
Diagnosis of Chromosome 15q-Terminal Deletion Syndrome through Elevated Fasting Serum Growth Hormone Levels

 and
and
Abstract
:1. Introduction
2. Case Presentation
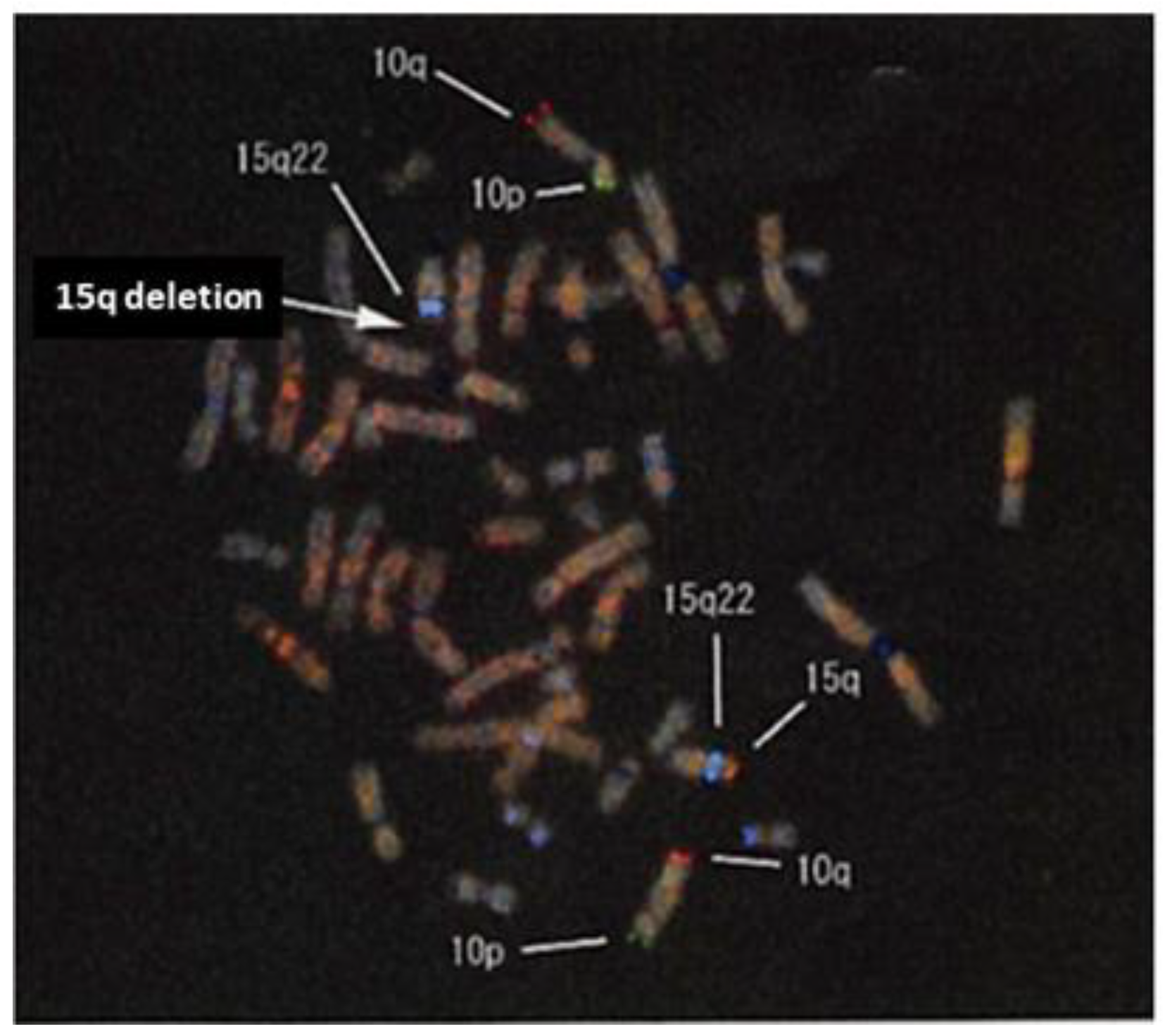
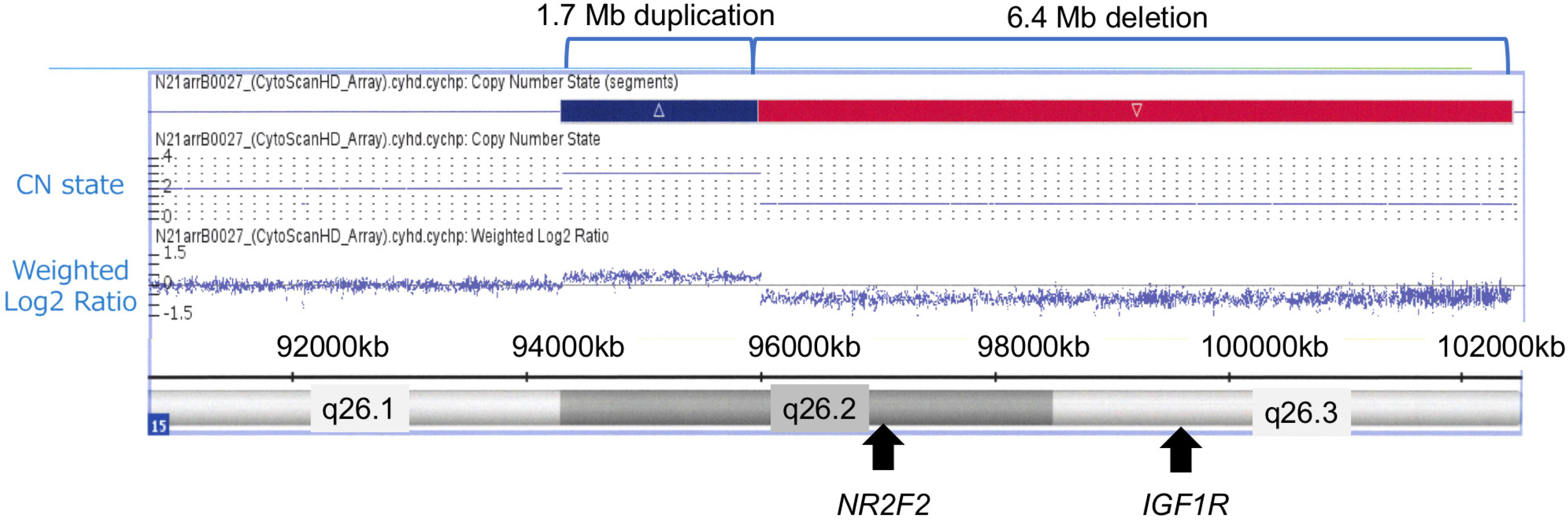
3. Diagnostic Assessment: Genetic Testing
4. Discussion

{kind=link}
{kind=link}
| IGF-1 (ng/mL) (Range * or SDS **) | Basal GH (ng/mL) | Peak GH (ng/mL) | IGF1R Abnormalities (Deletion Size) | Age | Sex | Authors (Reference Number) |
|---|---|---|---|---|---|---|
| 126 (+1.2) ** | 8.5–85.5 | NA | 15q26.2- > 15qter (6.4 Mb) | 5 m | F | Our case |
| 72 (33–102) * | NA | 7.6 | p.Asp1105Glu | 7 m | M | Solomon-Zemler et al. [21] |
| 231 (+2.9) ** | 8.0 | NA | p.Arg1256Ser | 1 y 5 m | M | Juanes et al. [22] |
| 211 (+1.7) ** | 1.2 | NA | p.Tyr865Cys | 1 y 9 m | F | Juanes et al. [22] |
| NA | 2.7 | 10.0 | 15q26.1- > 15qter (5 Mb) | 1 y 10 m | F | Okubo et al. [23] |
| 16 (−4.67) ** | 7.6 | NA | p.Tyr487Phe | 2 y | F | Labarta et al. [24] |
| 102 (51–303) * | 6.9 | 17.3 | p.Met1247Thr | 2 y 1 m | F | Yang et al. [25] |
| 185 (32–213) * | 10.5 | Normal | p.Trp1249X | 2 y 5 m | F | Fujimoto et al. [26] |
| 58 (+0.5) ** | 0.27 | NA | p.Asn359Tyr | 2 y 8 m | M | Juanes et al. [22] |
| 344 (+3.3) ** | 7.6 | NA | p.Arg431Leu | 3 y 0 m | F | Kawashima et al., 2012 [27] |
| 298.4 (+3.8) ** | NA | Normal | p.Gln1250X | 3 y 0 m | M | Fujimoto et al. [26] |
| 268 (+2.1) ** | 2.4 | 14.3 | 15q26.2- > 15qter | 3 y 1 m | F | Gonc et al. [7] |
| NA (−0.8 to +1.4) ** | NA | 9.8 | p.Arg59X | 3 y 2 m | M | Raile et al. [28] |
| 239 (+1.6) ** | NA | 62 | p.Gly1050Lys | 3 y 3 m | F | Walenkamp et al. [29] |
| 253 (54–161) * | High | High | p.Glu121lys/p.Glu234Lys | 3 y 4 m | M | Fang et al. [30] |
| Normal | Normal | Normal | 15q26.1- > 15qter (9.15 Mb) | 4 y | F | Benbouchta et al. [31] |
| 159 (+2.6) ** | NA | Normal | p.Tyr387X | 4 y | M | Mohn et al. [32] |
| 63 (NA) | NA | 51 | p.Arg108Gln/p.Lys115Asn | 4 y 5 m | F | Abuzzahab et al. [33] |
| 203 (+2.5) ** | 2.5–7.5 | NA | 15q26.2- > 15qter (5.2 Mb) | 4 y 5 m | F | Walenkamp et al. [34] |
| 222 (+2.3) ** | NA | 5.7–21.2 | p.Arg59X | 5 y 3 m | M | Abuzzahab et al. [33] |
| NA (+1.1) ** | 3.0 | 12.1 | p.Arg511Trp | 5 y 8 m | F | Leal et al. [35] |
| 344 (+3.3) ** | NA | Normal | p.Asp1105Glu | 6 y 0 m | F | Kawashima et al., 2014 [36] |
| 145 (−0.66) ** | NA | 3.72 | 15q26.2- >15qter (3.9 Mb) | 6 y 5 m | F | Yoon et al. [37] |
| 200 (+0.76) ** | 0.9–2.4 | Normal | p.Gly1125Ala | 7 y 5 m | F | Kruis et al. [38] |
