M−E hydrogels, with increasing strain rate, the tensile curves no longer overlap with each other and seem to slightly deviate from each other in the small deformation region, and then largely deviated in the large deformation region. The observed strong rate-dependent large deformation mechanical behaviors should result from the formation of dynamic and reversible sulfonate–Zr4+ coordination interactions in the PAMPS first network, which is strong evidence that the gels are viscoelastic [9]. At a low strain rate, the characterization relaxation time is smaller than the strain rate, where the dynamic bonds are easy to relax, resulting in negligible contribution to strength; at a high strain rate, the dynamic bonds do not have enough time to relax, and thereby act as permanent chemical bonds during tensile deformation, resulting in pronounced increase in strength and deviation from that at low strain rate [47]. Other research has also reported that the dynamic nature of metal coordination interactions imparts strong rate-dependent large deformation mechanical performance to a Zr4+ ion-toughened PAMPS single network hydrogel [41] and Fe3+ ion-toughened P(AAm-co-AAc) single network hydrogel [48]. The effect of deformation rate on the mechanical behaviors of the hydrogels reported was mainly attributed to the increase in the bonding strength with the deformation rate [48]. As shown in Figure 3b,c, the DN−Zr4+−0.1 M−E hydrogels show much stronger deformation rate dependence than the DN−Zr4+−0.1 M−AP hydrogels. This is attributed to the excess of Zr4+ ions in DN−Zr4+−0.1 M−AP hydrogels weakening the sulfonate–Zr4+ coordination interactions in the first PAMPS network; after the water-equilibrating process, the excess of Zr4+ ions will be dialyzed out from the gel, helping the formation of strong sulfonate−Zr4+ coordination in the first PAMPS network, which is consistent with the results deduced from time–temperature superposition.
Owing to the dynamic nature of the sulfonate–Zr4+ coordination, the Zr4+ ion-loaded DN hydrogels should have completely different internal fracture mechanisms compared to the virgin DN hydrogels. To examine the different internal fracture mechanisms, we perform cyclic tensile tests on various DN hydrogels to check their energy dissipation behaviors during the cyclic loading and unloading process. Figure 4d–f shows the sequential loading–unloading curves of the virgin DN hydrogels, DN−Zr4+−0.1 M−AP and DN−Zr4+−0.1 M−E hydrogels, respectively. As exhibited in Figure 4d, the virgin DN hydrogels only show irreversible hysteresis, similar to the results reported in previous studies [49,50]. For conventional DN hydrogels consisting of two chemically crosslinked elastic polymer networks, an abundance of covalent bonds in the densely crosslinked elastic first network sacrificially fractures to dissipate a large amount of energy upon deformation. Such covalent bonds cannot be recovered once bond scission occurs, resulting in irreversible mechanical hysteresis, as observed in Figure 4d. For the DN−Zr4+−0.1 M−AP and DN−Zr4+−0.1 M−E hydrogels (Figure 4e,f), in addition to the irreversible mechanical hysteresis, a certain amount of reversible mechanical hysteresis can also be observed between loading–unloading tensile cycles. The partially reversible mechanical hysteresis could be attributed to the dynamic nature of sulfonate–Zr4+ coordination, which can break and reform to some extent upon loading and unloading tensile cycles. Such results are consistent with the previous reports showing that when the first network contains noncovalent interactions as dynamic sacrificial bonds, both reversible and irreversible mechanical hysteresis appear in the cyclic tensile and compressive tests [11,32,50]. We have to mention here that the irreversible mechanical hysteresis observed for the DN−Zr4+−0.1 M−AP and DN−Zr4+−0.1 M−E hydrogels is dominant in the energy dissipation behaviors compared to the reversible part of mechanical hysteresis, meaning that the covalent bond scission still dominates the internal fracture mechanism for such Zr4+-loaded DN hydrogels upon deformation. In comparison to the DN−Zr4+−0.1 M−AP hydrogels, the DN−Zr4+−0.1 M−E hydrogels not only exhibit enhanced total mechanical hysteresis (the absolute energy dissipation density Wtotal), but also show enhanced reversible hysteresis Wre, the hysteresis recovery ratio Wre/Wtotal (Figure 4g–i). This result also suggests that for the DN−Zr4+−0.1 M−AP hydrogels, the amount or the strength of sulfonate–Zr4+ coordination is weakened due to the excess amount of Zr4+ ions, while the DN−Zr4+−0.1 M−E hydrogels can form relatively strong robust sulfonate−Zr4+ coordination interactions in the first PAMPS network by removing the excess amount of Zr4+ ions through dialysis of the hydrogels after the water-equilibrating process.
To investigate the specificity of Zr4+ ions to form robust coordination with the sulfonate groups in the PAMPS first network, we further evaluate the tensile behaviors and cyclic tensile behaviors for various Na+ and Fe3+ ion-loaded DN hydrogels. Figure 5a,b shows the rate-independent tensile behaviors of the DN−Na+−1.0 M−AP and DN−Na+−1.0 M−E hydrogels, respectively. They are exactly similar to those of the virgin DN hydrogels shown in Figure 4a. The DN−Na+−1.0 M−AP and DN−Na+−1.0 M−E hydrogels also exhibit irreversible mechanical hysteresis in the loading–unloading cycles, similar to the virgin DN hydrogels (Figure 5c,d). These pure elastic mechanical behaviors suggest that Na+ ions cannot form interactions with the first network in the DN hydrogels because monovalent Na+ ions lack coordination ability. Figure 6a,b show the tensile behaviors of the Fe3+ ion-loaded DN−Fe3+−0.1 M−AP and DN−Fe3+−0.1 M−E hydrogels, respectively. Interestingly, the DN−Fe3+−0.1 M−AP hydrogels show rate-independent tensile behaviors, while the DN−Fe3+−0.1 M−E hydrogels demonstrate slightly rate-dependent tensile behaviors. Additionally, it is found that the DN−Fe3+−0.1 M−AP hydrogels show completely irreversible hysteresis, while the DN−Fe3+−0.1 M−E hydrogels show partially reversible hysteresis in Figure 6c,d. These results suggest that the DN−Fe3+−0.1 M−AP hydrogels have a pure elastic first network with negligible metal coordination interactions, while DN−Fe3+−0.1 M−E hydrogels have a slightly viscoelastic first network with weak metal coordination interactions. The distinct difference between the DN−Fe3+−0.1 M−AP hydrogels and the DN−Fe3+−0.1 M−E hydrogels indicates that weak sulfonate–Fe3+ coordination interactions can only be formed after getting the excess amount of Fe3+ dialyzed out of the hydrogels.
2.4. Rescaled Mechanical Behaviors
The highly crosslinked first network, which is also in its highly stretching state due to the osmotic pressure-induced swelling effect, dominates the mechanical performance of the DN hydrogels [50,51,52,53]. To have a clear and quantitative understanding of the effect of various ions on the mechanical performance of various ion-loaded DN hydrogels, we need to normalize the strand density and pre-stretch level of the first network in different systems relative to its as-prepared state in order to remove the effect of osmotic pressure-induced swelling. Here, we used the normalization method for DN material systems by rescaling the nominal stress (σ) and stretch ratio (λ) with the pre-stretch ratio of the first network (λs) to the rescaled stress σλs2 and the rescaled stretch ratio λλs, respectively [50,52,53].
