Photochemical Dissipative Structuring of the Fundamental Molecules of Life †


Department of Nuclear Physics and Applications of Radiation, Instituto de Física, Universidad Nacional Autónoma de México, Circuito Interior de la Investigación Científica, Cuidad Universitaria, Mexico City C.P. 04510, Mexico
†
Presented at the 5th International Electronic Conference on Entropy and Its Applications, 18–30 November 2019; Available online: https://ecea-5.sciforum.net/.
Proceedings 2020, 46(1), 20; https://doi.org/10.3390/ecea-5-06692
Published: 18 November 2019
(This article belongs to the Proceedings of The 5th International Electronic Conference on Entropy and Its Applications)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:It has been conjectured that the origin of the fundamental molecules of life, their proliferation over the surface of Earth, and their complexation through time, are examples of photochemical dissipative structuring, dissipative proliferation, and dissipative selection, respectively, arising out of the nonequilibrium conditions created on Earth’s surface by the solar photon spectrum. Here I describe the nonequilibrium thermodynamics and the photochemical mechanisms involved in the synthesis and evolution of the fundamental molecules of life from simpler more common precursor molecules under the long wavelength UVC and UVB solar photons prevailing at Earth’s surface during the Archean. Dissipative structuring through photochemical mechanisms leads to carbon based UVC pigments with peaked conical intersections which endow them with a large photon disipative capacity (broad wavelength absorption and rapid radiationless dexcitation). Dissipative proliferation occurs when the photochemical dissipative structuring becomes autocatalytic. Dissipative selection arises when fluctuations lead the system to new stationary states (corresponding to different molecular concentration profiles) of greater dissipative capacity as predicted by the universal evolution criterion of Classical Irreversible Thermodynamic theory established by Onsager, Glansdorff, and Prigogine. An example of the UV photochemical dissipative structuring, proliferation, and selection of the nucleobase adenine from an aqueous solution of HCN under UVC light is given.
1. Introduction
Life, although incorporating equilibrium structures, is fundamentally a nonequilibrium process and therefore its existence is dependent on the dissipation of one or more thermodynamic potentials in its environment. Boltzmann recognized this almost 125 years ago [1] and suggested that the most important thermodynamic potential that life dissipates is the solar photon potential [2]. Present day life has evolved to dissipate other thermodynamic potentials accessible on Earth’s surface, for example, chemical potentials available in biological material or at hydrothermal vents. However, irrespective of the greater volcanic activity of a warmer Archean Earth at the origin of life, hydrothermal vents would have provided only a small fraction (∼0.1%) of the free energy available in the prevailing short wavelength solar photon flux [3]. There is, therefore, greater rational in the conjecture that the solar photon potential was responsible for the dissipative structuring of the fundamental molecules of life (common to all three domains). In fact, we show here that not only was the solar photon potential responsible for structuring but also for dissipative proliferation and dissipative selection, leading to evolution of greater overall photon dissipation of the incipient biosphere.
We identified the long wavelength part of the UVC plus the UVB solar photon spectrum as the thermodynamic potential which gave rise to the dissipative structuring, proliferation, and selection relevant to the origin of life [4,5]. This light prevailed at Earth’s surface from the Hadean, before the probable origin of life near the beginning of the Archean (∼3.9 Ga), and for at least 1000 million years [6,7,8] until the formation of an ozone layer when natural oxygen sinks (for example, free hydrogen and Fe) became overwhelmed by organisms performing oxygenic photosynthesis (see Figure 1). It has been estimated by Miller, Urey, and Oró that the solar light of wavelength less than 300 nm (Figure 1) provided approximately three orders of magnitude more free energy than electric discharges, radioactivity, and volcanic activity combined [9].
In contradistinction to the generally held view that UV wavelengths were dangerous to early life and thereby induced extreme selection pressure for mechanisms or behavioral traits that protected life from, or made life tolerable under, these photons [7,11,12], here I argue that these wavelengths were not only fundamental to the photochemical synthesis of life’s first molecules (as suggested with increasing sophistication by Oparin [13], Haldane [14], Urey [15], Sagan [16], and Mulkidjanian [12] and supported experimentally by Baly [17], Miller [18], Oro and Kimball [19], Ponnamperuma et al. [20,21,22], Ferris and Orgel [23], and Sagan and Khare [24] and others) but that this UV light was fundamental to the existence of the entire dissipative process known as early life: its synthesis, its proliferation, and its thermodynamic selection (see Section 2) leading to biosphere complexation with concomitant increases in dissipation over time. Rather than seeking refuge or procuring protection from this UV light, we believe that photon-induced molecular transformations provided innovations which allowed early life to maximize UV exposure, such as buoyancy at the ocean surface, larger molecular antennas for capturing this light, increases in the width of the wavelength absorption band, and peaked conical intersections providing extraordinarily low pigment dead-times; these would have been thermodynamically selected for. Indeed, there exists empirical evidence suggesting selection for traits optimizing UV exposure for particular amino acids complexed with their cognate codons, particularly for those amino acids displaying the strongest stereochemical affinity to their codons or anticodons [25]. This led us to suggest that UVC photon dissipation was the basis of the initial specificity in the amino acid–codon relation during an early stereochemical era [26].
The perspective taken here is, therefore, that the origin of life was not a scenario of organic material organization driven by natural selection leading to “better adapted” organisms or to greater chemically stability (e.g., UV resistant organisms) but rather a scenario of the dissipative structuring of material under the imposed UV solar photon potential leading to an organization of material in space and time such that this provided more efficient routes to the dissipation of this imposed photon potential. Through similar dissipative synthesis of photochemical catalysts and cofactors, more complex biosynthetic pathways were thermodynamically selected that could produce and maintain novel pigments for dissipating not only the fundamental UVC and UVB regions but eventually the entire short wavelength region of the solar photon spectrum up to the red-edge (∼700 nm).
2. Thermodynamic Foundations of Microscopic Dissipative Structuring, Proliferation, and Selection
Irreversible processes can be identified by the distribution (flow) of conserved quantities (e.g., energy, momentum, angular momentum, charge, etc.) over degrees of freedom, often spatial coordinate degrees of freedom. Corresponding to a given flow there exists a conjugate generalized thermodynamic force. For example, to the macroscopic flows of heat, matter, and charge, over coordinate space, there corresponds the conjugate forces of a gradient of the inverse of temperature, gradient of mass density (concentration gradient), and the gradient of the electric charge density (the electrostatic potential) respectively. Flows of the conserved quantities, however, can also occur over microscopic degrees of freedom, such as over electronic or vibrational coordinates, spin coordinates, and reaction coordinates (ionizations, deprotonations, disassociations, isomerizations, tautomerizations, rotations around covalent bonds, sigmatrophic shifts, etc.), obeying statistical quantum mechanical rules. The corresponding conjugate forces to the microscopic flows involved in life processes are electromagnetic in nature, for example, the chemical and photochemical potentials. Because, for covalently bound organic material, access to these microscopic degrees of freedom usually requires the deposition of a large amount of the conserved quantity (e.g., energy) locally (e.g., on a particular region of a molecule), such flow, and any resulting dissipative structuring, occurring before the invention of complex biosynthetic pathways, was necessarily associated with ultraviolet photon absorption.