| NA (+1.2 to +2.2) ** | NA | 4.4–21 | p.Arg59X | 8 y 5 m | M | Raile et al. [28] |
| 164 (123–275) * | 0.42 | 7.6–9.1 | p.Met1123Argfs * 14 | 9 y 6 m | M | Fang et al. [5] |
| 509 (51–303) * | 0.36 | 15.4 | p.Cys248Trp | 10 y 8 m | F | Yang et al. [25] |
| 291 (+1.53) ** | 0.15 | 11.2 | 15q26.3- > 15qter | 12 y 8 m | M | Gonc et al. [7] |
| 350 (+0.6) ** | 3.7 | 12.9 | p.Ile678Val | 13 y | M | Gonc et al. [7] |
| 950 (+7) ** | 0.86 | NA | p.Arg10Leu | 13 y 5 m | F | Gannagé-Yared et al. [39] |
| 100 (−1.11) ** | 0.46 | 10 | p.Arg461His | 13 y 5 m | M | Gonc et al. [7] |
| 404.3 (165–300) * | 7.4 | 10.6 | p.Arg481Gln | 13 y 6 m | F | Inagaki et al. [40] |
| 671 (+2.13) ** | 25.3 | NA | p.Thr79Met | 13 y 8 m | F | Gonc et al. [7] |
| 112 (–2.44) ** | 0.63 | 20.3 | p.Lys704Arg | 15 y | M | Gonc et al. [7] |
| 389.8 (244–787) * | NA | 18.8 | p.Ala140Glyfs * 5 | 15 y | F | Wang et al. [41] |
| 537 (+0.85) ** | 0.7 | 11.7 | p.Leu19Profs * 27 | 16 y 3 m | M | Gonc et al. [7] |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rudaks, L.I.; Nicholl, J.K.; Bratkovic, D.; Barnett, C.P. Short stature due to 15q26 microdeletion involving IGF1R: Report of an additional case and review of the literature. Am. J. Med. Genet. A 2011, 155, 3139–3143. [Google Scholar] [CrossRef] [PubMed]
- Pinson, L.; Perrin, A.; Plouzennec, C.; Parent, P.; Metz, C.; Collet, M.; Le Bris, M.J.; Douet-Guilbert, N.; Morel, F.; De Braekeleer, M. Detection of an unexpected subtelomeric 15q26.2→qter deletion in a little girl: Clinical and cytogenetic studies. Am. J. Med. Genet. A 2005, 138, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Poot, M.; Eleveld, M.J.; van’t Slot, R.; van Genderen, M.M.; Verrijn Stuart, A.A.; Hochstenbach, R.; Beemer, F.A. Proportional growth failure and oculocutaneous albinism in a girl with a 6.87 Mb deletion of region 15q26.2→qter. Eur. J. Med. Genet. 2007, 50, 432–440. [Google Scholar] [CrossRef]
- Tönnies, H.; Schulze, I.; Hennies, H.; Neumann, L.M.; Keitzer, R.; Neitzel, H. De novo terminal deletion of chromosome 15q26.1 characterised by comparative genomic hybridisation and FISH with locus specific probes. J. Med. Genet. 2001, 38, 617–621. [Google Scholar] [CrossRef] [Green Version]
