
Evaluating the Utility of Five Gene Fragments for Genetic Diversity Analyses of Mytella strigata Populations
 and
and
Abstract
: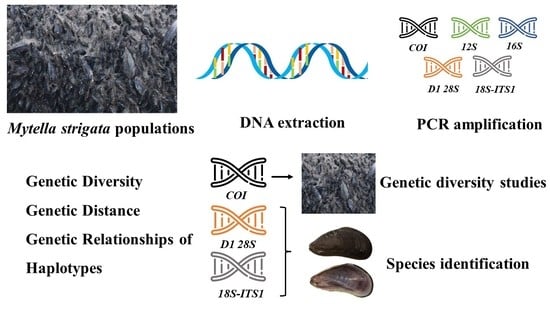
1. Introduction
2. Materials and Methods
3. Results
3.1. Genetic Diversity Analyses
3.2. Genetic Distance Analyses
3.3. Genetic Relationships of Haplotypes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Encarnação, J.; Teodósio, M.A.; Morais, P. Citizen science and biological invasions: A review. Front. Environ. Sci. 2021, 8, 602980. [Google Scholar] [CrossRef]
- Rytwinski, T.; Taylor, J.J.; Donaldson, L.A.; Britton, J.R.; Browne, D.R.; Gresswell, R.E.; Lintermans, M.; Prior, K.A.; Pellatt, M.G.; Vis, C.; et al. The effectiveness of non-native fish removal techniques in freshwater ecosystems: A systematic review. Environ. Rev. 2019, 27, 71–94. [Google Scholar] [CrossRef]
- Simberloff, D.; Martin, J.-L.; Genovesi, P.; Maris, V.; Wardle, D.A.; Aronson, J.; Courchamp, F.; Galil, B.; Garcia-Berthou, E.; Pascal, M.; et al. Impacts of biological invasions: What’s what and the way forward. Trends Ecol. Evol. 2013, 28, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, F.T.; Briski, E. An overview of recent research in marine biological invasions. Mar. Biol. 2017, 164, 121. [Google Scholar] [CrossRef] [Green Version]
- He, W.M. Biological invasions: Are their impacts precisely knowable or not? Biodiversity Sci. 2020, 28, 253–255. [Google Scholar]
- Bergman, J.N.; Raby, G.D.; Neigel, K.L.; Rennie, C.D.; Balshine, S.; Bennett, J.R.; Fisk, A.T.; Cooke, S.J. Tracking the early stages of an invasion with biotelemetry: Behaviour of round goby (Neogobius melanostomus) in Canada’s historic Rideau Canal. Biol. Invasions 2022, 24, 1149–1173. [Google Scholar] [CrossRef]
- Davidson, I.; Cahill, P.; Hinz, A.; Kluza, D.; Scianni, C.; Georgiades, E. A review of biofouling of ships’ internal seawater systems. Front. Mar. Sci. 2021, 8, 761531. [Google Scholar] [CrossRef]
- Iacarella, J.C.; Burke, L.; Davidson, I.C.; DiBacco, C.; Therriault, T.W.; Dunham, A. Unwanted networks: Vessel traffic heightens the risk of invasions in marine protected areas. Biol. Conserv. 2020, 245, 108553. [Google Scholar] [CrossRef]
- Robins, P.E.; Neill, S.P.; Giménez, L.; Jenkins, S.R.; Malham, S.K. Physical and biological controls on larval dispersal and connectivity in a highly energetic shelf sea. Limnol. Oceanogr. 2013, 58, 505–524. [Google Scholar] [CrossRef]
- Casillas, S.; Barbadilla, A. Molecular population genetics. Genetics 2019, 213, 721–722. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.Y.; Tay, T.S.; Lim, C.S.; Lee, S.S.C.; Teo, S.L.M.; Tan, K.S. Mytella strigata (Bivalvia: Mytilidae): An alien mussel recently introduced to Singapore and spreading rapidly. Molluscan Res. 2018, 38, 170–186. [Google Scholar] [CrossRef]