The existence of any macroscopic flow, or equivalently any unbalanced generalized thermodynamic force, necessarily implies that the system is not in thermodynamic equilibrium. Under the assumption of local thermodynamic equilibrium (e.g., local Maxwell–Bolzmann distribution of particle velocities or excited vibrational states), Onsager, Prigogine, Glansdorff, Nicolis, and others developed the mathematical framework to treat out-of-equilibrium phenomena known as Classical Irreversible Thermodynamics (CIT) [27]. In this framework, the total internal (to the system) entropy production P per unit volume is of all irreversible processes occurring within the system due to n generalized thermodynamic forces is simply the sum of all forces (where are the affinities and T is the temperature) multiplied by their conjugate flows . This sum, by the local formulation of the second law of thermodynamics [27], is positive definite for irreversible processes and equal to zero for reversible processes (those occurring in thermodynamic equilibrium),
The assumption of local equilibrium for the case studied here of molecular photochemical dissipative structuring requires that the absorbed energy (of the photon) becomes distributed with Boltzmann statistics over the nuclear vibrational degrees of freedom responsible for molecular transformations. Organic materials in the condensed phase are generally soft materials in the sense that their electronic degrees of freedom couple significantly to their nuclear vibrational degrees of freedom (unlike in the case of inorganic material). This nonadiabatic coupling is mediated by conical intersections which allow for ultra-fast equilibration of the photon energy over vibrational degrees of freedom of the molecule, often on femtosecond time scales, leaving the molecule with an effective vibrational temperature of 2000–4000 K. This time for vibrational equilibration is generally less than the time required for a typical chemical reaction and therefore the irreversible process of molecular dissipative photochemical structuring can be justifiably treated under the CIT framework in the nonlinear regime. Indeed, Prigogine showed that electronic ground state chemical reactions can be treated successfully under CIT theory as long as the reactants retain a Maxwell–Boltzmann distribution of their velocities, which is the case for all but very explosive reactions [27].
The time change of the total entropy production P for any out-of-equilibrium system can be split into two parts, one depending on the time change of the forces X and the other on the time change of the flows J,
where, for a continuous system,
For the case of constant external constraints over the system, for example, when affinities ; are externally imposed and held constant, CIT theory indicates that the system will eventually come to a stationary state in which its thermodynamic state variables (for example, the internal energy E, entropy S, and entropy production ) become time invariant. For flows linearly related to their forces, it is easy to show that there is only one stationary state and that the entropy production in this stationary state takes on its minimal value with respect to variation of the free affinities ; in the system [27]. This principle of minimum dissipation for linear systems was first proposed by Lord Rayleigh in 1873 [28].
However, if the flows are nonlinearly related to the forces (e.g., for chemical reactions the rates of the reaction (flows) are proportional to reactant concentration differences while the affinities (forces) are proportional to the logarithm of the concentration ratios), then, depending on the number of degrees of freedom and how nonlinear the system is, at a certain value of a variable of the system (e.g., overall affinity), labeled a critical point, the system becomes unstable and new, possibly many, different stationary states become available, each with a possibly different value of internal entropy production . In this case, stationary states are only locally stable in some variables of the system, or even unstable in all variables. The nonlinear dynamics is such that different stationary states, corresponding to different sets of flows , , etc. conjugate to their sets of free affinities , , etc. become available through current fluctuations , , etc., at the critical instability point (or bifurcation point) along a particular variable of the system, because, unlike in the equilibrium or in the linear nonequilibrium regimes, in the nonlinear nonequilibrium regime these microscopic fluctuations can be amplified through feedback (e.g., autocatalysis) into new macroscopic flows [27].
Since for such a system, under an externally imposed thermodynamic force in the nonlinear regime, multiple stationary states are available, an interesting question arises concerning the stability of the system and how the system will evolve over time. Because the system harbors critical points at which microscopic fluctuations can be amplified into macroscopic flows leading the system to a new stationary state, it cannot be expected that there exists a potential for the system whose optimization would predict evolution. What could be hoped for, however, is a statistical rule giving relative probabilities for the different evolutionary trajectories.
Prigogine and coworkers have shown that although in general no total differential (thermodynamic potential) exists for these nonlinear systems, there does, however, exist a nontotal differential; the time variation of the entropy production with respect to the time variation of the free forces (see Equation (2)), which always has a definite sign,
This is the most general result so far obtained from CIT theory, valid in the whole domain of its applicability, independent of the nature of the relation between the flows and forces. It is known as the universal evolution criterion and is sometimes called the Glansdoff-Prigogine criterion. This criterion indicates that the free forces always arrange themselves within the system such that this rearrangement contributes to a decrease in the entropy production. However, in general, there is no such restriction on the total entropy production of the system because this also includes a component due to the corresponding rearrangement of the flows (see Equation (2)) which has no definite sign. The total entropy production may either increase or decrease during evolution in the nonlinear regime, depending on the relative sizes and signs of the two terms in Equation (2). In the regime of linear phenomenological relations (a linear relation between the flows and forces), it is easy to show [27] that and thus the universal evolutionary criterion, Equation (4), correctly gives the theorem of minimum entropy production alluded to above, . The asymptotic stability of the unique stationary state in this linear regime is guaranteed by the fact that the entropy production is a Lyapunov function (i.e., and .
Given that, in the nonlinear regime, bifurcations can be reached leading to multiple stationary states (which for the case studied here of the dissipative structuring of the fundamental molecules of life corresponds to different concentration profiles of distinct molecular configurations, each with a different rate of dissipation of the applied photon potential), it is pertinent to inquire if there indeed exists a statistical law (as alluded to above) regarding the direction of evolution of the system. In this nonlinear regime with multiple stationary states, the local stability of each stationary state has to be evaluated individually and the probability of evolution governed by Equation (4) from one state to another has to be evaluated through a local stability analysis which ultimately concerns the size of the fluctuations, the size of the barriers in , and the size of the catchment basins of neighboring stationary states in a generalized phase space. For the dissipative synthesis of the fundamental molecules of life, the size of these catchment basins is related to the number of conical intersections associated with a particular molecular photochemical transformation and the accessibility of these is related to the size of the energy barriers on route to the conical intersections from the excited state nuclear coordinates in the Frank–Condon region (i.e., unperturbed from the ground state).
