
Update on the Pathology of Pediatric Liver Tumors: A Pictorial Review
 , , and
, , and
Abstract
:1. Introduction
2. Mesenchymal Tumors in Children
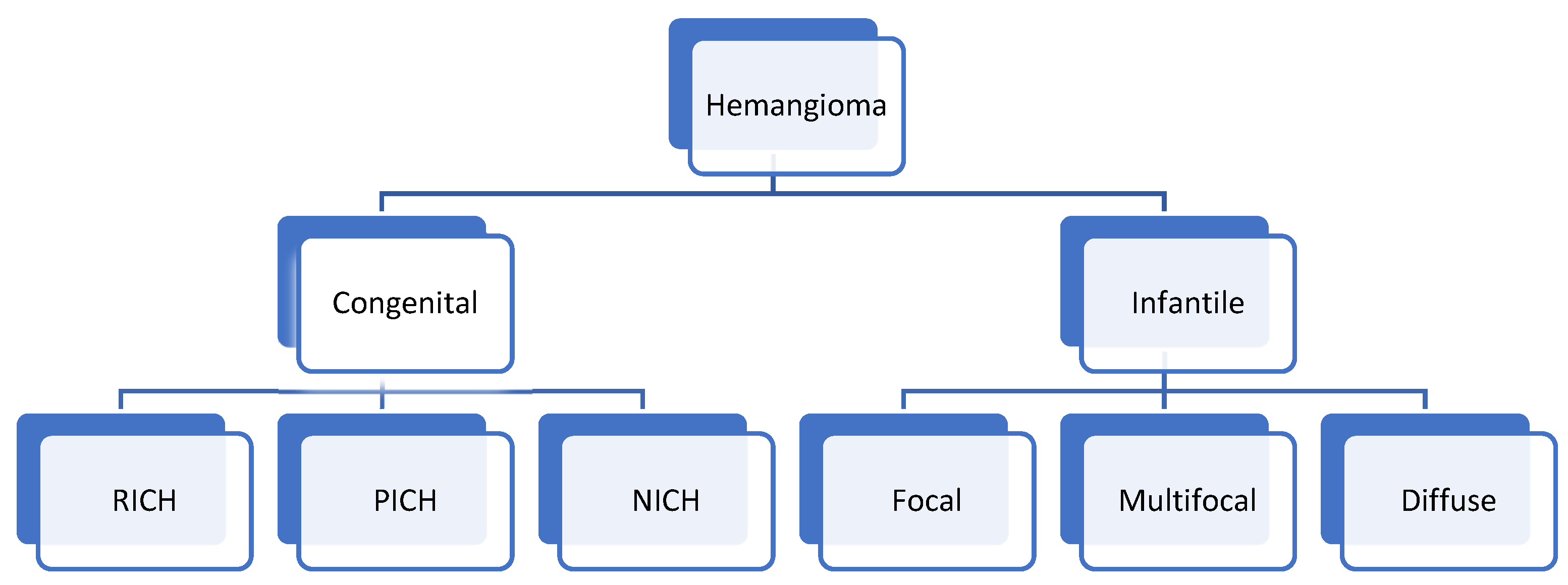
Hemangioma
3. Hepatic Congenital Hemangioma
3.1. Clinical Presentation
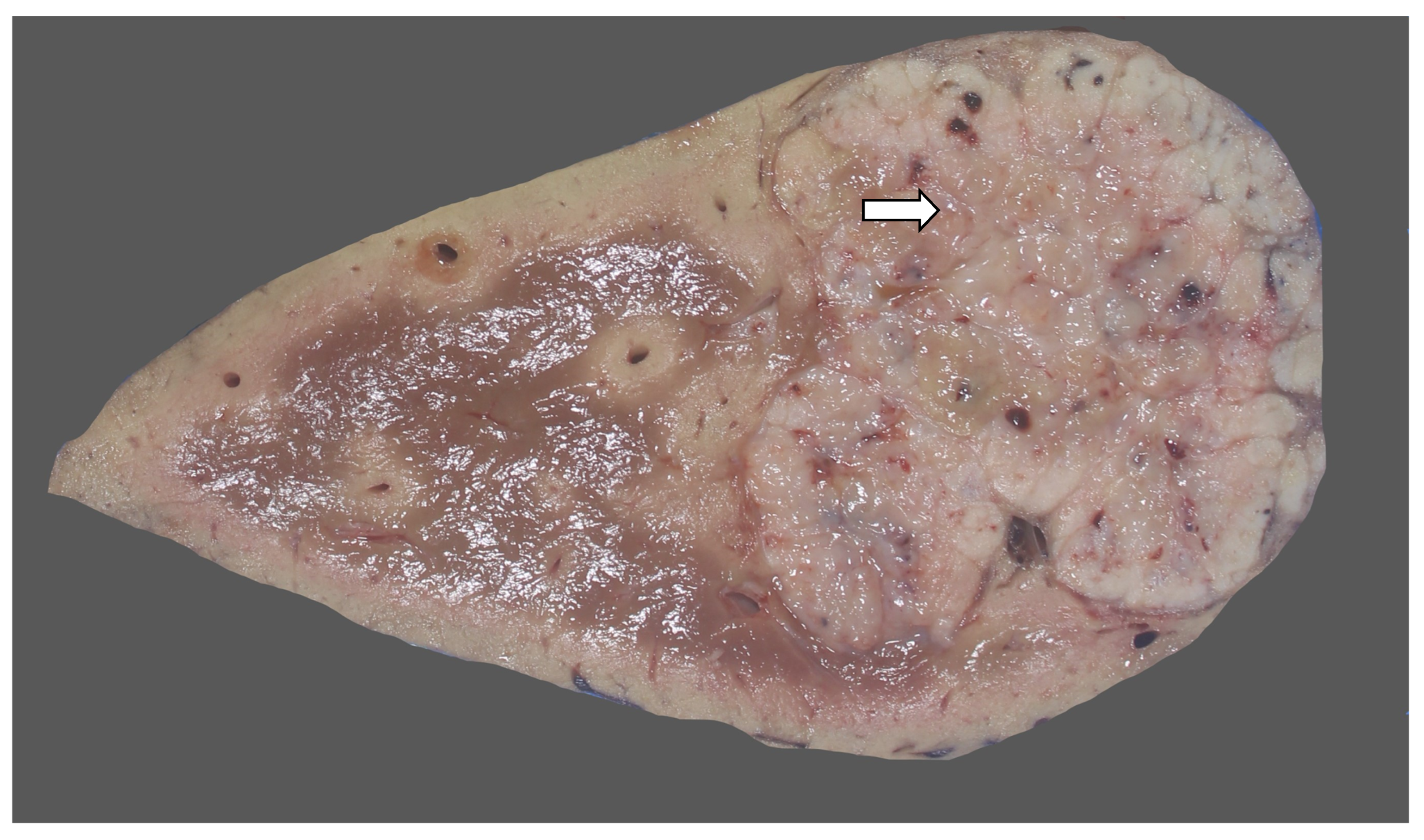