Figure 7 shows the rescaled stress σλs2–rescaled stretch ratio λλs curves for various DN hydrogels loaded with Zr4+, Fe3+, and Na+ as metal ions under different deformation rates ranging from 10 to 1000 mm/min in comparison to the virgin DN hydrogels. As shown in Figure 7a, these perfectly overlapped rescaled stress curves for the DN−Na+−1.0 M−AP hydrogels and DN−Na+−1.0 M−E hydrogels under different deformation rates indicate the negligible effect of Na+ ions due to the lack of coordination ability between monovalent Na+ ions and sulfonate groups in the PAMPS first network. The DN−Fe3+−0.1 M−AP hydrogels, as shown in Figure 6b, show perfectly overlapped rate-independent rescaled tensile curves with those of the virgin DN hydrogels; the Na+ ion-loaded DN hydrogels are similar, suggesting the negligible influence of Fe3+ ions in the DN−Fe3+−0.1 M−AP hydrogels. Given that Fe3+ ions have coordination ability with the sulfonate groups in the PAMPS first network, such negligible influence of Fe3+ ions in the DN−Fe3+−0.1 M−AP hydrogels can be explained by the fact that the excess amount of Fe3+ ions weakens the Fe3+-coordination interactions. Note that the ion concentration of Fe3+ ions in the solution (Cm = 0.1 M) is larger than the effective concentration of sulfonate functional groups (~0.05 M). As opposed to the DN−Fe3+−0.1 M−AP hydrogels, the water-equilibrated DN−Fe3+−0.1 M−E hydrogels show slightly enhanced stress with a slight rate dependency in the rescaled curves compared to these of virgin DN hydrogels (Figure 6b), suggesting that the formation of weak sulfonate–Fe3+ coordination interactions in the water-equilibrated DN−Fe3+−0.1 M−E hydrogels only contributes slightly to the mechanical enhancement of the DN hydrogels after the excess amount of Fe3+ has been dialyzed out of the hydrogels.
For the Zr4+–coordinated DN hydrogels, as exhibited in Figure 7c, both the DN−Zr4+−0.1 M−AP hydrogels and the DN−Zr4+−0.1 M−E hydrogels deviate largely from the virgin DN hydrogels in the rescaled stress curves, indicating a strong rate dependency. The mechanical enhancement of Zr4+ ions is more significant in the DN−Zr4+−0.1 M−E hydrogels than in the DN−Zr4+−0.1 M−AP hydrogels. This result clearly shows that the Zr4+ ions in the DN−Zr4+−0.1 M−E hydrogels can form relatively robust sulfonate–Zr4+ coordination in the first PAMPS network, contributing to the mechanical enhancement. Meanwhile, for the DN−Zr4+−0.1 M−AP hydrogels, the enhancement is weakened due to an excess amount of Zr4+ ions. Although the mechanical enhancement of Zr4+ ions in the DN−Zr4+−0.1 M−AP hydrogels is comparatively weakened from that in the DN−Zr4+−0.1 M−AP hydrogels, the enhancement is more obvious than the Fe3+ ions in the DN−Fe3+−0.1 M−E hydrogels. This suggests that the bond strength of sulfonate–Zr4+ coordination is stronger than sulfonate–Fe3+ coordination interactions. This has also been verified in a previous report showing that the PAMPS hydrogels cannot be effectively toughened by Fe3+ ions but can be remarkably toughened by Zr4+ ions [41]. This specific effect of Zr4+ ions has been generally explained by the hard–soft acid–base theory [41,54]. Based on the hard–soft acid–base theory, the Zr4+ ions with four positive charges and a small radius of 0.72 Å belong to hard acids, which can accept non-bonding electron pairs [55]; the sulfonate groups can be seen as hard bases owing to the high electronegativity of the oxygen atom, serving as multidentate ligands to coordinate with Zr4+ ions. Moreover, the roughly spherical shape of the electron density of the sulfonate groups has more coordinative flexibility to form robust metal coordination bonds [56].
Next, we systematically discuss the effect of ion types on the Young’s modulus E, the yielding stretch ratio λy, and the yielding stress σy. Figure 8a,b depicts the dependence of the Young’s modulus E and the rescaled Young’s modulus Eλs on strain rate for various DN hydrogels. Except for the DN−Zr4+−0.1 M−E hydrogels, all series have a Young’s modulus E and a rescaled Young’s modulus Eλs that are comparable with those of virgin DN hydrogels, and show almost no rate dependency. The DN−Zr4+−0.1 M−E hydrogels show a distinctively enhanced Young’s modulus E and rescaled Young’s modulus Eλs, and exhibit a slightly increasing tendency with the strain rate. The robust sulfonate–Zr4+ coordination stiffens the polymer network by increasing the effective crosslinking density, because these strong dynamic bonds can serve as temporary crosslinks to restrict the movements of polymer strands.
As shown in Figure 7, all series of hydrogels except for the DN−Zr4+−0.1 M−E hydrogels exhibit the typical tensile behaviors accompanied by a distinct stress-yielding, remarkable necking, and subsequent strain-hardening phenomena [10]. It has already been revealed in the toughening mechanisms of DN hydrogels that the yielding corresponds to the catastrophic fracture of the brittle first network into discontinuous fragments [51]. Thus, the rescaled yielding stretch ratio λyλs possibly represents the stretching limit of the brittle first network strands [51]. Although the DN−Zr4+−0.1 M−E hydrogels do not show apparent stress-yielding behavior, we also denote the transition point between two stress-regions, showing contrasting slopes as the yielding point. Figure 8c,d depicts the dependence of the yielding stretch ratio λy and the rescaled yielding stretch ratio λyλs on strain rate. Interestingly, we find that the DN−Zr4+−0.1 M−AP hydrogels with excess amounts of Zr4+ ions show a much more enhanced rescaled yielding stretch ratio λyλs compared to other DNs, including the virgin DN hydrogels as well as the water-equilibrated DN–Zr4+–0.1 M–E hydrogels. This suggests that the first network in the DN−Zr4+−0.1 M−AP hydrogels has a much improved stretching limit of the brittle first network strands. The brittle first network strands have a wide distribution of strand lengths due to their inhomogeneity; the short strands in first network would break at a small stretching limit, while the long strands would break at a large stretching limit. This improvement of stretching limit by excessive Zr4+ ions in the DN−Zr4+−0.1 M−AP hydrogels may be explained by the fact that the sulfonate–Zr4+ coordination interactions may homogenize the distribution of strand lengths by adding self-adjustable dynamic crosslinks in the inhomogeneous first network strands; this reduces the effective fracture of these short strands. This homogenization seems to be effective only when the metal coordination is neither strong enough nor weak enough, which is also observed for the DN−Fe3+−0.1 M−E hydrogels, having relatively weak sulfonate–Fe3+ coordination interactions. Moreover, with increasing strain rate, the DN−Zr4+−0.1 M−AP hydrogels, DN−Zr4+−0.1 M−E hydrogels, and DN−Fe3+−0.1 M−E hydrogels show a decreasing tendency. This is reasonable because the dynamic nature of metal coordination determines that the bond strength increases with the deformation rate [41,48].