Even though is not a total differential, it can still be used to determine the nature and local stability of each stationary state, not only in the linear regime as shown above but also in the nonlinear regime. To illustrate the utility of the general evolution criterion () in determining probabilities of paths of evolution among multiple stationary states, consider the case of the synthesis of adenine from 5 HCN molecules in water under an impressed UVC photon spectrum like that of Figure 1. A photochemical synthesis of adenine from 5HCN (see Figure 2) was first discovered experimentally by Ferris and Orgel in 1966 [23] and we first suggested that this may be an example of dissipative structuring in reference [29]. Here we will study the nonequilibrium thermodynamics and kinetics of a simplified version of this synthesis, showing how the evolution of the concentration profile (over different stationary states) leads to an optimization of photon dissipation.
3. The Dissipative Structuring of Adenine
Our photochemical reaction system consists of ocean surface water absorbing HCN from the atmosphere under the constant UVC photon flux of Figure 1. HCN is very soluble in water. The process leading from 4HCN to the most stable tetramer, cis-DAMN (1), is exothermic so would occur through normal thermal chemistry, but relatively high free energy barriers imply that a hot ocean surface would have accelerated the process. Step (2) of Figure 2 which transforms cis-DAMN into trans-DAMN, requires a rotation around a double covalent carbon bond and as such requires the absorption of a high energy photon ( eV) to overcome the large energy barrier for rotation. Under equilibrium conditions, cis-DAMN is more stable than trans-DAMN which is 0.61 kcal·mol higher in energy, implying its equilibrium population at normal temperatures is only about 1/10 that of cis-DAMN [30]. However, under nonequilibrium conditions of a continuous flux of UVC photons, we will see how the population of trans-DAMN can become greater than that of cis-DAMN, thereby providing a proficient pathway to the synthesis of adenine. The autocatalytic nature of trans-DAMN in aiding in the thermal reactions will be seen to lead to the proliferation of it, and therefore adenine, under the UVC flux. The final product of adenine (8) will be seen to have the greatest absorption and dissipative efficacy of all these precursor and intermediate molecules (Figure 2) in the UVC region presented in Figure 1.
The following symbols are assigned to the concentrations of the participating species for the photochemical reaction leading to adenine shown in Figure 2; = [flux of HCN from the atmosphere into the ocean surface, assumed constant], H = [HCN] at ocean surface (1), F = [formamide], C = [cis-DAMN] (2), T = [trans-DAMN] (3), J = [AIAC] (4), I = [imidazole AICN] (7), A = [adenine] (8), = [UVC photons at wavelength y].
The flux of the UVC photons at the ocean surface where adenine is presumed to be synthesized is taken to be constant and homogeneous and we assume a Beer–Lambert law for photon absorption at depth x due to the particular absorbing molecule of molar extinction and water absorption (which are functions of wavelength). The following chemical and photochemical reactions will occur and are described in detail below:
The reaction steps are;
(5) There is a flow of HCN from the external environment (atmosphere), , into the ocean surface microlayer dependent on the diffusion constant of HCN in water.
(6) Hydrolysis of HCN at the ocean surface leads to formamide (F), HNCOH.
(7)+(8) HCN can polymerize into its most stable tetramer cis-DAM (7) and to a lesser extent (1/10) into trans-DAMN (8). The tetramization of 4HCN into cis-DAMN is a thermal reaction and occurs most rapidly at a solvent pH at its pKa value, which decreases with increasing temperature (pKa at 60 C and at 100 C) [30]. Note that the tetramization of HCN into DAMN is not elementary but involves successive polymerization of HCN with H and CN ions [30] so is second order in the concentration of HCN. The rate constants for hydrolysis and polymerization are similar for concentrations of HCN between approximately 0.01 M and 0.1 M. At lower concentrations, hydrolysis dominates while at higher concentrations polymerization dominates [30].
(9)+(10) Trans-DAMN produced through the thermal reaction, Equation (), or through the UV photon-induced transformation of cis-DAMN, Equation () acts as a catalyst for the tetramization of 4HCN into cis-DAMN (Equation ()) or into trans-DAMN (Equation ()) [33], while cis-DAMN does not act as a catalyst for their production [30].
(11) Absorption of a photon at 298 nm excites cis-DAMN.
(12) Excited cis-DAMN decays to the ground state through internal conversion.
(13) Excited cis-DAMN transforms into trans-DAMN through rotation around a double covalent carbon-carbon bond.
(14) Absorption of a photon at 313 nm excites trans-DAMN.
(15) Excited trans-DAMN decays to the ground state through internal conversion.
(16) Excited trans-DAMN transforms into AIAC [32].
(17) AIAC goes into its excited state by absorbing a photon at 275 nm.
(18) Excited AIAC decays to the ground state through internal conversion.
(19) Excited AIAC transforms into the imidazole AICN [32].
(20) AICN absorbs at 250 nm into its excited state.
(21) Excited AICN decays through internal conversion to its ground state.
(22) The final coupling of a fifth HCN to AICN to form adenine is a very exothermal reaction, kcal/mol, but the first step of this thermal reaction is rate limiting since it has a high energy barrier 29.6 kcal/mol but can be catalyzed by water molecules which reduce the barrier from a value in the gas phase of 39.7 kcal/mol [34].
(23) By absorbing a photon at 260 nm, adenine goes to its first excited S state.
(24) Adenine decays to its ground state through internal conversion.
(25) Adenine, largely unable to undergo further photochemical transformation, diffuses into the environment.
Backward reaction rates are assumed negligible. The concentrations are calculated at a given depth under the surface of microns and we assume that there is a similar net outflow due to diffusion for each component equal to , where s and represents the concentration of the particular component. Furthermore, we assume that the molecules absorb at their maximum molar extinction only within a 10 nm wavelength region at their wavelength of maximum absorption to simplify calculation. Note that this is a cross-catalytic reaction where the trans-DAMN (T) acts as a catalyst for the thermal reactions.
The kinetic equations for the concentrations are thus:
where and are the diffusion coeficients for HCN and adenine, cm·s and cm·s respectively, and , and are the photon intensities at the specified wavelength taken from Figure 1. and are the coefficients of molar extinction for the molecules and the water absorption coefficients at the corresponding photon wavelengths, respectively. The tetramization rate of HCN increases as the square of its concentration and increases linearly with temperature [30].
4. Results and Discussion
In Figure 3, Figure 4, Figure 5 and Figure 6 we present the concentrations of the relevant molecules obtained by solving simultaneously the differential kinetic equations given above for different initial conditions and diffusion rates. For the dissipative structuring of adenine from HCN under UVC light, the flows (reaction rates) are nonlinear with respect to the forces (affinities over the temperature) and therefore our system could be expected to harbor numerous stationary states. We have discovered two such stationary states for our particular system, one in which the accumulation of adenine is almost zero and the other in which the concentration becomes large (∼1.4 M). Dissipative proliferation occurs because our system is both auto- and cross-catalytic. For these auto- or cross-catalytic systems, the universal evolution criterion indicates that nature will tend to amplify (select) fluctuations leading to greater dissipation and for our case this corresponds to the stationary state with a large concentration of adenine (see Figure 7).