- Fang, P.; Schwartz, I.D.; Johnson, B.D.; Derr, M.A.; Roberts, C.T., Jr.; Hwa, V.; Rosenfeld, R.G. Familial short stature caused by haploinsufficiency of the insulin-like growth factor I receptor due to nonsense-mediated messenger ribonucleic acid decay. J. Clin. Endocrinol. Metab. 2009, 94, 1740–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klammt, J.; Pfäffle, R.; Werner, H.; Kiess, W. IGF signaling defects as causes of growth failure and IUGR. Trends Endocrinol. Metab. 2008, 19, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Gonc, E.N.; Ozon, Z.A.; Oguz, S.; Kabacam, S.; Taskiran, E.Z.; Kiper, P.O.S.; Utine, G.E.; Alikasifoglu, A.; Kandemir, N.; Boduroglu, O.K.; et al. Genetic IGF1R defects: New cases expand the spectrum of clinical features. J. Endocrinol. Investig. 2020, 43, 1739–1748. [Google Scholar] [CrossRef]
- Giabicani, E.; Willems, M.; Steunou, V.; Chantot-Bastaraud, S.; Thibaud, N.; Abi Habib, W.; Azzi, S.; Lam, B.; Bérard, L.; Bony-Trifunovic, H.; et al. Increasing knowledge in IGF1R defects: Lessons from 35 new patients. J. Med. Genet. 2020, 57, 160–168. [Google Scholar] [CrossRef]
- Ester, W.A.; van Duyvenvoorde, H.A.; de Wit, C.C.; Broekman, A.J.; Ruivenkamp, C.A.; Govaerts, L.C.; Wit, J.M.; Hokken-Koelega, A.C.; Losekoot, M. Two short children born small for gestational age with insulin-like growth factor 1 receptor haploinsufficiency illustrate the heterogeneity of its phenotype. J. Clin. Endocrinol. Metab. 2009, 94, 4717–4727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isojima, T.; Shimatsu, A.; Yokoya, S.; Chihara, K.; Tanaka, T.; Hizuka, N.; Teramoto, A.; Tatsumi, K.I.; Tachibana, K.; Katsumata, N.; et al. Standardized centile curves and reference intervals of serum insulin-like growth factor-I (IGF-I) levels in a normal Japanese population using the LMS method. Endocr. J. 2012, 59, 771–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, X.H.; Wang, Q.; Wang, J.; Liu, X.Y.; Xu, Y.J.; Huang, R.T.; Xue, S.; Li, Y.J.; Zhang, M.; Qu, X.K.; et al. A novel NR2F2 loss-of-function mutation predisposes to congenital heart defect. Eur. J. Med. Genet. 2018, 61, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Weng, L.; Kavaslar, N.; Ustaszewska, A.; Doelle, H.; Schackwitz, W.; Hébert, S.; Cohen, J.C.; McPherson, R.; Pennacchio, L.A. Lack of MEF2A mutations in coronary artery disease. J. Clin. Investig. 2005, 115, 1016–1020. [Google Scholar] [CrossRef] [Green Version]
- Walenkamp, M.J.E.; Robers, J.M.L.; Wit, J.M.; Zandwijken, G.R.J.; Van Duyvenvoorde, H.A.; Oostdijk, W.; Hokken-Koelega, A.C.S.; Kant, S.G.; Losekoot, M. Phenotypic Features and Response to GH Treatment of Patients with a Molecular Defect of the IGF-1 Receptor. J. Clin. Endocrinol. Metab. 2019, 104, 3157–3171. [Google Scholar] [CrossRef]