We also check the dependence of the yielding stress σy and the rescaled yielding stress σyλs2 on strain rate for various DN hydrogels in Figure 8e,f. The yielding stress is typically interpreted as the total load sustained by the first network strands when catastrophic fracture occurs in first network. The DN−Zr4+−0.1 M−AP hydrogels, DN−Zr4+−0.1 M−E hydrogels, and DN−Fe3+−0.1 M−E hydrogels show remarkably enhanced yielding stress σy and rescaled yielding stress σyλs2 compared to the virgin DN hydrogels. This result suggests that the metal-coordination interactions can significantly increase the total load sustained by the first network strands when catastrophic fracture occurs in first network. Notably, the bond strength of metal coordination has the following order: DN−Zr4+−0.1 M−E > DN−Zr4+−0.1 M−AP > DN−Fe3+−0.1 M−E > DN−Fe3+−0.1 M−AP hydrogels. In addition, with increasing the strain rate, the DN−Zr4+−0.1 M−AP hydrogels, DN−Zr4+−0.1 M−E hydrogels, and DN−Fe3+−0.1 M−E hydrogels show an increasing tendency in the yielding stress σy and rescaled yielding stress σyλs2. This can be ascribed to the increased bond strength with the deformation rate. The rescaled yielding stress σyλs2 is proportional to the area density of the first network at the reference state (v1st2/3) times the bond-breaking force per strand (fb), [57]. Thus, we have because all DN hydrogels are prepared with the same PAMPS network (the area density of the first network at the reference state (v1st2/3) should be constant), where the fb and fb,0 represent the breaking force of first network in the ion-coordinated DN hydrogels and virgin DN gels, respectively. By normalizing all the rescaled yielding stress σyλs2 with (σyλs2)virgin, we can have a quantitative understanding of how much the breaking force of first network strands would be enhanced. Figure 9 depicts the ratio of breaking force of first network strands fb/fb,0 for various DN hydrogels relative to the virgin DN gels. The result suggests that the breaking force of first network strands can be significantly enhanced to 1.5 to 2-folds in the DN−Zr4+−0.1 M−E hydrogels, owing to the strong sulfonate–Zr4+ coordination interactions compared to the virgin DN gels.
The huge difference in the strength and stretchability of the brittle PAMPS first network and the stretchable PAAm second network dominates the load-transfer mechanisms upon deformation, thereby affecting the fracture behaviors at break of DN gels [43,58]; the maximum stretch ratio λb and fracture stress at break σb should reflect the relative strength/stretchability of the two networks. We next discuss the mechanical behaviors at break for various DN hydrogels. Figure 10 depicts the λb, σb, and work of extension Wb. All series of DN hydrogels have the same stretchable PAAm network which is hardly affected by the different ions; thus, the λb and σb of the PAMPS first network for different series of DN hydrogels should be comparable. Interestingly, the DN−Zr4+−0.1 M−E hydrogels show a significantly reduced maximum stretch ratio at break λb, but remarkably enhanced fracture stress at break σb, as shown in Figure 10a,b. This result indicates that the sulfonate–Zr4+ coordination formed in the DN−Zr4+−0.1 M−E hydrogels increases the relative strength of first network while decreasing its relative stretchability. On the other hand, the DN−Zr4+−0.1 M−AP hydrogels demonstrate slightly enhanced fracture stress at break σb, accompanied by the hardly reduced maximum stretch ratio at break λb compared to the virgin DN hydrogels. This also suggests that the relatively weak sulfonate–Zr4+ coordination interactions will slightly increase the relative strength of first network without sacrificing its relative stretchability, thereby effectively increasing the work of extension Wb (even enhanced compared to the DN−Zr4+−0.1 M−E hydrogels). The work of extension Wb can also be used to characterize the toughness of the soft materials; the slightly increased work of extension Wb of the DN−Zr4+−0.1 M−AP hydrogels indicates that the bond strength can be neither strong nor weak to enhance the overall toughness upon deformation.
3. Conclusions
In summary, we presented a facile strategy for transforming the rate-independent elastic DN hydrogels to rate-dependent viscoelastic DN hydrogels via metal-coordinated with sulfonate functional groups in the PAMPS first network. The effect of metal coordination on the small strain rheology, yielding behaviors, and large strain hysteresis was systematically studied. Our results show that the viscoelasticization of DN gels can be achieved via metal ions (Fe3+, Zr4+) which have a coordinated ability with sulfonate. In the DN−Zr4+−0.1 M−AP gels, the excessive ions impart a metal coordination bond with homogeneous bong strength (Ea: 80.8 kJ/mol). Meanwhile, the dialysis process differentiates the bond strength into two values (48.6 kJ/mol and 160.9 kJ/mol), which is possibly attributed to the inhomogeneous redistribution of coordinated chains. In our observation time scale, we found that stronger bond strength leads to a larger yielding strength and a larger hysteresis ratio, which originates from the enhancement strength of first network strand via the metal coordination formed in first network chain. Based on quantitative analysis, we deduced that the breaking force of first network strands could be increased by 1.5 to 2-folds in the DN−Zr4+−0.1 M−E hydrogels compared to the virgin DN gels. The maximum toughness of the metal-coordinated DN hydrogels is achieved with a moderate dynamic bond strength, which is a synergy of increasing breaking force of polymer chains to dissipate energy, and keeping deformability due to structural homogenization.
4. Materials and Methods
4.1. Materials
The 2-acrylamido-2-methylpropanesulfonic acid, acrylamide (AAm, purity ≥ 99.8%), N,N′-methylenebis(acrylamide) (MBAA, purity ≥ 99.0%), and α-ketoglutaric acid (α-keto, purity: 99%) were purchased from Shanghai Aladdin Bio-Chem Technology Co., Ltd. (Shanghai, China) and used as received. DI water (resistivity: 18.3 MΩ·cm) was used in all experiments.
4.2. Synthesis of the Virgin PAMPS/PAAm DN Hydrogels
The poly(2-acrylamido-2-methylpropanesulfonic acid)/polyacrylamide (PAMPS/PAAm) DN hydrogels were synthesized by a two-step sequential network formation technique following the literature [43]. The first PAMPS network of the DN hydrogels was synthesized from an aqueous solution of 1.0 M AMPS containing 3 mol% crosslinking agent, MBAA, and 1 mol% initiator, α-keto. The solution was purged in an argon atmosphere to remove dissolved oxygen and then poured into a reaction cell consisting of a pair of glass plates with 0.5 mm spacing. The reaction cell was irradiated with UV light (365 nm) for 8 h. These gels (first network) were then immersed in an aqueous solution of 2.0 M AAm, containing 0.01 mol% MBAA and 0.01 mol% α-keto, for one day until swelling equilibrium was reached. The polymerization was performed again by 365 nm UV irradiation for 8 h. The as-prepared DN gels were then immersed in pure water to reach equilibrium to obtain the virgin DN gels for further experiments.