3.2. Imaging Findings
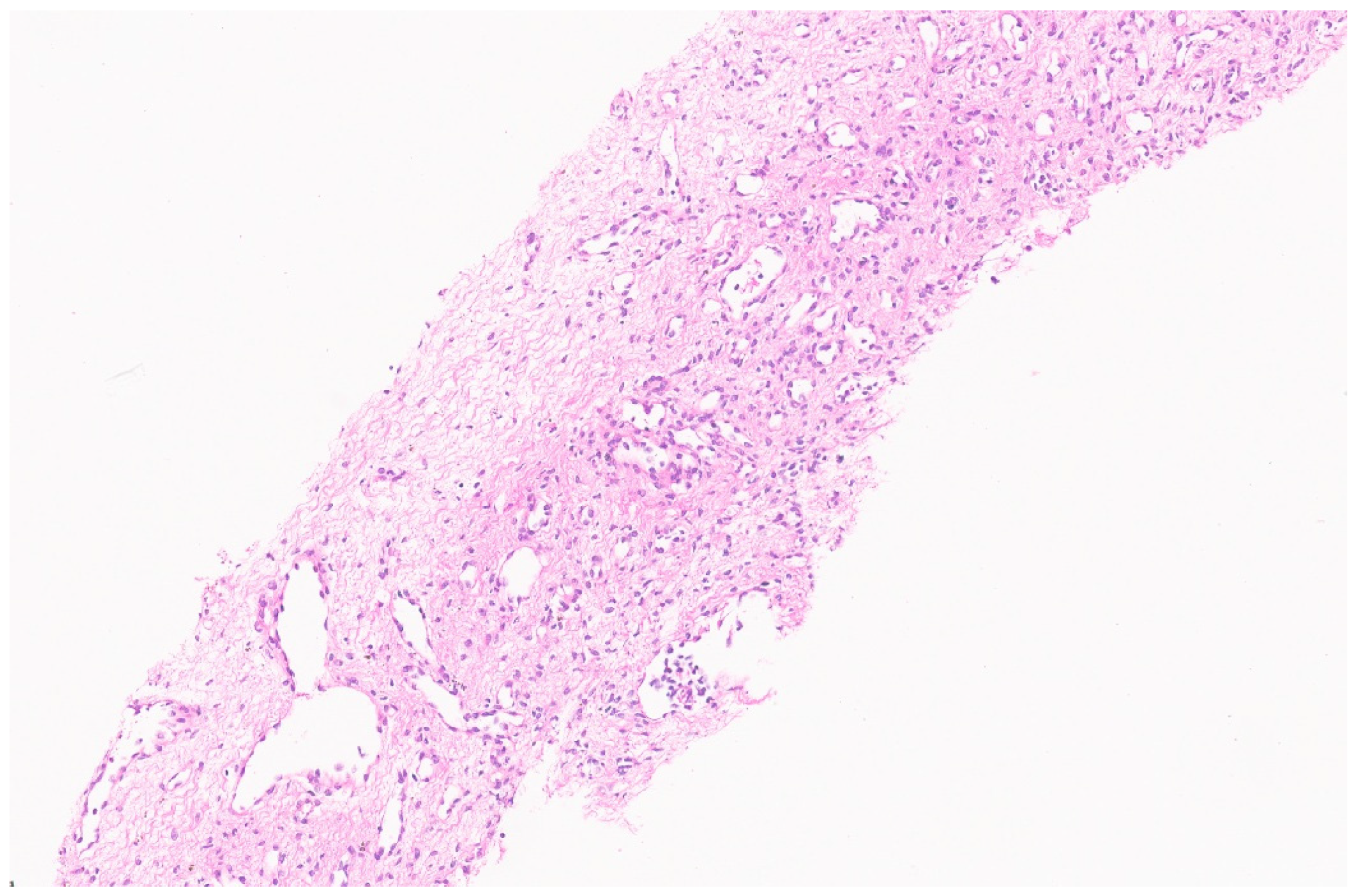
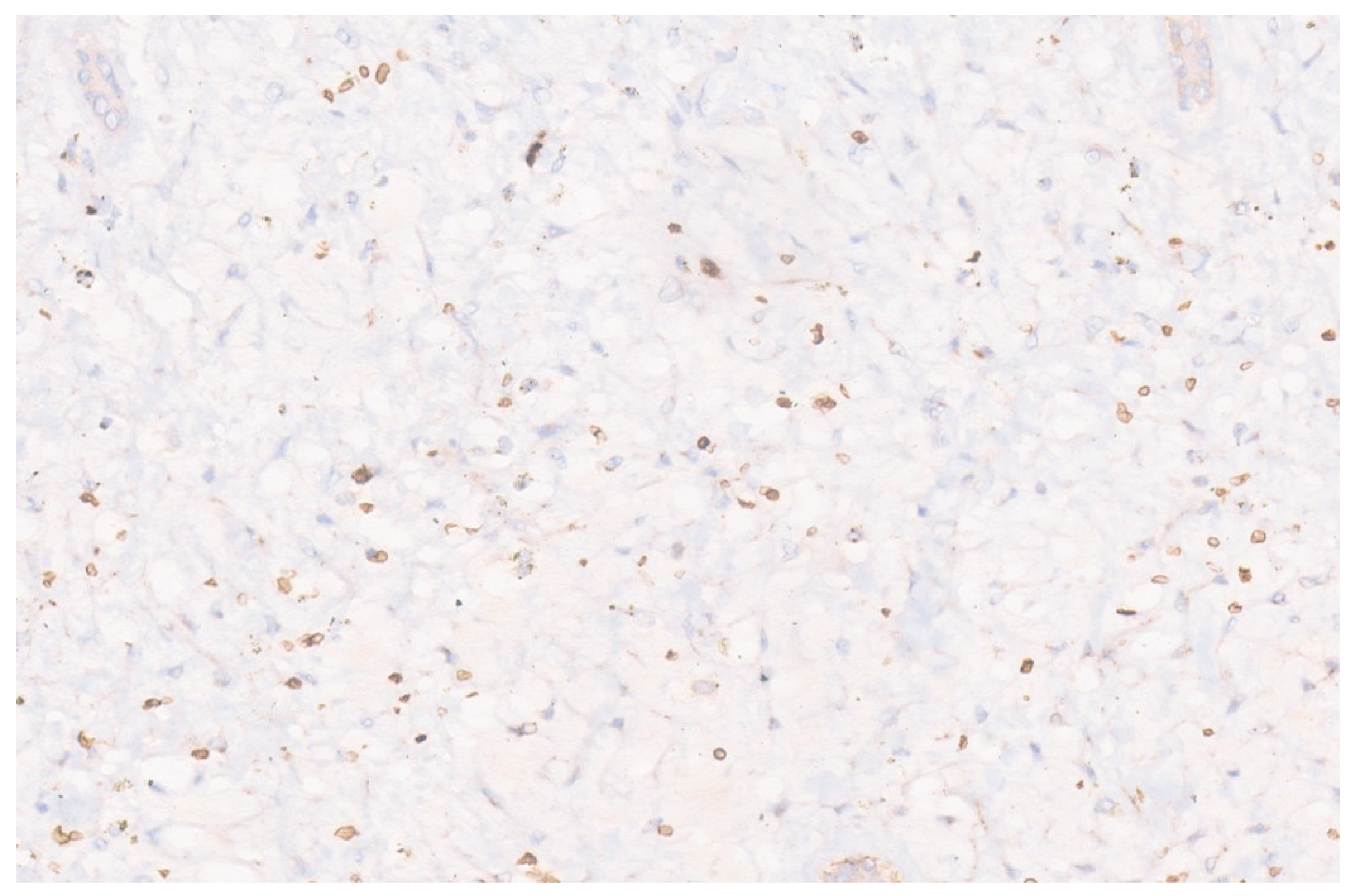
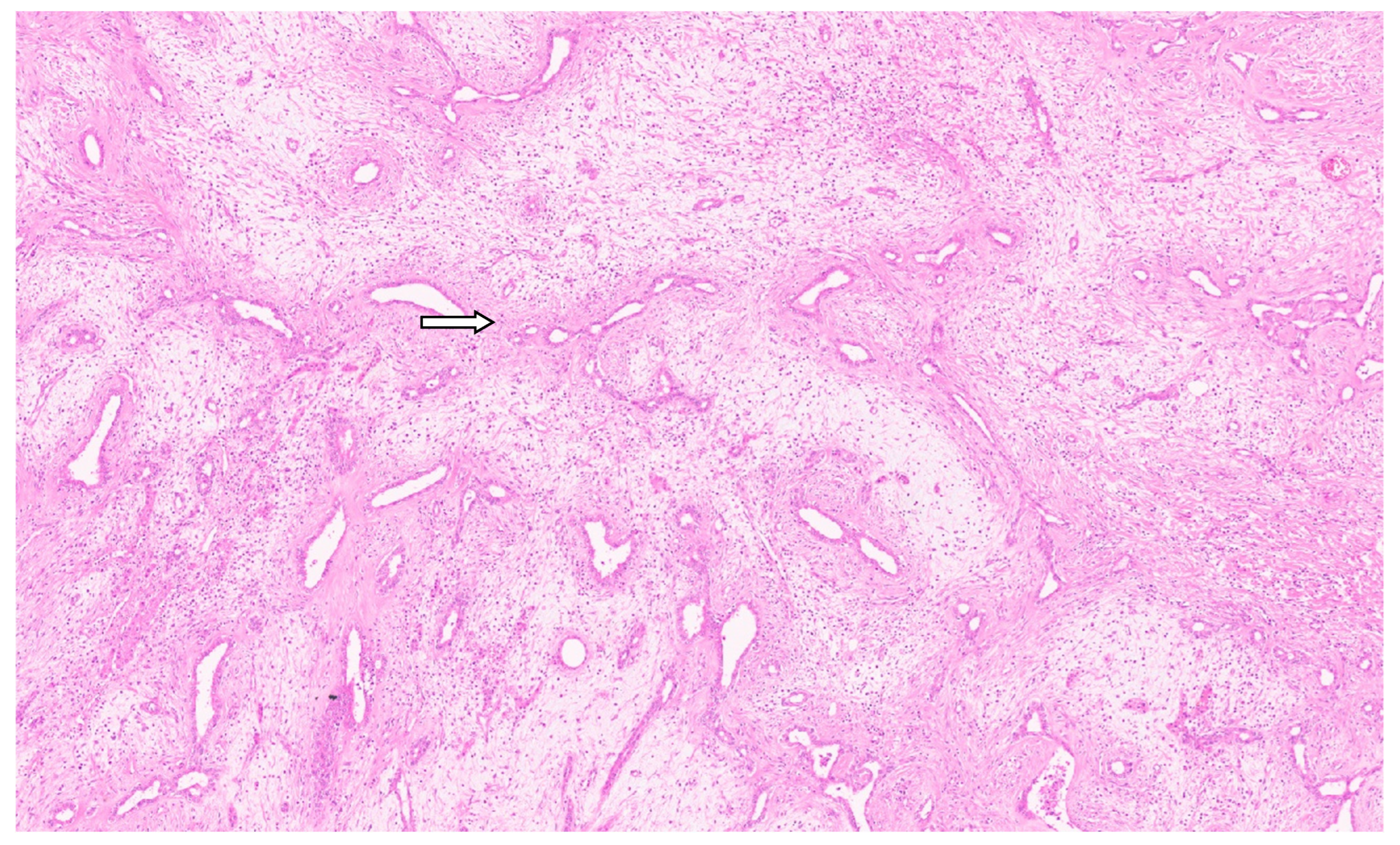
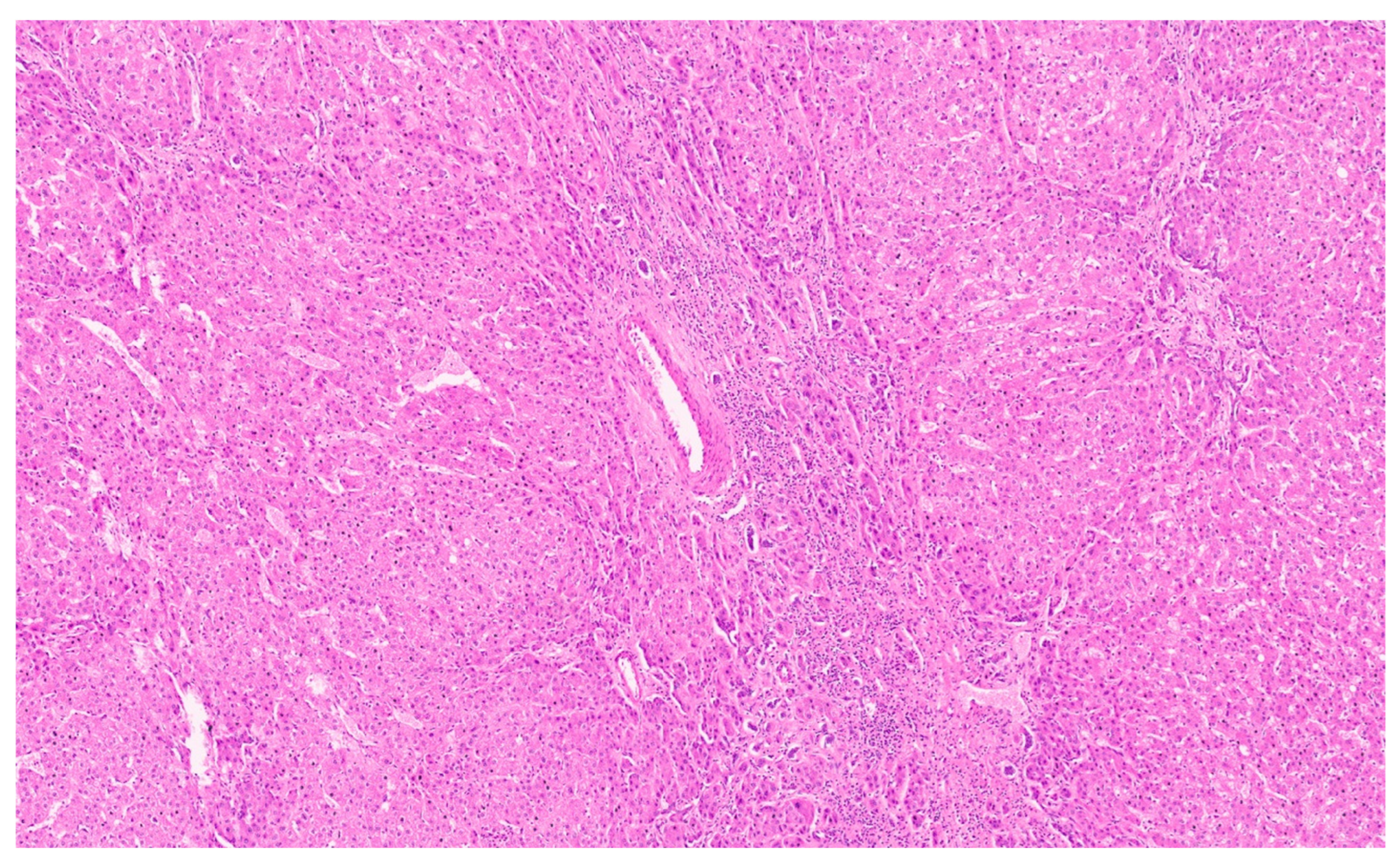
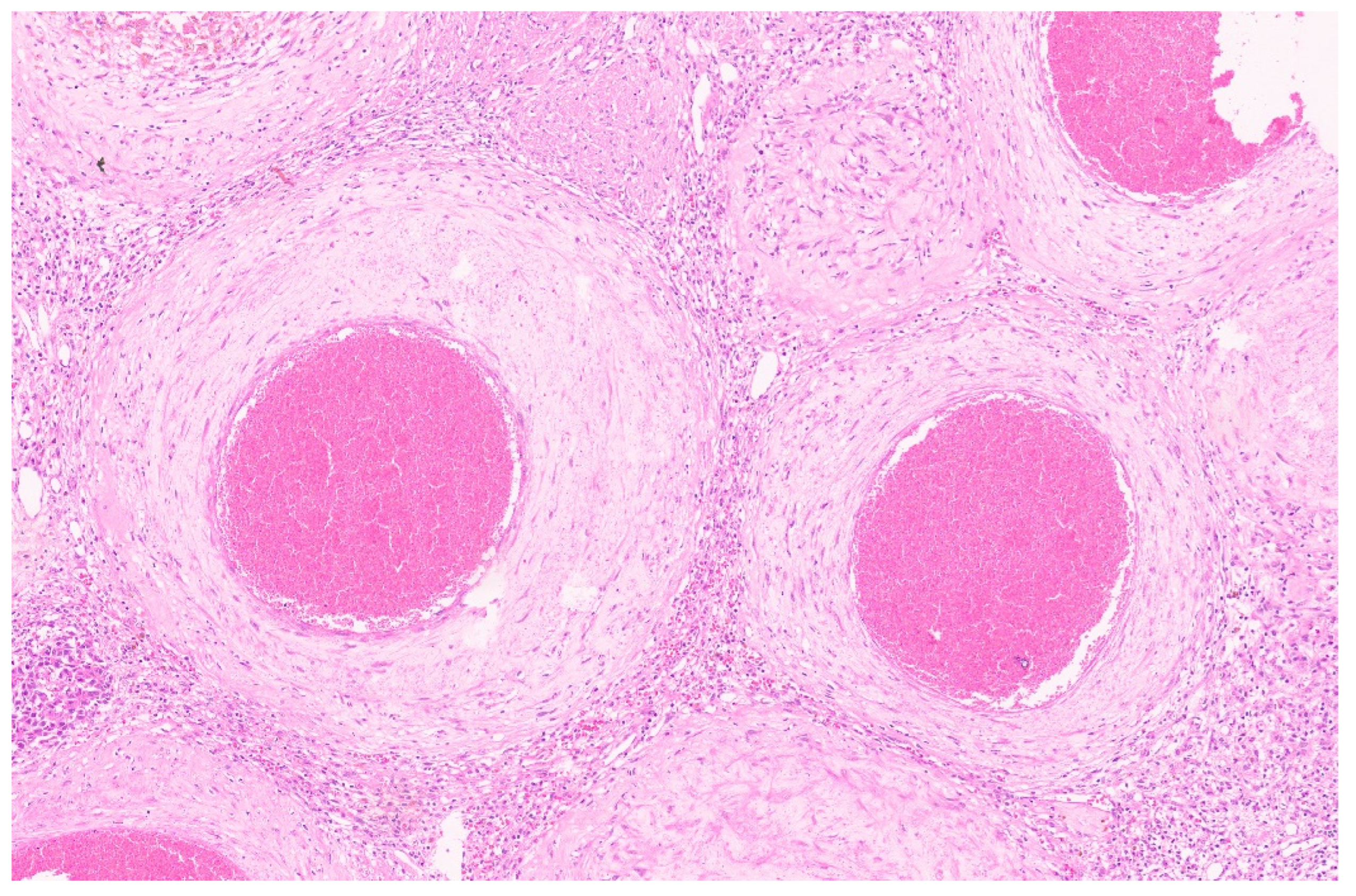
3.3. Pathology
4. Hepatic Infantile Hemangioma
