
Protein Hydrolysis and Glycosylation as Strategies to Produce Bioactive Ingredients from Unmarketable Prawns

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Preparation of Shrimp Hydrolysate and Maillard Glycation
2.3. Molecular Weight Distribution
2.4. Determination of Protein Content
2.5. Degree of Hydrolysis (DH), Free Amino Acids, and Browning
2.6. Measurement of Fluorescence
2.7. Total Phenols (TPs)
2.8. Fourier Transform Infrared Spectroscopy (FTIR)
2.9. Antioxidant Activity
2.9.1. ABTS•+ Radical Cation Scavenging Activity (ABTS•+)
2.9.2. Oxygen Radical Absorbance Capacity (ORAC)
2.9.3. DPPH· Radical Scavenging Activity
2.9.4. Ferric-Reducing Antioxidant Power (FRAP)
2.10. Prolyl Endopeptidase (PEP)-Inhibiting Activity
2.11. Statistical Analysis
3. Results and Discussion
3.1. Effect of Reaction Time on DH and Free Amino Acid Content of Shrimp
3.2. Evolution of the Average Molecular Weight (Mw) during Protein Hydrolysis
3.3. Evolution of the Average Molecular Weight during the Maillard Reaction
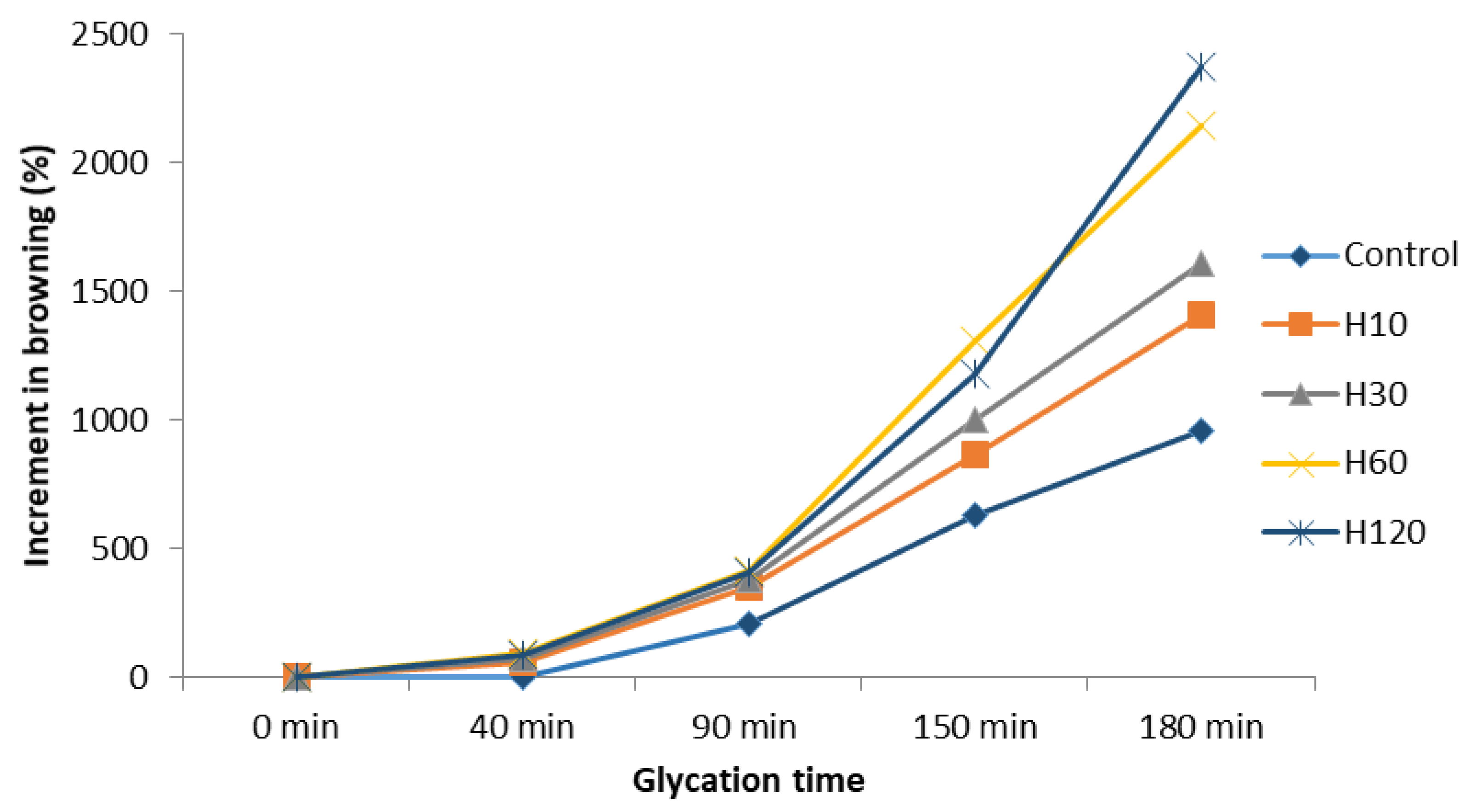
3.4. Evolution of Browning during the Maillard Reaction
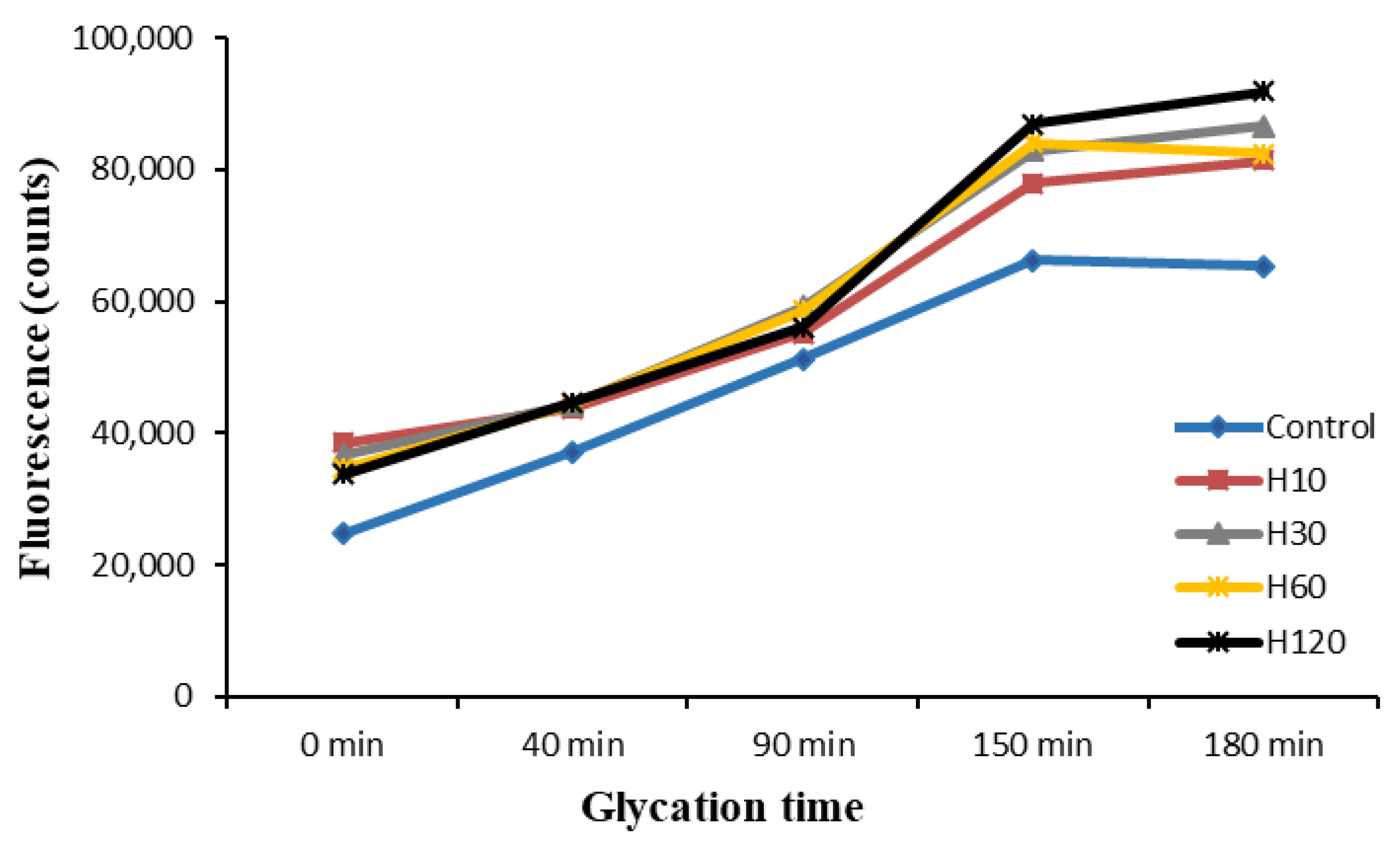
3.5. Evolution of Fluorescence during the Maillard Reaction
3.6. Phenol Content
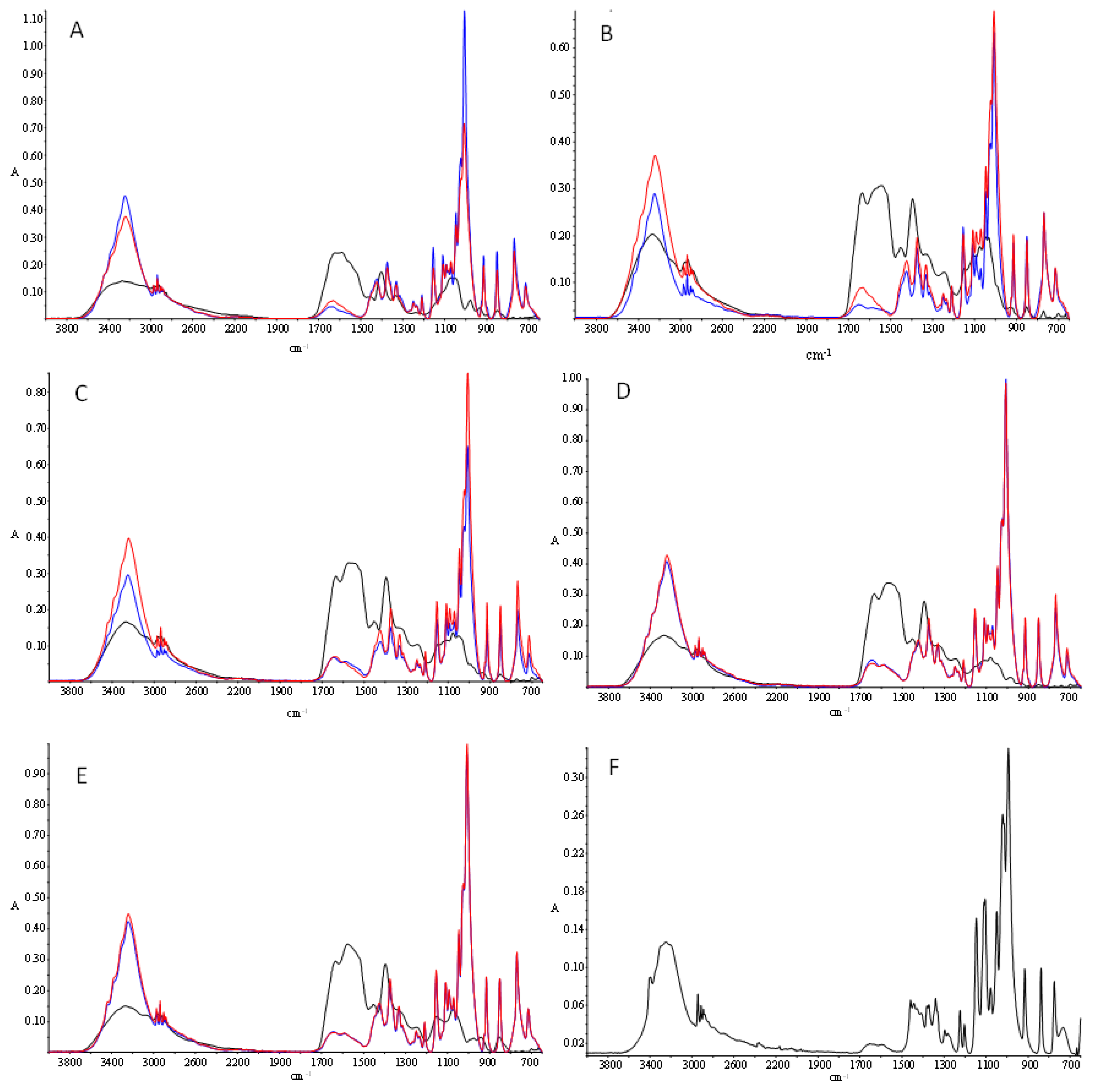
3.7. Fourier Transform Infrared Spectroscopy (FTIR)
3.8. Prolyl Endopeptidase-Inhibiting Activity (PEP)
3.9. Antioxidant Activity
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Baisier, W.M.; Labuza, T.P. Maillard browning kinetics in a liquid model system. J. Agric. Food Chem. 1992, 40, 707–713. [Google Scholar] [CrossRef]
- Benzie, I.F.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “Antioxidant Power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. LWT Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Cael, J.J.; Koenig, J.L.; Blackwell, J. Infrared and Raman spectroscopy of carbohydrates. Part IV. Identification of configuration- and conformation-sensitive modes for D-glucose by normal coordinate analysis. Carbohydr. Res. 1974, 32, 79–91. [Google Scholar] [CrossRef]
- Cai, L.; Li, D.; Dong, Z.; Cao, A.; Lin, H.; Li, J. Change regularity of the characteristics of Maillard reaction products derived from xylose and Chinese shrimp waste hydrolysates. LWT Food Sci. Technol. 2016, 65, 908–916. [Google Scholar] [CrossRef]
- Chen, H.M.; Muramoto, K.; Yamauchi, F.; Fujimoto, K.; Nokihara, K. Antioxidative properties of histidine-containing peptides designed from peptide fragments found in the digests of a soybean protein. J. Agric. Food Chem. 1998, 46, 49–53. [Google Scholar] [CrossRef]
- Delgado-Andrade, C.; Rufián-Henares, J.A.; Morales, F.J. Assessing the antioxidant activity of melanoidins from coffee brews by different antioxidant methods. J. Agric. Food Chem. 2005, 53, 7832–7836. [Google Scholar] [CrossRef] [Green Version]
