Integrating Signals from Sperm Methylome Analysis and Genome-Wide Association Study for a Better Understanding of Male Fertility in Cattle

 , , , , ,
, , , , ,
Abstract
:1. Introduction
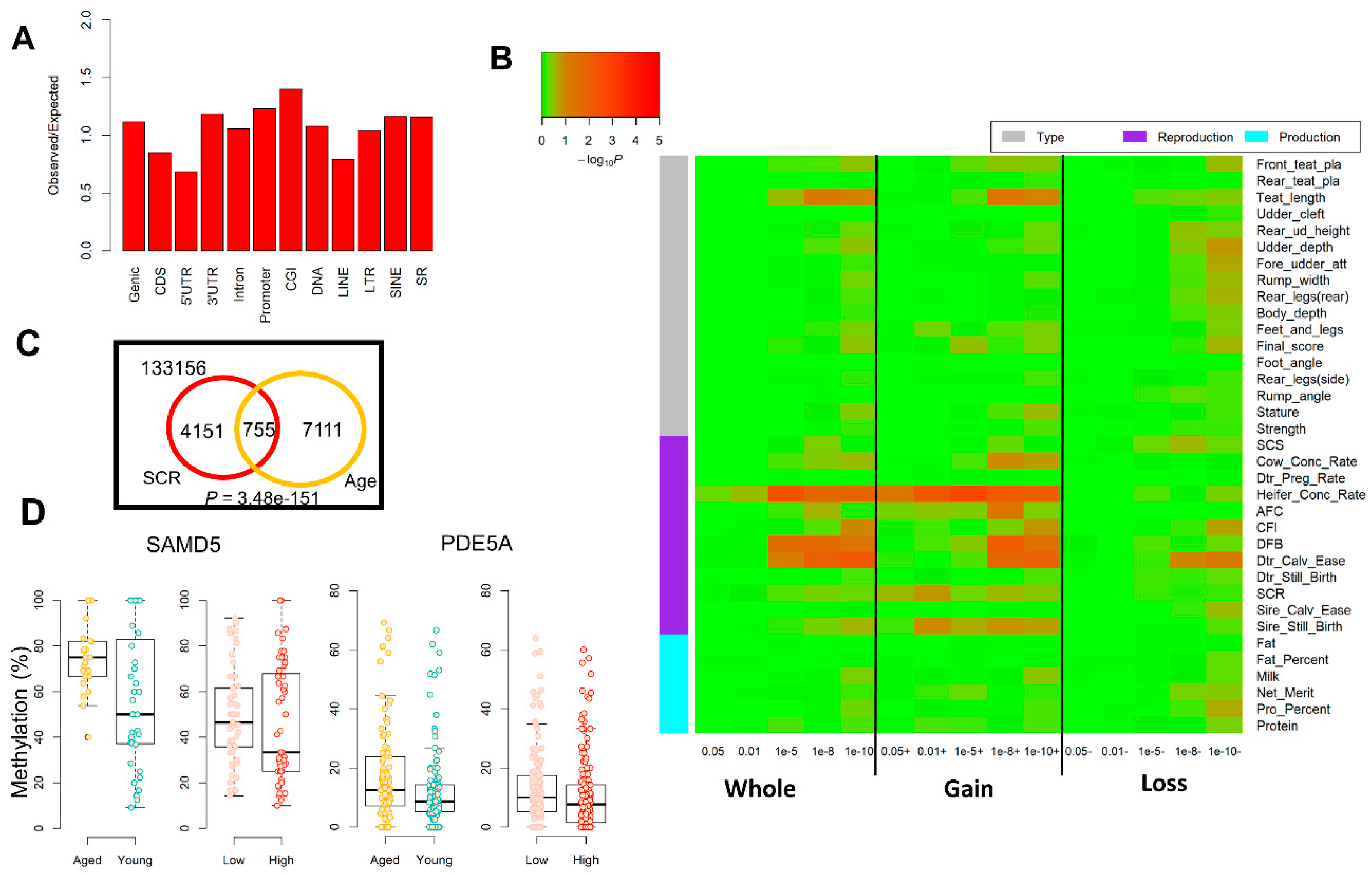
2. Results
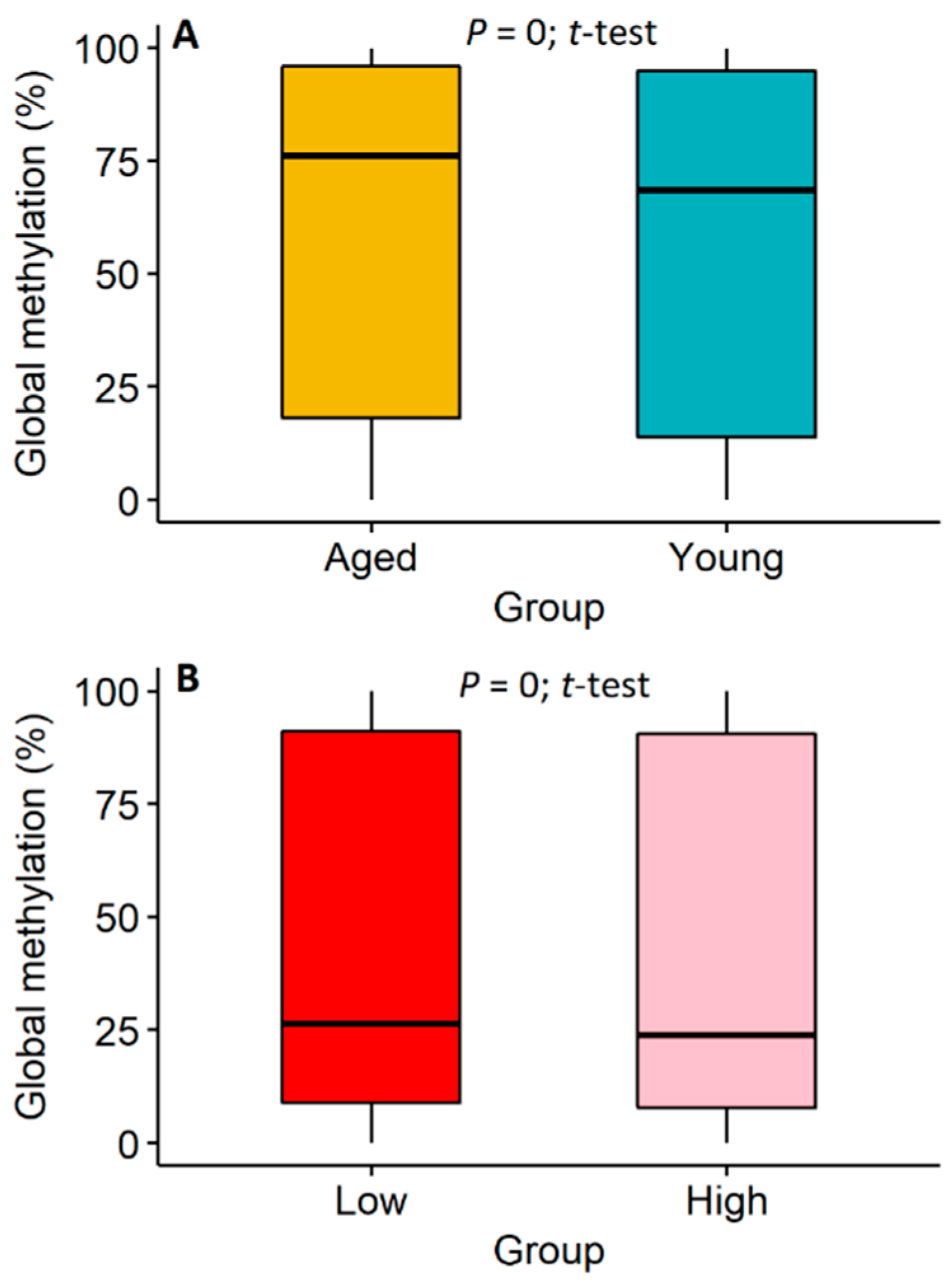
2.1. Analyses of Global Methylome in Cattle Sperm
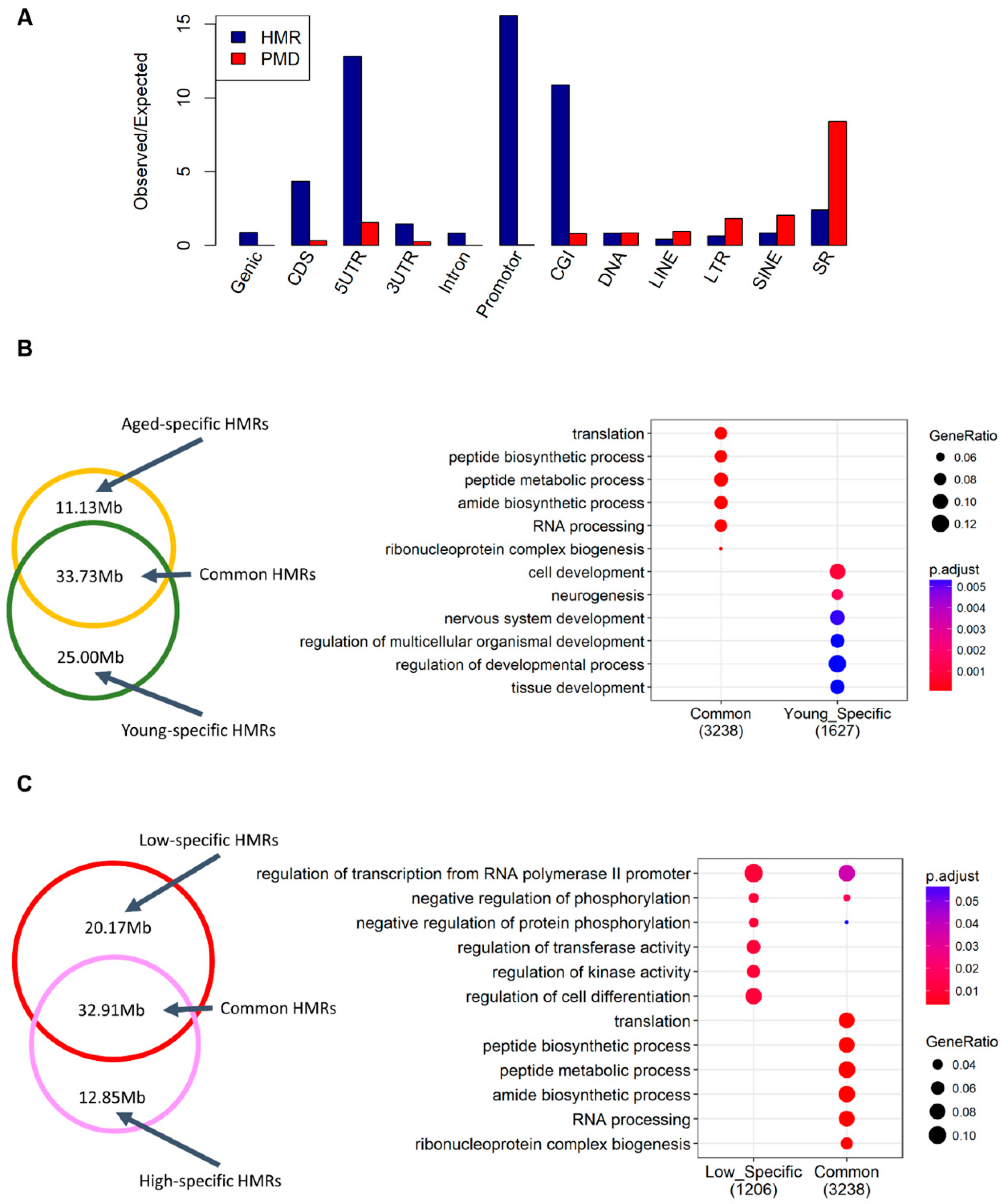
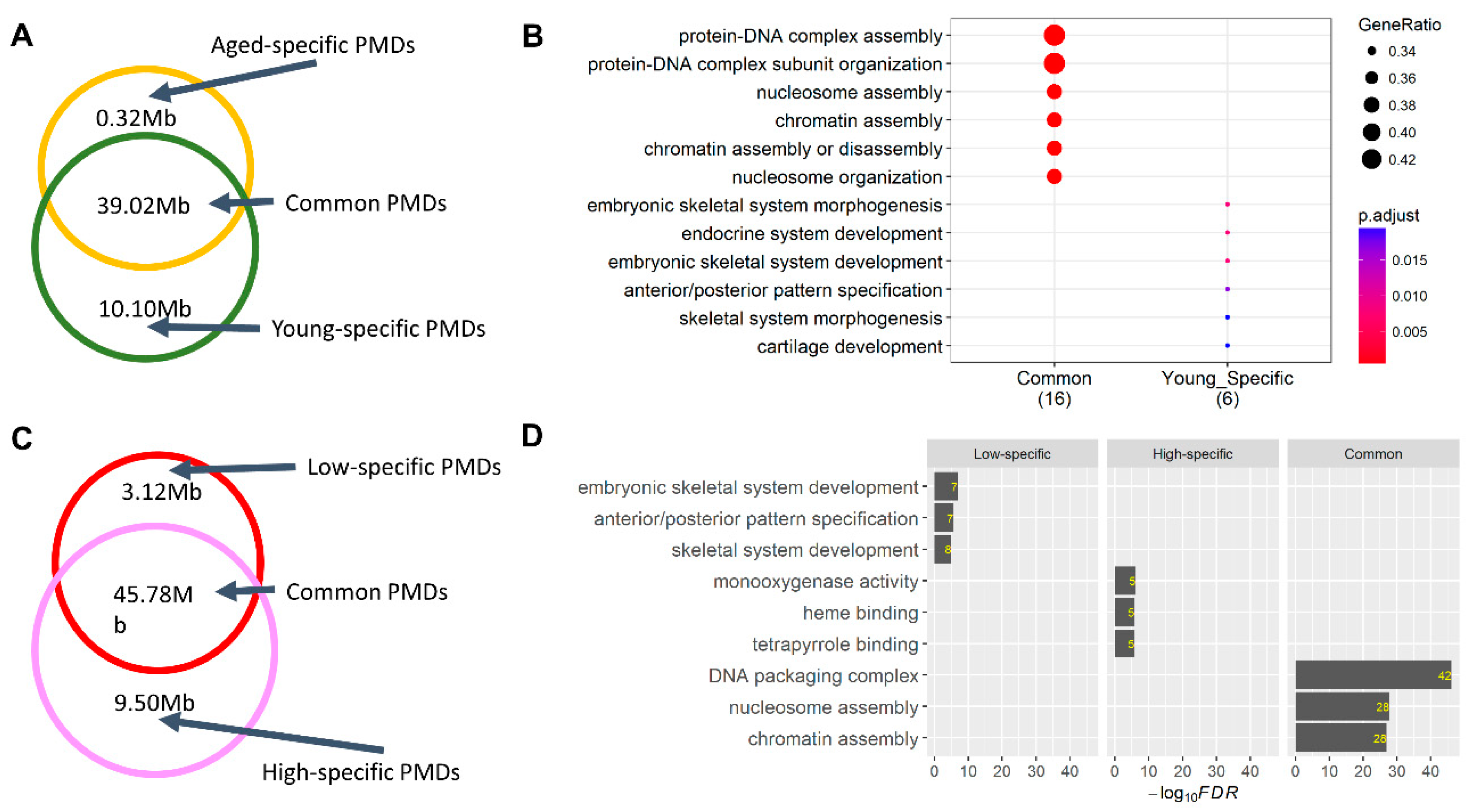
2.2. Alterations in Sperm Hypomethylated Regions (HMRs) and Partially Methylated Domains (PMDs)
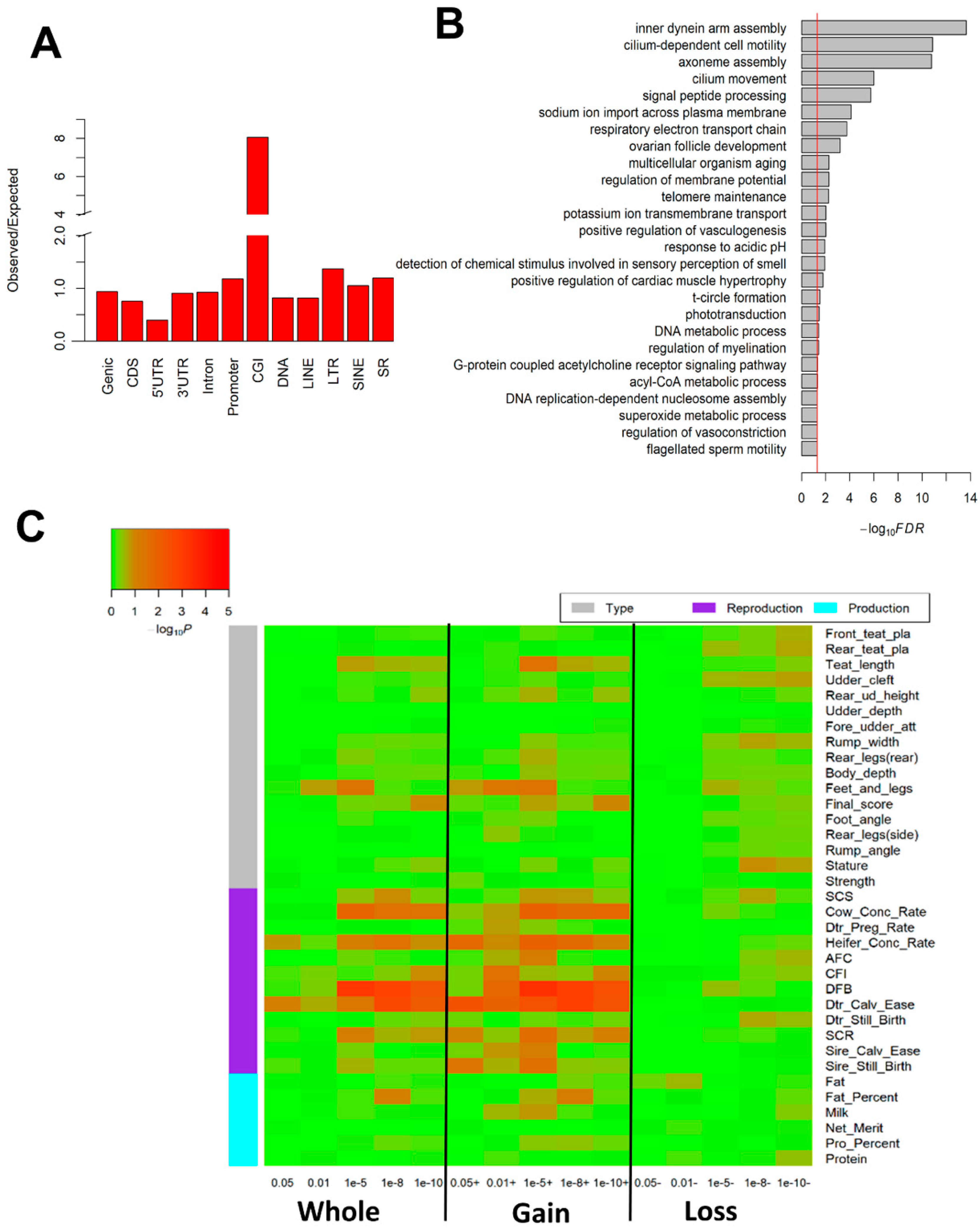
2.3. Genome-Wide Differentially Methylated Regions (DMRs)
3. Discussion
4. Materials and Methods
4.1. Sample Description and Whole-Genome Bisulfite Sequencing (WGBS)
4.2. Raw Data Profiling and DNA Methylation Calling
4.3. Detection of Differentially Methylated Regions (DMRs) and Differentially Methylated Cytosines (DMCs)
4.4. PMD and HMR Identification
4.5. GWAS Summary Statistics
4.6. GWAS Signal Enrichment Analysis for Differentially Methylated Regions (DMRs)
4.7. DMR/DMC-Set Enrichment Analysis Based on GO Database
4.8. Gene-Set Functional Enrichment Analysis
4.9. Availability of Data and Materials
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HMR | Hypomethylated regions |
| PMD | Partially methylated domains |
| GWAS | Genome-wide association study |
| SCR | Sire-conception-rate |
| DMR | Differentially methylated region |
| DMC | Differentially methylated cytosine |
| GO | Gene Ontology |
| CGI | CpG islands |
References
- Levine, H.; Jørgensen, N.; Martino-Andrade, A.; Mendiola, J.; Weksler-Derri, D.; Mindlis, I.; Pinotti, R.; Swan, S.H. Temporal trends in sperm count: A systematic review and meta-regression analysis. Hum. Reprod. Update 2017, 23, 646–659. [Google Scholar] [CrossRef]
- Taylor, J.F.; Schnabel, R.D.; Sutovsky, P. Identification of genomic variants causing sperm abnormalities and reduced male fertility. Anim. Reprod. Sci. 2018, 194, 57–62. [Google Scholar] [CrossRef]