- Boudreaux, M.L.; Walters, L.J. Mytella charruana (Bivalvia: Mytilidae): A new, invasive bivalve in Mosquito Lagoon, Florida. Nautilus 2006, 120, 34–36. [Google Scholar]
- Ma, P.Z.; Li, H.M.; Liu, Y.M.; Li, C.; Zhang, Z.; Wang, H.Y. First confirmed occurrence of the invasive mussel Mytella strigata (Hanley, 1843) in Guangdong and Hainan, China, and Indo-West Pacific regions. BioInvasions Rec. 2022, 11, 947–963. [Google Scholar] [CrossRef]
- Rice, M.A.; Rawson, P.D.; Salinas, A.D.; Rosario, W.R. Identification and salinity tolerance of the western hemisphere mussel Mytella charruana (d’orbigny, 1842) in the Philippines. J. Shellfish Res. 2016, 35, 865–873. [Google Scholar] [CrossRef]
- Sanpanichl, K.; Wells, F.E. Mytella strigata (Hanley, 1843) emerging as an invasive marine threat in Southeast Asia. BioInvasions Rec. 2019, 8, 343–356. [Google Scholar] [CrossRef]
- Jayachandran, P.R.; Aneesh, B.P.; Oliver, P.G.; Philomina, J.; Jima, M.; Harikrishnan, K.; Nandan, S.B. First record of the alien invasive biofouling mussel Mytella strigata (Hanley, 1843) (Mollusca: Mytilidae) from Indian waters. BioInvasions Rec. 2019, 8, 828–837. [Google Scholar] [CrossRef]
- Huang, Y.C.; Li, Z.K.; Chen, W.L.; Chan, C.C.; Hsu, H.Y.; Lin, Y.T.; Huang, Y.S.; Han, Y.S. First record of the invasive biofouling mussel Mytella strigata (Hanley, 1843) (Bivalvia: Mytilidae) from clam ponds in Taiwan. BioInvasions Rec. 2021, 10, 304–312. [Google Scholar] [CrossRef]
- Ma, P.Z.; Zuo, C.X.; Li, H.M.; Wang, H.Y.; Wang, Q.H.; Zhang, Z. The basic biology and biological invasion of Mytella strigata. Acta Ecol. Sin. 2022, 43. [Google Scholar] [CrossRef]
- Gillis, N.K.; Walters, L.J.; Fernandes, F.C.; Hoffman, E.A. Higher genetic diversity in introduced than in native populations of the mussel Mytella charruana: Evidence of population admixture at introduction sites. Divers. Distrib. 2009, 15, 784–795. [Google Scholar] [CrossRef]
- Gusman, A.; Lecomte, S.; Stewart, D.T.; Passamonti, M.; Breton, S. Pursuing the quest for better understanding the taxonomic distribution of the system of doubly uniparental inheritance of mtDNA. PeerJ 2016, 4, e2760. [Google Scholar] [CrossRef] [Green Version]
- Zouros, E. Biparental inheritance through uniparental transmission: The doubly uniparental inheritance (DUI) of mitochondrial DNA. Evol. Biol. 2012, 40, 1–31. [Google Scholar] [CrossRef]
- Zouros, E.; Ball, A.O.; Saavedra, C.; Freeman, K.R. An unusual type of mitochondrial DNA inheritance in the blue mussel Mytilus. Proc. Natl. Acad. Sci. USA 1994, 91, 7463–7467. [Google Scholar] [CrossRef] [PubMed]