Finally, the concentration of HCN may be kept high at the ocean surface (and thus allow the copious production of adenine in the second stationary state) not only due to its diffusion constant being dependent on the concentration of other molecules at the surface but also because of the coupling between reaction and diffusion in the nonlinear regime breaking the spatial symmetry (e.g., the Belousov–Zhabotinsky reaction). We are presently investigating this possibility.
Dissipative structuring, dissipative proliferation, and dissipative selection of the fundamental molecules from common precursor molecules at the ocean surface under UVC light are the necessary and sufficient elements to explain in physical-chemical terms the origin and evolution of life. Non-linear out-of-equilibrium systems have numerous stationary states. Amplification of fluctuations, due to the non-linearity (autocatalytic) nature of the system, allow it to leave one stationary state and evolve towards another. The universal evolutionary criterion of Glansdorff and Prigogine, along with the fact that the system is autocatalytic, imply that the direction of evolution is generally towards that of increasing photon dissipation (entropy production). In the case studied here, this corresponds to a stationary state with a much greater concentration of adenine which then dissipates more strongly the imposed UVC photon spectrum. Over evolutionary history, life has evolved, through dissipative structuring, ever more complex biosynthetic pathways which has allowed it to dissipate not only UVC photons, but also UVB, UVA, and visible photons (up to the red-edge nm) where the intensity is much greater but photon energies are not sufficient to break and remake carbon covalent bonds directly.
Funding
This research was funded by DGAPA-UNAM grant number PAPIIT IN104920.
Conflicts of Interest
The author declares no conflict of interest.
References
- Boltzmann, L. Ludwig Boltzmann: Theoretical Physics and Philosophical Problems: Selected Writings; Springer: Berlin, Germany, 1974. [Google Scholar]
- Michaelian, K. Thermodynamic Dissipation Theory of the Origina and Evolution of Life: Salient characteristics of RNA and DNA and other fundamental molecules suggest an origin of life driven by UV-C light; Self-published; Printed by CreateSpace: Mexico City, Mexico, 2016; ISBN 9781541317482. [Google Scholar]
- Oró, J.; Miller, S.L.; Lazcano, A. The origin and early evolution of life on Earth. Ann. Rev. Earth Planet. Sci. 1990, 18, 317–356. [Google Scholar] [CrossRef]
- Michaelian, K. Thermodynamic origin of life. ArXiv 2009, arXiv:0907.0042. [Google Scholar]
- Michaelian, K. Thermodynamic dissipation theory for the origin of life. Earth Syst. Dyn. 2011, 224, 37–51. [Google Scholar] [CrossRef]
- Berkner, L.; Marshall, L. Origin and Evolution of the Oceans and Atmosphere; John Wiley and Sons: Hoboken, NJ, USA, 1964; pp. 102–126. [Google Scholar]
- Sagan, C. Ultraviolet Selection Pressure on the Earliest Organisms. J. Theor. Biol. 1973, 39, 195–200. [Google Scholar] [CrossRef]
- Cnossen, I.; Sanz-Forcada, J.; Favata, F.; Witasse, O.; Zegers, T.; Arnold, N.F. The habitat of early life: Solar X-ray and UV radiation at Earth’s surface 4–3.5 billion years ago. J. Geophys. Res. 2007, 112, E02008. [Google Scholar] [CrossRef]
- Miller, S.L.; Urey, H.C.; Oró, J. Origin of Organic Compounds on the Primitive Earth and in Meteorites. J. Mol. Evol. 1976, 9, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Michaelian, K.; Simeonov, A. Fundamental molecules of life are pigments which arose and co-evolved as a response to the thermodynamic imperative of dissipating the prevailing solar spectrum. Biogeosciences 2015, 12, 4913–4937. [Google Scholar] [CrossRef]
- Cockell, C.S. The ultraviolet history of the terrestrial planets — implications for biological evolution. Planetary Space Sci. 2000, 48, 203–214. [Google Scholar] [CrossRef]
- Mulkidjanian, A.Y.; Cherepanov, D.A.; Galperin, M.Y. Survival of the fittest before the beginning of life: selection of the first oligonucleotide-like polymers by UV light. BMC Evol. Biol. 2003, 3, 12. [Google Scholar] [CrossRef]
- Oparin, A.I. Proiskhozhdenie zhizny (The origin of life). In The Origin of Life; Bernal, J.D., Ed.; Weidenfeld and Nicholson: London, UK, 1924. [Google Scholar]
- Haldane, J.B.S. Origin of life. Ration. Annu. 1929, 148, 3–10. [Google Scholar]
- Urey, H.C. On the Early Chemical History of the Earth and the Origin of Life. Proc. Natl. Acad. Sci. USA 1952, 38, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Sagan, C. Radiation and the Origin of the Gene. Evolution 1957, 11, 40–55. [Google Scholar] [CrossRef]
- Baly, E.C.C. Light and Life. J. Soc. Dyers Colourists 1927, 43, 387–390. Available online: http://xxx.lanl.gov/abs/https://onlinelibrary.wiley.com/doi/pdf/10.1111/j.1478-4408.1927.tb01410.x (accessed on 14 March 2020). [CrossRef]
- Miller, S.L. A Production of Amino Acids Under Possible Primitive Earth Conditions. Science 1953, 117, 528–529. [Google Scholar] [CrossRef] [PubMed]
- Oró, J.; Kimball, A. Synthesis of purines under possible primitive earth conditions: II. Purine intermediates from hydrogen cyanide. Arch. Biochem. Biophys. 1962, 96, 293–313. [Google Scholar] [CrossRef]