- Djellouli, M.; López-Caballero, M.E.; Arancibia, M.Y.; Karam, N.; Martínez-Alvarez, O. Antioxidant and antimicrobial enhancement by reaction of protein hydrolysates derived from shrimp by-products with glucosamine. Waste Biomass Valorization 2020, 11, 2491–2505. [Google Scholar] [CrossRef]
- Fay, L.B.; Brevard, H. Contribution of mass spectrometry to the study of the Maillard reaction in food. Mass Spectrom. Rev. 2005, 24, 487–507. [Google Scholar] [CrossRef]
- García-Horsman, J.A.; Männistö, P.T.; Venäläinen, J.I. On the role of prolyl oligopeptidase in health and disease. Neuropeptides 2007, 41, 1–24. [Google Scholar] [CrossRef]
- Gómez-Estaca, J.; Montero, P.; Gómez-Guillén, M.C. Shrimp (Litopenaeus vannamei) muscle proteins as source to develop edible films. Food Hydrocoll. 2014, 41, 86–94. [Google Scholar] [CrossRef] [Green Version]
- Gu, F.-L.; Kim, J.M.; Abbas, S.; Zhang, X.-M.; Xia, S.-Q.; Chen, Z.-X. Structure and antioxidant activity of high molecular weight Maillard reaction products from casein–glucose. Food Chem. 2010, 120, 505–511. [Google Scholar] [CrossRef]
- Guerard, F.; Sumaya-Martinez, M.T.; Laroque, D.; Chabeaud, A.; Dufossé, L. Optimization of free radical scavenging activity by response surface methodology in the hydrolysis of shrimp processing discards. Process Biochem. 2007, 42, 1486–1491. [Google Scholar] [CrossRef]
- Hodge, J.E. Dehydrated foods, chemistry of browning reactions in model systems. J. Agric. Food Chem. 1953, 1, 928–943. [Google Scholar] [CrossRef]
- Hofmann, T. Studies on the relationship between molecular weight and the color potency of fractions obtained by thermal treatment of glucose/amino acid and glucose/protein solutions by using ultracentrifugation and color dilution techniques. J. Agric. Food Chem. 1998, 46, 3891–3895. [Google Scholar] [CrossRef]
- Horvat, S.; Jakas, A. Peptide and amino acid glycation: New insights into the Maillard reaction. J. Pept. Sci. 2004, 10, 119–137. [Google Scholar] [CrossRef]
- Ibrahim, M.; Alaam, M.; El-Haes, H.; Jalbout, A.F.; De Leon, A. Analysis of the structure and vibrational spectra of glucose and fructose. Eclet. Quim. 2006, 31, 15–21. [Google Scholar] [CrossRef]
- Jiang, Z.; Rai, D.K.; O’Connor, P.M.; Brodkorb, A. Heat-induced Maillard reaction of the tripeptide IPP and ribose: Structural characterization and implication on bioactivity. Food Res. Int. 2013, 50, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Ketnawa, S.; Benjakul, S.; Martínez-Alvarez, O.; Rawdkuen, S. Physical, chemical, and microbiological properties of fish tofu containing shrimp hydrolysate. Fish. Sci. 2016, 82, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Ketnawa, S.; Martínez-Alvarez, O.; Gómez-Estaca, J.; Gómez-Guillén, M.C.; Benjakul, S.; Rawdkuen, S. Obtaining of functional components from cooked shrimp (Penaeus vannamei) by enzymatic hydrolysis. Food Biosci. 