- Hawkes, C.P.; Grimberg, A. Measuring growth hormone and insulin-like growth factor-I in infants: What is normal? Pediatr. Endcrinol. Rev. 2013, 11, 126–146. [Google Scholar]
- Hwa, V.; Fujimoto, M.; Zhu, G.; Gao, W.; Foley, C.; Kumbaji, M.; Rosenfeld, R.G. Genetic causes of growth hormone insensitivity beyond GHR. Rev. Endocr. Metab. Disord. 2021, 22, 43–58. [Google Scholar] [CrossRef]
- Ogilvy-Stuart, A.L. Growth hormone deficiency (GHD) from birth to 2 years of age: Diagnostic specifics of GHD during the early phase of life. Horm. Res. 2003, 60, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Leger, J.; Noel, M.; Limal, J.M.; Czernichow, P. Growth factors and intrauterine growth retardation. II. Serum growth hormone, insulin-like growth factor (IGF) I, and IGF-binding protein 3 levels in children with intrauterine growth retardation compared with normal control subjects: Prospective study from birth to two years of age. Study Group of IUGR. Pediatr. Res. 1996, 40, 101–107. [Google Scholar] [PubMed] [Green Version]
- Klaassens, M.; Galjaard, R.J.; Scott, D.A.; Bruggenwirth, H.T.; van Opstal, D.; Fox, M.V.; Higgins, R.R.; Cohen-Overbeek, T.E.; Schoonderwaldt, E.M.; Lee, B.; et al. Prenatal Detection and Outcome of Congenital Diaphragmatic Hernia (CDH) Associated with Deletion of Chromosome 15q26: Two Patients and Review of the Literature. Am. J. Med. Genet. A 2007, 143, 2204–2212. [Google Scholar] [CrossRef]
- Genesio, R.; De Brasi, D.; Conti, A.; Borghese, A.; Di Micco, P.; Di Costanzo, P.; Paladini, D.; Ungaro, P.; Nitsch, L. Inverted duplication of 15q with terminal deletion in a multiple malformed newborn with intrauterine growth failure and lethal phenotype. Am. J. Med. Genet. A 2004, 128, 422–428. [Google Scholar]
- Upadia, J.; Gonzales, P.R.; Robin, N.H. Novel de novo pathogenic variant in the NR2F2 gene in a boy with congenital heart defect and dysmorphic features. Am. J. Med. Genet. A 2018, 176, 1423–1426. [Google Scholar] [CrossRef]
- Solomon-Zemler, R.; Basel-Vanagaite, L.; Steier, D.; Yakar, S.; Mel, E.; Phillip, M.; Bazak, L.; Bercovich, D.; Werner, H.; de Vries, L. A novel heterozygous IGF-1 receptor mutation associated with hypoglycemia. Endocr. Connect. 2017, 6, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Juanes, M.; Guercio, G.; Marino, R.; Berensztein, E.; Warman, D.M.; Ciaccio, M.; Gil, S.; Bailez, M.; Rivarola, M.A.; Belgorosky, A. Three novel IGF1R mutations in microcephalic patients with prenatal and postnatal growth impairment. Clin. Endocrinol. 2015, 82, 704–711. [Google Scholar] [CrossRef]