4.1. Clinical Presentation
4.2. Imaging Findings
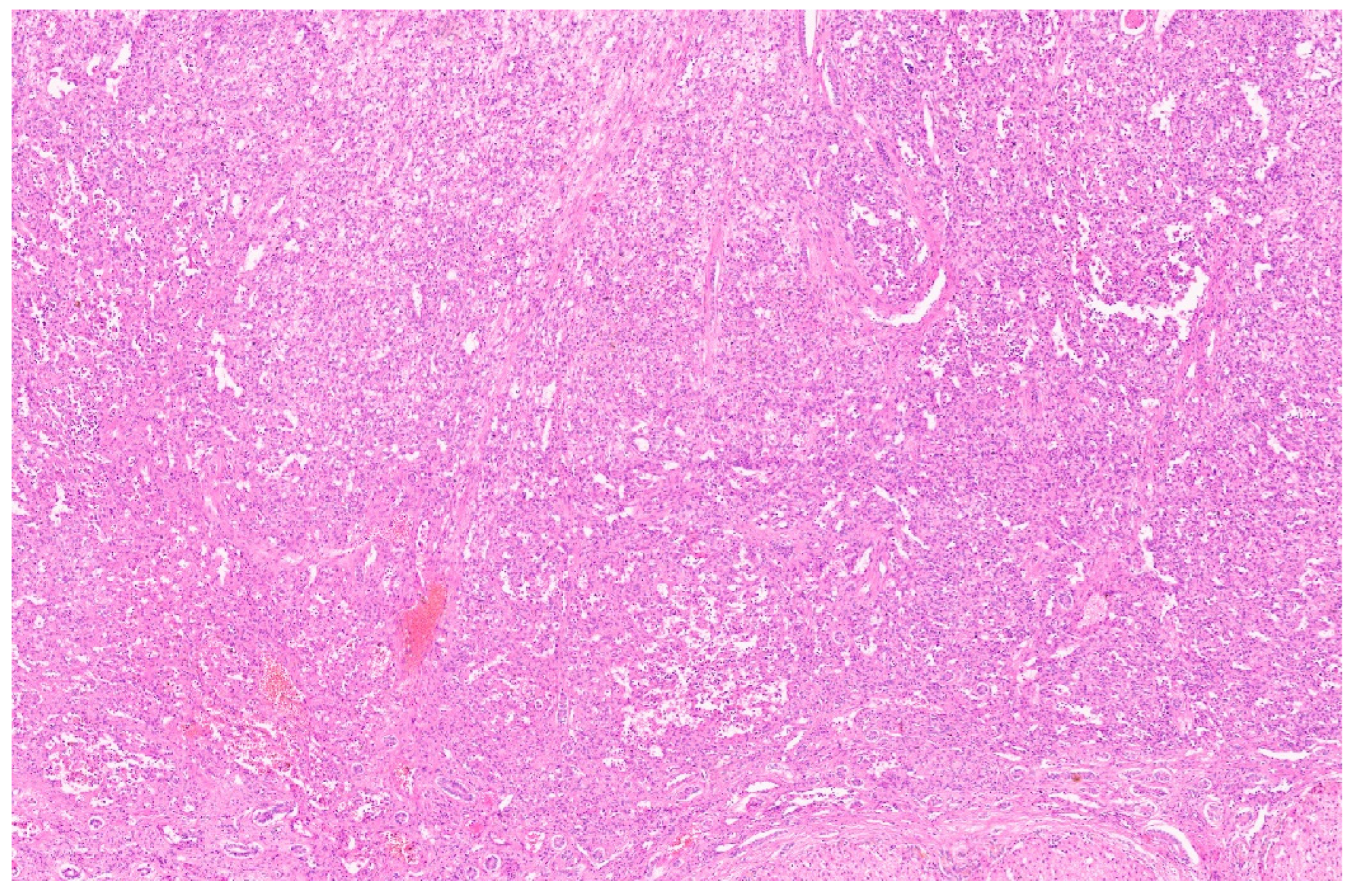
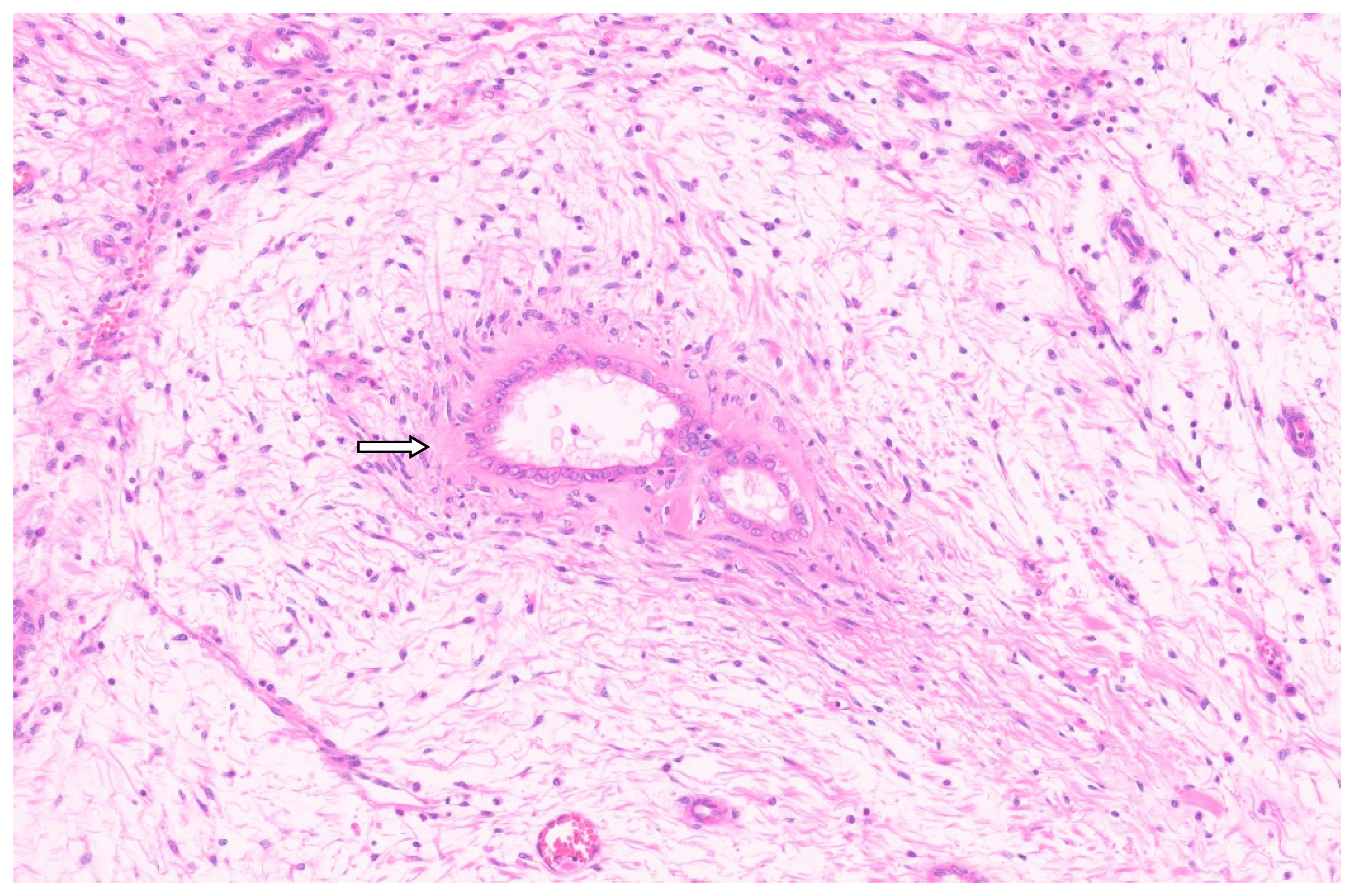
4.3. Pathology
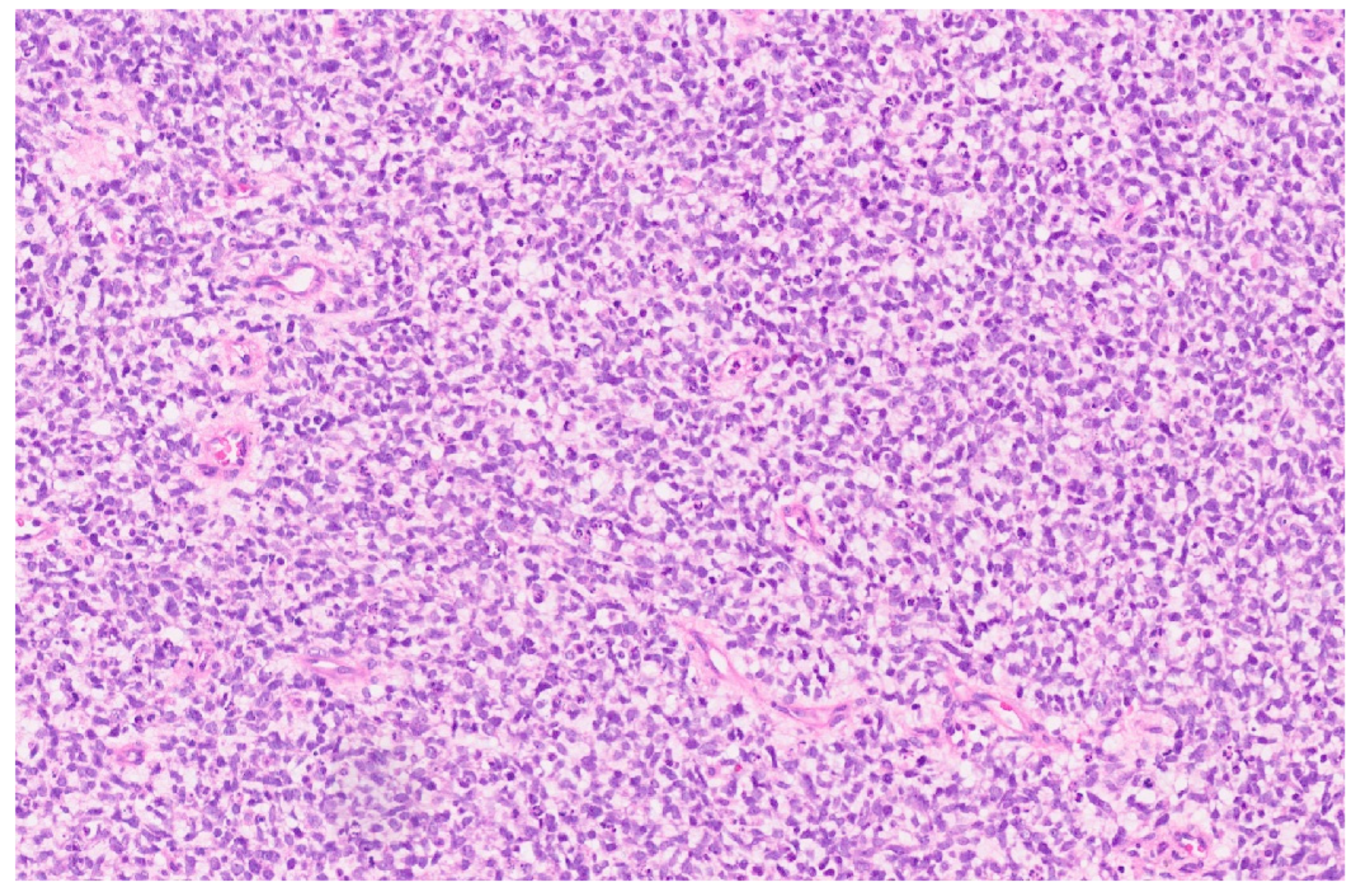
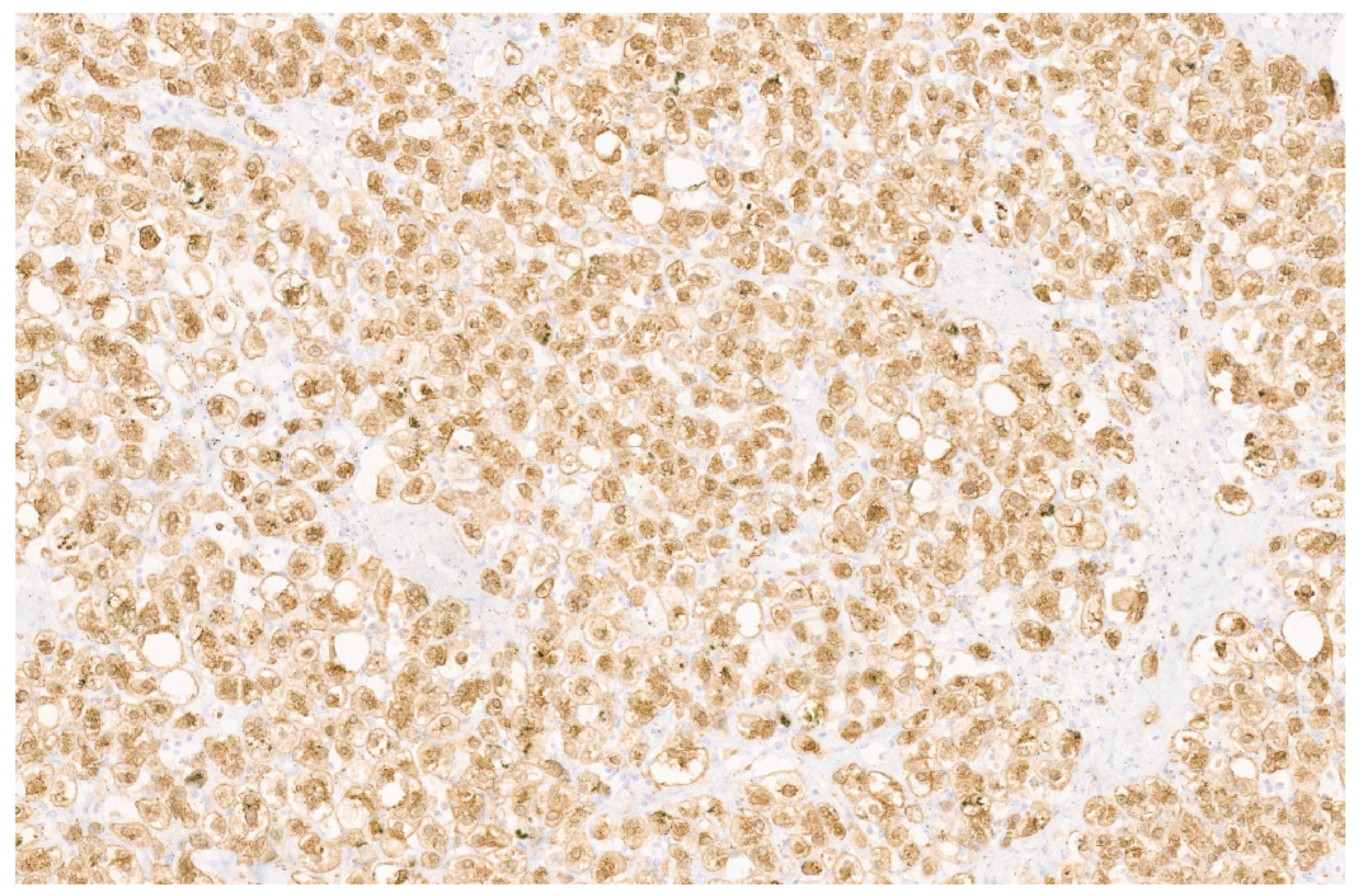
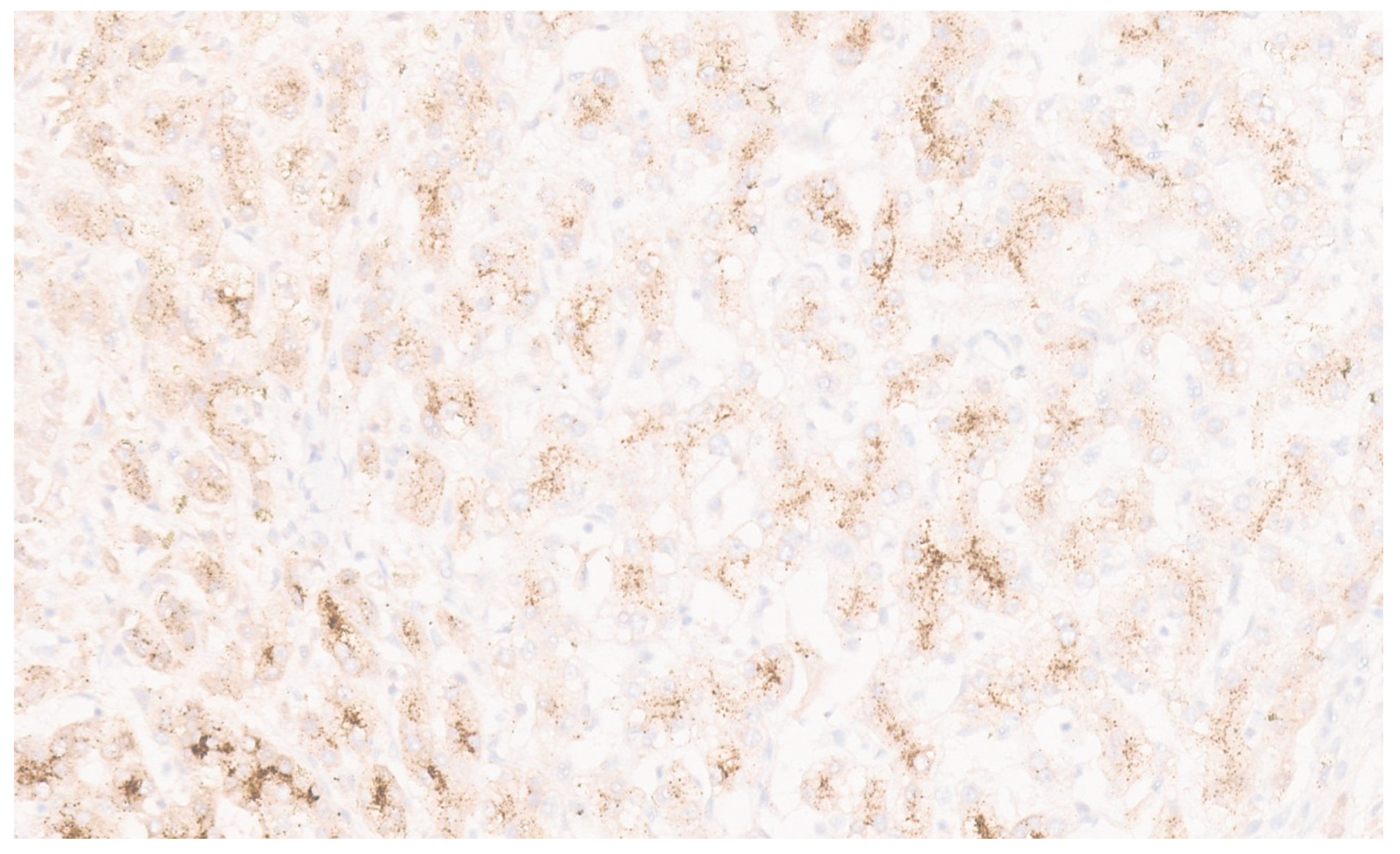
5. Epithelioid Hemangioendothelioma