4.3. Preparation of the “DN−Mn+−Cm−AP” and “DN−Mn+−Cm−E” Hydrogels
The metal ion-loaded DN hydrogels were prepared first through immersion in metal–ion solutions and subsequently through an equilibrium approach. For simplification, the as-prepared DN hydrogels were first immersed in a metal–ion solution (in the as-prepared state) and subsequently transferred to DI water for equilibrium. For the sulfonate−Zr coordination, the reported paper shows that the atomic ratio of S to Zr is about 0.5, suggesting that one Zr cluster coordinated with two sulfonate groups [41]. For simplicity, the Zr4+ ion is used to show the coordination with sulfonate groups. For the sulfonate−Fe coordination, the Fe3+ ion can form with sulfonate in mono-, bis-, and tris-complex [42]; here, we use the simple notation “Fe3+−sulfonate” to represent the coordination bonds. Here, the DN hydrogels balanced in metal–ion solution and DI water were denoted as “DN−Mn+−Cm−AP” and “DN−Mn+−Cm−E” hydrogels, respectively, where Mn+ represents Zr4+, Fe3+ and Na+ ions, and Cm (M) represents the metal–ion concentration in the solution. Typically, to prepare the DN−Zr4+−0.1 M−AP hydrogels, the virgin DN hydrogels were first soaked in 200 mL zirconium (IV) chloride (ZrCl4) solution (Cm = 0.1 M) at ambient temperature for one week to reach a swelling equilibrium in the solution. The obtained DN−Zr4+−0.1 M−AP hydrogels were then immersed in adequate DI water for at least one week to remove the excessive ions dialyzed from the hydrogels and to reach an equilibrium state in water (DN−Zr4+−0.1 M−E hydrogels). DI water was changed twice a day to ensure a sufficient dialysis process.
4.4. Rheological Test
Dynamic rheological tests were performed with an ARES rheometer (Advanced Rheometric Expansion System, Rheometric Scientific Inc.). A disk-shaped sample with a diameter of 15.0 mm was fixed between aluminum plates. The various DN hydrogel samples were surrounded by liquid paraffin during measurement to prevent water evaporation and ion diffusion from the DN hydrogels. Before each measurement, the DN hydrogel samples were held at the set temperature for 300 s to reach the temperature equilibrium. For the DN gels coordinated with Zr4+, the rheological measurements were performed over a frequency range of 0.1–100 rad/s with a shear strain of 0.2% at a temperature range of 8−88 °C. The data were processed into master curves of storage modulus G′, loss modulus G″, and loss factor tan δ via time−temperature superposition (TTS) shifts at a reference temperature of 24 °C. Meanwhile, for the virgin DN gels and gels coordinated with Fe3+ and Na+, the frequency sweep was only performed at reference temperature due to their weak temperature dependence of rheological behaviors.
4.5. Tensile Test
The tensile mechanical properties of various DN hydrogels were measured with a commercial test machine in the air (4466, Instron Instruments, Inc., Norwood, MA, USA). To prevent hydrogel dehydration, a humidifier was used during the tests. The samples were cut into dumbbell shapes standardized as JISK6251-7 size (gauge length 12 mm, width 2 mm) with a gel cutting machine. The nominal stress σ-stretch ratio λ curves were recorded while the sample gels were stretched at different constant velocities of 10, 100, and 1000 mm/min (corresponding to strain rates of 0.014, 0.14, and 1.4 s−1). The Young’s modulus, E, was calculated from the initial slope of the stress–strain curves at the stain within 10%. The work of tension of the samples during the tests, Wt, was calculated by integrating the area under the stress–strain curves by the equation: , where and were the stress and stretching ratio, respectively, and was the stretching ratio at breaking.
4.6. Cyclic Tensile Test
Cyclic tensile tests were performed using the same experimental setup as the tensile tests. The samples were stretched to a pre-set stretch ratio of 50%, 75%, 100%, 125%, 150%, 175%, 200%, 225%, 250%, 300%, 350%, 400%, 450%, 500%, 550%, 600%, 650%, 700%, 750%, 800%, 850%, and 900%, respectively, followed by unloading at the same strain rate, and the sequential loading−unloading cycles with increased stretch ratios were performed without waiting time. The stretching velocity was fixed at 100 mm/min.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/gels9020145/s1, Figure S1: Frequency dependence of (a) storage moduli G′, (b) loss moduli G″ and (c) loss factor tan δ for DN−Na+−1.0 M−AP and DN−Na+−1.0 M−E hydrogels.
Author Contributions
Conceptualization, S.Z., Y.Y. and Y.Z.; methodology, W.Y., F.Z. and Q.Z.; data acquisition, Y.W., Z.W. and L.C.; writing—review and editing, Y.Y. and Y.Z.; supervision, Q.Z.; project administration, Q.Z.; funding acquisition, Q.Z., W.Y. and F.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by National Natural Science Foundation of China (32173026, 52103019), the Shanxi-Zheda Institute of Advanced Materials and Chemical Engineering (2021SX-TD001, 2021SX-TD010), the 2021 Major Science and Technology Program of Taiyuan, and the Fund for Shanxi “1331 Project”.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Simha, N.; Carlson, C.; Lewis, J. Evaluation of fracture toughness of cartilage by micropenetration. J. Mater. Sci. Mater. Med. 2004, 15, 631–639. [Google Scholar] [PubMed]
- Kerin, A.; Wisnom, M.; Adams, M. The compressive strength of articular cartilage. Proc. Inst. Mech. Eng. Part H J. Eng. Med. 1998, 212, 273–280. [Google Scholar]
- Zhao, X.; Chen, X.; Yuk, H.; Lin, S.; Liu, X.; Parada, G. Soft Materials by Design: Unconventional Polymer Networks Give Extreme Properties. Chem. Rev. 2021, 121, 4309–4372. [Google Scholar]
- Sakai, T.; Matsunaga, T.; Yamamoto, Y.; Ito, C.; Yoshida, R.; Suzuki, S.; Sasaki, N.; Shibayama, M.; Chung, U.-I. Design and fabrication of a high-strength hydrogel with ideally homogeneous network structure from tetrahedron-like macromonomers. Macromolecules 2008, 41, 5379–5384. [Google Scholar] [CrossRef]