- Okubo, Y.; Siddle, K.; Firth, H.; O’Rahilly, S.; Wilson, L.C.; Willatt, L.; Fukushima, T.; Takahashi, S.; Petry, C.J.; Saukkonen, T.; et al. Cell Proliferation Activities on Skin Fibroblasts from a Short Child with Absence of One Copy of the Type 1 Insulin-Like Growth Factor Receptor (IGF1R) Gene and a Tall Child with Three Copies of the IGF1R Gene. J. Clin. Endocrinol. Metab. 2003, 88, 5981–5988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labarta, J.I.; Barrio, E.; Audí, L.; Fernández-Cancio, M.; Andaluz, P.; de Arriba, A.; Puga, B.; Calvo, M.T.; Mayayo, E.; Carrascosa, A.; et al. Familial short stature and intrauterine growth retardation associated with a novel mutation in the IGF-I receptor (IGF1R) gene. Clin. Endocrinol. 2013, 78, 255–262. [Google Scholar] [CrossRef]
- Yang, L.; Xu, D.D.; Sun, C.J.; Wu, J.; Wei, H.Y.; Liu, Y.; Zhang, M.Y.; Luo, F.H. IGF1R variants in patients with growth impairment: Four novel variants and genotype-phenotype correlations. J. Clin. Endocrinol. Metab. 2018, 103, 3939–3944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, M.; Kawashima Sonoyama, Y.; Hamajima, N.; Hamajima, T.; Kumura, Y.; Miyahara, N.; Nishimura, R.; Adachi, K.; Nanba, E.; Hanaki, K.; et al. Heterozygous nonsense mutations near the C-terminal region of IGF1R in two patients with small-for-gestational-age-related short stature. Clin. Endocrinol. 2015, 83, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, Y.; Higaki, K.; Fukushima, T.; Hakuno, F.; Nagaishi, J.; Hanaki, K.; Nanba, E.; Takahashi, S.; Kanzaki, S. Novel missense mutation in the IGF-I receptor L2 domain results in intrauterine and postnatal growth retardation. Clin. Endocrinol. 2012, 77, 246–254. [Google Scholar] [CrossRef]
- Raile, K.; Klammt, J.; Schneider, A.; Keller, A.; Laue, S.; Smith, R.; Pfäffle, R.; Kratzsch, J.; Keller, E.; Kiess, W. Clinical and functional characteristics of the human Arg59Ter insulin-like growth factor I receptor (IGF1R) mutation: Implications for a gene dosage effect of the human IGF1R. J. Clin. Endocrinol. Metab. 2006, 91, 2264–2271. [Google Scholar] [CrossRef] [Green Version]
- Walenkamp, M.J.; van der Kamp, H.J.; Pereira, A.M.; Kant, S.G.; van Duyvenvoorde, H.A.; Kruithof, M.F.; Breuning, M.H.; Romijn, J.A.; Karperien, M.; Wit, J.M. A variable degree of intrauterine and postnatal growth retardation in a family with a missense mutation in the insulin-like growth factor I receptor. J. Clin. Endocrinol. Metab. 2006, 91, 3062–3070. [Google Scholar] [CrossRef] [Green Version]