- Kropp, J.; Carrillo, J.A.; Namous, H.; Daniels, A.; Salih, S.M.; Song, J.; Khatib, H. Male fertility status is associated with DNA methylation signatures in sperm and transcriptomic profiles of bovine preimplantation embryos. BMC Genom. 2017, 18, 280. [Google Scholar] [CrossRef]
- Barlow, D.P. Methylation and imprinting: From host defense to gene regulation? Science 1993, 260, 309–311. [Google Scholar] [CrossRef]
- Bestor, T.H. Cytosine methylation and the unequal developmental potentials of the oocyte and sperm genomes. Am. J. Hum. Genet. 1998, 62, 1269–1273. [Google Scholar] [CrossRef]
- Bourc’his, D.; Bestor, T.H. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 2004, 431, 96. [Google Scholar] [CrossRef]
- Sharp, A.J.; Stathaki, E.; Migliavacca, E.; Brahmachary, M.; Montgomery, S.B.; Dupre, Y.; Antonarakis, S.E. DNA methylation profiles of human active and inactive X chromosomes. Genome Res. 2011, 21, 1592–1600. [Google Scholar] [CrossRef]
- Jenkins, T.G.; Carrell, D.T. The sperm epigenome and potential implications for the developing embryo. Reproduction 2012, 143, 727–734. [Google Scholar] [CrossRef]
- Hammoud, S.S.; Nix, D.A.; Zhang, H.; Purwar, J.; Carrell, D.T.; Cairns, B.R. Distinctive chromatin in human sperm packages genes for embryo development. Nature 2009, 460, 473. [Google Scholar] [CrossRef]
- Jenkins, T.G.; Aston, K.I.; Pflueger, C.; Cairns, B.R.; Carrell, D.T. Age-associated sperm DNA methylation alterations: Possible implications in offspring disease susceptibility. PLoS Genet. 2014, 10, e1004458. [Google Scholar] [CrossRef]
- Aston, K.I.; Uren, P.J.; Jenkins, T.G.; Horsager, A.; Cairns, B.R.; Smith, A.D.; Carrell, D.T. Aberrant sperm DNA methylation predicts male fertility status and embryo quality. Fertil. Steril. 2015, 104, 1388–1397. [Google Scholar] [CrossRef]
- Atsem, S.; Reichenbach, J.; Potabattula, R.; Dittrich, M.; Nava, C.; Depienne, C.; Böhm, L.; Rost, S.; Hahn, T.; Schorsch, M. Paternal age effects on sperm FOXK1 and KCNA7 methylation and transmission into the next generation. Hum. Mol. Genet. 2016, 25, 4996–5005. [Google Scholar]