5.1. Clinical Presentation
5.2. Imaging Findings
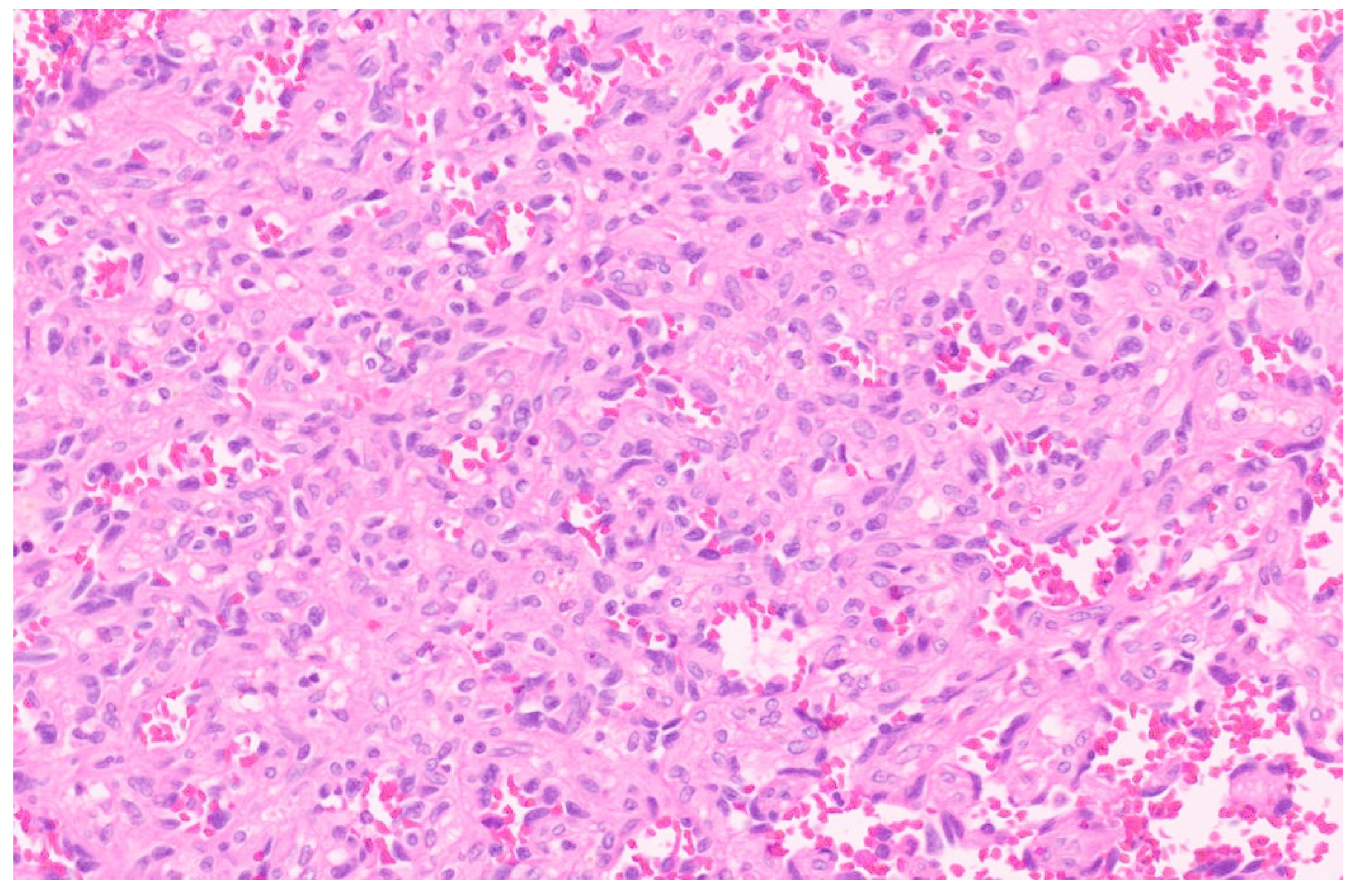
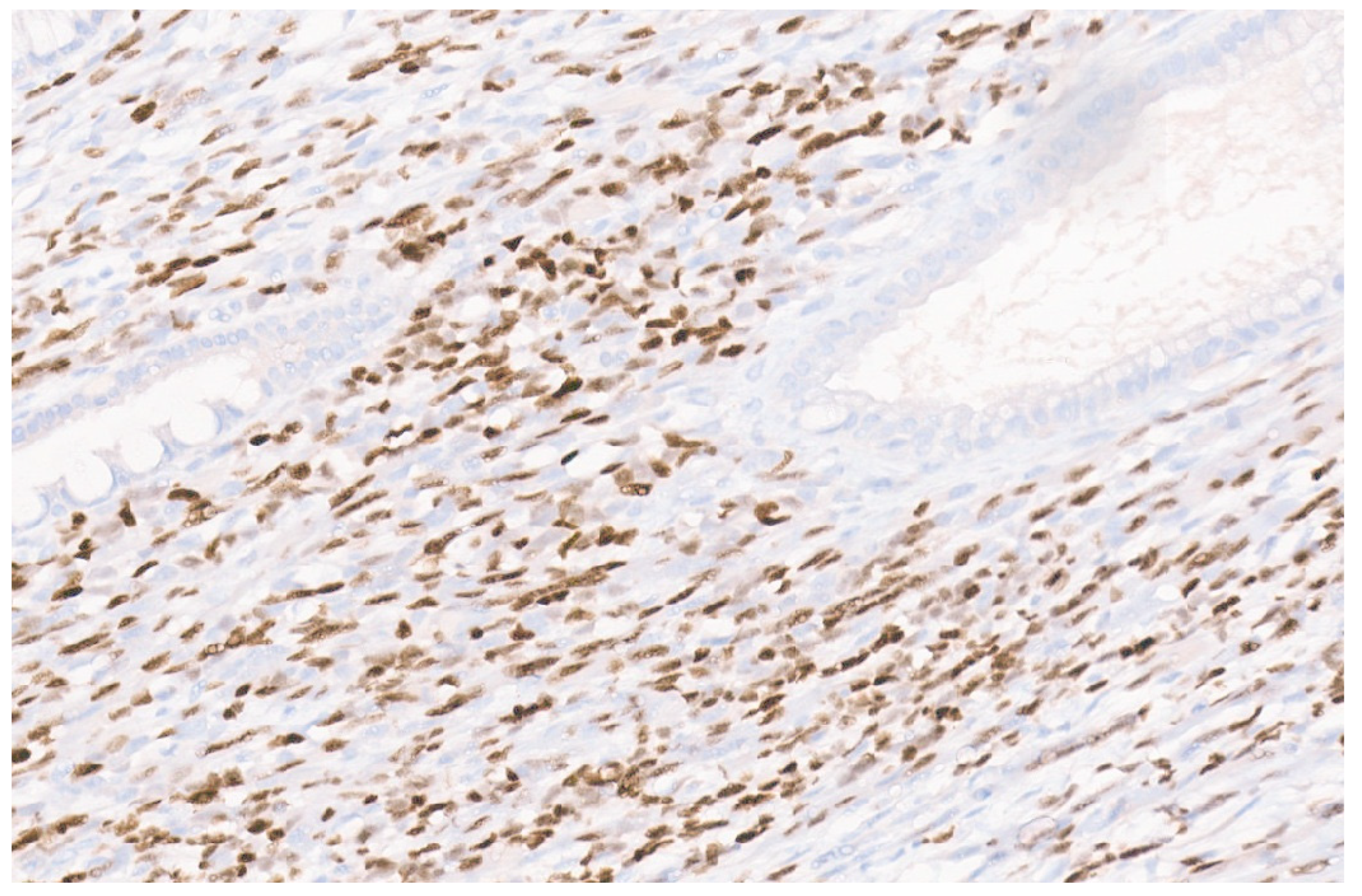
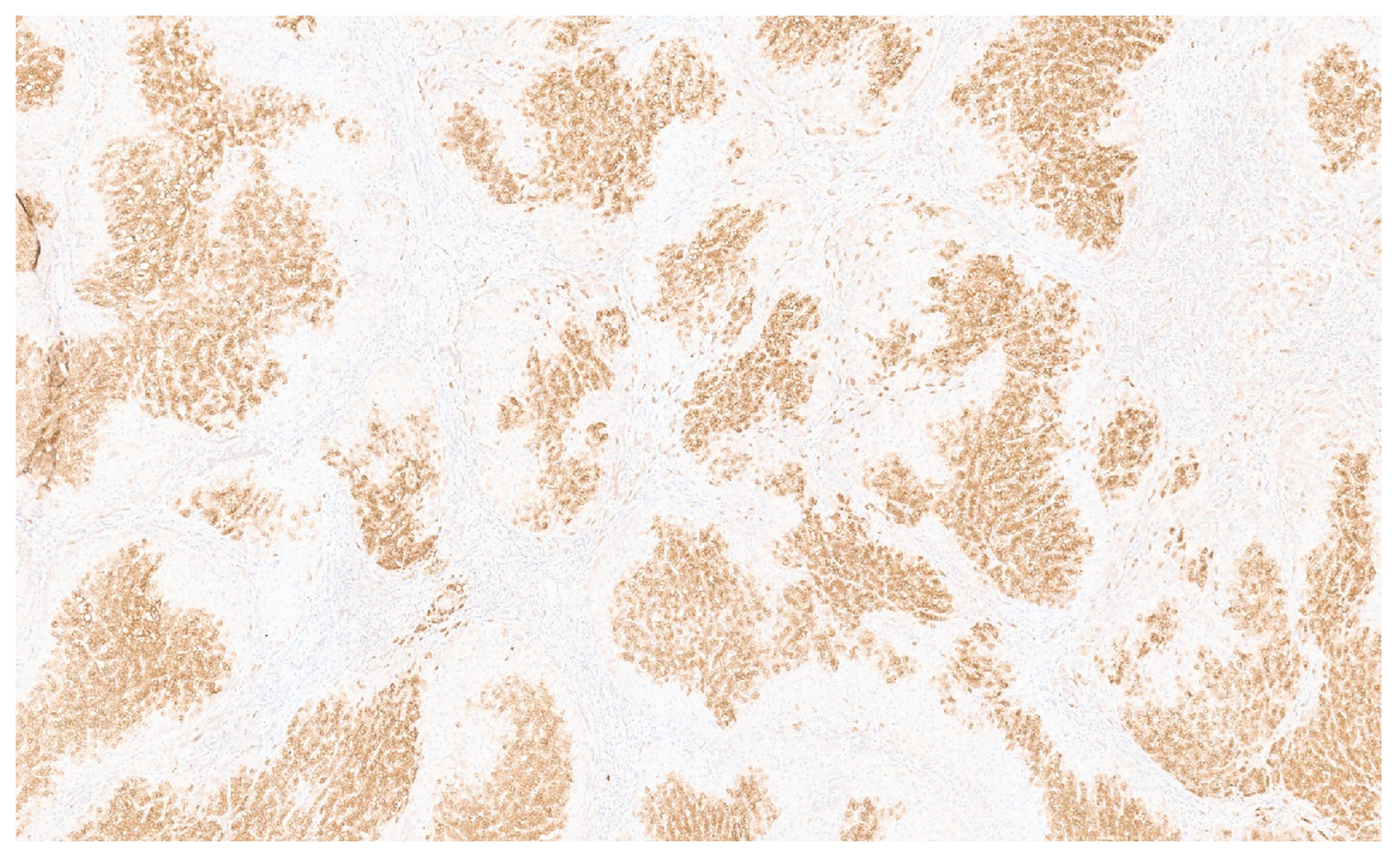
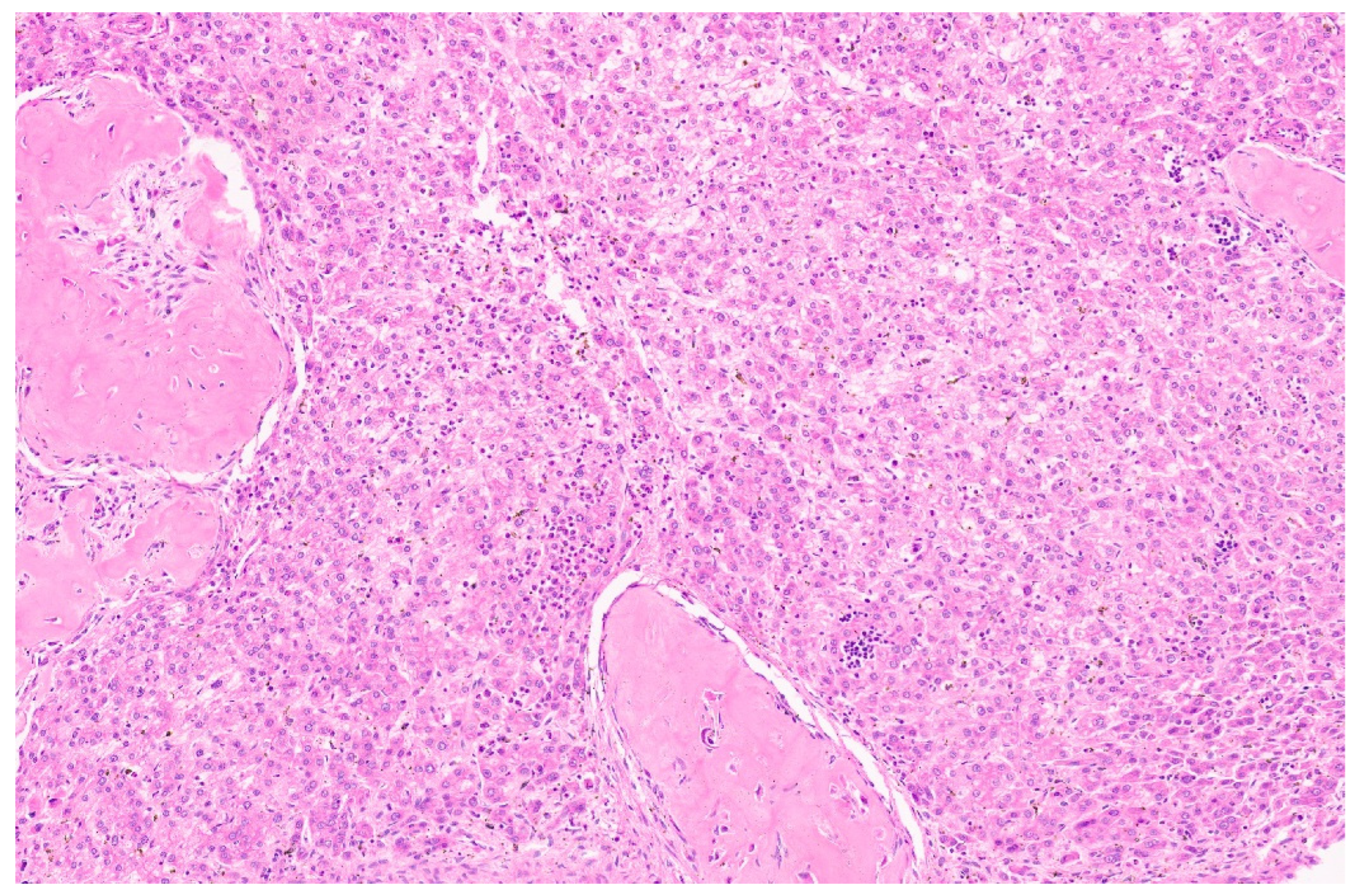
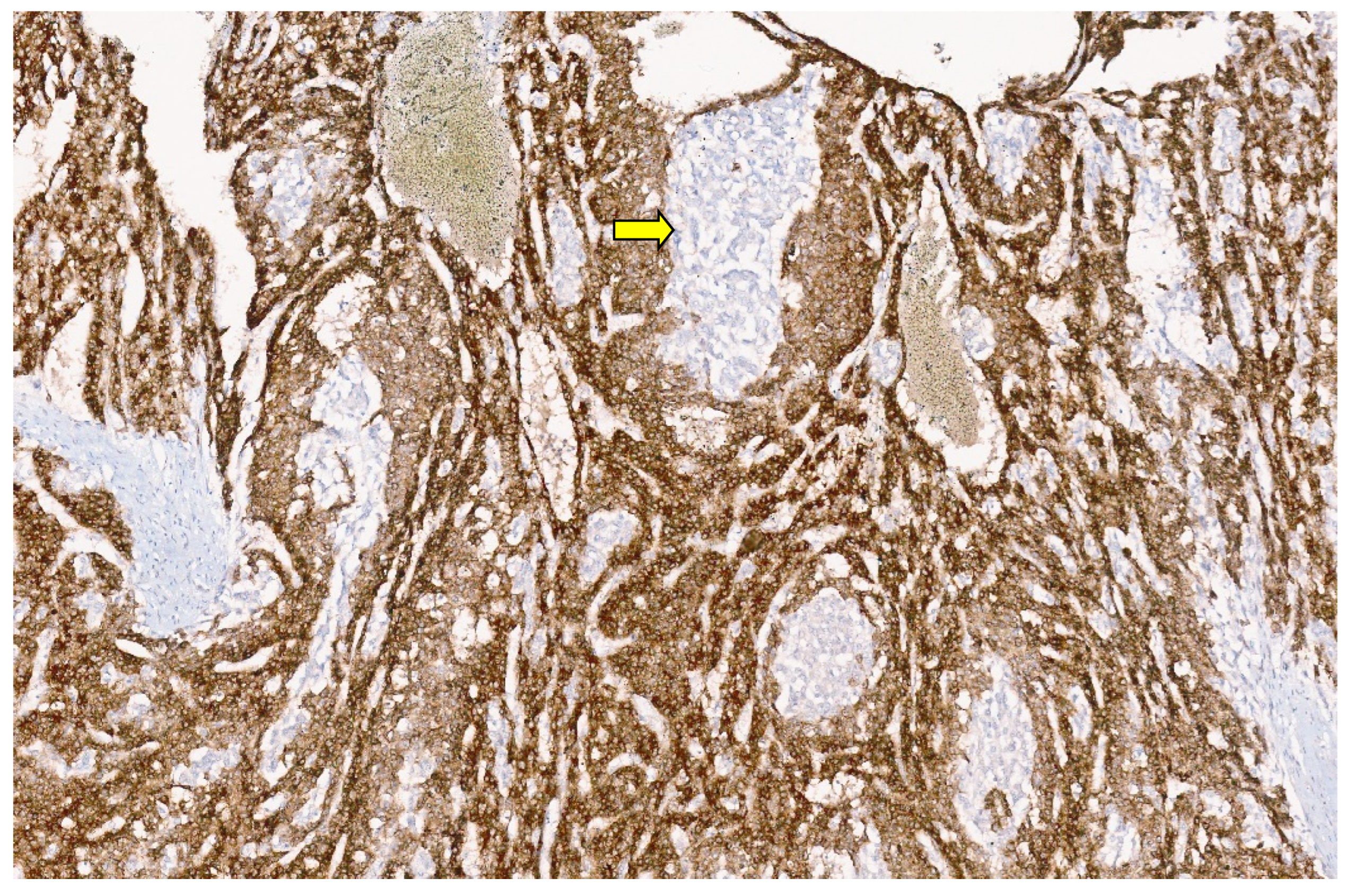