- Ponnamperuma, C.; Sagan, C.; Mariner, R. Synthesis of adenosine triphosphate under possible primitive Earth conditions. Nature 1963, 199, 222–226. [Google Scholar] [CrossRef]
- Ponnamperuma, C.; Mariner, R. Formation of ribose and deoxyribose by ultraviolet irradiation of formaldehyde in water. Rad. Res. 1963, 19, 183. [Google Scholar]
- Ponnamperuma, C.; Mariner, R.; Sagan, C. Formation of Adenosine by Ultra-violet Irradiation of a Solution of Adenine and Ribose. Nature 1963, 198, 1199–1200. [Google Scholar] [CrossRef]
- Ferris, J.P.; Orgel, L.E. An Unusual Photochemical Rearrangement in the Synthesis of Adenine from Hydrogen Cyanide. J. Am. Chem. Soc. 1966, 88, 1074. [Google Scholar] [CrossRef]
- Sagan, C.; Khare, B.N. Long-Wavelength Ultraviolet Photoproduction of Amino Acids on the Primitive Earth. Science 1971, 173, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Mejía, J.; Michaelian, K. Information Encoding in Nucleic Acids through a Dissipation-Replication Relation. arXiv 2018, arXiv:1804.05939. [Google Scholar]
- Yarus, M.; Widmann, J.; Knight, R. RNA-Amino Acid Binding: A Stereochemecal Era for the Genetic Code. J. Mol. Evol. 2009, 69, 406–429. [Google Scholar] [CrossRef] [PubMed]
- Prigogine, I. Introduction to Thermodynamics Of Irreversible Processes, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 1967. [Google Scholar]
- Rayleigh. Some general theorems relating to vibrations. Proc. Math. Soc. Lond. 1873, 4, 357–368. [Google Scholar]
- Michaelian, K. Microscopic Dissipative Structuring and Proliferation at the Origin of Life. Heliyon 2017, 3, e00424. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.A.; Ferbis, J.P.; Orgel, L.E. Studies in Prebiodc Synthesis II: Synthesis of Purine Precursors and Amino Acids from Aqueous Hydrogen Cyanide. J. Mol. Biol. 1967, 80, 223–253. [Google Scholar]
- Koch, T.; Rodehorst, R. Quantitative investigation of the photochemical conversion of diaminomaleonitrile to diaminofumaronitrile and 4-amino-5-cyanoimidazole. J. Am. Chem. Soc. 1974, 96, 6707–6710. [Google Scholar] [CrossRef]
- Boulanger, E.; Anoop, A.; Nachtigallova, D.; Thiel, W.; Barbatti, M. Photochemical Steps in the Prebiotic Synthesis of Purine Precursors from HCN. Angew. Chem. Int. 2013, 52, 8000–8003. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.A.; Ferbis, J.P.; Orgel, L.E. Studies in Prebiodc Synthesis IV: Conversion of 4-Aminoimidazole-5-carbonitrile Derivatives to Purines. J. Mol. Biol. 1968, 38, 121–128. [Google Scholar] [CrossRef]
- Roy, D.; Najafian, K.; von Rague Schleyer, P. Chemical evolution: The mechanism of the formation of adenine under prebiotic conditions. Proc. Natl. Acad. Sci. USA 2007, 104, 17272–17277. [Google Scholar] [CrossRef]
Figure 1.
Maxima in the absorption of many of the fundamental molecules of life coincide with the predicted atmospheric window which existed from before the origin of life at approximately 3.85 Ga and until at least 2.9 Ga (curves black and red respectively). CO and probably some HS were responsible for absorption at wavelengths shorter than ∼205 nm and atmospheric aldehydes (common photochemical products of CO and water) absorbed between approximately 285 and 310 nm [7]. Around 2.2 Ga (yellow curve), UVC light at Earth’s surface was extinguished by oxygen and ozone resulting from organisms performing oxygenic photosynthesis. The green curve corresponds to the present surface spectrum. Energy fluxes are for the sun at the zenith. The font size of the letter is roughly proportional to the relative size of the molar extinction coefficient of the indicated fundamental molecule. Adapted from (Michaelian and Simeonov, 2015) [10].
Figure 1.
Maxima in the absorption of many of the fundamental molecules of life coincide with the predicted atmospheric window which existed from before the origin of life at approximately 3.85 Ga and until at least 2.9 Ga (curves black and red respectively). CO and probably some HS were responsible for absorption at wavelengths shorter than ∼205 nm and atmospheric aldehydes (common photochemical products of CO and water) absorbed between approximately 285 and 310 nm [7]. Around 2.2 Ga (yellow curve), UVC light at Earth’s surface was extinguished by oxygen and ozone resulting from organisms performing oxygenic photosynthesis. The green curve corresponds to the present surface spectrum. Energy fluxes are for the sun at the zenith. The font size of the letter is roughly proportional to the relative size of the molar extinction coefficient of the indicated fundamental molecule. Adapted from (Michaelian and Simeonov, 2015) [10].

Figure 2.
The photochemical synthesis of adenine from 5 molecules of hydrogen cyanide in water is a dissipative structuring process which involves the absorption of at least three photons in the UVC and UVB regions of the Archean spectrum.
Figure 2.
The photochemical synthesis of adenine from 5 molecules of hydrogen cyanide in water is a dissipative structuring process which involves the absorption of at least three photons in the UVC and UVB regions of the Archean spectrum.

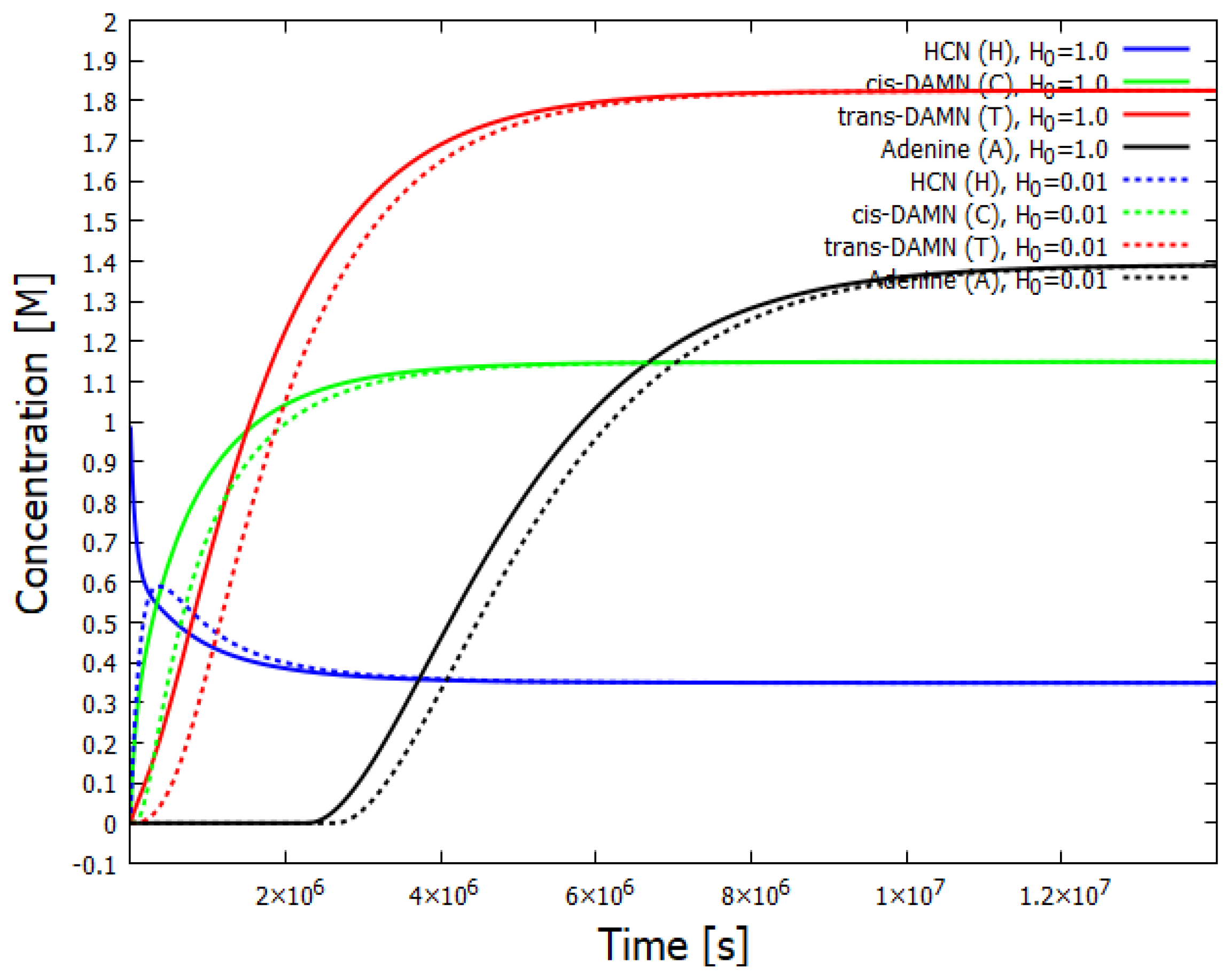
Figure 3.