- Jenkins, T.G.; Aston, K.I.; Meyer, T.D.; Hotaling, J.M.; Shamsi, M.B.; Johnstone, E.B.; Cox, K.J.; Stanford, J.B.; Porucznik, C.A.; Carrell, D.T. Decreased fecundity and sperm DNA methylation patterns. Fertil. Steril. 2016, 105, 51–57.e3. [Google Scholar] [CrossRef]
- Milekic, M.; Xin, Y.; O’donnell, A.; Kumar, K.; Bradley-Moore, M.; Malaspina, D.; Moore, H.; Brunner, D.; Ge, Y.; Edwards, J. Age-related sperm DNA methylation changes are transmitted to offspring and associated with abnormal behavior and dysregulated gene expression. Mol. Psychiatry 2015, 20, 995–1001. [Google Scholar] [CrossRef]
- Lambert, S.; Blondin, P.; Vigneault, C.; Labrecque, R.; Dufort, I.; Sirard, M.A. Spermatozoa DNA methylation patterns differ due to peripubertal age in bulls. Theriogenology 2018, 106, 21–29. [Google Scholar] [CrossRef]
- Donkin, I.; Barrès, R. Sperm epigenetics and influence of environmental factors. Mol. Metab. 2018, 14, 1–11. [Google Scholar] [CrossRef]
- Zhou, Y.; Connor, E.E.; Bickhart, D.M.; Li, C.; Baldwin, R.L.; Schroeder, S.G.; Rosen, B.D.; Yang, L.; Van Tassell, C.P.; Liu, G.E. Comparative whole genome DNA methylation profiling of cattle sperm and somatic tissues reveals striking hypomethylated patterns in sperm. GigaScience 2018, 7, giy039. [Google Scholar] [CrossRef]
- Perrier, J.-P.; Sellem, E.; Prézelin, A.; Gasselin, M.; Jouneau, L.; Piumi, F.; Al Adhami, H.; Weber, M.; Fritz, S.; Boichard, D.; et al. A multi-scale analysis of bull sperm methylome revealed both species peculiarities and conserved tissue-specific features. BMC Genom. 2018, 19, 404. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Berman, B.P.; Weisenberger, D.J.; Aman, J.F.; Hinoue, T.; Ramjan, Z.; Liu, Y.; Noushmehr, H.; Lange, C.P.; van Dijk, C.M.; Tollenaar, R.A.; et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 2011, 44, 40–46. [Google Scholar] [CrossRef]