2016, 15, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Khantaphant, S.; Benjakul, S.; Kishimura, H. Antioxidative and ACE inhibitory activities of protein hydrolysates from the muscle of brownstripe red snapper prepared using pyloric caeca and commercial proteases. Process Biochem. 2011, 46, 318–327. [Google Scholar] [CrossRef]
- Kim, J.-S.; Lee, Y.-S. Study of Maillard reaction products derived from aqueous model systems with different peptide chain lengths. Food Chem. 2009, 116, 846–853. [Google Scholar] [CrossRef]
- Kim, J.S.; Lee, Y.S. FT-IR and X-RAY diffraction characterization of melanoidins formed from glucose and fructose with amino acid enantiomers in the maillard reaction. Food Sci. Biotechnol. 2009, 18, 546–551. [Google Scholar]
- Kong, J.; Yu, S. Fourier transform infrared spectroscopic analysis of protein secondary structures. Acta Bioch. Bioph. Sin. 2007, 39, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lajmi, K.; Gómez-Estaca, J.; Hammami, M.; Martínez-Alvarez, O. Upgrading collagenous smooth hound by-products: Effect of hydrolysis conditions, in vitro gastrointestinal digestion and encapsulation on bioactive properties. Food Biosci. 2019, 28, 99–108. [Google Scholar] [CrossRef]
- Lan, X.; Liu, P.; Xia, S.; Jia, C.; Mukunzi, D.; Zhang, X.; Xia, W.; Tian, H.; Xiao, Z. Temperature effect on the non-volatile compounds of Maillard reaction products derived from xylose–soybean peptide system: Further insights into thermal degradation and cross-linking. Food Chem. 2010, 120, 967–972. [Google Scholar] [CrossRef]
- Laroque, D.; Inisan, C.; Berger, C.; Vouland, E.; Dufosse, L.; Guerard, F. Kinetic study on the Maillard reaction. Consid. Sugar React. Food Chem. 2008, 111, 1032–1042. [Google Scholar] [CrossRef]
- Li-Chan, E.C.Y. Bioactive peptides and protein hydrolysates: Research trends and challenges for application as nutraceuticals and functional food ingredients. Curr. Opin. Food Sci. 2015, 1, 28–37. [Google Scholar] [CrossRef] [Green Version]
- Martin-Diana, A.B.; Izquierdo, N.; Albertos, I.; Sanchez, M.S.; Herrero, A.; Sanz, M.A.; Rico, D. Valorization of Carob’s germ and seed peel as natural antioxidant ingredients in gluten-free crackers. J. Food Process. Preserv. 2017, 41, e12770. [Google Scholar] [CrossRef]
- Matiacevich, S.B.; Santagapita, P.R.; Buera, M.P. Fluorescence from the Maillard reaction and its potential applications in food science. Crit. Rev. Food Sci. Nutr. 2005, 45, 483–495. [Google Scholar] [CrossRef]