- Alves, F.A.d.S.; Beasley, C.R.; Hoeh, W.R.; Rocha, R.M.d.; Simone, L.R.L.d.; Tagliaro, C.H. Detection of mitochondrial DNA heteroplasmy suggests a doubly uniparental inheritance pattern in the mussel Mytella charruana. Braz. J. Biol. Sci. 2012, 10, 176–185. [Google Scholar]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [PubMed]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene-sequences and a compilation of conserved polymerase chain-reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Lovejoy, N.R.; Lester, K.; Crampton, W.G.R.; Marques, F.P.L.; Albert, J.S. Phylogeny, biogeography, and electric signal evolution of Neotropical knifefishes of the genus Gymnotus (Osteichthyes: Gymnotidae). Mol. Phylogenet. Evol. 2010, 54, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Barrandeguy, M.E.; García, M.V. Quantifying genetic diversity: The starting point for population genetic studies using molecular markers. J. Genet. 2014, 93, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, A.E.; Loonam, G.A.; Blakeslee, A.M.H. Population structure and demography of non-indigenous Japanese mystery snails in freshwater habitats of Virginia and Washington, DC, USA. Aquat. Invasions 2022, 17, 415–430. [Google Scholar] [CrossRef]
- Paolacci, S.; Bog, M.; Lautenschlager, U.; Bonfield, R.; Appenroth, K.-J.; Oberprieler, C.; Jansen, M.A.K. Clonal diversity amongst island populations of alien, invasive Lemna minuta Kunth. Biol. Invasions 2021, 23, 2649–2660. [Google Scholar] [CrossRef]
- Young, R.G.; Mitterboeck, T.F.; Loeza-Quintana, T.; Adamowicz, S.J. Rates of molecular evolution and genetic diversity in European vs. North American populations of invasive insect species. Eur. J. Entomol. 2018, 115, 718–728. [Google Scholar] [CrossRef]
- Han, G.D.; Wang, W.; Dong, Y.W. Effects of balancing selection and microhabitat temperature variations on heat tolerance of the intertidal black mussel Septifer virgatus. Integr. Zool. 2020, 15, 416–427. [Google Scholar] [CrossRef]
- Brown, W.M.; George, M., Jr.; Wilson, A.C. Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. USA. 1979, 76, 1967–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, M.; Koskella, B.; Schaack, S. Mutation pressure and the evolution of organelle genomic architecture. Science 2006, 311, 1727–1730. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Pérez, J.; Froufe, E.; Nantón, A.; Gaspar, M.B.; Méndez, J. Genetic diversity and population genetic analysis of Donax vittatus (Mollusca: Bivalvia) and phylogeny of the genus with mitochondrial and nuclear markers. Estuar. Coast. Shelf Sci. 2017, 197, 126–135. [Google Scholar] [CrossRef]
- Lubośny, M.; Śmietanka, B.; Arculeo, M.; Burzyński, A. No evidence of DUI in the Mediterranean alien species Brachidontes pharaonis (P. Fisher, 1870) despite mitochondrial heteroplasmy. Sci. Rep. 2022, 12, 8569. [Google Scholar] [CrossRef]