- Hon, G.C.; Hawkins, R.D.; Caballero, O.L.; Lo, C.; Lister, R.; Pelizzola, M.; Valsesia, A.; Ye, Z.; Kuan, S.; Edsall, L.E.; et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 2012, 22, 246–258. [Google Scholar] [CrossRef]
- Schroeder, D.I.; Blair, J.D.; Lott, P.; Yu, H.O.; Hong, D.; Crary, F. The human placenta methylome. Proc. Natl. Acad. Sci. USA 2013, 110, 6037–6042. [Google Scholar] [CrossRef]
- Schroeder, D.I.; Jayashankar, K.; Douglas, K.C.; Thirkill, T.L.; York, D.; Dickinson, P.J.; Williams, L.E.; Samollow, P.B.; Ross, P.J.; Bannasch, D.L. Early developmental and evolutionary origins of gene body DNA methylation patterns in mammalian placentas. PLoS Genet. 2015, 11, e1005442. [Google Scholar] [CrossRef]
- Kubo, N.; Toh, H.; Shirane, K.; Shirakawa, T.; Kobayashi, H.; Sato, T.; Sone, H.; Sato, Y.; Tomizawa, S.-I.; Tsurusaki, Y.; et al. DNA methylation and gene expression dynamics during spermatogonial stem cell differentiation in the early postnatal mouse testis. BMC Genom. 2015, 16, 624. [Google Scholar] [CrossRef]
- Jiang, J.; Cole, J.M.; Da, Y.; VanRaden, P.M.; Ma, L. Fast Bayesian fine-mapping of 35 production, reproduction and body conformation traits with imputed sequences of 27K Holstein bulls. bioRxiv 2018, 428227. [Google Scholar] [CrossRef]
- Plas, E.; Berger, P.; Hermann, M.; Pflüger, H. Effects of aging on male fertility? Exp. Gerontol. 2000, 35, 543–551. [Google Scholar] [CrossRef]
- Harris, I.D.; Fronczak, C.; Roth, L.; Meacham, R.B. Fertility and the aging male. Rev. Urol. 2011, 13, e184. [Google Scholar]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef]
- Wagner, J.R.; Busche, S.; Ge, B.; Kwan, T.; Pastinen, T.; Blanchette, M. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol. 2014, 15, R37. [Google Scholar] [CrossRef]
- Salhab, A.; Nordström, K.; Kattler, K.; Ebert, P.; Ramirez, F.; Arrigoni, L.; Müller, F.; Cadenas, C.; Hengstler, J.; Lengauer, T. Partially methylated domains are hallmarks of a cell specific epigenome topology. bioRxiv 2018, 249334. [Google Scholar] [CrossRef]