Concentration versus time for the various molecular components involved in the dissipative structuring of adenine. A stationary state is reached with a concentration of about 1.3 M for adenine after about seconds. Arrival at the stationary state is seen for two different initial conditions of HCN, 1 M (solid lines) and 0.01 M (dotted lines).
Figure 3.
Concentration versus time for the various molecular components involved in the dissipative structuring of adenine. A stationary state is reached with a concentration of about 1.3 M for adenine after about seconds. Arrival at the stationary state is seen for two different initial conditions of HCN, 1 M (solid lines) and 0.01 M (dotted lines).

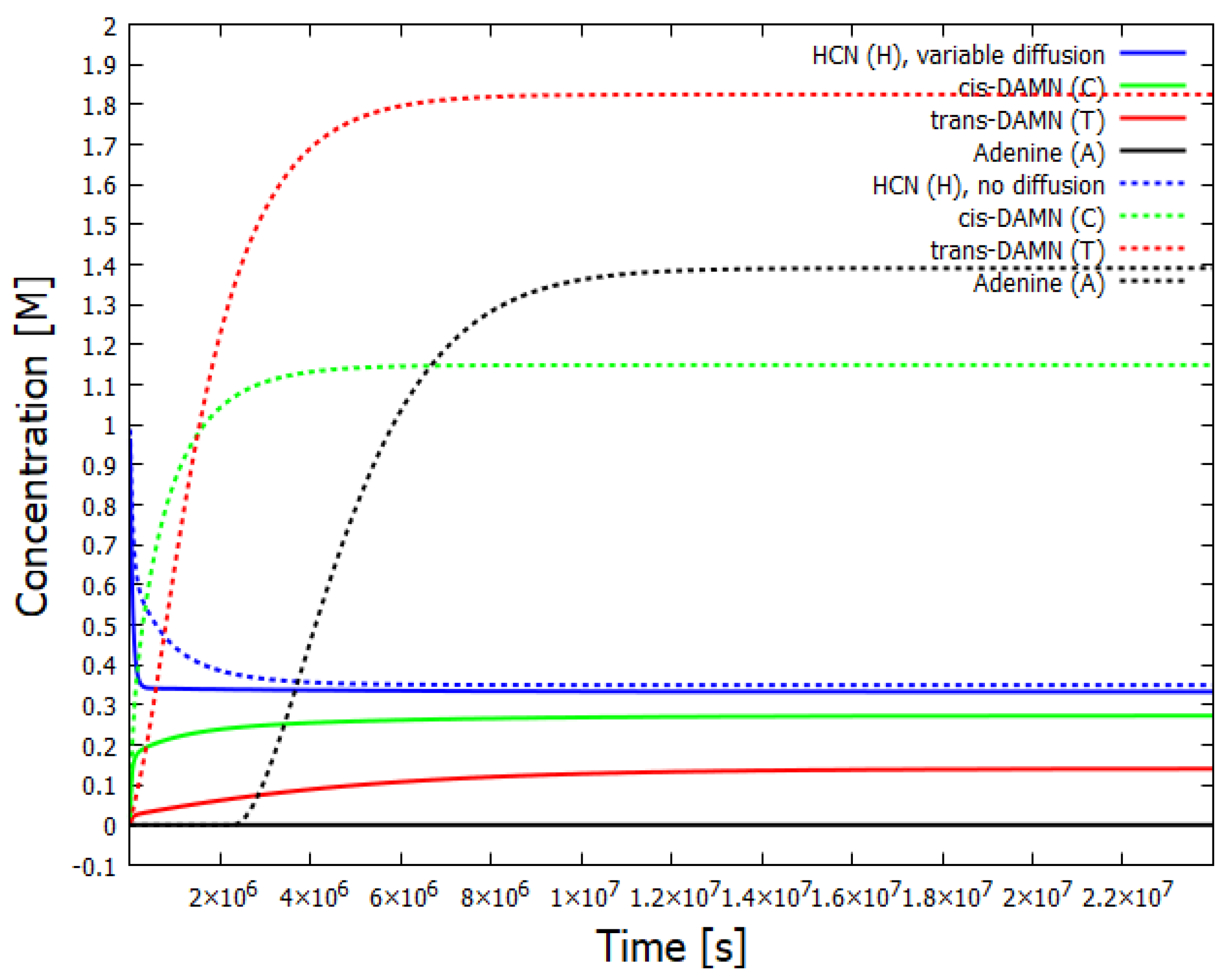
Figure 4.
The concentrations of the relevant components as a function of time for two different values of the diffusion constant for HCN. The case of no diffusion, or low diffusion, is given by the dotted lines and corresponds to the stationary state of Figure 3. For HCN diffusion dependent on the concentration of adenine (solid lines), a stationary state is reached but there is no production of adenine even though the concentration of HCN (solid blue line) is only slightly lower than the case of no diffusion (dotted blue line). Note that the production of trans-DAMN becomes greater than that of cis-DAMN for the stationary state of the higher concentration of HCN, but this is not the case for the stationary state of slightly lower concentration of HCN.
Figure 4.
The concentrations of the relevant components as a function of time for two different values of the diffusion constant for HCN. The case of no diffusion, or low diffusion, is given by the dotted lines and corresponds to the stationary state of Figure 3. For HCN diffusion dependent on the concentration of adenine (solid lines), a stationary state is reached but there is no production of adenine even though the concentration of HCN (solid blue line) is only slightly lower than the case of no diffusion (dotted blue line). Note that the production of trans-DAMN becomes greater than that of cis-DAMN for the stationary state of the higher concentration of HCN, but this is not the case for the stationary state of slightly lower concentration of HCN.