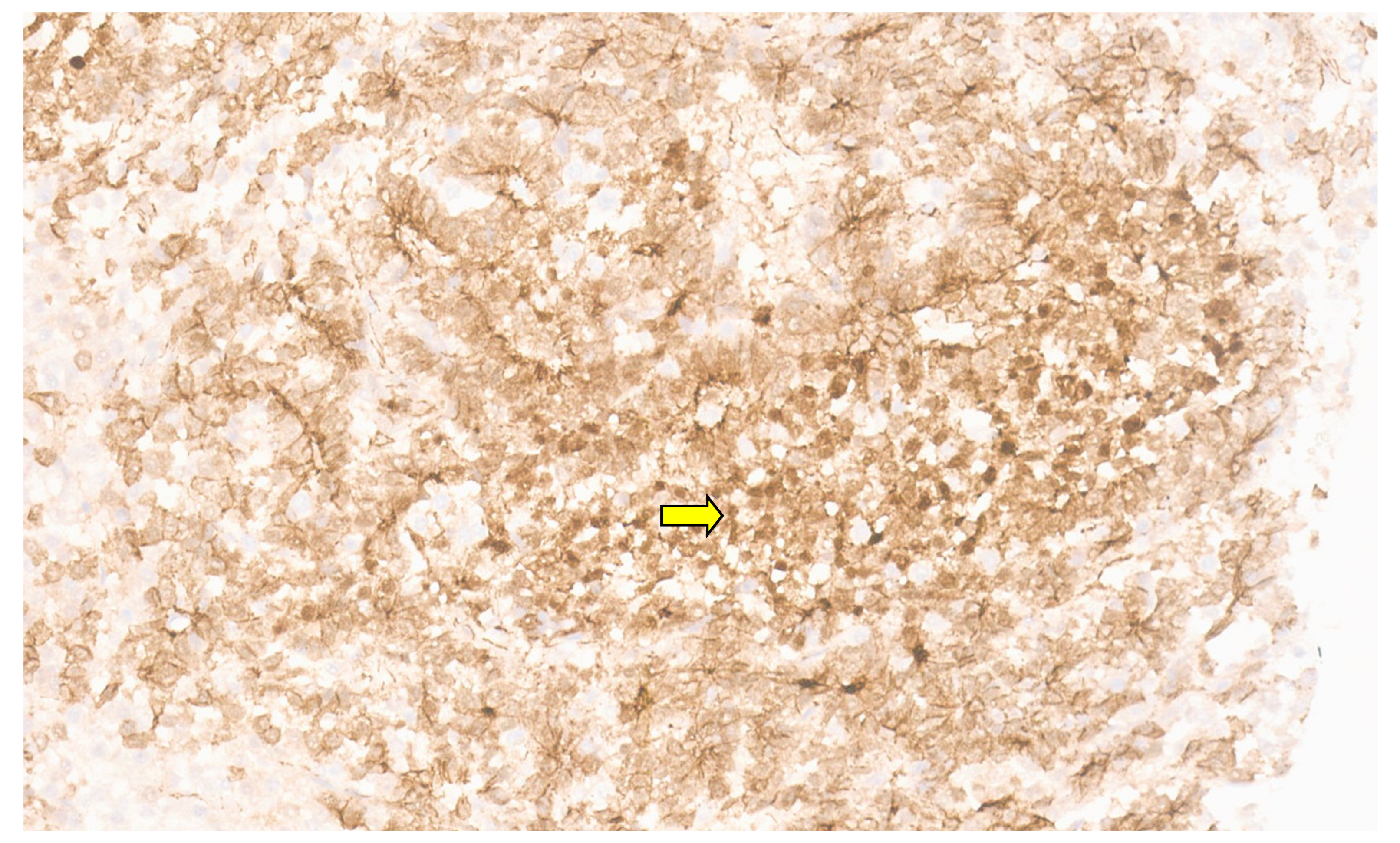
5.3. Pathology
6. Hepatic Angiosarcoma
6.1. Clinical Presentation
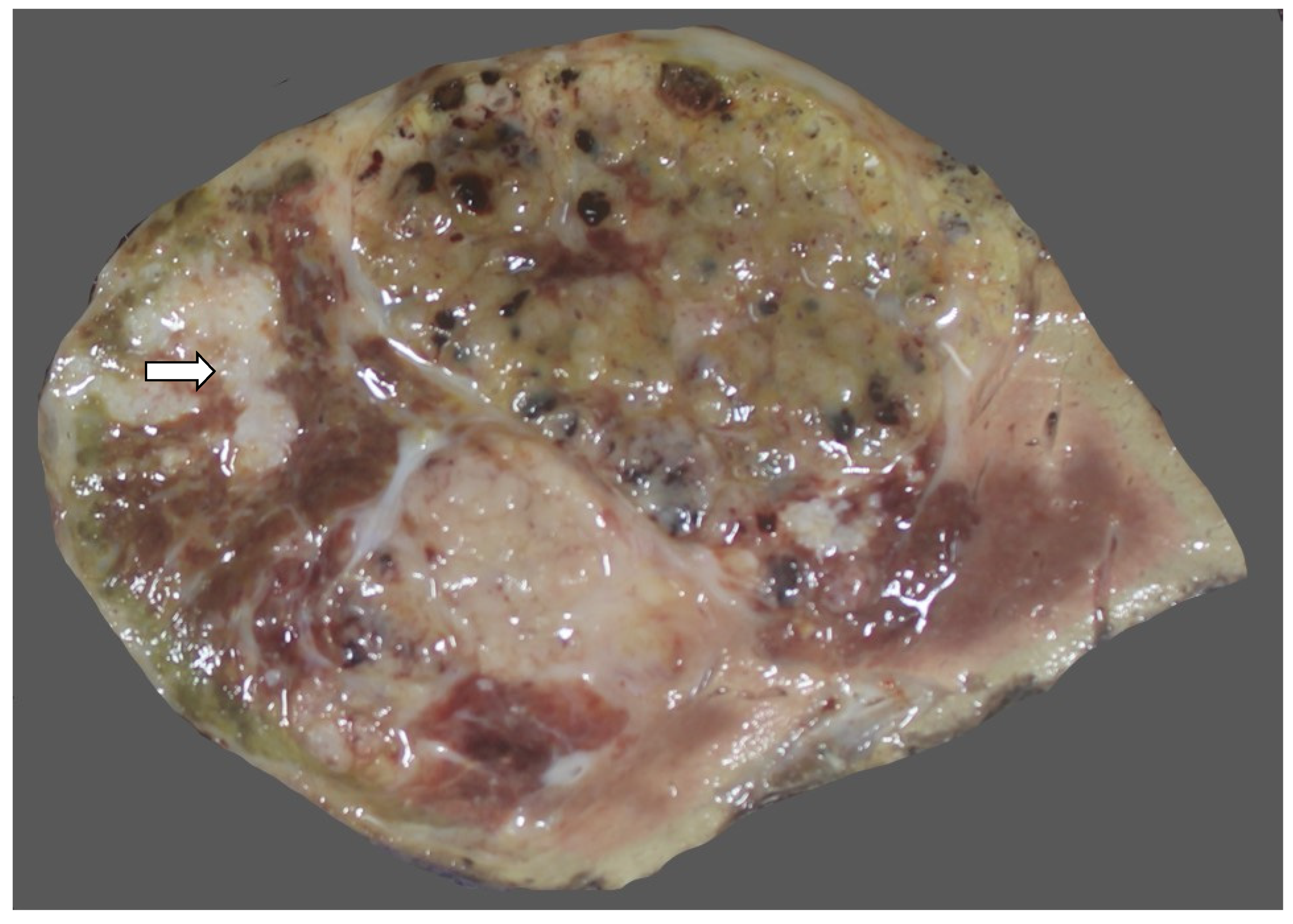
6.2. Imaging Findings
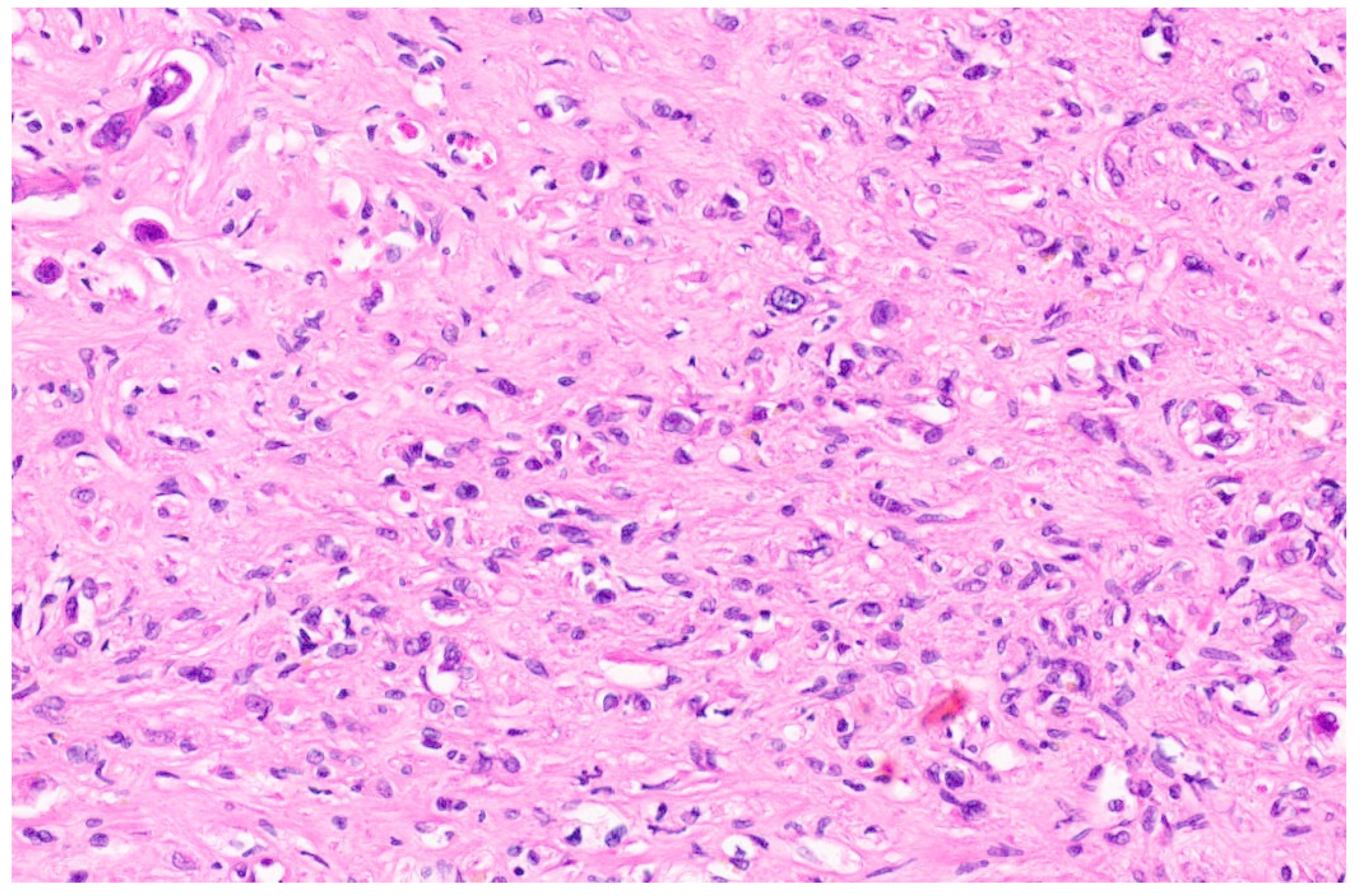
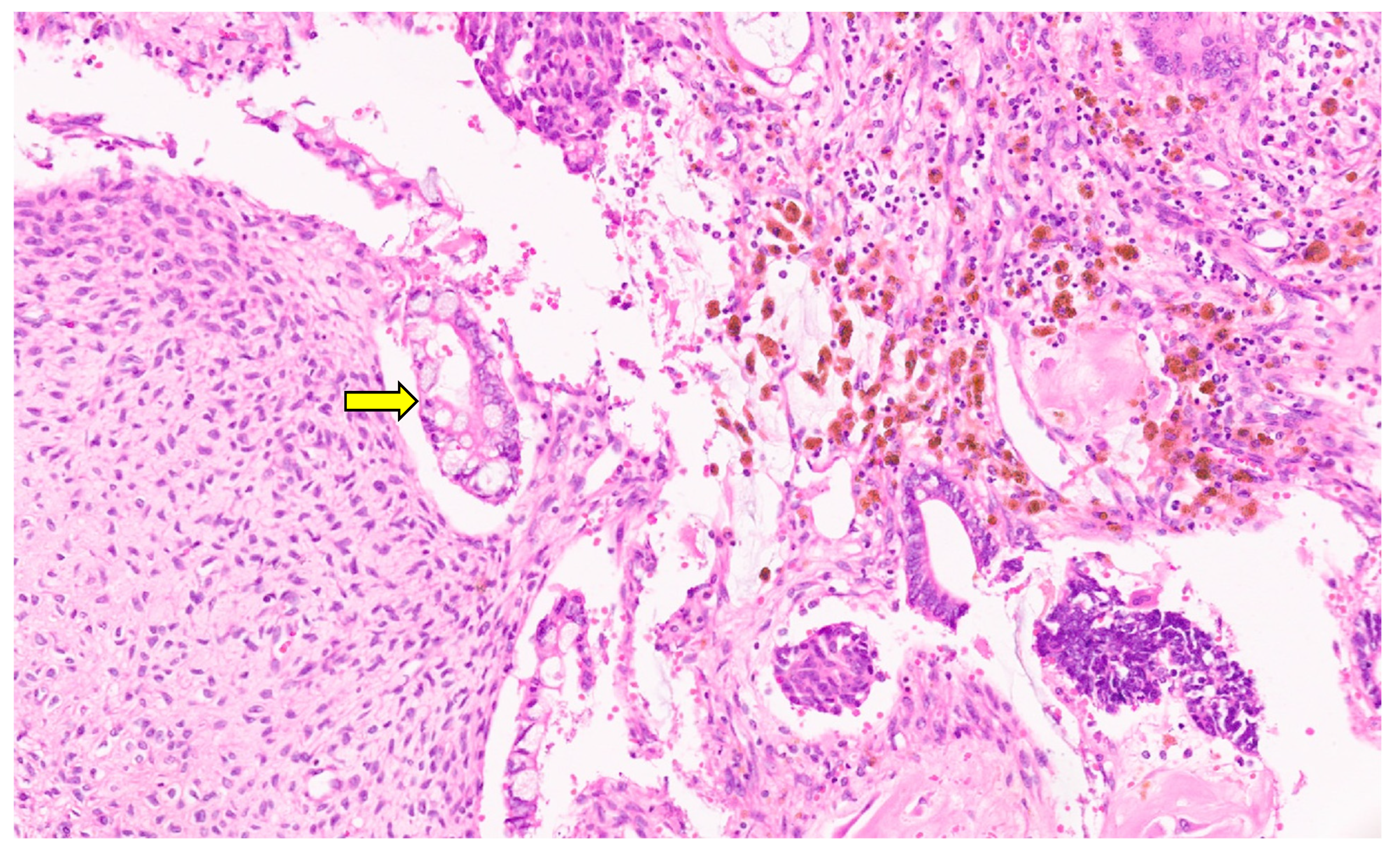
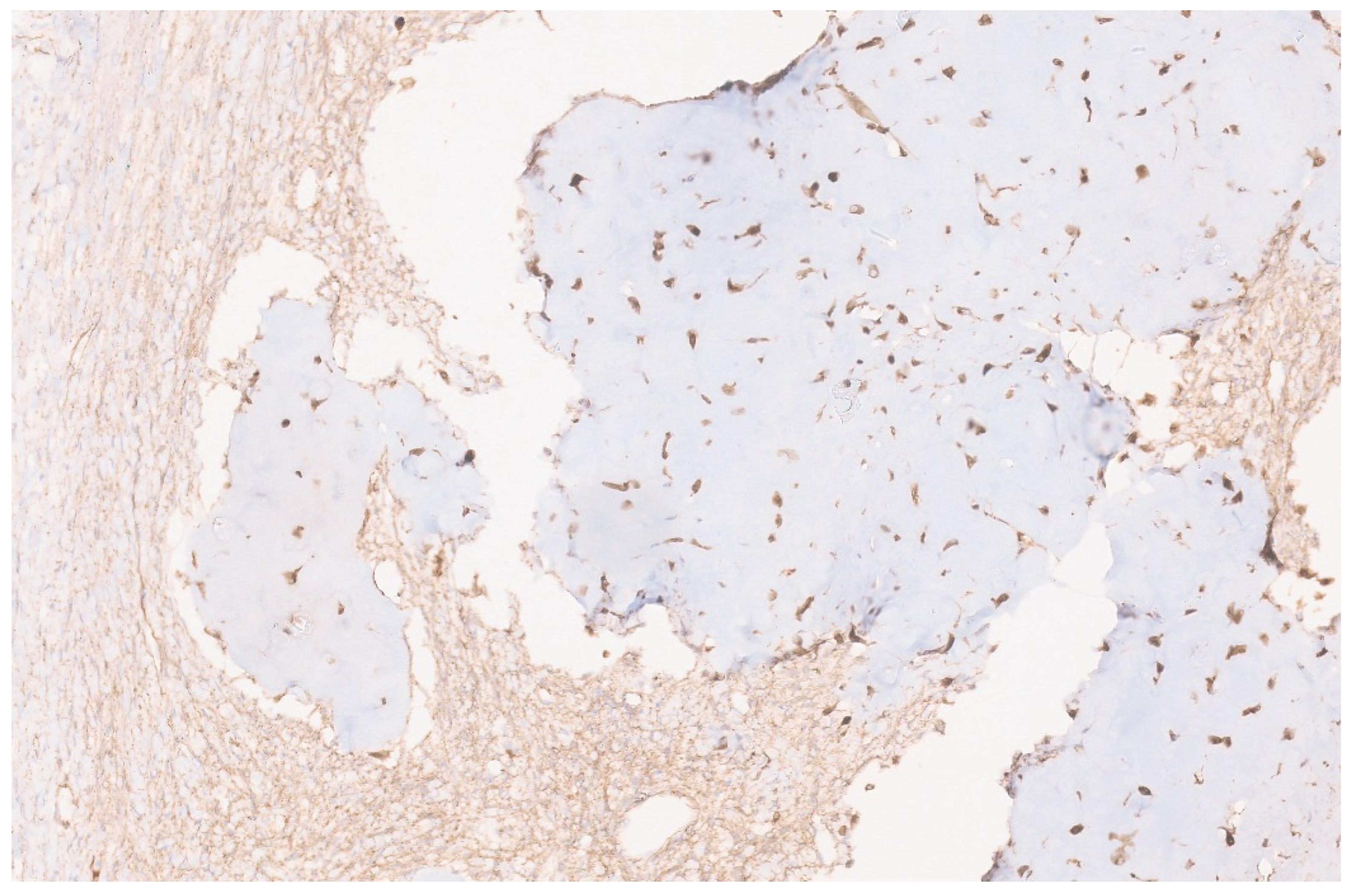
6.3. Pathology