- Tourtellotte, W.G.; Nagarajan, R.; Auyeung, A.; Mueller, C.; Milbrandt, J. Infertility associated with incomplete spermatogenic arrest and oligozoospermia in Egr4-deficient mice. Development 1999, 126, 5061–5071. [Google Scholar]
- Lin, C.; Chow, S.; Lau, A.; Tu, R.; Lue, T. Human PDE5A gene encodes three PDE5 isoforms from two alternate promoters. Int. J. Impot. Res. 2002, 14, 15. [Google Scholar] [CrossRef]
- Kalsi, J.; Bahadur, G.; Muneer, A.; Ozturk, O.; Christopher, N.; Ralph, D.; Minhas, S. Novel PDE5 inhibitors for the treatment of male erectile dysfunction. Reprod. Biomed. Online 2003, 7, 456–461. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J.; Duan, J.; Gao, X.; Zhu, W.; Lu, X.; Yang, L.; Zhang, J.; Li, G.; Ci, W. Programming and inheritance of parental DNA methylomes in mammals. Cell 2014, 157, 979–991. [Google Scholar] [CrossRef]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Relton, C.L. Statistical and integrative system-level analysis of DNA methylation data. Nat. Rev. Genet. 2018, 19, 129. [Google Scholar] [CrossRef]
- Gao, X.; Jia, M.; Zhang, Y.; Breitling, L.P.; Brenner, H. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: A systematic review of DNA methylation studies. Clin. Epigenetics 2015, 7, 113. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef]
- Wang, H.-Q.; Tuominen, L.K.; Tsai, C.-J. SLIM: A sliding linear model for estimating the proportion of true null hypotheses in datasets with dependence structures. Bioinformatics 2010, 27, 225–231. [Google Scholar] [CrossRef]
- Guo, S.; Diep, D.; Plongthongkum, N.; Fung, H.-L.; Zhang, K.; Zhang, K. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat. Genet. 2017, 49, 635. [Google Scholar] [CrossRef]
- Eckhardt, F.; Lewin, J.; Cortese, R.; Rakyan, V.K.; Attwood, J.; Burger, M.; Burton, J.; Cox, T.V.; Davies, R.; Down, T.A. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 2006, 38, 1378. [Google Scholar] [CrossRef]