- Fang, P.; Cho, Y.H.; Derr, M.A.; Rosenfeld, R.G.; Hwa, V.; Cowell, C.T. Severe short stature caused by novel compound heterozygous mutations of the insulin-like growth factor 1 receptor (IGF1R). J. Clin. Endocrinol. Metab. 2012, 97, E243–E247. [Google Scholar] [CrossRef] [Green Version]
- Benbouchta, Y.; De Leeuw, N.; Amasdl, S.; Sbiti, A.; Smeets, D.; Sadki, K.; Sefiani, A. 15q26 deletion in a patient with congenital heart defect, growth restriction and intellectual disability: Case report and literature review. Ital. J. Pediatr. 2021, 47, 188. [Google Scholar] [CrossRef] [PubMed]
- Mohn, A.; Marcovecchio, M.L.; de Giorgis, T.; Pfaeffle, R.; Chiarelli, F.; Kiess, W. An insulin-like growth factor-I receptor defect associated with short stature and impaired carbohydrate homeostasis in an Italian pedigree. Horm Res. Paediatr. 2011, 76, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Abuzzahab, M.J.; Schneider, A.; Goddard, A.; Grigorescu, F.; Lautier, C.; Keller, E.; Kiess, W.; Klammt, J.; Kratzsch, J.; Osgood, D.; et al. IGF-I Receptor Mutations Resulting in Intrauterine and Postnatal Growth Retardation. N. Engl. J. Med. 2003, 349, 2211–2222. [Google Scholar] [CrossRef] [PubMed]
- Walenkamp, M.J.; de Muinck Keizer-Schrama, S.M.; de Mos, M.; Kalf, M.E.; van Duyvenvoorde, H.A.; Boot, A.M.; Kant, S.G.; White, S.J.; Losekoot, M.; Den Dunnen, J.T.; et al. Successful long-term growth hormone therapy in a girl with haploinsufficiency of the insulin-like growth factor-I receptor due to a terminal 15q26.2→qter deletion detected by multiplex ligation probe amplification. J. Clin. Endocrinol. Metab. 2008, 93, 2421–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal, A.C.; Montenegro, L.R.; Saito, R.F.; Ribeiro, T.C.; Coutinho, D.C.; Mendonca, B.B.; Arnhold, I.J.; Jorge, A.A. Analysis of the insulin-like growth factor 1 receptor gene in children born small for gestational age: In vitro characterization of a novel mutation (p.Arg511Trp). Clin. Endocrinol. 2013, 78, 558–563. [Google Scholar] [CrossRef]
- Kawashima, Y.; Hakuno, F.; Okada, S.; Hotsubo, T.; Kinoshita, T.; Fujimoto, M.; Nishimura, R.; Fukushima, T.; Hanaki, K.; Takahashi, S.; et al. Familial short stature is associated with a novel dominant-negative heterozygous insulin-like growth factor 1 receptor (IGF1R) mutation. Clin. Endocrinol. 2014, 81, 312–314. [Google Scholar] [CrossRef]
- Yoon, J.S.; Hwang, I.T. Microdeletion in the IGF-1 receptor gene of a patient with short stature and obesity: A case report. J. Pediatr. Endocrinol. Metab. 2021, 34, 255–259. [Google Scholar] [CrossRef]