7. Mesenchymal Hamartoma
7.1. Clinical Presentation
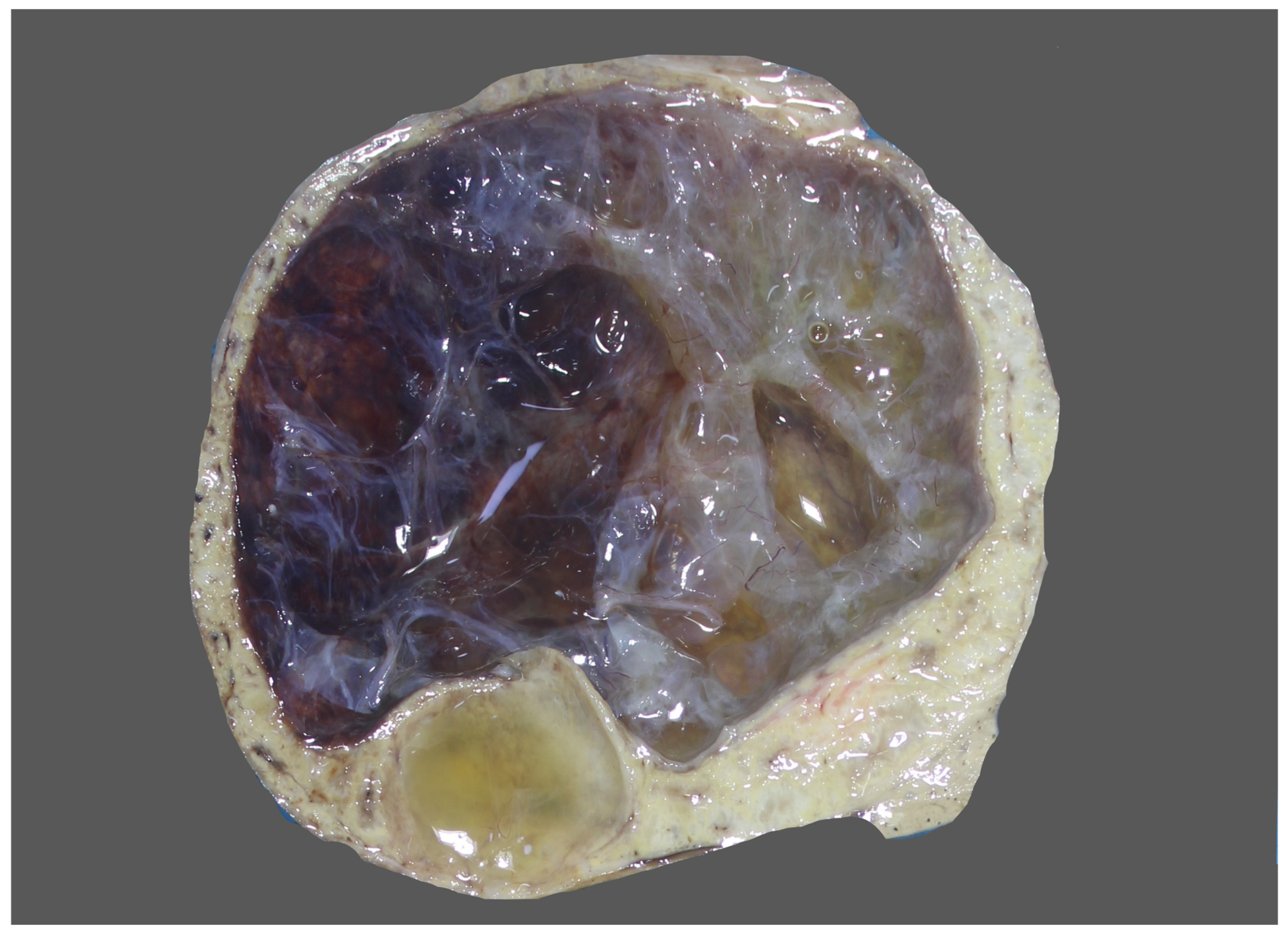
7.2. Imaging Findings
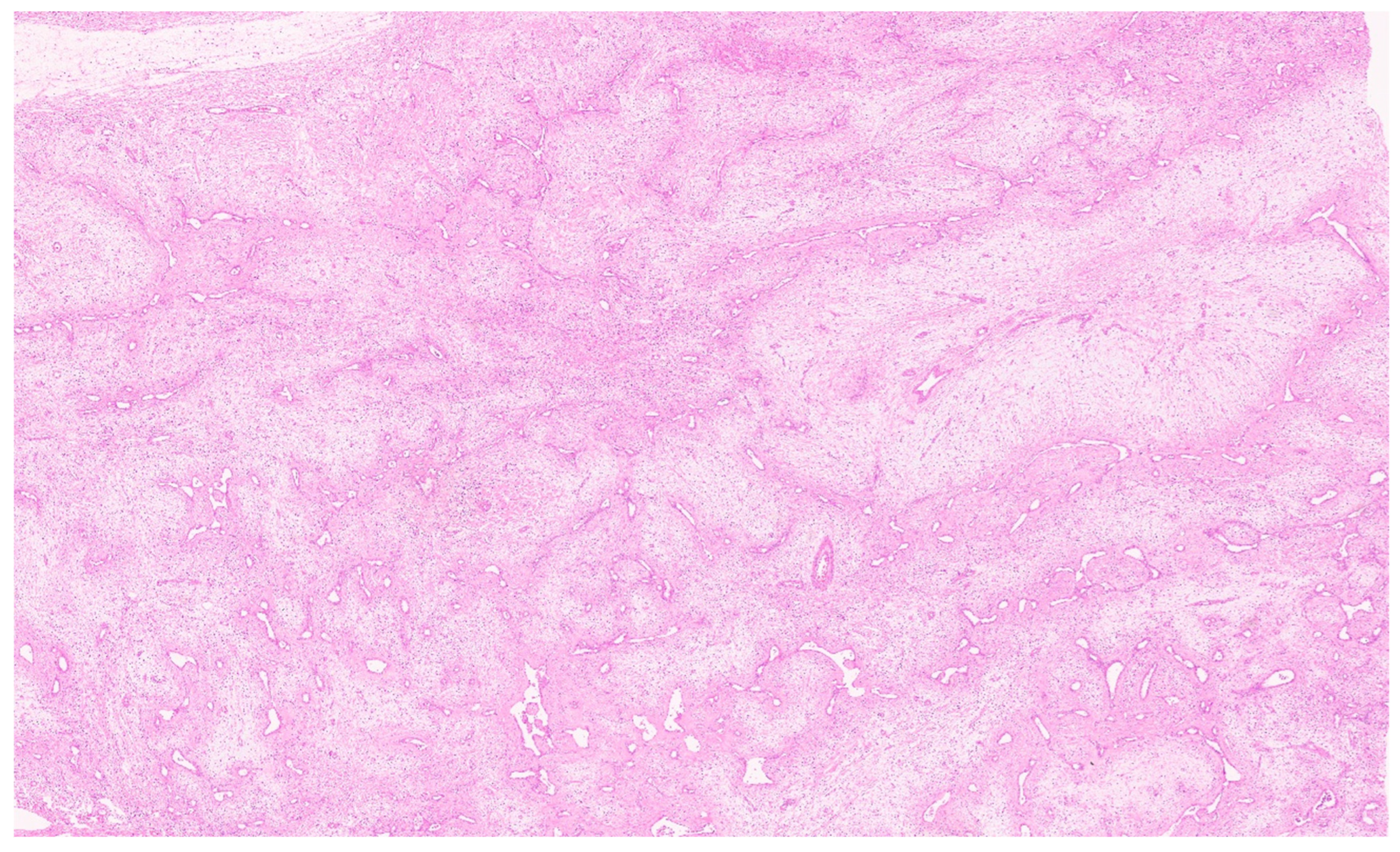
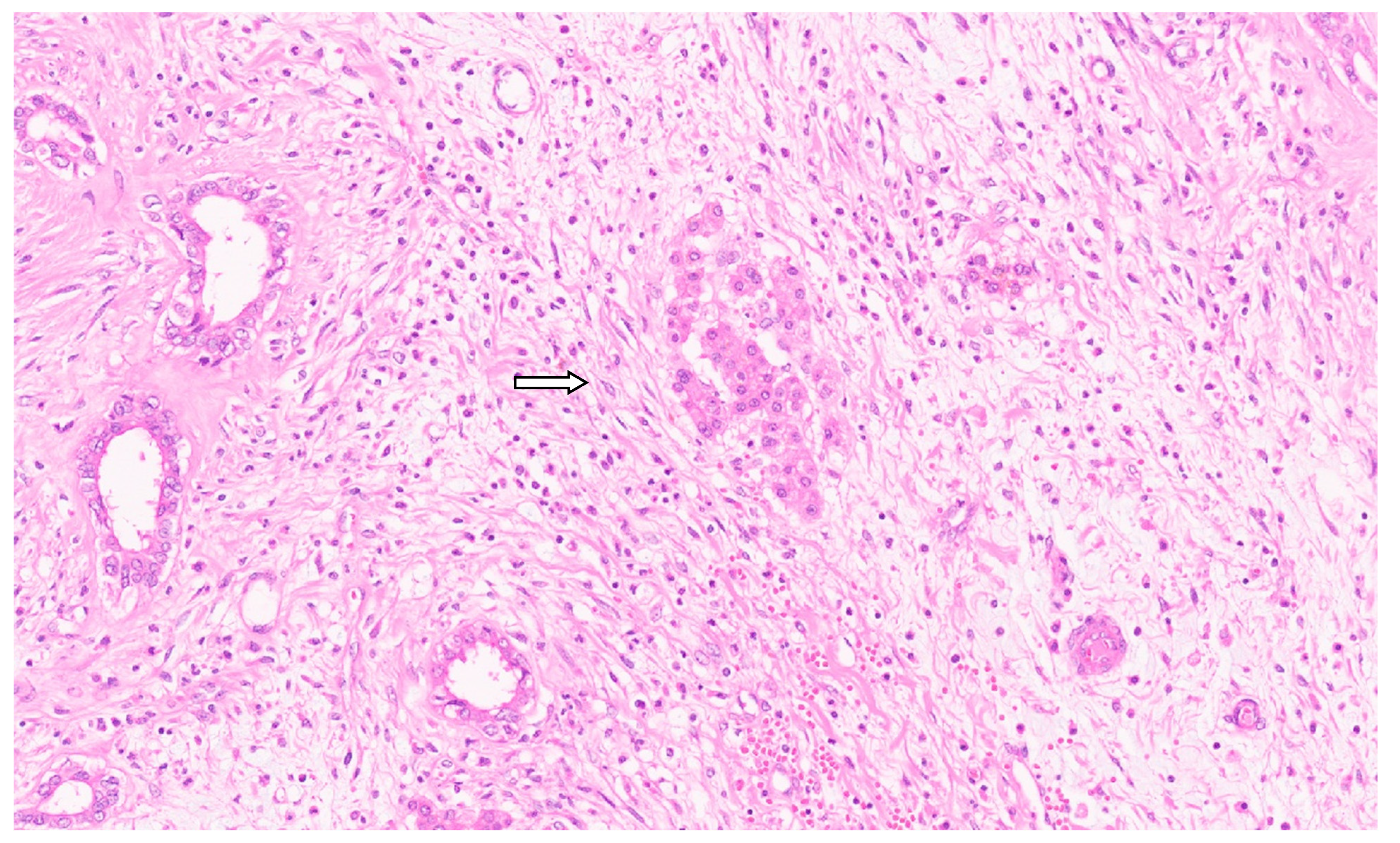
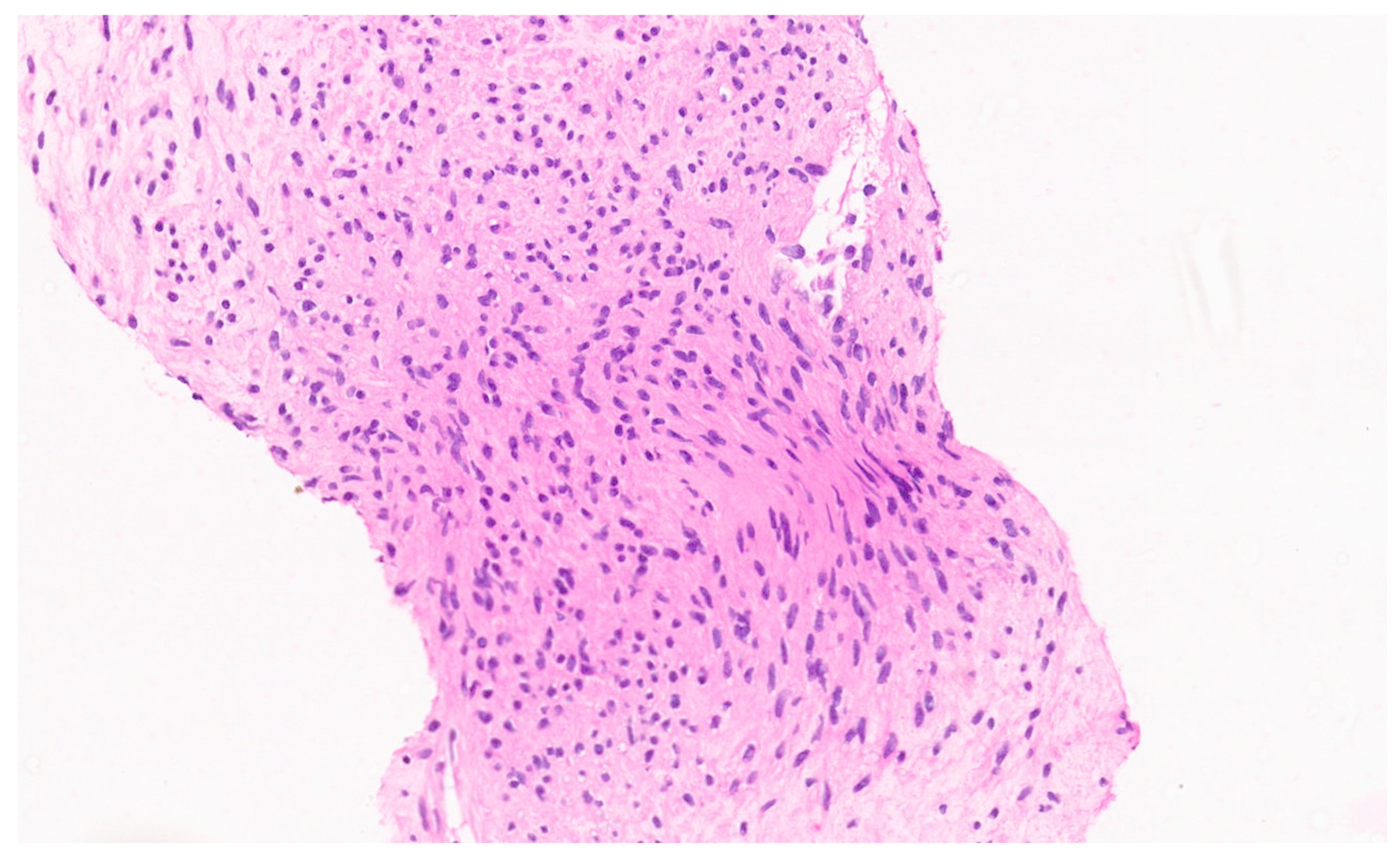
8. Pathology
8.1. Inflammatory Myofibroblastic Tumors