- Libertini, E.; Heath, S.C.; Hamoudi, R.A.; Gut, M.; Ziller, M.J.; Czyz, A.; Ruotti, V.; Stunnenberg, H.G.; Frontini, M.; Ouwehand, W.H. Information recovery from low coverage whole-genome bisulfite sequencing. Nat. Commun. 2016, 7, 11306. [Google Scholar] [CrossRef]
- Libertini, E.; Heath, S.C.; Hamoudi, R.A.; Gut, M.; Ziller, M.J.; Herrero, J.; Czyz, A.; Ruotti, V.; Stunnenberg, H.G.; Frontini, M. Saturation analysis for whole-genome bisulfite sequencing data. Nat. Biotechnol. 2016, 34, 691. [Google Scholar] [CrossRef]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An expanded view of complex traits: From polygenic to omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef]
- Rohde, P.D.; Demontis, D.; Cuyabano, B.C.D.; Børglum, A.D.; Sørensen, P. Covariance Association Test (CVAT) Identifies Genetic Markers Associated with Schizophrenia in Functionally Associated Biological Processes. Genetics 2016, 203, 1901–1913. [Google Scholar] [CrossRef]
- Sørensen, I.F.; Edwards, S.M.; Rohde, P.D.; Sørensen, P. Multiple Trait Covariance Association Test Identifies Gene Ontology Categories Associated with Chill Coma Recovery Time in Drosophila melanogaster. Sci. Rep. 2017, 7, 2413. [Google Scholar] [CrossRef]
- Sarup, P.; Jensen, J.; Ostersen, T.; Henryon, M.; Sørensen, P. Increased prediction accuracy using a genomic feature model including prior information on quantitative trait locus regions in purebred Danish Duroc pigs. BMC Genet. 2016, 17, 11. [Google Scholar] [CrossRef]
- Fang, L.; Sahana, G.; Ma, P.; Su, G.; Yu, Y.; Zhang, S.; Lund, M.S.; Sørensen, P. Exploring the genetic architecture and improving genomic prediction accuracy for mastitis and milk production traits in dairy cattle by mapping variants to hepatic transcriptomic regions responsive to intra-mammary infection. Genet. Sel. Evol. 2017, 49, 44. [Google Scholar] [CrossRef]