- Mohsin, G.F.; Schmitt, F.J.; Kanzler, C.; Dirk Epping, J.; Flemig, S.; Hornemann, A. Structural characterization of melanoidin formed from D-glucose and L-alanine at different temperatures applying FTIR, NMR, EPR, and MALDI-ToF-MS. Food Chem. 2018, 245, 761–767. [Google Scholar] [CrossRef]
- Morales, F.J. Assessing the non-specific hydroxyl radical scavenging properties of melanoidins in a Fenton-type reaction system. Anal. Chim. Acta 2005, 534, 171–176. [Google Scholar] [CrossRef]
- Morales, F.J.; Jiménez-Pérez, S. Free radical scavenging capacity of Maillard reaction products as related to colour and fluorescence. Food Chem. 2001, 72, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Morales, F.J.; Romero, C.; Jiménez-Pérez, S. Fluorescence associated with Maillard reaction in milk and milk-resembling systems. Food Chem. 1996, 57, 423–428. [Google Scholar] [CrossRef]
- Murakami, M.; Shigeeda, A.; Danjo, K.; Yamaguchi, T.; Takamura, H.; Matoba, T. Radical-scavenging activity and brightly colored pigments in the early stage of the Maillard reaction. J. Food Sci. 2002, 67, 93–96. [Google Scholar] [CrossRef]
- Nielsen, P.; Petersen, D.; Dambmann, C. Improved method for determining food protein degree of hydrolysis. J. Food Sci. 2001, 66, 642–646. [Google Scholar] [CrossRef]
- Nirmal, N.P.; Santivarangkna, C.; Rajput, M.S.; Benjakul, S. Trends in shrimp processing waste utilization: An industrial prospective. Trends Food Sci. Technol. 2020, 103, 20–35. [Google Scholar] [CrossRef]
- Nooshkam, M.; Varidi, M.; Verma, D.K. Functional and biological properties of Maillard conjugates and their potential application in medical and food: A review. Food Res. Int. 2020, 131, 109003. [Google Scholar] [CrossRef] [PubMed]
- Ou, B.; Hampsch-Woodill, M.; Prior, R.L. Development and validation of an improved oxygen radical absorbance capacity assay using fluorescein as the fluorescent probe. J. Agric. Food Chem. 2001, 49, 4619–4626. [Google Scholar] [CrossRef] [PubMed]
- Richards-Rajadurai, N. Shrimp Waste Utilisation/Compiled by Roelof Schoemaker and Edited by Nirmala Richards-Rajadurai; INFOFISH Technical Handbook; INFOFISH: Kuala Lumpur, Malaysia, 1991. [Google Scholar]
- Rustad, T. Utilization of marine by-product. Electron. J. Environ. Agric. Food Chem. 2003, 2, 458–463. [Google Scholar]
- Sila, A.; Martinez-Alvarez, O.; Haddar, A.; Gómez-Guillén, M.C.; Nasri, M.; Montero, M.P.; Bougatef, A. Recovery, viscoelastic and functional properties of Barbel skin gelatine: Investigation of anti-DPP-IV and anti-prolyl endopeptidase activities of generated gelatine polypeptides. Food Chem. 2015, 168, 478–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slinkard, K.; Singleton, V.L. Total phenol analysis: Automation and comparison with manual methods. Am. J. Enol. Vitic. 1977, 28, 49–55. [Google Scholar]