8.2. Clinical Presentation
8.3. Imaging Findings
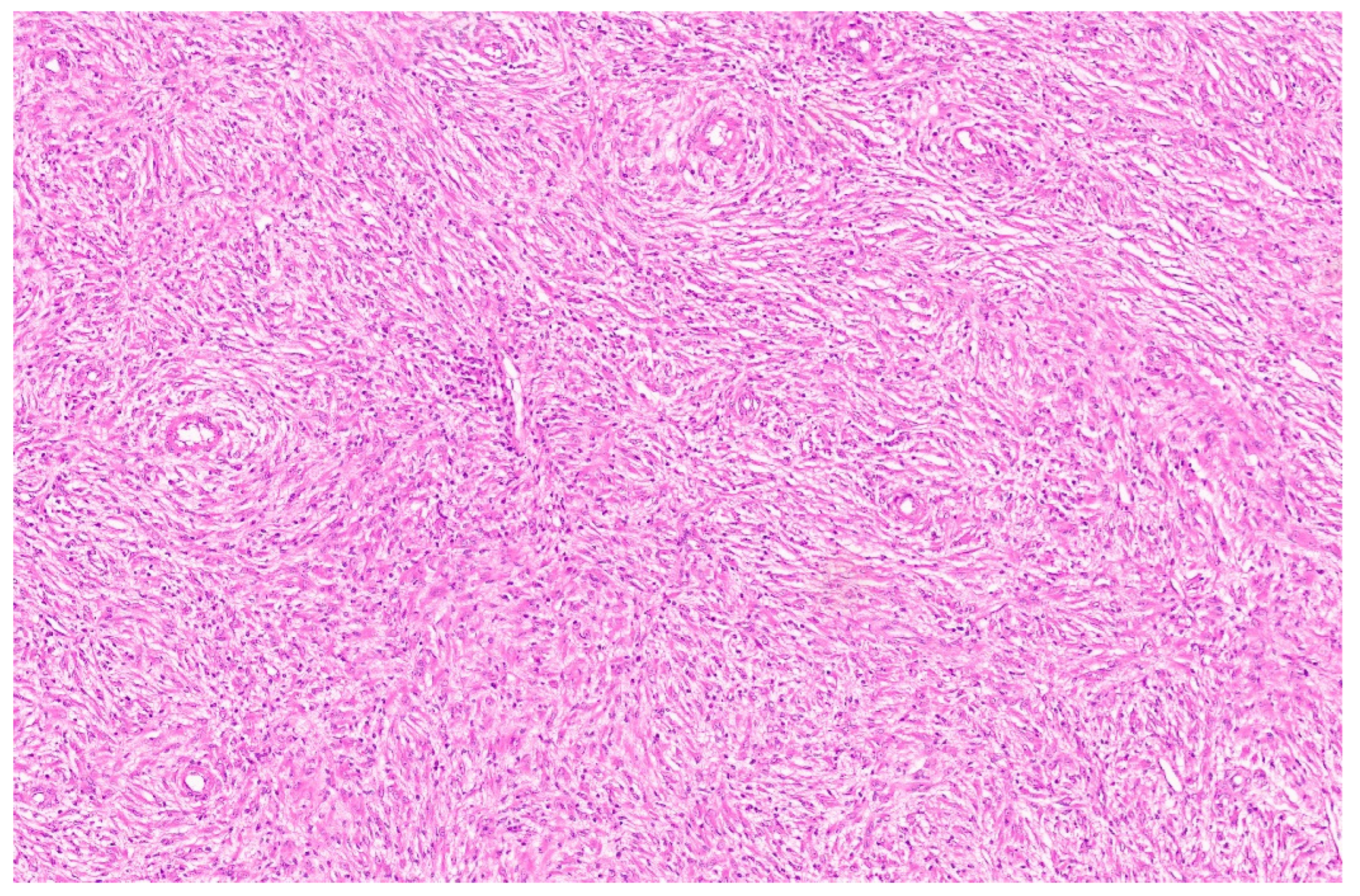
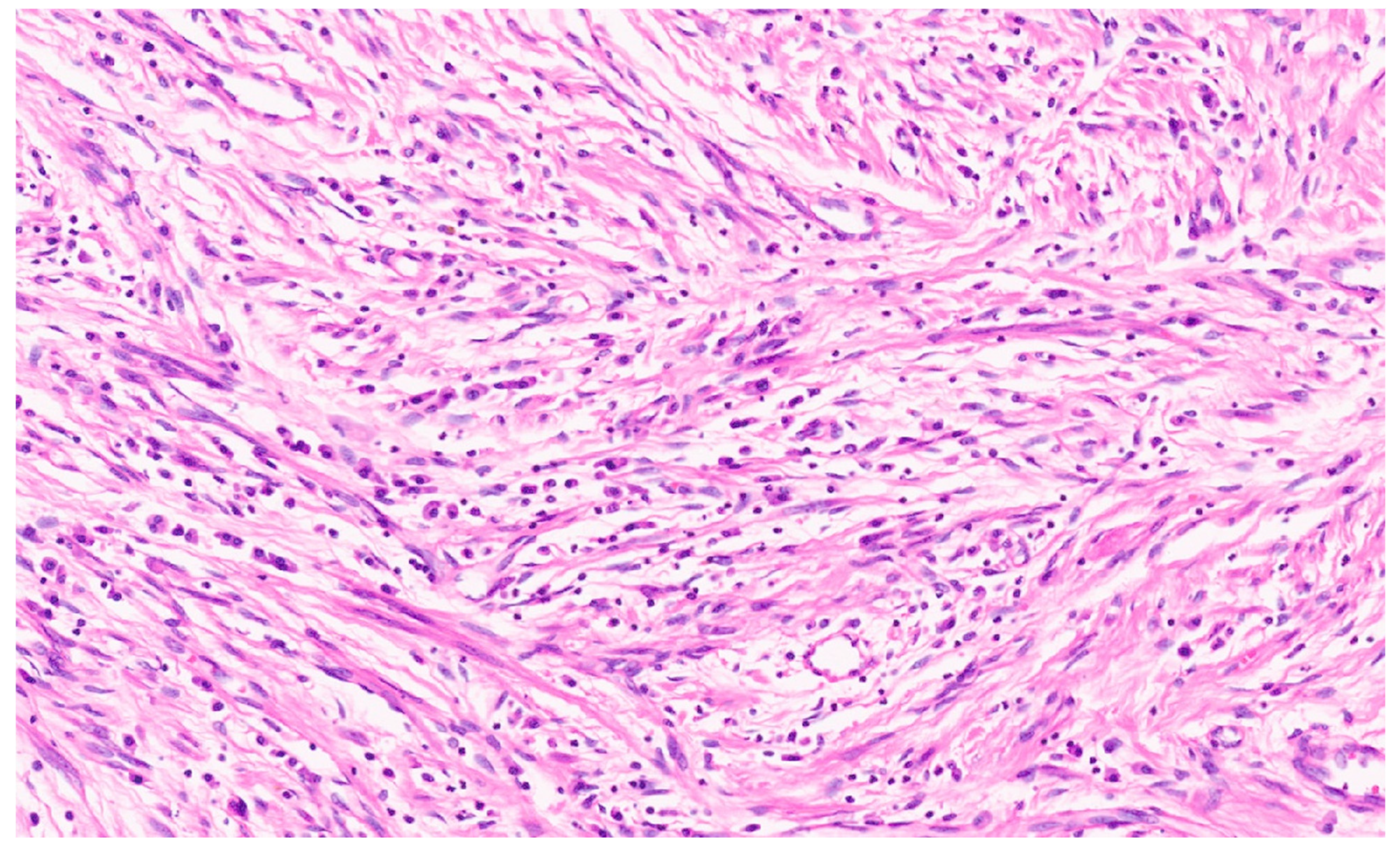
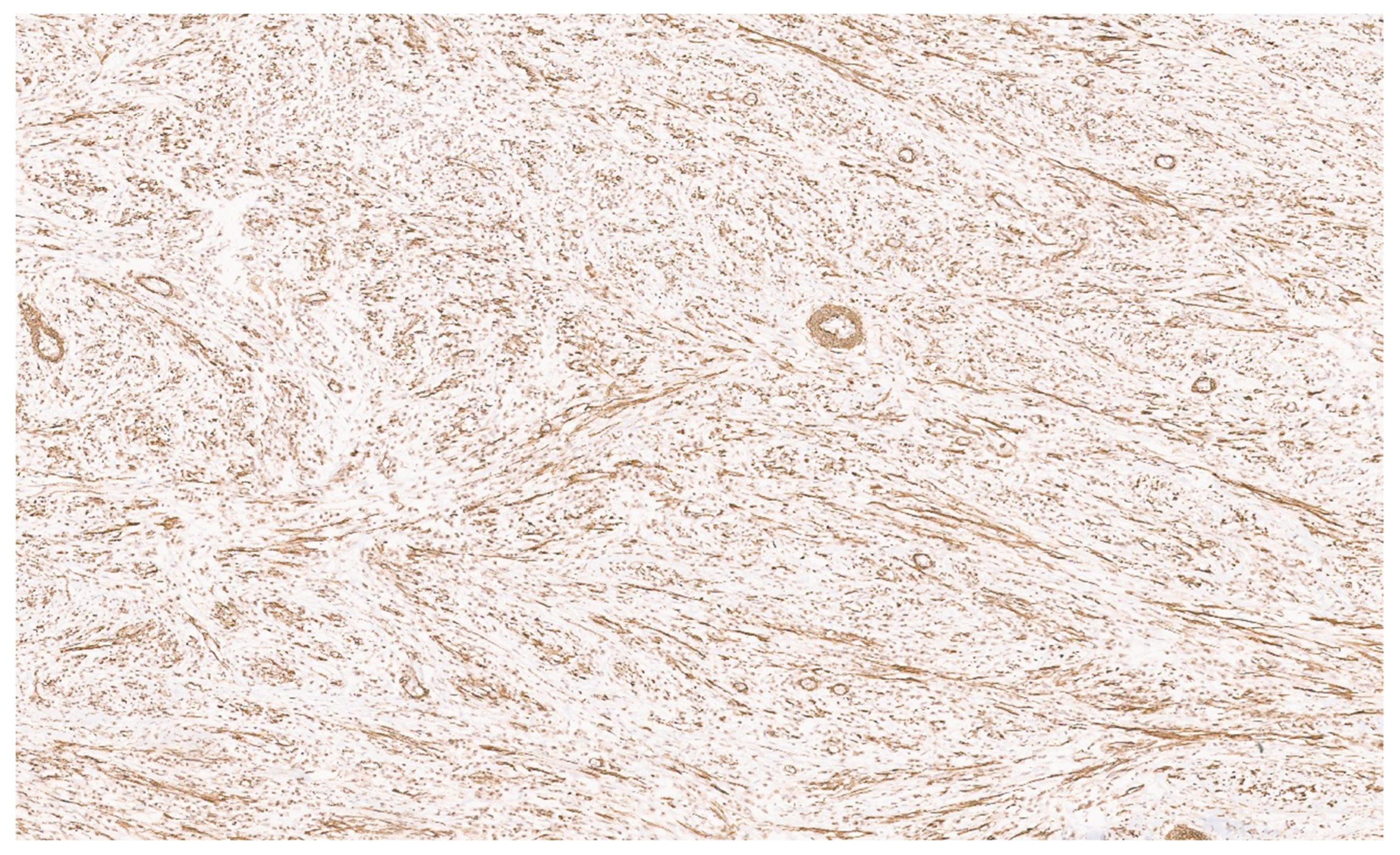
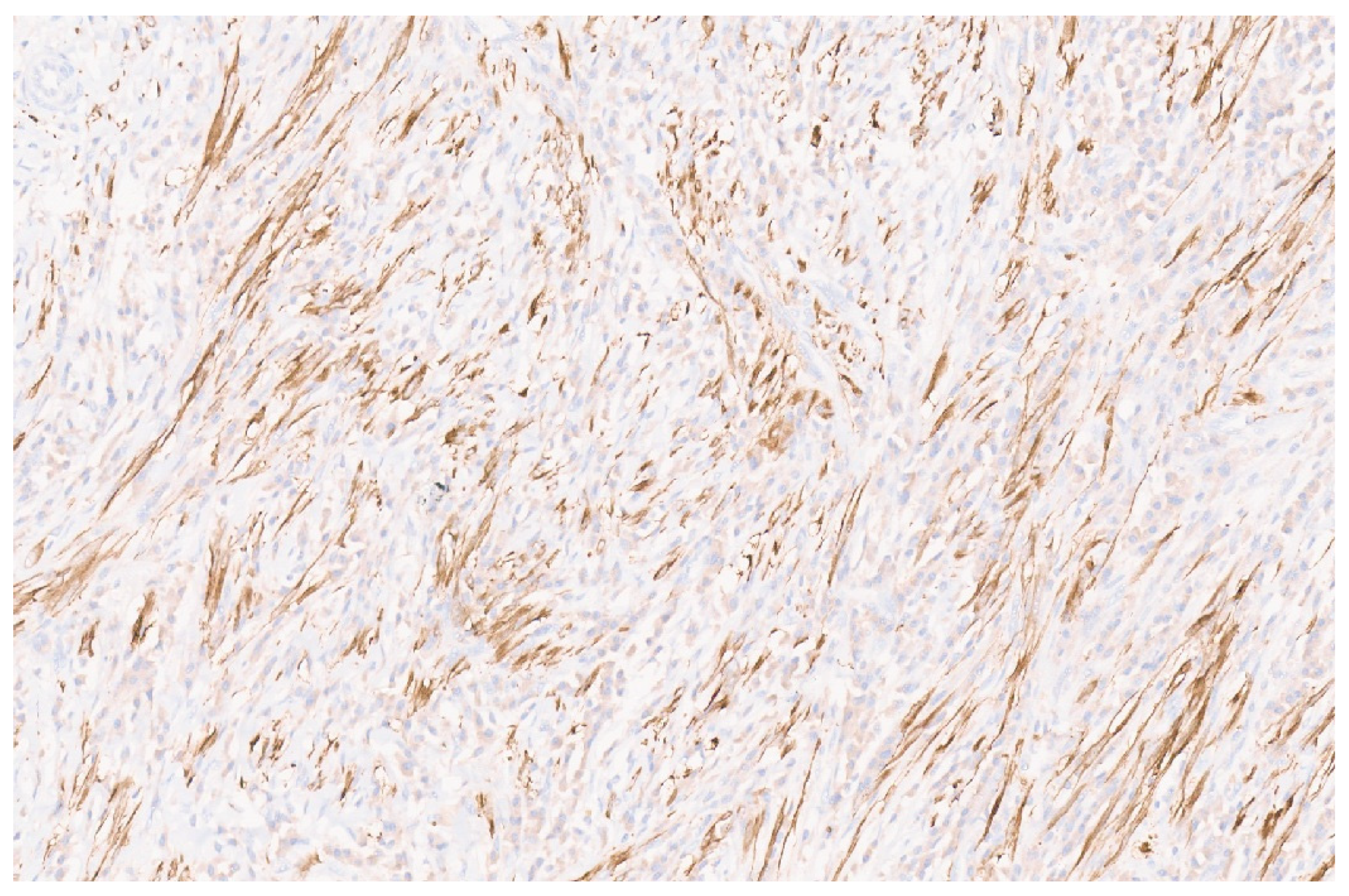
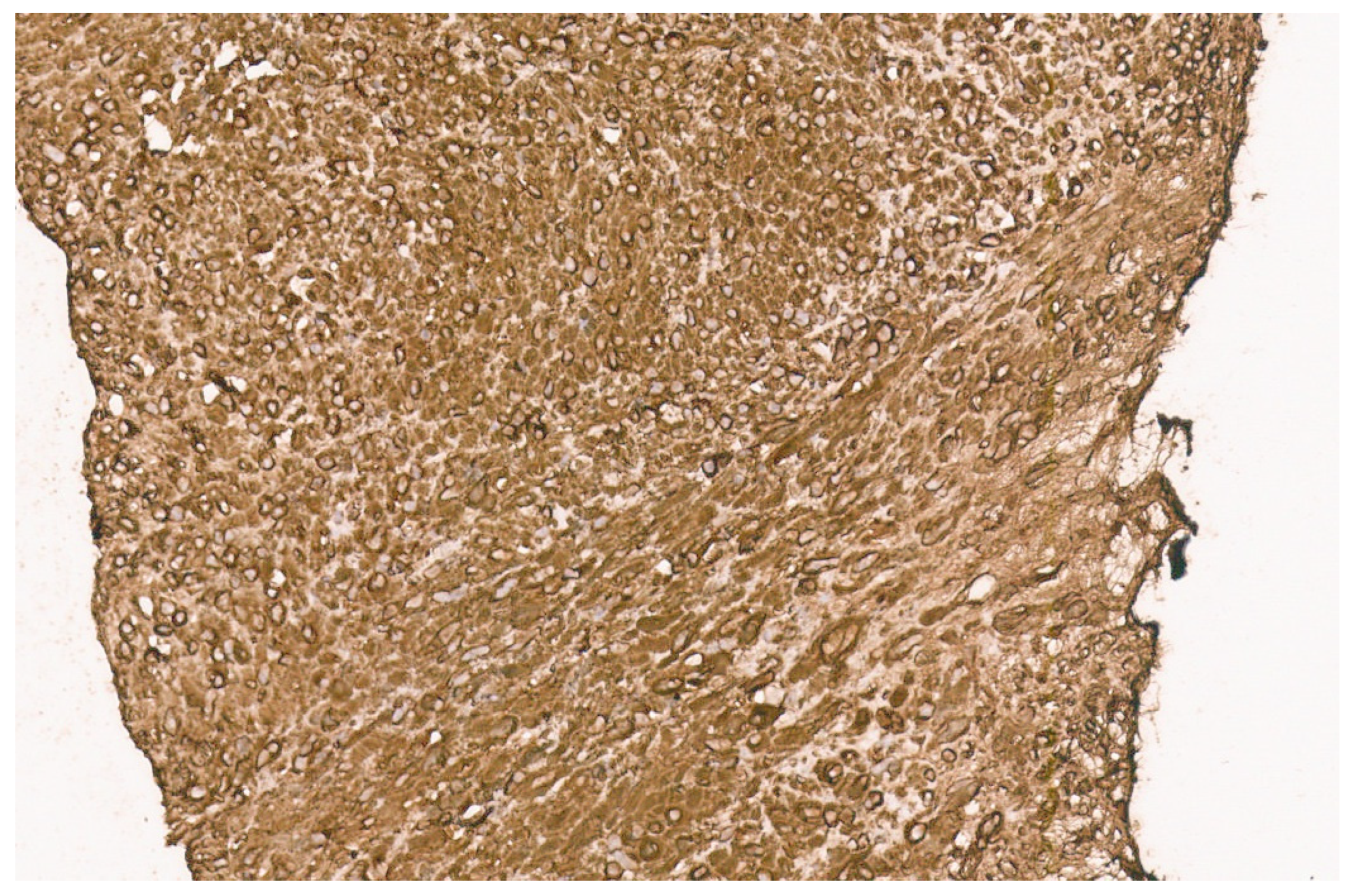
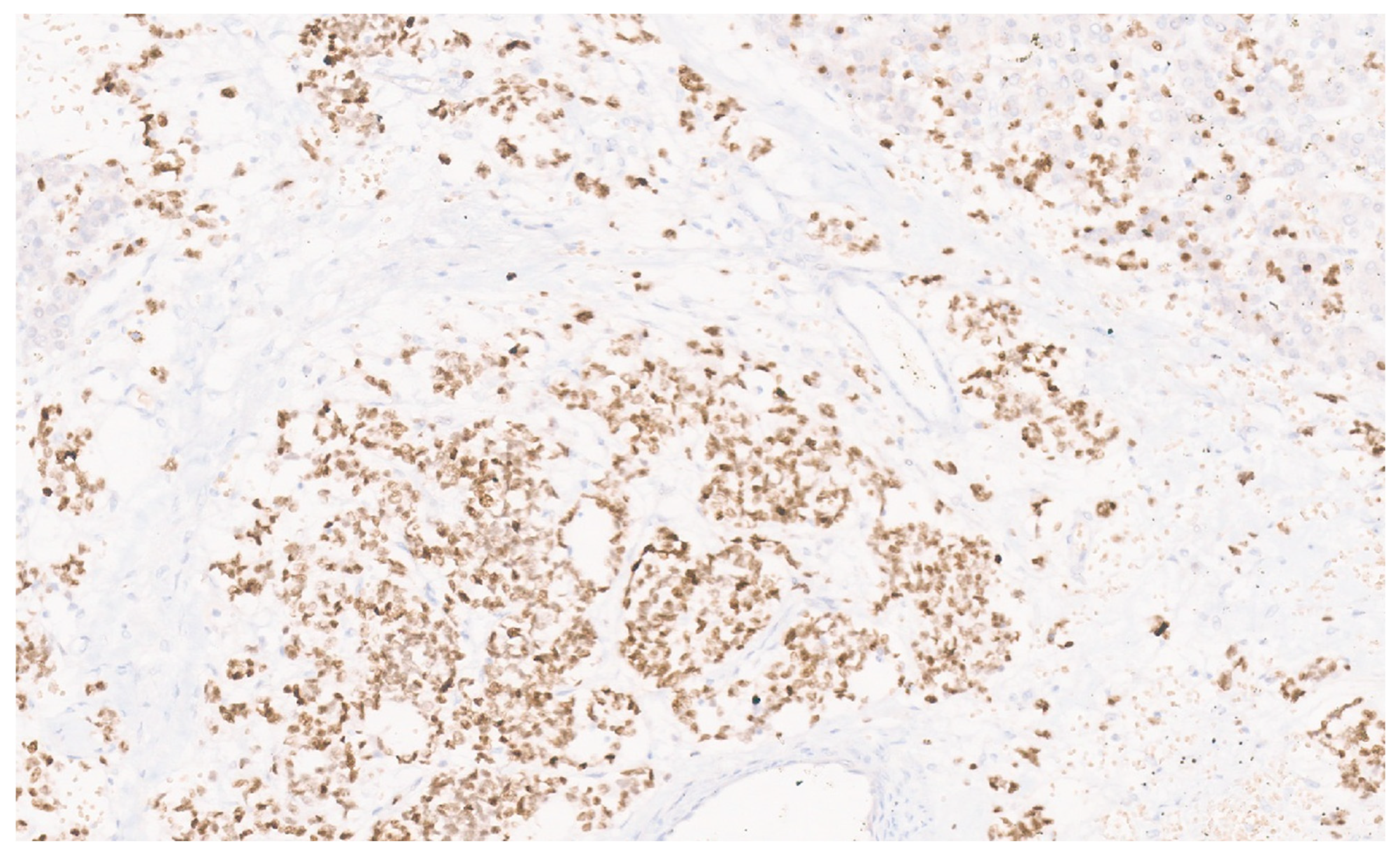