- Gong, J.P.; Katsuyama, Y.; Kurokawa, T.; Osada, Y. Double-network hydrogels with extremely high mechanical strength. Adv. Mater. 2003, 15, 1155–1158. [Google Scholar] [CrossRef]
- Okumura, Y.; Ito, K. The polyrotaxane gel: A topological gel by figure-of-eight cross-links. Adv. Mater. 2001, 13, 485–487. [Google Scholar] [CrossRef]
- Creton, C. 50th anniversary perspective: Networks and gels: Soft but dynamic and tough. Macromolecules 2017, 50, 8297–8316. [Google Scholar]
- Nakajima, T. Generalization of the sacrificial bond principle for gel and elastomer toughening. Polym. J. 2017, 49, 477–485. [Google Scholar]
- Sun, T.L.; Kurokawa, T.; Kuroda, S.; Ihsan, A.B.; Akasaki, T.; Sato, K.; Haque, M.A.; Nakajima, T.; Gong, J.P. Physical hydrogels composed of polyampholytes demonstrate high toughness and viscoelasticity. Nat. Mater. 2013, 12, 932–937. [Google Scholar] [CrossRef]
- Gong, J.P. Why are double network hydrogels so tough? Soft Matter 2010, 6, 2583–2590. [Google Scholar]
- Chen, Q.; Zhu, L.; Zhao, C.; Wang, Q.; Zheng, J. A robust, one-pot synthesis of highly mechanical and recoverable double network hydrogels using thermoreversible sol-gel polysaccharide. Adv. Mater. 2013, 25, 4171–4176. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, K.; Takehisa, T. Nanocomposite hydrogels: A unique organic–inorganic network structure with extraordinary mechanical, optical, and swelling/de-swelling properties. Adv. Mater. 2002, 14, 1120–1124. [Google Scholar] [CrossRef]
- Haraguchi, K.; Takehisa, T.; Fan, S. Effects of clay content on the properties of nanocomposite hydrogels composed of poly (N-isopropylacrylamide) and clay. Macromolecules 2002, 35, 10162–10171. [Google Scholar]
- Illeperuma, W.R.; Sun, J.-Y.; Suo, Z.; Vlassak, J.J. Fiber-reinforced tough hydrogels. Extrem. Mech. Lett. 2014, 1, 90–96. [Google Scholar]
- Lin, S.; Cao, C.; Wang, Q.; Gonzalez, M.; Dolbow, J.E.; Zhao, X. Design of stiff, tough and stretchy hydrogel composites via nanoscale hybrid crosslinking and macroscale fiber reinforcement. Soft Matter 2014, 10, 7519–7527. [Google Scholar]
- Lin, W.-C.; Fan, W.; Marcellan, A.; Hourdet, D.; Creton, C. Large strain and fracture properties of poly (dimethylacrylamide)/silica hybrid hydrogels. Macromolecules 2010, 43, 2554–2563. [Google Scholar]
- Utech, S.; Boccaccini, A.R. A review of hydrogel-based composites for biomedical applications: Enhancement of hydrogel properties by addition of rigid inorganic fillers. J. Mater. Sci. 2016, 51, 271–310. [Google Scholar] [CrossRef]
- Nonoyama, T.; Gong, J.P. Double-network hydrogel and its potential biomedical application: A review. Proc. Inst. Mech. Eng. Part H J. Eng. Med. 2015, 229, 853–863. [Google Scholar] [CrossRef]
- Nonoyama, T.; Wada, S.; Kiyama, R.; Kitamura, N.; Mredha, M.T.I.; Zhang, X.; Kurokawa, T.; Nakajima, T.; Takagi, Y.; Yasuda, K. Double-network hydrogels strongly bondable to bones by spontaneous osteogenesis penetration. Adv. Mater. 2016, 28, 6740–6745. [Google Scholar]
- Yasuda, K.; Gong, J.P.; Katsuyama, Y.; Nakayama, A.; Tanabe, Y.; Kondo, E.; Ueno, M.; Osada, Y. Biomechanical properties of high-toughness double network hydrogels. Biomaterials 2005, 26, 4468–4475. [Google Scholar]
- Azuma, C.; Yasuda, K.; Tanabe, Y.; Taniguro, H.; Kanaya, F.; Nakayama, A.; Chen, Y.M.; Gong, J.P.; Osada, Y. Biodegradation of high-toughness double network hydrogels as potential materials for artificial cartilage. J. Biomed. Mater. Res. Part A Off. J. Soc. Biomater. Jpn. Soc. Biomater. Aust. Soc. Biomater. Korean Soc. Biomater. 2007, 81, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Arakaki, K.; Kitamura, N.; Fujiki, H.; Kurokawa, T.; Iwamoto, M.; Ueno, M.; Kanaya, F.; Osada, Y.; Gong, J.P.; Yasuda, K. Artificial cartilage made from a novel double-network hydrogel: In vivo effects on the normal cartilage and ex vivo evaluation of the friction property. J. Biomed. Mater. Res. Part A Off. J. Soc. Biomater. Jpn. Soc. Biomater. Aust. Soc. Biomater. Korean Soc. Biomater. 2010, 93, 1160–1168. [Google Scholar]
- Yasuda, K.; Kitamura, N.; Gong, J.P.; Arakaki, K.; Kwon, H.J.; Onodera, S.; Chen, Y.M.; Kurokawa, T.; Kanaya, F.; Ohmiya, Y. A novel double-network hydrogel induces spontaneous articular cartilage regeneration in vivo in a large osteochondral defect. Macromol. Biosci. 2009, 9, 307–316. [Google Scholar] [PubMed]
- Kitamura, N.; Yasuda, K.; Ogawa, M.; Arakaki, K.; Kai, S.; Onodera, S.; Kurokawa, T.; Gong, J.P. Induction of spontaneous hyaline cartilage regeneration using a double-network gel: Efficacy of a novel therapeutic strategy for an articular cartilage defect. Am. J. Sport. Med. 2011, 39, 1160–1169. [Google Scholar]
- Cacopardo, L.; Guazzelli, N.; Nossa, R.; Mattei, G.; Ahluwalia, A. Engineering hydrogel viscoelasticity. J. Mech. Behav. Biomed. Mater. 2019, 89, 162–167. [Google Scholar]
- Drozdov, A.D.; Christiansen, J.d. Tuning the viscoelastic response of hydrogel scaffolds with covalent and dynamic bonds. J. Mech. Behav. Biomed. Mater. 2022, 130, 105179. [Google Scholar]
- Cameron, A.R.; Frith, J.E.; Gomez, G.A.; Yap, A.S.; Cooper-White, J.J. The effect of time-dependent deformation of viscoelastic hydrogels on myogenic induction and Rac1 activity in mesenchymal stem cells. Biomaterials 2014, 35, 1857–1868. [Google Scholar] [CrossRef]
- Chaudhuri, O.; Gu, L.; Darnell, M.; Klumpers, D.; Bencherif, S.A.; Weaver, J.C.; Huebsch, N.; Mooney, D.J. Substrate stress relaxation regulates cell spreading. Nat. Commun. 2015, 6, 6365. [Google Scholar] [CrossRef]
- Chaudhuri, O.; Gu, L.; Klumpers, D.; Darnell, M.; Bencherif, S.A.; Weaver, J.C.; Huebsch, N.; Lee, H.-p.; Lippens, E.; Duda, G.N. Hydrogels with tunable stress relaxation regulate stem cell fate and activity. Nat. Mater. 2016, 15, 326–334. [Google Scholar]
- Chaudhuri, O.; Cooper-White, J.; Janmey, P.A.; Mooney, D.J.; Shenoy, V.B. Effects of extracellular matrix viscoelasticity on cellular behaviour. Nature 2020, 584, 535–546. [Google Scholar]