- Sohn, M.; Ho, C.-T. Ammonia generation during thermal degradation of amino acids. J. Agric. Food Chem. 1995, 43, 3001–3003. [Google Scholar] [CrossRef]
- Su, G.; Zheng, L.; Cui, C.; Yang, B.; Ren, J.; Zhao, M. Characterization of antioxidant activity and volatile compounds of Maillard reaction products derived from different peptide fractions of peanut hydrolysate. Food Res. Int. 2011, 44, 3250–3258. [Google Scholar] [CrossRef]
- Su, J.F.; Huang, Z.; Yuan, X.Y.; Wang, X.Y.; Li, M. Structure and properties of carboxymethyl cellulose/soy protein isolate blend edible films crosslinked by Maillard reactions. Carbohydr. Polym. 2010, 79, 145–153. [Google Scholar] [CrossRef]
- Sumaya-Martinez, M.T.; Thomas, S.; Linard, B.; Binet, A.; Guerard, F. Effect of Maillard reaction conditions on browning and antiradical activity of sugar-tuna stomach hydrolysate model system. Food Res. Int. 2005, 38, 1045–1050. [Google Scholar] [CrossRef]
- Verzelloni, E.; Tagliazucchi, D.; Conte, A. From balsamic to healthy: Traditional balsamic vinegar melanoidins inhibit lipid peroxidation during simulated gastric digestion of meat. Food Chem. Toxicol. 2010, 48, 2097–2102. [Google Scholar] [CrossRef]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): An overview of their mechanisms of formation. Free Radic. Res. 2013, 47 (Suppl. S1), 3–27. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-Y.; Qian, H.; Yao, W.-R. Melanoidins produced by the Maillard reaction: Structure and biological activity. Food Chem. 2011, 128, 573–584. [Google Scholar] [CrossRef]
- Wiriyaphan, C.; Chitsomboon, B.; Yongsawadigul, J. Antioxidant activity of protein hydrolysates derived from threadfin bream surimi byproducts. Food Chem. 2012, 132, 104–111. [Google Scholar] [CrossRef]
- Yang, Y.; Cui, S.W.; Gong, J.; Guo, Q.; Wang, Q.; Hua, Y. A soy protein-polysaccharides Maillard reaction product enhanced the physical stability of oil-in-water emulsions containing citral. Food Hydrocoll. 2015, 48, 155–164. [Google Scholar] [CrossRef]
- Yu, X.; Zhao, M.; Hu, J.; Zeng, S.; Bai, X. Correspondence analysis of antioxidant activity and UV–Vis absorbance of Maillard reaction products as related to reactants. LWT Food Sci. Technol. 2012, 46, 1–9. [Google Scholar] [CrossRef]
- Zamora, R.; Hidalgo, F.J. Coordinate contribution of lipid oxidation and Maillard reaction to the nonenzymatic food browning. Crit. Rev. Food Sci. Nutr. 2005, 45, 49–59. [Google Scholar] [CrossRef]
- Zeng, Y.; Zhang, X.; Guan, Y.; Sun, Y. Enzymatic hydrolysates from tuna backbone and the subsequent Maillard reaction with different ketohexoses. Int. J. Food Sci. Technol. 2012, 47, 1293–1301. [Google Scholar] [CrossRef]