- Theologidis, I.; Fodelianakis, S.; Gaspar, M.B.; Zouros, E. Doubly uniparental inheritance (DUI) of mitochondrial dna in Donax trunculus (Bivalvia: Donacidae) and the problem of its sporadic detection in Bivalvia. Evolution 2008, 62, 959–970. [Google Scholar] [CrossRef]
- Cao, L.; Kenchington, E.; Zouros, E. Differential segregation patterns of sperm mitochondria in embryos of the blue mussel (Mytilus edulis). Genetics 2004, 166, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Kakoi, S.; Kin, K.; Miyazaki, K.; Wada, H. Early development of the Japanese spiny oyster (Saccostrea kegaki): Characterization of some genetic markers. Zool. Sci. 2008, 25, 455–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zouros, E.; Rodakis, G.C. Doubly uniparental inheritance of mtDNA: An unappreciated defiance of a general rule. Adv. Anat. Embyrol. Cell Biol. 2019, 231, 25–49. [Google Scholar]
- Degletagne, C.; Abele, D.; Held, C. A distinct mitochondrial genome with DUI-like inheritance in the ocean quahog Arctica islandica. Mol. Biol. Evol. 2016, 33, 375–383. [Google Scholar] [CrossRef]
- Boyle, E.E.; Etter, R.J. Heteroplasmy in a deep-sea protobranch bivalve suggests an ancient origin of doubly uniparental inheritance of mitochondria in Bivalvia. Mar. Biol. 2013, 160, 413–422. [Google Scholar] [CrossRef]
- Breton, S.; Burger, G.; Stewart, D.T.; Blier, P.U. Comparative analysis of gender-associated complete mitochondrial genomes in marine mussels (Mytilus spp.). Genetics 2006, 172, 1107–1119. [Google Scholar] [CrossRef] [Green Version]
- Doucet-Beaupré, H.; Breton, S.; Chapman, E.G.; Blier, P.U.; Bogan, A.E.; Stewart, D.T.; Hoeh, W.R. Mitochondrial phylogenomics of the Bivalvia (Mollusca): Searching for the origin and mitogenomic correlates of doubly uniparental inheritance of mtDNA. BMC Evol. Biol. 2010, 10, 50. [Google Scholar] [CrossRef] [Green Version]
- Ladoukakis, E.D.; Saavedra, C.; Magoulas, A.; Zouros, E. Mitochondrial DNA variation in a species with two mitochondrial genomes: The case of Mytilus galloprovincialis from the Atlantic, the Mediterranean and the Black Sea. Mol. Ecol. 2002, 11, 755–769. [Google Scholar] [CrossRef]
- Vargas, J.; Pérez, M.; Toro, J.; Astorga, M.P. Presence of two mitochondrial genomes in the mytilid Perumytilus purpuratus: Phylogenetic evidence for doubly uniparental inheritance. Genet. Mol. Biol. 2015, 38, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Grant, W.S.; Bowen, B.W. Shallow population histories in deep evolutionary lineages of marine fishes: Insights from sardines and anchovies and lessons for conservation. J. Hered. 1998, 89, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Sung, C.H.; Lin, C.H.; Huang, C.W.; Wang, L.J. Characterization and phylogenetic analysis of the complete mitochondrial genome of Mytella strigata (Hanley 1843) (Bivalvia: Mytiloida: Mytilidae). Mitochondrial DNA B 2021, 6, 2345–2347. [Google Scholar] [CrossRef] [PubMed]