- Charrier, E.E.; Pogoda, K.; Wells, R.G.; Janmey, P.A. Control of cell morphology and differentiation by substrates with independently tunable elasticity and viscous dissipation. Nat. Commun. 2018, 9, 449. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.-Y.; Zhao, X.; Illeperuma, W.R.; Chaudhuri, O.; Oh, K.H.; Mooney, D.J.; Vlassak, J.J.; Suo, Z. Highly stretchable and tough hydrogels. Nature 2012, 489, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.J.; Sun, T.L.; Zhang, A.K.; Ikura, Y.; Nakajima, T.; Nonoyama, T.; Kurokawa, T.; Ito, O.; Ishitobi, H.; Gong, J.P. Tough physical double-network hydrogels based on amphiphilic triblock copolymers. Adv. Mater. 2016, 28, 4884–4890. [Google Scholar] [PubMed]
- Luo, F.; Sun, T.L.; Nakajima, T.; King, D.R.; Kurokawa, T.; Zhao, Y.; Ihsan, A.B.; Li, X.; Guo, H.; Gong, J.P. Strong and tough polyion-complex hydrogels from oppositely charged polyelectrolytes: A comparative study with polyampholyte hydrogels. Macromolecules 2016, 49, 2750–2760. [Google Scholar]
- Choi, S.; Choi, Y.; Kim, J. Anisotropic hybrid hydrogels with superior mechanical properties reminiscent of tendons or ligaments. Adv. Funct. Mater. 2019, 29, 1904342. [Google Scholar] [CrossRef]
- Xie, Y.; Gao, S.; Ling, Z.; Lai, C.; Huang, Y.; Wang, J.; Wang, C.; Chu, F.; Xu, F.; Dumont, M.-J. A multiscale biomimetic strategy to design strong, tough hydrogels by tuning the self-assembly behavior of cellulose. J. Mater. Chem. A 2022, 10, 13685–13696. [Google Scholar] [CrossRef]
- Fan, X.; Fang, Y.; Zhou, W.; Yan, L.; Xu, Y.; Zhu, H.; Liu, H. Mussel foot protein inspired tough tissue-selective underwater adhesive hydrogel. Mater. Horiz. 2021, 8, 997–1007. [Google Scholar]
- Han, L.; Yan, L.; Wang, K.; Fang, L.; Zhang, H.; Tang, Y.; Ding, Y.; Weng, L.-T.; Xu, J.; Weng, J. Tough, self-healable and tissue-adhesive hydrogel with tunable multifunctionality. NPG Asia Mater. 2017, 9, e372. [Google Scholar] [CrossRef]
- Song, G.; Zhang, L.; He, C.; Fang, D.-C.; Whitten, P.G.; Wang, H. Facile fabrication of tough hydrogels physically cross-linked by strong cooperative hydrogen bonding. Macromolecules 2013, 46, 7423–7435. [Google Scholar]
- Luo, F.; Sun, T.L.; Nakajima, T.; Kurokawa, T.; Zhao, Y.; Sato, K.; Ihsan, A.B.; Li, X.; Guo, H.; Gong, J.P. Oppositely charged polyelectrolytes form tough, self-healing, and rebuildable hydrogels. Adv. Mater. 2015, 27, 2722–2727. [Google Scholar]
- Yu, H.C.; Zheng, S.Y.; Fang, L.; Ying, Z.; Du, M.; Wang, J.; Ren, K.F.; Wu, Z.L.; Zheng, Q. Reversibly transforming a highly swollen polyelectrolyte hydrogel to an extremely tough one and its application as a tubular grasper. Adv. Mater. 2020, 32, 2005171. [Google Scholar]
- Huang, Y.; Xiao, L.; Zhou, J.; Liu, T.; Yan, Y.; Long, S.; Li, X. Strong Tough Polyampholyte Hydrogels via the Synergistic Effect of Ionic and Metal–Ligand Bonds. Adv. Funct. Mater. 2021, 31, 2103917. [Google Scholar]
- Ahmed, S.; Nakajima, T.; Kurokawa, T.; Haque, M.A.; Gong, J.P. Brittle–ductile transition of double network hydrogels: Mechanical balance of two networks as the key factor. Polymer 2014, 55, 914–923. [Google Scholar] [CrossRef] [Green Version]
- Urzhumtsev, Y.S. Time-temperature superposition. Review. Polym. Mech. 1975, 11, 57–72. [Google Scholar] [CrossRef]
- Rubinstein, M.; Colby, R.H. Polymer Physics; Oxford University Press: New York, NY, USA, 2003; Volume 23. [Google Scholar]
- Cui, K.; Sun, T.L.; Liang, X.; Nakajima, K.; Ye, Y.N.; Chen, L.; Kurokawa, T.; Gong, J.P. Multiscale energy dissipation mechanism in tough and self-healing hydrogels. Phys. Rev. Lett. 2018, 121, 185501. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.N.; Cui, K.; Hong, W.; Li, X.; Yu, C.; Hourdet, D.; Nakajima, T.; Kurokawa, T.; Gong, J.P. Molecular mechanism of abnormally large nonsoftening deformation in a tough hydrogel. Proc. Natl. Acad. Sci. 2021, 118, e2014694118. [Google Scholar] [CrossRef]
- Zheng, S.Y.; Ding, H.; Qian, J.; Yin, J.; Wu, Z.L.; Song, Y.; Zheng, Q. Metal-coordination complexes mediated physical hydrogels with high toughness, stick–slip tearing behavior, and good processability. Macromolecules 2016, 49, 9637–9646. [Google Scholar]
- Nakajima, T.; Kurokawa, T.; Ahmed, S.; Wu, W.-l.; Gong, J.P. Characterization of internal fracture process of double network hydrogels under uniaxial elongation. Soft Matter 2013, 9, 1955–1966. [Google Scholar] [CrossRef]
- Zheng, Y.; Kiyama, R.; Matsuda, T.; Cui, K.; Li, X.; Cui, W.; Guo, Y.; Nakajima, T.; Kurokawa, T.; Gong, J.P. Nanophase separation in immiscible double network elastomers induces synergetic strengthening, toughening, and fatigue resistance. Chem. Mater. 2021, 33, 3321–3334. [Google Scholar] [CrossRef]
- Matsuda, T.; Nakajima, T.; Fukuda, Y.; Hong, W.; Sakai, T.; Kurokawa, T.; Chung, U.-I.; Gong, J.P. Yielding criteria of double network hydrogels. Macromolecules 2016, 49, 1865–1872. [Google Scholar]
- Millereau, P.; Ducrot, E.; Clough, J.M.; Wiseman, M.E.; Brown, H.R.; Sijbesma, R.P.; Creton, C. Mechanics of elastomeric molecular composites. Proc. Natl. Acad. Sci. USA 2018, 115, 9110–9115. [Google Scholar] [PubMed]
- Ducrot, E.; Creton, C. Characterizing large strain elasticity of brittle elastomeric networks by embedding them in a soft extensible matrix. Adv. Funct. Mater. 2016, 26, 2482–2492. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and soft acids and bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A Cryst. Phys. Diffr. Theor. Gen. Crystallogr. 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Côté, A.P.; Shimizu, G.K. The supramolecular chemistry of the sulfonate group in extended solids. Coord. Chem. Rev. 2003, 245, 49–64. [Google Scholar] [CrossRef]
- Wang, Z.J.; Jiang, J.; Mu, Q.; Maeda, S.; Nakajima, T.; Gong, J.P. Azo-Crosslinked Double-Network Hydrogels Enabling Highly Efficient Mechanoradical Generation. J. Am. Chem. Soc. 2022, 144, 3154–3161. [Google Scholar] [CrossRef]
- Fukao, K.; Nakajima, T.; Nonoyama, T.; Kurokawa, T.; Kawai, T.; Gong, J.P. Effect of Relative Strength of Two Networks on the Internal Fracture Process of Double Network Hydrogels As Revealed by in Situ Small-Angle X-ray Scattering. Macromolecules 2020, 53, 1154–1163. [Google Scholar] [CrossRef]
Figure 1.