- Fang, L.; Sørensen, P.; Sahana, G.; Panitz, F.; Su, G.; Zhang, S.; Yu, Y.; Li, B.; Ma, L.; Liu, G. MicroRNA-guided prioritization of genome-wide association signals reveals the importance of microRNA-target gene networks for complex traits in cattle. Sci. Rep. 2018, 8, 9345. [Google Scholar] [CrossRef] [PubMed]
- Finucane, H.K.; Bulik-Sullivan, B.; Gusev, A.; Trynka, G.; Reshef, Y.; Loh, P.-R.; Anttila, V.; Xu, H.; Zang, C.; Farh, K. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015, 47, 1228. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CHR | DMR Start | DMR End | q (Age) | Meth. Diff. (Age) | q (SCR) | Meth. Diff. (SCR) | Gene | Associated Traits | Top SNP Position | p-Value |
|---|---|---|---|---|---|---|---|---|---|---|
| 9 | 85957501 | 85958000 | 1.11 × 10−7 | 31.92 | 1.24 × 10−3 | 14.72 | SAMD5 | SCR | 85988014 | 4.74 × 10−7 |
| 6 | 7012001 | 7012500 | 1.41 × 10−6 | 5.51 | 9.38 × 10−12 | 5.42 | PDE5A | SCR | 6880354 | 6.44 × 10−6 |
| 5 | 117709001 | 117709500 | 4.88 × 10−3 | 8.90 | 5.96 × 10−3 | 7.10 | GTSE1 | SCR | 117689528 | 6.19 × 10−6 |
| Sire_Calv_Ease | 117691137 | 4.89 × 10−6 | ||||||||
| 27 | 30563001 | 30563500 | 1.13 × 10−4 | 6.98 | 1.22 × 10−6 | 6.73 | UNC5D | SCR | 30756178 | 5.71 × 10−6 |
| 1 | 147329501 | 147330000 | 3.92 × 10−5 | 34.09 | 1.09 × 10−7 | 25.52 | PCBP3 | AFC | 147188444 | 6.37 × 10−6 |
| 22 | 30725501 | 30726000 | 4.05 × 10−3 | 27.20 | 5.19 × 10−3 | 12.16 | FOXP1 | Sir_Calv_Ease | 30678479 | 4.86 × 10−6 |
| 8 | 92536501 | 92537000 | 2.03 × 10−5 | 6.15 | 4.82 × 10−6 | 5.22 | PLPPR1 | Dtr_Still_Birth | 92354263 | 5.14 × 10−7 |
| Sire_Still_Birth | 92503882 | 9.70 × 10−7 | ||||||||
| 28 | 7153001 | 7153500 | 9.68 × 10−3 | 19.49 | 4.03 × 10−3 | 22.46 | SLC35F3 | Sir_Still_Birth | 6894934 | 5.02 × 10−10 |
| 6 | 115334001 | 115334500 | 8.21 × 10−3 | 31.99 | 4.58 × 10−3 | 10.88 | C1QTNF7 | Dtr_Calv_Ease | 115292546 | 1.24 × 10−7 |
| 28 | 29788001 | 29788500 | 3.99 × 10−4 | 5.53 | 1.64 × 10−3 | 5.07 | MYOZ1 | Cow_Conc_Rate | 29798384 | 2.92 × 10−6 |
| 6 | 89203501 | 89204000 | 6.86 × 10−3 | 6.41 | 8.49 × 10−3 | 5.38 | ADAMTS3 | CFI | 89297757 | 9.55 × 10−9 |