- Kruis, T.; Klammt, J.; Galli-Tsinopoulou, A.; Wallborn, T.; Schlicke, M.; Müller, E.; Kratzsch, J.; Körner, A.; Odeh, R.; Kiess, W.; et al. Heterozygous mutation within a kinase-conserved motif of the insulin-like growth factor I receptor causes intrauterine and postnatal growth retardation. J. Clin. Endocrinol. Metab. 2010, 95, 1137–1142. [Google Scholar] [CrossRef] [Green Version]
- Gannagé-Yared, M.H.; Klammt, J.; Chouery, E.; Corbani, S.; Meǵarbané, H.; Abou Ghoch, J.; Choucair, N.; Pfäffle, R.; Mégarbané, A. Homozygous mutation of the IGF1 receptor gene in a patient with severe pre- and postnatal growth failure and congenital malformations. Eur. J. Endocrinol. 2012, 168, K1–K7. [Google Scholar] [CrossRef]
- Inagaki, K.; Tiulpakov, A.; Rubtsov, P.; Sverdlova, P.; Peterkova, V.; Yakar, S.; Terekhov, S.; LeRoith, D. A familial insulin-like growth factor-I receptor mutant leads to short stature: Clinical and biochemical characterization. J. Clin. Endocrinol. Metab. 2007, 92, 1542–1548. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.R.; Carmichael, H.; Andrew, S.F.; Miller, T.C.; Moon, J.E.; Derr, M.A.; Hwa, V.; Hirschhorn, J.N.; Dauber, A. Large-scale pooled next-generation sequencing of 1077 genes to identify genetic causes of short stature. J. Clin. Endocrinol. Metab. 2013, 98, E1428–E1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]


| Age in Months | 3 Months | 4 Months | 5 Months |
|---|---|---|---|
| Serum GH (ng/mL) | 85.5 | 8.5 | 26.1 |
| IGF-1 (ng/mL) (SDS) * | NA | 75 (+0.16) | 126 (+1.2) |
| Blood glucose (mg/dL) | 57 | NA | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ono, M.; Tanaka, M.; Hiroshima, S.; Sawano, K.; Ogawa, Y.; Nagasaki, K.; Saitoh, A. Diagnosis of Chromosome 15q-Terminal Deletion Syndrome through Elevated Fasting Serum Growth Hormone Levels. Endocrines 2022, 3, 92-99. https://doi.org/10.3390/endocrines3010008
Ono M, Tanaka M, Hiroshima S, Sawano K, Ogawa Y, Nagasaki K, Saitoh A. Diagnosis of Chromosome 15q-Terminal Deletion Syndrome through Elevated Fasting Serum Growth Hormone Levels. Endocrines. 2022; 3(1):92-99. https://doi.org/10.3390/endocrines3010008
Chicago/Turabian StyleOno, Masato, Masato Tanaka, Shota Hiroshima, Kentaro Sawano, Yohei Ogawa, Keisuke Nagasaki, and Akihiko Saitoh. 2022. "Diagnosis of Chromosome 15q-Terminal Deletion Syndrome through Elevated Fasting Serum Growth Hormone Levels" Endocrines 3, no. 1: 92-99. https://doi.org/10.3390/endocrines3010008