- Zhang, Q.; Ames, J.M.; Smith, R.D.; Baynes, J.W.; Metz, T.O. A perspective on the Maillard reaction and the analysis of protein glycation by mass spectrometry: Probing the pathogenesis of chronic disease. J. Proteome Res. 2009, 8, 754–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Sample | Time of Hydrolysis (min) | DH (%) | Free Amino Acids Content (mEq Ser NH2/g Protein) | |
|---|---|---|---|---|
| Before Glycation | After Glycation | |||
| Control | 0 | 0 | 1.52 ± 0.07 | 0.97 ± 0.05 * (−36%) |
| H10 | 10 | 6.09 | 2.56 ± 0.05 | 1.33 ± 0.06 * (−48%) |
| H30 | 30 | 10.47 | 2.43 ± 0.30 | 3.11 ± 0.09 * (+27%) |
| H60 | 60 | 12.07 | 3.42 ± 0.05 | 2.87 ± 0.38 * (−16%) |
| H120 | 120 | 12.08 | 4.39 ± 0.18 | 2.71 ± 0.37 * (−38%) |
| Glycation Time | |||||
|---|---|---|---|---|---|
| 214 nm | 0 min | 40 min | 90 min | 150 min | 180 min |
| Control | 2836 | 6455 (8%) | 7146 (8%) | 6749 (7%) | 7065 (7%) |
| 585 (9%) | 656 (8%) | 585 (10%) | 585 (11%) | ||
| 337 (36%) | 335 (37%) | 335 (40%) | 332 (33%) | ||
| 194 (47%) | 211 (47%) | 213 (43%) | 222 (50%) | ||
| (>1, 1–0.5, <0.5 kDa) | (75, 14, 11%) | (23, 8, 69%) | (26, 8, 67%) | (23, 10, 67%) | (22, 10, 67%) |
| 10 min | 949 (24%) | 767 (12%) | 769 (26%) | 772 (13%) | 769 (14%) |
| 574 (29%) | 344 (59%) | 432 (42%) | 343 (59%) | 343 (58%) | |
| 279 (28%) | 255 (29%) | 262 (32%) | 254 (29%) | 253 (28%) | |
| <110 (20%) | |||||
| (>1, 1–0.5, <0.5 kDa) | (32, 25, 43%) | (23, 15, 62%) | (29, 23, 48%) | (21, 16, 63%) | (22, 16, 61%) |
| 30 min | 902 (23%) | 748 (12%) | 722 (10%) | 836 (14%) | 713 (12%) |
| 582 (28%) | 342 (57%) | 343 (59%) | 343 (51%) | 332 (58%) | |
| 263 (34%) | 239 (33%) | 256 (31%) | 230 (35%) | 250 (30%) | |
| <110 (15%) | |||||
| (>1, 1–0.5, <0.5 kDa) | (25, 28, 46%) | (17, 14, 70%) | (29, 36, 35%) | (33, 34, 33%) | (18, 16, 66%) |
| 60 min | 876 (21%) | 714 (9%) | 693 (10%) | 715 (12%) | 699 (12%) |
| 570 (29%) | 343 (57%) | 343 (56%) | 343 (56%) | 341 (56%) | |
| 282 (29%) | 245 (33%) | 246 (34%) | 240 (32%) | 251 (32%) | |
| <110 (21%) | |||||
| (>1, 1–0.5, <0.5 kDa) | (23, 26, 52%) | (13, 12, 75%) | (14, 13, 73%) | (15, 15, 70%) | (15, 15, 70%) |
| 120 min | 857 (21%) | 716 (8%) | 688 (9%) | 693 (22%) | 665 (12%) |
| 565 (32%) | 341 (56%) | 342 (56%) | 437 (42%) | 341 (55%) | |
| 274 (28%) | 234 (36%) | 238 (34%) | 254 (36%) | 240 (33%) | |
| <110 (19%) | |||||
| (>1, 1–0.5, <0.5 kDa) | (22, 27, 51%) | (10, 11, 79%) | (12, 12, 76%) | (15, 22, 63%) | (14, 15, 71%) |
| 280 nm | 40 min | 90 min | 150 min | 180 min | |
| Control | 460 (22%) | 480 (15%) | 479 (17%) | 481 (18%) | |
| 235 (78%) | 321 (35%) | 328 (37%) | 326 (36%) | ||