- Lushai, G.; Smith, D.A.S.; Goulson, D.; Allen, J.A.; Maclean, N. Mitochondrial DNA clocks and the phylogeny of Danaus butterflies. Int. J. Trop. Insect Sci. 2003, 23, 309–315. [Google Scholar] [CrossRef] [Green Version]
- Calazans, C.S.H.; Walters, L.J.; Fernandes, F.C.; Ferreira, C.E.L.; Hoffman, E.A. Genetic structure provides insights into the geographic origins and temporal change in the invasive charru mussel (Sururu) in the southeastern United States. PLoS ONE 2018, 13, e0195159. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Collection Date | Geographical Coordinates | Quantity | COI | 12S | 16S | D1 28S | 18S-ITS1 |
|---|---|---|---|---|---|---|---|---|
| Jimei | 21 October 2021 | 24°34′891″ N, 118°6′578″ E | 35 | 33(1) | 27(1) | 32 | 35 | 35 |
| Shanwei | 31 October 2021 | 22°49′354″ N, 115°33′91″ E | 35 | 29(3) | 25(2) | 33 | 35 | 35 |
| Xuwen | 17 August 2022 | 20°24′739″ N, 109°56′15″ E | 36 | 36(6) | 35(5) | 34 | 36 | 36 |
| Beihai | 12 October 2021 | 21°24′6″ N, 109°10′9″ E | 24 | 20(3) | 21(2) | 21 | 24 | 24 |
| Zhanjiang | 27 November 2020 | 20°55′53″ N, 110°31′43″ E | 35 | 30(1) | 31(1) | 34 | 35 | 35 |
| Hainan | 6 December 2020 | 19°59′7″ N, 110°35′7″ E | 26 | 23(8) | 19(3) | 23 | 26 | 26 |
| 6 December 2020 | 19°37′35″ N, 110°49′43″ E | |||||||
| Total | - | - | 191 | 171(22) | 158(14) | 177 | 191 | 191 |
| Markers | Primers | PCR Programs |
|---|---|---|
| COI | LCO 1490 5′-GGTCAACAAATCATAAAGATATTGG-3′ HCO 2198 5′-TAAACTTCAGGGTGACCAAAAAATCA-3′ [24] | 95 °C 3 min, 32× (95 °C 30 s, 48 °C 1 min, 72 °C 1 min), 72 °C 5 min |
| 12S | 12S-SR-J14197 5′-GTACAYCTACTATGTTACGACTT-3′ 12S-SR-N14745 5′-GTGCCAGCAGYYGCGGTTANAC-3′ [25] | 95 °C 3 min, 32× (95 °C 30 s, 48 °C 1 min, 72 °C 1 min), 72 °C 5 min |
| 16S | 16Sar 5′-CGCCTGTTTATCAAAAACAT-3′ 16Sbr 5′-CCGGTCTGAACTCAGATCACGT-3′ [26] | 95 °C 3 min, 32× (95 °C 30 s, 48 °C 1 min, 72 °C 1 min), 72 °C 5 min |
| D1 28S | LSU5b 5′-ACCCGCTGAAYTTAAGCA-3′ D1R 5′-AACTCTCTCMTTCARAGTTC-3′ [11] | 95 °C 5 min, 49 °C 45 s, 72 °C 1 min, 34× (95 °C 30 s, 52 °C 45 s 72 °C 1 min), 95 °C 30 s, 52 °C 45 s, 72 °C 5 min |
| 18S-ITS1 | ITS1A-sal 5′-AAAAAGCTTTTGTACACACCGCCCGTCGC-3′ ITS1B-sal 5′-AGCTTGCTGCGTTCTTCATCGA-3′ [11] | 95 °C 3 min, 35× (95 °C 1 min, 45.5 °C 1 min, 72 °C 1.5 min), 72 °C 7 min |
| F-COI | F-12S | 16S | D1 28S | 18S-ITS1 | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| h | S | Hd | Pi | K | h | S | Hd | Pi | K | h | S | Hd | Pi | K | h | S | Hd | Pi | K | h | S | Hd | Pi | K | |
| JM | 10 | 12 | 0.847 | 0.00454 | 2.756 | 4 | 4 | 0.665 | 0.00303 | 1.526 | 3 | 2 | 0.446 | 0.00115 | 0.474 | 2 | 1 | 0.420 | 0.00142 | 0.420 | 1 | 0 | 0.000 | 0.00000 | 0.000 |
| SW | 10 | 13 | 0.871 | 0.00547 | 3.323 | 7 | 8 | 0.791 | 0.00315 | 1.589 | 6 | 5 | 0.606 | 0.00172 | 0.708 | 2 | 1 | 0.514 | 0.00174 | 0.514 | 1 | 0 | 0.000 | 0.00000 | 0.000 |