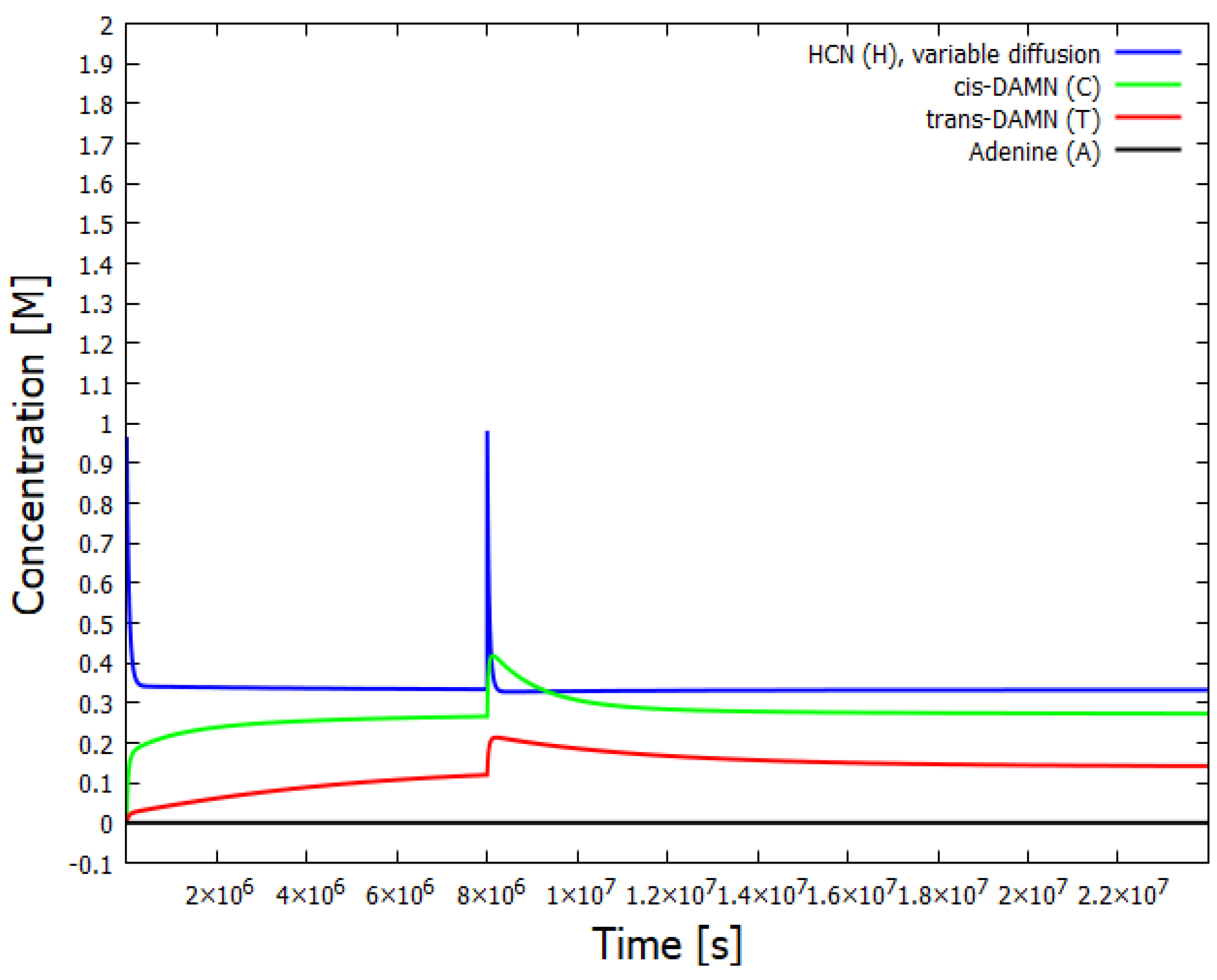
Figure 5.
The concentrations of the relevant components as a function of time for a diffusion rate of HCN dependent on the concentration of adenine. An instantaneous perturbation of HCN to 1 M concentration is performed at s but the system relaxes back to its original stationary state with practically zero adenine production.
Figure 5.
The concentrations of the relevant components as a function of time for a diffusion rate of HCN dependent on the concentration of adenine. An instantaneous perturbation of HCN to 1 M concentration is performed at s but the system relaxes back to its original stationary state with practically zero adenine production.

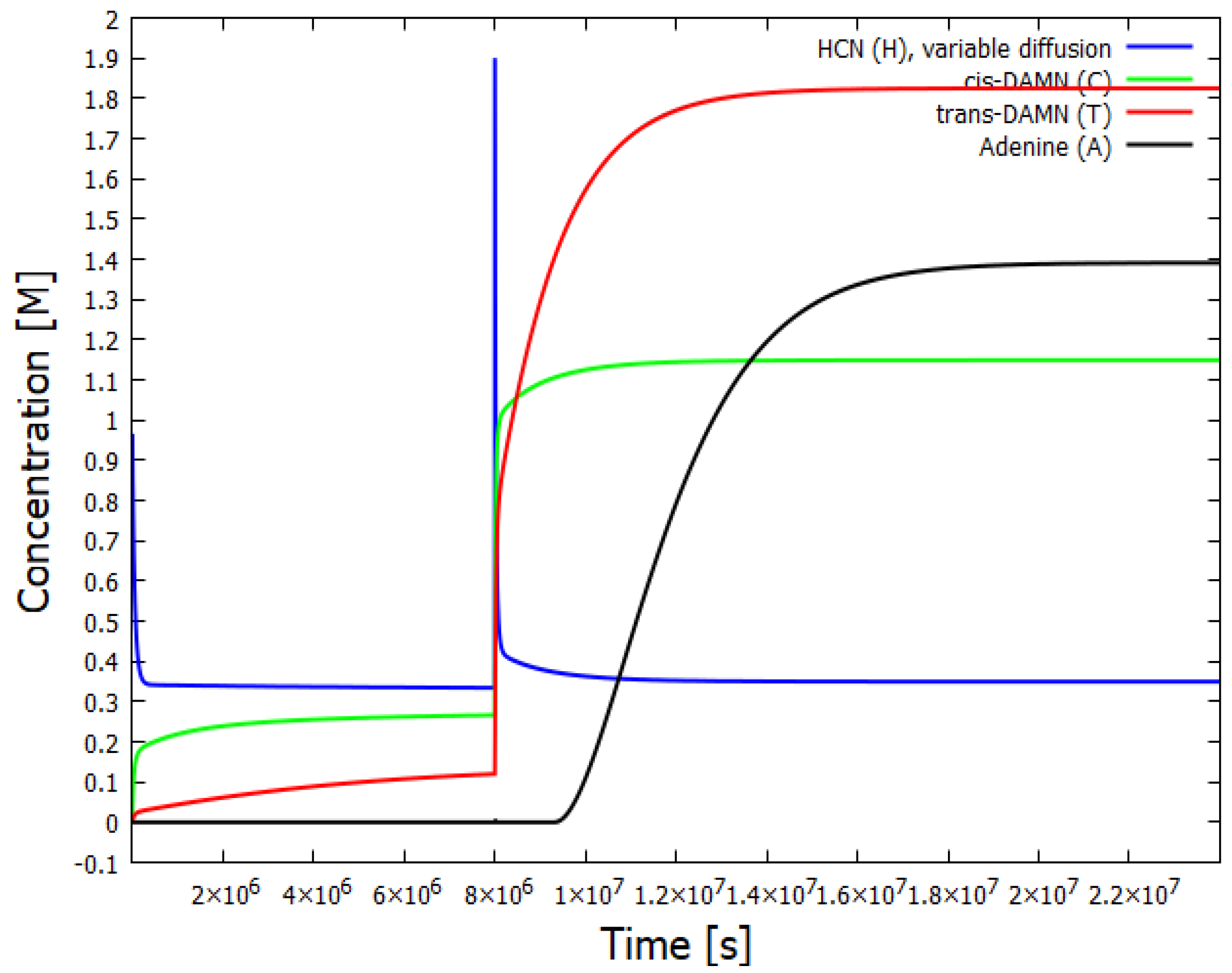
Figure 6.