| 241 (50%) | 242 (46%) | 244 (46%) | |||
| (>1, 1–0.5, <0.5 kDa) | (2, 7, 91%) | (5, 9, 87%) | (8, 12, 80%) | (8, 13, 79%) | |
| 10 min | 342 (60%) | 421 (41%) | 341 (59%) | 341 (60%) | |
| 245 (40%) | 247 (59%) | 245 (41%) | 245 (40%) | ||
| (>1, 1–0.5, <0.5 kDa) | (12, 11, 77%) | (18, 17, 65%) | (13, 13, 74%) | (15, 13, 72%) | |
| 30 min | 341 (61%) | 341 (59%) | 340 (61%) | 339 (60%) | |
| 234 (39%) | 245 (41%) | 239 (39%) | 245 (40%) | ||
| (>1, 1–0.5, <0.5 kDa) | (8, 9, 83%) | (9, 10, 81%) | (13, 12, 75%) | (12, 11, 77%) | |
| 60 min | 341 (58%) | 341 (58%) | 341 (60%) | 339 (60%) | |
| 241 (42%) | 244 (42%) | 243 (40%) | 249 (40%) | ||
| (>1, 1–0.5, <0.5 kDa) | (5, 8, 87%) | (7, 10, 83%) | (10, 12, 78%) | (11, 13, 76%) | |
| 120 min | 339 (58%) | 340 (58%) | 432 (43%) | 339 (61%) | |
| 241 (42%) | 242 (42%) | 246 (57%) | 245 (39%) | ||
| (>1, 1–0.5, <0.5 kDa) | (5, 7, 88%) | (6, 10, 84%) | (10, 19, 71%) | (10, 13, 77%) | |
| 420 nm | 40 min | 90 min | 150 min | 180 min | |
| Control | 496 (20%) | 510 (21%) | 560 (28%) | 551 (26%) | |
| 327 (37%) | 358 (32%) | ||||
| 281 (43%) | 282 (47%) | 262 (72%) | 277 (74%) | ||
| (>1, 1–0.5, <0.5 kDa) | (4, 21, 75%) | (9, 21, 70%) | (14, 22, 64%) | (11, 23, 66%) | |
| 10 min | 526 (18%) | ||||
| 291 | 282 | 283 | 286 (82%) | ||
| (>1, 1–0.5, <0.5 kDa) | (17, 16, 67%) | (16, 16, 68%) | (18, 16, 66%) | (20, 16, 64%) | |
| 30 min | 693 (16%) | 555 (15%) | |||
| 298 | 284 | 299 (84%) | 280 (85%) | ||
| (>1, 1–0.5, <0.5 kDa) | (16, 15, 69%) | (13, 15, 71%) | (21, 15, 64%) | (19, 16, 65%) | |
| 60 min | 487 (14%) | 530 (12%) | |||
| 286 | 283 | 286 (86%) | 280 (88%) | ||
| (>1, 1–0.5, <0.5 kDa) | (8, 15, 74%) | (12, 16, 72%) | (15, 15, 70%) | (16, 15, 69%) | |
| 120 min | 483 (12%) | 667 (16%) | |||
| 284 | 281 | 283 (88%) | 281 (84%) | ||
| (>1, 1–0.5, <0.5 kDa) | (10, 15, 75%) | (12, 15, 73%) | (13, 15, 71%) | (17, 16, 67%) | |
| Hydrolysis/ Heating Time | Assay | 0 min | 40 min | 90 min | 150 min | 180 min |
|---|---|---|---|---|---|---|
| Control | TP | 117.54 ± 0.5 Aa | 1375.31 ± 9.64 Ac | 1448.11 ± 11.54 Ac | 710.86 ± 0.24 Ab | 2405.63 ± 96.95 Ad |
| H10 | 432.34 ± 0.2 Ba | 1593.26 ± 3.35 Bb | 4220.61 ± 127.81 Bc | 3553.15 ± 65.62 Bc | 11,161.73 ± 6.85 Bd | |
| H30 | 640.81 ± 8.33 Da | 3851.77 ± 124.01 Eb | 4144.05 ± 137.38 Bb | 5808.61 ± 85.36 Ec | 9718.77 ± 63.36 Cd | |
| H60 | 588.3 ± 5.7 Ca | 2723.46 ± 37.31 Cb | 4951.76 ± 16.57 Cc | 4518.19 ± 104.63 Dc | 10,875.64 ± 17.91 Cd | |
| H120 | 585.9 ± 4.1 Ca | 3214.33 ± 96.06 Db | 5034.13 ± 43.66 Cd | 4053.64 ± 62.00 Cc | 12,865.95 ± 487.75 De | |
| Control | ABTS•+ | 34.0 ± 6.2 Aa | 425.8 ± 8.5 Ac | 421.6 ± 3.7 Ac | 211.4 ± 0.6 Ab | 3045.18 ± 25.2 Ad |