Schematics of design and transformation of the virgin elastic DN hydrogels to viscoelastic materials via metal-coordination complex. As-prepared DN hydrogels in metal–ion solutions are denoted as “DN−Mn+−Cm−AP”, while the DN hydrogels equilibrated in water afterward are denoted as “DN−Mn+−Cm−E”, where the notations “AP” and “E” mean “as-prepared” and “equilibrated”, respectively, and Cm (M) represents the metal–ion concentration in solution. The metal–ions used in this work are Na+, Fe3+, and Zr4+.
Figure 1.
Schematics of design and transformation of the virgin elastic DN hydrogels to viscoelastic materials via metal-coordination complex. As-prepared DN hydrogels in metal–ion solutions are denoted as “DN−Mn+−Cm−AP”, while the DN hydrogels equilibrated in water afterward are denoted as “DN−Mn+−Cm−E”, where the notations “AP” and “E” mean “as-prepared” and “equilibrated”, respectively, and Cm (M) represents the metal–ion concentration in solution. The metal–ions used in this work are Na+, Fe3+, and Zr4+.

Figure 2.
Linear dynamic mechanical behavior of virgin DN, DN−Zr4+−0.1 M−AP and DN−Zr4+−0.1 M−E hydrogels. Frequency dependence of (a) storage moduli G′, (b) loss moduli G″ and (c) loss factor tan δ for virgin DN hydrogels. Frequency dependence of (d) storage moduli G′, (e) loss moduli G″ and (f) loss factor tan δ for DN−Zr4+−0.1 M−AP; frequency dependence of (g) storage moduli G′, (h) loss moduli G″ and (i) loss factor tan δ for DN−Zr4+−0.1 M−E hydrogels.
Figure 2.
Linear dynamic mechanical behavior of virgin DN, DN−Zr4+−0.1 M−AP and DN−Zr4+−0.1 M−E hydrogels. Frequency dependence of (a) storage moduli G′, (b) loss moduli G″ and (c) loss factor tan δ for virgin DN hydrogels. Frequency dependence of (d) storage moduli G′, (e) loss moduli G″ and (f) loss factor tan δ for DN−Zr4+−0.1 M−AP; frequency dependence of (g) storage moduli G′, (h) loss moduli G″ and (i) loss factor tan δ for DN−Zr4+−0.1 M−E hydrogels.

Figure 3.
Master curves of the storage modulus G′, loss modulus G″, and loss factor tanδ of DN−Zr4+−0.1 M−AP hydrogels (a) and DN−Zr4+−0.1 M−E hydrogels (b). The measurements were performed from 0.1 to 100 rad/s at a shear strain of 0.2%, at different temperatures from 8 °C to 88 °C with an interval temperature of 8 °C, and the results were obtained by performing classical time–temperature superposition shifts at a reference temperature of at 24 °C, deduced from the data in Figure 2 using horizontal shift factor aT and without vertical shift. Arrhenius plot depicting the temperature dependence of the shift factors aT used for generating master curves of DN−Zr4+−0.1 M−AP hydrogels (c) and DN−Zr4+−0.1 M−E hydrogels (d). The apparent activation energy values were calculated from the slope of the curves.
Figure 3.
Master curves of the storage modulus G′, loss modulus G″, and loss factor tanδ of DN−Zr4+−0.1 M−AP hydrogels (a) and DN−Zr4+−0.1 M−E hydrogels (b). The measurements were performed from 0.1 to 100 rad/s at a shear strain of 0.2%, at different temperatures from 8 °C to 88 °C with an interval temperature of 8 °C, and the results were obtained by performing classical time–temperature superposition shifts at a reference temperature of at 24 °C, deduced from the data in Figure 2 using horizontal shift factor aT and without vertical shift. Arrhenius plot depicting the temperature dependence of the shift factors aT used for generating master curves of DN−Zr4+−0.1 M−AP hydrogels (c) and DN−Zr4+−0.1 M−E hydrogels (d). The apparent activation energy values were calculated from the slope of the curves.

Figure 4.
Tensile behaviors and cyclic tensile behaviors for various DN hydrogels with Zr4+ as metal ions for the metal coordination complex. (a) Rate-independent elastic tensile behaviors of the virgin DN hydrogels under different deformation rates ranging from 10 to 1000 mm/min. (b) Rate-dependent viscoelastic tensile behaviors of the DN−Zr4+−0.1 M−AP hydrogels under different deformation rates ranging from 10 to 1000 mm/min. (c) Rate-dependent viscoelastic tensile behaviors of the DN−Zr4+−0.1 M−E hydrogels under different deformation rates ranging from 10 to 1000 mm/min. (d–f) Sequential loading–unloading cycles for the virgin DN gels (d), DN−Zr4+−0.1 M−AP hydrogels (e), and DN−Zr4+−0.1 M−E hydrogels (f) at a deformation rate of 100 mm/min. The virgin DN hydrogels show completely irreversible hysteresis, while the rate-dependent viscoelastic DN−Zr4+−0.1 M−AP hydrogels and DN−Zr4+−0.1 M−E hydrogels show partially reversible hysteresis. The total mechanical hysteresis Wtotal (g), the reversible mechanical hysteresis Wre (h), and the recovery ratio Wre/Wtotal (i) as functions of tensile strain for various DN hydrogels.
Figure 4.