The same as for Figure 5 except that the instantaneous perturbation of HCN is increased to 2 M. The resulting dynamics is now very different; the system relaxes to a new stationary state in which the production of adenine is very large.
Figure 6.
The same as for Figure 5 except that the instantaneous perturbation of HCN is increased to 2 M. The resulting dynamics is now very different; the system relaxes to a new stationary state in which the production of adenine is very large.

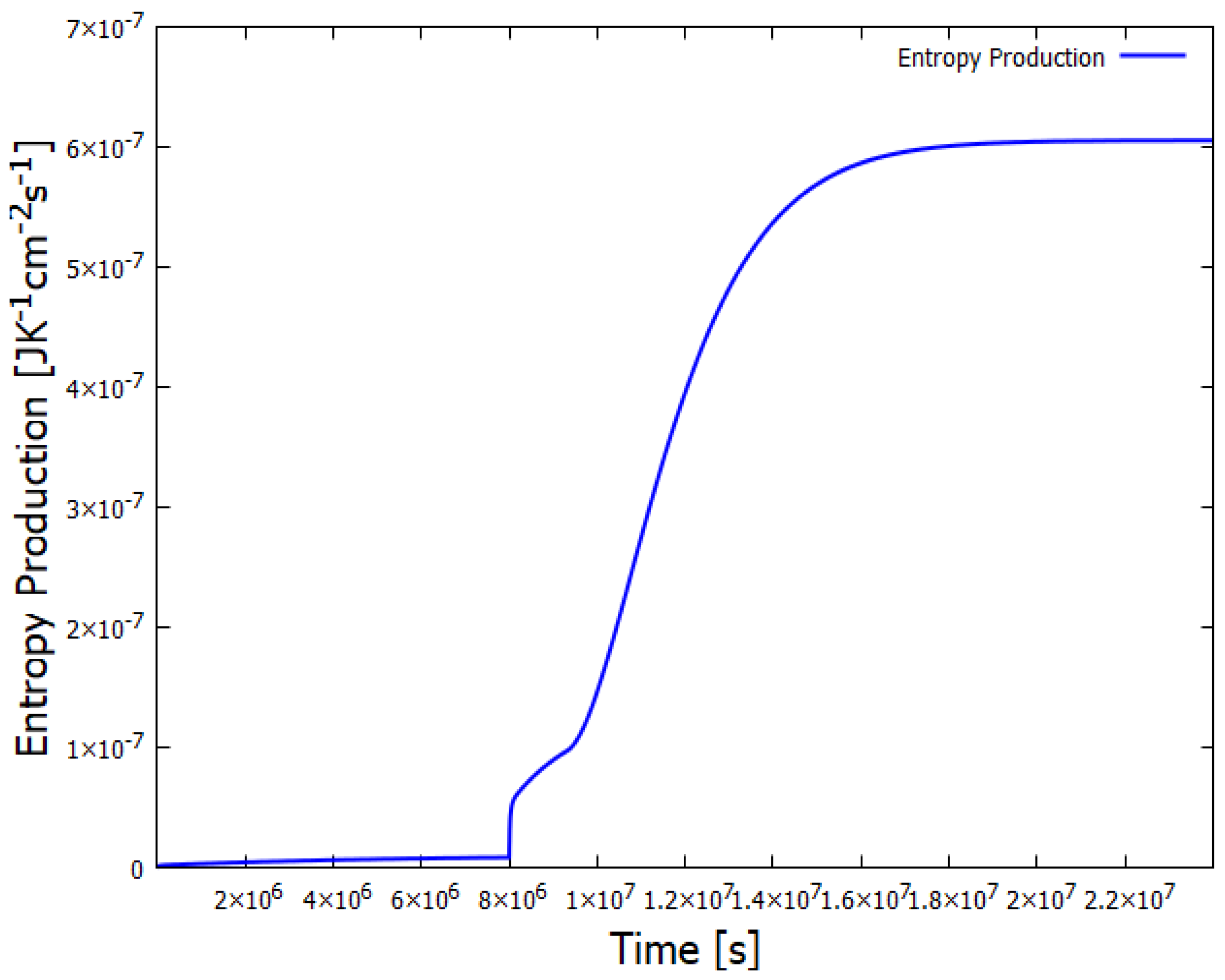
Figure 7.
The entropy production of the system for the case of Figure 6 in which an instantaneous fluctuation of HCN to 2 M shifts the system to a stationary state with large adenine production. The universal evolutionary criterion of Glansdorff and Prigogine suggests that nature is more likely to amplify those fluctuations at the bifurcation leading to greater entropy production for auto- or cross-catalytic systems.
Figure 7.
The entropy production of the system for the case of Figure 6 in which an instantaneous fluctuation of HCN to 2 M shifts the system to a stationary state with large adenine production. The universal evolutionary criterion of Glansdorff and Prigogine suggests that nature is more likely to amplify those fluctuations at the bifurcation leading to greater entropy production for auto- or cross-catalytic systems.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Michaelian, K. Photochemical Dissipative Structuring of the Fundamental Molecules of Life. Proceedings 2020, 46, 20. https://doi.org/10.3390/ecea-5-06692
AMA Style
Michaelian K. Photochemical Dissipative Structuring of the Fundamental Molecules of Life. Proceedings. 2020; 46(1):20. https://doi.org/10.3390/ecea-5-06692
Chicago/Turabian StyleMichaelian, Karo. 2020. "Photochemical Dissipative Structuring of the Fundamental Molecules of Life" Proceedings 46, no. 1: 20. https://doi.org/10.3390/ecea-5-06692