| H10 | 107.3 ± 23.1 Ba | 890.7 ± 18.2 Bb | 1235.1 ± 80.0 BCc | 1083.5 ± 21.2 Bc | 9749.84 ± 75.7 Bd | |
| H30 | 188.4 ± 12.3 Ca | 1163.3 ± 55.7 Cb | 1122.8 ± 104.3 Bb | 1601.1 ± 65.1 Cc | 9256.53 ± 34.8 Bd | |
| H60 | 111.4 ± 1.6 Ba | 742.9 ± 7.5 Db | 804.5 ± 109.6 Db | 1136.3 ± 133.1 Bb | 10,090.7 ± 62.1 Cc | |
| H120 | 178.3 ± 2.8 Ca | 936.6 ± 17.8 Bb | 1358.9 ± 81.6 Cc | 902.8 ± 150.5 Bd | 11,126.3 ± 64.9 Dd | |
| Control | ORAC | 6.56 ± 0.28 Aa | 79.10 ± 6.6 Abc | 102.68 ± 6.17 Ac | 74.50 ± 1.2 Ab | 411.5 ± 18.8 Ad |
| H10 | 62.23 ± 4.35 Ba | 478.19 ± 8.9 Db | 537.47 ± 10.98 Bc | 521.09 ± 32.6 Bc | 1966.0 ± 46.8 Cd | |
| H30 | 94.73 ± 6.44 Ca | 517.28 ± 18.78 Eb | 517.66 ± 16.46 Bb | 1066.95 ± 35.0 Dc | 1827.4 ± 17.2 Bd | |
| H60 | 84.83 ± 7.11 BCa | 359.50 ± 24.41 Bb | 654.81 ± 133.33 Bc | 632.92 ± 30.3 Cc | 1983.5 ± 14.9 Cd | |
| H120 | 70.55 ± 7.15 Ba | 450.86 ± 7.21 Cb | 871.75 ± 65.67 Cd | 619.58 ± 29.3 Cc | 2105.1 ± 40.2 De | |
| Control | DPPH | 0.02 ± 0.00 Aa | 0.12 ± 0.00 Ad | 0.06 ± 0.00 Ac | 0.00 ± 0.00 Ab | 0.12 ± 0.00 Ad |
| H10 | 0.06 ± 0.00 Aa | 0.11 ± 0.00 Bb | 0.25 ± 0.00 Cc | 0.24 ± 0.01 Dc | 0.41 ± 0.01 BCd | |
| H30 | 0.03 ± 0.00 Aab | 0.35 ± 0.01 Cab | 0.37 ± 0.02 Eb | 0.28 ± 0.01 Ea | 0.40 ± 0.00 Bc | |
| H60 | 0.07 ± 0.01 Aa | 0.24 ± 0.00 Dc | 0.29 ± 0.01 Dd | 0.22 ± 0.02 Cb | 0.43 ± 0.00 Ce | |
| H120 | 0.06 ± 0.00 Aa | 0.27 ± 0.00 Ed | 0.22 ± 0.00 Bc | 0.19 ± 0.00 Bb | 0.46 ± 0.01 De | |
| Control | FRAP | 0.03 ± 0.00 Aa | 0.39 ± 0.02 Ac | 0.52 ± 0.01 Ad | 0.18 ± 0.00 Ab | 1.22 ± 0.02 Ae |
| H10 | 0.07 ± 0.00 Ba | 0.67 ± 0.012 Bb | 1.89 ± 0.10 Dc | 0.80 ± 0.01 Bb | 2.05 ± 0.06 Bd | |
| H30 | 0.13 ± 0.00 Ca | 0.84 ± 0.02 Cc | 0.77 ± 0.01 Bb | 1.27 ± 0.00 Ed | 2.51 ± 0.02 De | |
| H60 | 0.20 ± 0.00 Ca | 0.58 ± 0.00 Db | 1.08 ± 0.00 Cd | 1.01 ± 0.005 Dc | 2.25 ± 0.01 Ce | |
| H120 | 0.12 ± 0.00 Da | 0.53 ± 0.00 Eb | 1.18 ± 0.00 Cd | 0.92 ± 0.05 Cc | 2.80 ± 0.09 Ee |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Estaca, J.; Albertos, I.; Martín-Diana, A.B.; Rico, D.; Martínez-Álvarez, Ó. Protein Hydrolysis and Glycosylation as Strategies to Produce Bioactive Ingredients from Unmarketable Prawns. Foods 2021, 10, 2844. https://doi.org/10.3390/foods10112844
Gómez-Estaca J, Albertos I, Martín-Diana AB, Rico D, Martínez-Álvarez Ó. Protein Hydrolysis and Glycosylation as Strategies to Produce Bioactive Ingredients from Unmarketable Prawns. Foods. 2021; 10(11):2844. https://doi.org/10.3390/foods10112844
Chicago/Turabian StyleGómez-Estaca, Joaquín, Irene Albertos, Ana Belén Martín-Diana, Daniel Rico, and Óscar Martínez-Álvarez. 2021. "Protein Hydrolysis and Glycosylation as Strategies to Produce Bioactive Ingredients from Unmarketable Prawns" Foods 10, no. 11: 2844. https://doi.org/10.3390/foods10112844