| XW | 10 | 14 | 0.832 | 0.00455 | 2.761 | 3 | 3 | 0.600 | 0.00259 | 1.303 | 5 | 4 | 0.439 | 0.00116 | 0.476 | 2 | 1 | 0.464 | 0.00157 | 0.464 | 1 | 0 | 0.000 | 0.00000 | 0.000 |
| BH | 6 | 8 | 0.654 | 0.00344 | 2.088 | 3 | 3 | 0.602 | 0.00261 | 1.310 | 3 | 2 | 0.452 | 0.00116 | 0.476 | 2 | 1 | 0.464 | 0.00157 | 0.464 | 1 | 0 | 0.000 | 0.00000 | 0.000 |
| ZJ | 8 | 11 | 0.766 | 0.00489 | 2.966 | 3 | 3 | 0.625 | 0.00252 | 1.269 | 7 | 9 | 0.629 | 0.00226 | 0.927 | 3 | 2 | 0.528 | 0.00190 | 0.561 | 1 | 0 | 0.000 | 0.00000 | 0.000 |
| HN | 8 | 11 | 0.790 | 0.00559 | 3.390 | 3 | 3 | 0.592 | 0.00154 | 0.775 | 6 | 8 | 0.696 | 0.00294 | 1.209 | 2 | 1 | 0.508 | 0.00172 | 0.508 | 1 | 0 | 0.000 | 0.00000 | 0.000 |
| Total | 22 | 25 | 0.844 | 0.00491 | 2.981 | 8 | 9 | 0.674 | 0.00284 | 1.432 | 10 | 13 | 0.557 | 0.00172 | 0.790 | 3 | 2 | 0.490 | 0.00168 | 0.496 | 1 | 0 | 0.000 | 0.00000 | 0.000 |
| SW | ZJ | JM | HN | BH | XW | |
|---|---|---|---|---|---|---|
| SW | 0.00317 | |||||
| ZJ | 0.00307 | 0.00253 | ||||
| JM | 0.00333 | 0.00273 | 0.00305 | |||
| HN | 0.00239 | 0.00278 | 0.00301 | 0.00154 | ||
| BH | 0.00334 | 0.00251 | 0.00276 | 0.00318 | 0.00261 | |
| XW | 0.00332 | 0.00252 | 0.00276 | 0.00314 | 0.00250 | 0.00260 |
| SW | ZJ | JM | HN | BH | XW | Ecuador | Colombia | USA | |
|---|---|---|---|---|---|---|---|---|---|
| SW | 0.00587 | ||||||||
| ZJ | 0.00556 | 0.00523 | |||||||
| JM | 0.00559 | 0.00525 | 0.00486 | ||||||
| HN | 0.00604 | 0.00575 | 0.00652 | 0.00577 | |||||
| BH | 0.00492 | 0.00454 | 0.00429 | 0.00576 | 0.00369 | ||||
| XW | 0.00537 | 0.00501 | 0.00478 | 0.00613 | 0.00409 | 0.00476 | |||
| Ecuador | 0.01686 | 0.01700 | 0.01666 | 0.01763 | 0.01588 | 0.01649 | 0.00522 | ||
| Colombia | 0.00561 | 0.00538 | 0.00498 | 0.00644 | 0.00444 | 0.00495 | 0.01624 | 0.00515 | |
| USA | 0.01069 | 0.01084 | 0.01044 | 0.01146 | 0.00970 | 0.01037 | 0.01399 | 0.01008 | 0.00880 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuo, C.; Zhang, T.; Zhang, C.; Zhao, D.; Zhu, Y.; Ma, X.; Wang, H.; Ma, P.; Zhang, Z. Evaluating the Utility of Five Gene Fragments for Genetic Diversity Analyses of Mytella strigata Populations. Fishes 2023, 8, 34. https://doi.org/10.3390/fishes8010034
Zuo C, Zhang T, Zhang C, Zhao D, Zhu Y, Ma X, Wang H, Ma P, Zhang Z. Evaluating the Utility of Five Gene Fragments for Genetic Diversity Analyses of Mytella strigata Populations. Fishes. 2023; 8(1):34. https://doi.org/10.3390/fishes8010034
Chicago/Turabian StyleZuo, Chenxia, Tingting Zhang, Chenchen Zhang, Daotan Zhao, Yi Zhu, Xiaojie Ma, Haiyan Wang, Peizhen Ma, and Zhen Zhang. 2023. "Evaluating the Utility of Five Gene Fragments for Genetic Diversity Analyses of Mytella strigata Populations" Fishes 8, no. 1: 34. https://doi.org/10.3390/fishes8010034