Tensile behaviors and cyclic tensile behaviors for various DN hydrogels with Zr4+ as metal ions for the metal coordination complex. (a) Rate-independent elastic tensile behaviors of the virgin DN hydrogels under different deformation rates ranging from 10 to 1000 mm/min. (b) Rate-dependent viscoelastic tensile behaviors of the DN−Zr4+−0.1 M−AP hydrogels under different deformation rates ranging from 10 to 1000 mm/min. (c) Rate-dependent viscoelastic tensile behaviors of the DN−Zr4+−0.1 M−E hydrogels under different deformation rates ranging from 10 to 1000 mm/min. (d–f) Sequential loading–unloading cycles for the virgin DN gels (d), DN−Zr4+−0.1 M−AP hydrogels (e), and DN−Zr4+−0.1 M−E hydrogels (f) at a deformation rate of 100 mm/min. The virgin DN hydrogels show completely irreversible hysteresis, while the rate-dependent viscoelastic DN−Zr4+−0.1 M−AP hydrogels and DN−Zr4+−0.1 M−E hydrogels show partially reversible hysteresis. The total mechanical hysteresis Wtotal (g), the reversible mechanical hysteresis Wre (h), and the recovery ratio Wre/Wtotal (i) as functions of tensile strain for various DN hydrogels.

Figure 5.
Tensile behaviors and cyclic tensile behaviors for various DN hydrogels with Na+ as metal ions. (a,b) Rate-independent tensile behaviors of the DN−Na+−1.0 M−AP (a) and DN−Na+−1.0 M−E (b) hydrogels under different deformation rates ranging from 10 to 1000 mm/min. (c,d) Sequential loading–unloading cycles for the DN−Na+−1.0 M−AP hydrogels (c) and DN−Na+−1.0 M−E hydrogels (d) at a deformation rate of 100 mm/min. The rate-independent elastic DN−Na+−1.0 M−AP and DN−Na+−1.0 M−E hydrogels show completely irreversible hysteresis.
Figure 5.
Tensile behaviors and cyclic tensile behaviors for various DN hydrogels with Na+ as metal ions. (a,b) Rate-independent tensile behaviors of the DN−Na+−1.0 M−AP (a) and DN−Na+−1.0 M−E (b) hydrogels under different deformation rates ranging from 10 to 1000 mm/min. (c,d) Sequential loading–unloading cycles for the DN−Na+−1.0 M−AP hydrogels (c) and DN−Na+−1.0 M−E hydrogels (d) at a deformation rate of 100 mm/min. The rate-independent elastic DN−Na+−1.0 M−AP and DN−Na+−1.0 M−E hydrogels show completely irreversible hysteresis.

Figure 6.
Tensile behaviors and cyclic tensile behaviors for various DN hydrogels coordinated with Fe3+ ions. (a,b) Rate-independent tensile behaviors of the DN−Fe3+−0.1 M−AP (a) and rate-dependent tensile behaviors of the DN−Fe3+−0.1 M−E (b) hydrogels under different deformation rates ranging from 10 to 1000 mm/min. (c,d) Sequential loading–unloading cycles for the DN−Fe3+−0.1 M−AP hydrogels (c) and DN−Fe3+−0.1 M−E hydrogels (d) at a deformation rate of 100 mm/min. The rate-independent elastic DN−Fe3+−0.1 M−AP hydrogels show irreversible hysteresis, while the rate-dependent viscoelastic DN−Fe3+−0.1 M−E hydrogels show partially reversible hysteresis owing to the metal coordination interactions.
Figure 6.
Tensile behaviors and cyclic tensile behaviors for various DN hydrogels coordinated with Fe3+ ions. (a,b) Rate-independent tensile behaviors of the DN−Fe3+−0.1 M−AP (a) and rate-dependent tensile behaviors of the DN−Fe3+−0.1 M−E (b) hydrogels under different deformation rates ranging from 10 to 1000 mm/min. (c,d) Sequential loading–unloading cycles for the DN−Fe3+−0.1 M−AP hydrogels (c) and DN−Fe3+−0.1 M−E hydrogels (d) at a deformation rate of 100 mm/min. The rate-independent elastic DN−Fe3+−0.1 M−AP hydrogels show irreversible hysteresis, while the rate-dependent viscoelastic DN−Fe3+−0.1 M−E hydrogels show partially reversible hysteresis owing to the metal coordination interactions.

Figure 7.
(a–c) The rescaled stress σλs2–rescaled stretch ratio λλs curves for DN hydrogels coordinated with Na+ (a), Fe3+ (b), and Zr4+ (c) as metal ions under different deformation rates ranging from 10 to 1000 mm/min.
Figure 7.
(a–c) The rescaled stress σλs2–rescaled stretch ratio λλs curves for DN hydrogels coordinated with Na+ (a), Fe3+ (b), and Zr4+ (c) as metal ions under different deformation rates ranging from 10 to 1000 mm/min.

Figure 8.
(a,b) Dependence of the Young’s modulus E (a) and the rescaled Young’s modulus Eλs (b) on strain rate for various DN hydrogels. (c,d) Dependence of the yielding stretch ratio λy (c) and the rescaled yielding stretch ratio λyλs (d) on strain rate for various DN hydrogels. (e,f) Dependence of the yielding stress σy (c) and the rescaled yielding stress σyλs2 (d) on strain rate for various DN hydrogels.
Figure 8.
(a,b) Dependence of the Young’s modulus E (a) and the rescaled Young’s modulus Eλs (b) on strain rate for various DN hydrogels. (c,d) Dependence of the yielding stretch ratio λy (c) and the rescaled yielding stretch ratio λyλs (d) on strain rate for various DN hydrogels. (e,f) Dependence of the yielding stress σy (c) and the rescaled yielding stress σyλs2 (d) on strain rate for various DN hydrogels.

Figure 9.
The ratio of breaking force of first network strands fb/fb,0 for various DN hydrogels relative to the virgin DN gels. The fb and fb,0 represent the breaking force of first network in various DN hydrogels and the virgin DN gels, respectively.
Figure 9.
The ratio of breaking force of first network strands fb/fb,0 for various DN hydrogels relative to the virgin DN gels. The fb and fb,0 represent the breaking force of first network in various DN hydrogels and the virgin DN gels, respectively.

Figure 10.
(a–c) Dependence of the stretch ratio at break λb (a), the stress at break σb (b), and work of extension Wb (c) on strain rate for various DN hydrogels.
Figure 10.
(a–c) Dependence of the stretch ratio at break λb (a), the stress at break σb (b), and work of extension Wb (c) on strain rate for various DN hydrogels.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhu, S.; Wang, Y.; Wang, Z.; Chen, L.; Zhu, F.; Ye, Y.; Zheng, Y.; Yu, W.; Zheng, Q. Metal-Coordinated Dynamics and Viscoelastic Properties of Double-Network Hydrogels. Gels 2023, 9, 145. https://doi.org/10.3390/gels9020145
AMA Style
Zhu S, Wang Y, Wang Z, Chen L, Zhu F, Ye Y, Zheng Y, Yu W, Zheng Q. Metal-Coordinated Dynamics and Viscoelastic Properties of Double-Network Hydrogels. Gels. 2023; 9(2):145. https://doi.org/10.3390/gels9020145
Chicago/Turabian StyleZhu, Shilei, Yan Wang, Zhe Wang, Lin Chen, Fengbo Zhu, Yanan Ye, Yong Zheng, Wenwen Yu, and Qiang Zheng. 2023. "Metal-Coordinated Dynamics and Viscoelastic Properties of Double-Network Hydrogels" Gels 9, no. 2: 145. https://doi.org/10.3390/gels9020145
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.