| Dtr_Preg_Rate | 89436001 | 1.04 × 10−7 | ||||||||
| Dtr_Still_Birth | 89227960 | 6.15 × 10−7 | ||||||||
| 9 | 50054001 | 50054500 | 2.51 × 10−3 | 8.90 | 1.86 × 10−3 | 8.68 | ASCC3 | AFC | 49838161 | 2.18 × 10−6 |
| Sire_Calv_Ease | 49839702 | 5.74 × 10−6 | ||||||||
| 13 | 26858001 | 26858500 | 8.21 × 10−3 | 20.41 | 2.55 × 10−3 | 8.13 | MYO3A | Sire_Still_Birth | 26887898 | 1.52 × 10−6 |
| 19 | 12575001 | 12575500 | 9.96 × 10−11 | 31.08 | 4.82 × 10−6 | 16.16 | BCAS3 | Heifer_Conc_Rate | 12459490 | 1.18 × 10−9 |
| Sire_Calv_Ease | 12432636 | 5.06 × 10−7 | ||||||||
| Sire_Still_Birth | 12432710 | 3.56 × 10−7 | ||||||||
| 3 | 101902501 | 101903000 | 4.66 × 10−3 | 16.05 | 1.77 × 10−3 | 8.81 | C1orf228 | CFI | 10191696 | 7.16 × 10−6 |
| Sire_Still_Birth | 101936626 | 7.16 × 10−6 | ||||||||
| 4 | 57398001 | 57398500 | 3.21 × 10−7 | 5.68 | 7.47 × 10−10 | 5.25 | IMMP2L | Sire_Still_Birth | 57140432 | 3.90 × 10−7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, L.; Zhou, Y.; Liu, S.; Jiang, J.; Bickhart, D.M.; Null, D.J.; Li, B.; Schroeder, S.G.; Rosen, B.D.; Cole, J.B.; et al. Integrating Signals from Sperm Methylome Analysis and Genome-Wide Association Study for a Better Understanding of Male Fertility in Cattle. Epigenomes 2019, 3, 10. https://doi.org/10.3390/epigenomes3020010
Fang L, Zhou Y, Liu S, Jiang J, Bickhart DM, Null DJ, Li B, Schroeder SG, Rosen BD, Cole JB, et al. Integrating Signals from Sperm Methylome Analysis and Genome-Wide Association Study for a Better Understanding of Male Fertility in Cattle. Epigenomes. 2019; 3(2):10. https://doi.org/10.3390/epigenomes3020010
Chicago/Turabian StyleFang, Lingzhao, Yang Zhou, Shuli Liu, Jicai Jiang, Derek M. Bickhart, Daniel J. Null, Bingjie Li, Steven G. Schroeder, Benjamin D. Rosen, John B. Cole, and et al. 2019. "Integrating Signals from Sperm Methylome Analysis and Genome-Wide Association Study for a Better Understanding of Male Fertility in Cattle" Epigenomes 3, no. 2: 10. https://doi.org/10.3390/epigenomes3020010