8.4. Pathology
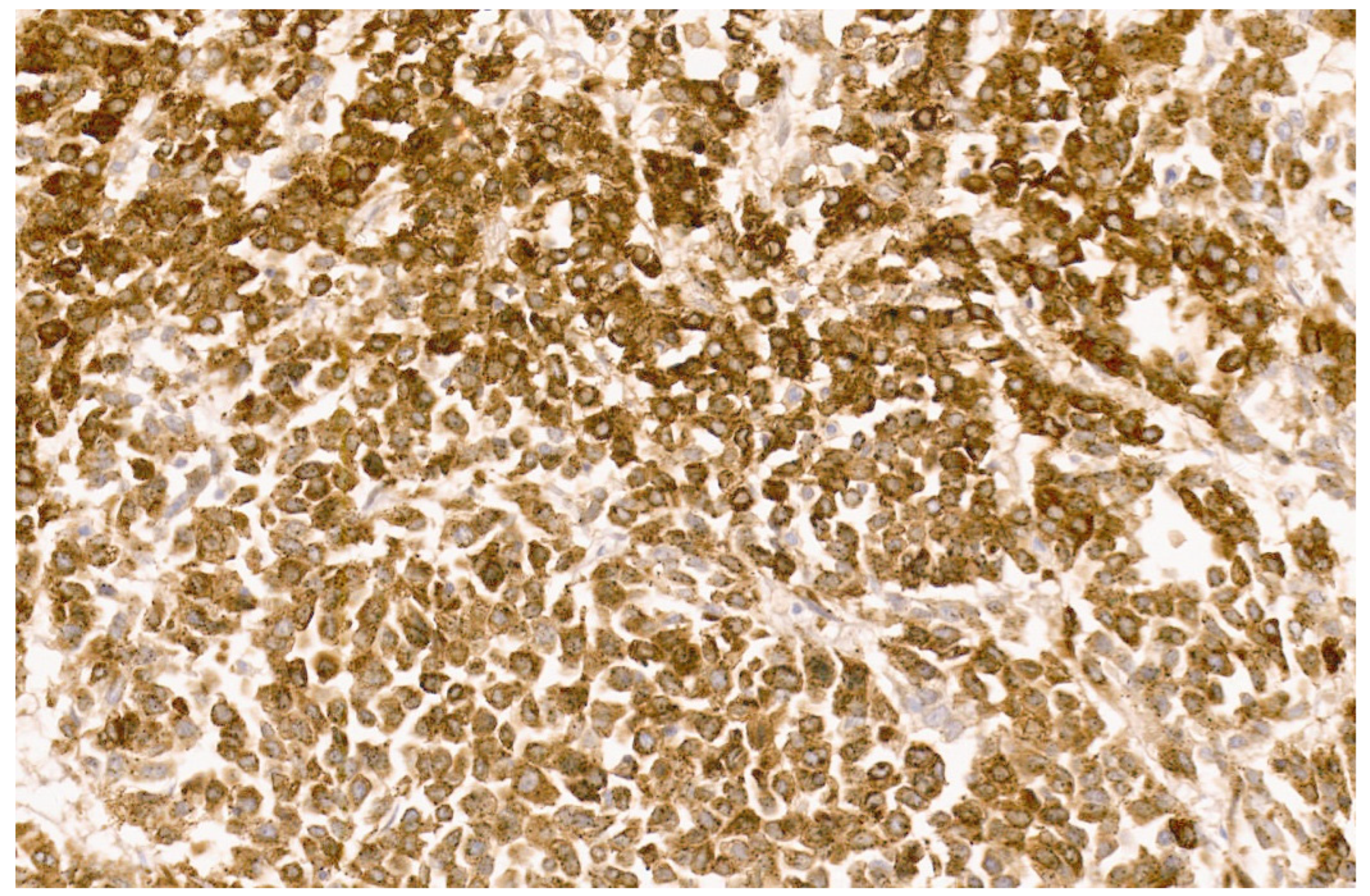
9. Malignant Rhabdoid Tumor
9.1. Clinical Presentation
9.2. Imaging Features
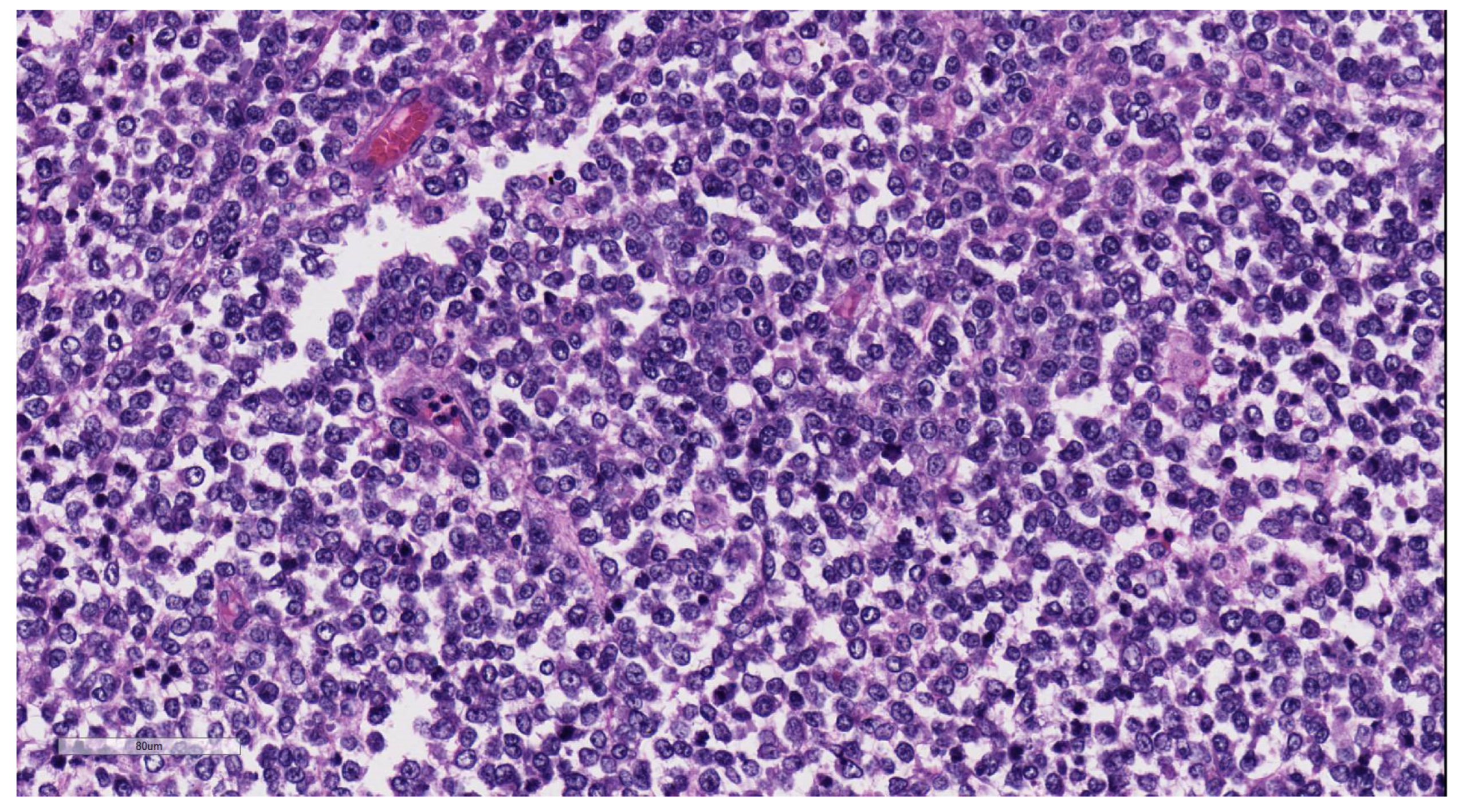
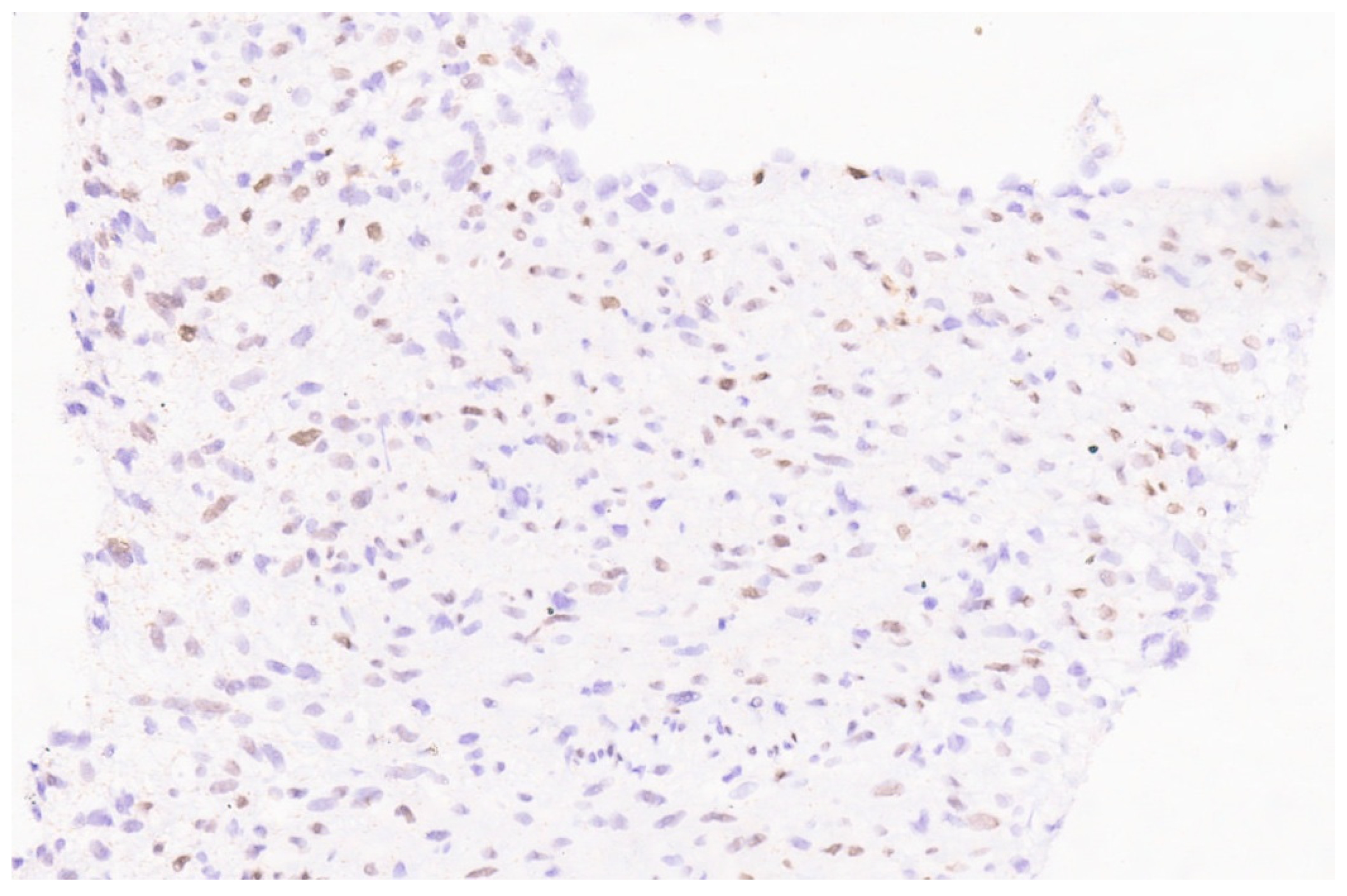
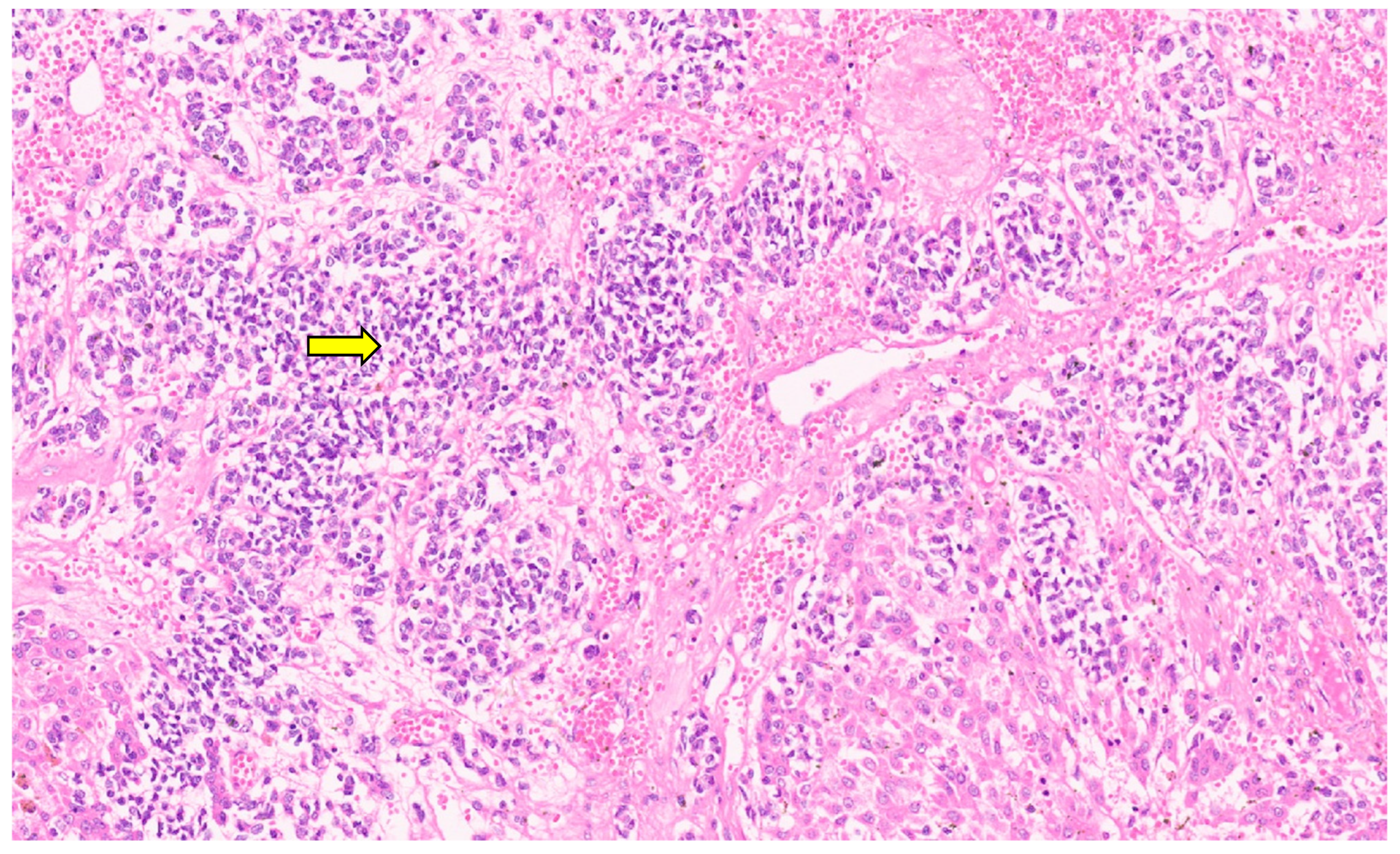
9.3. Pathology
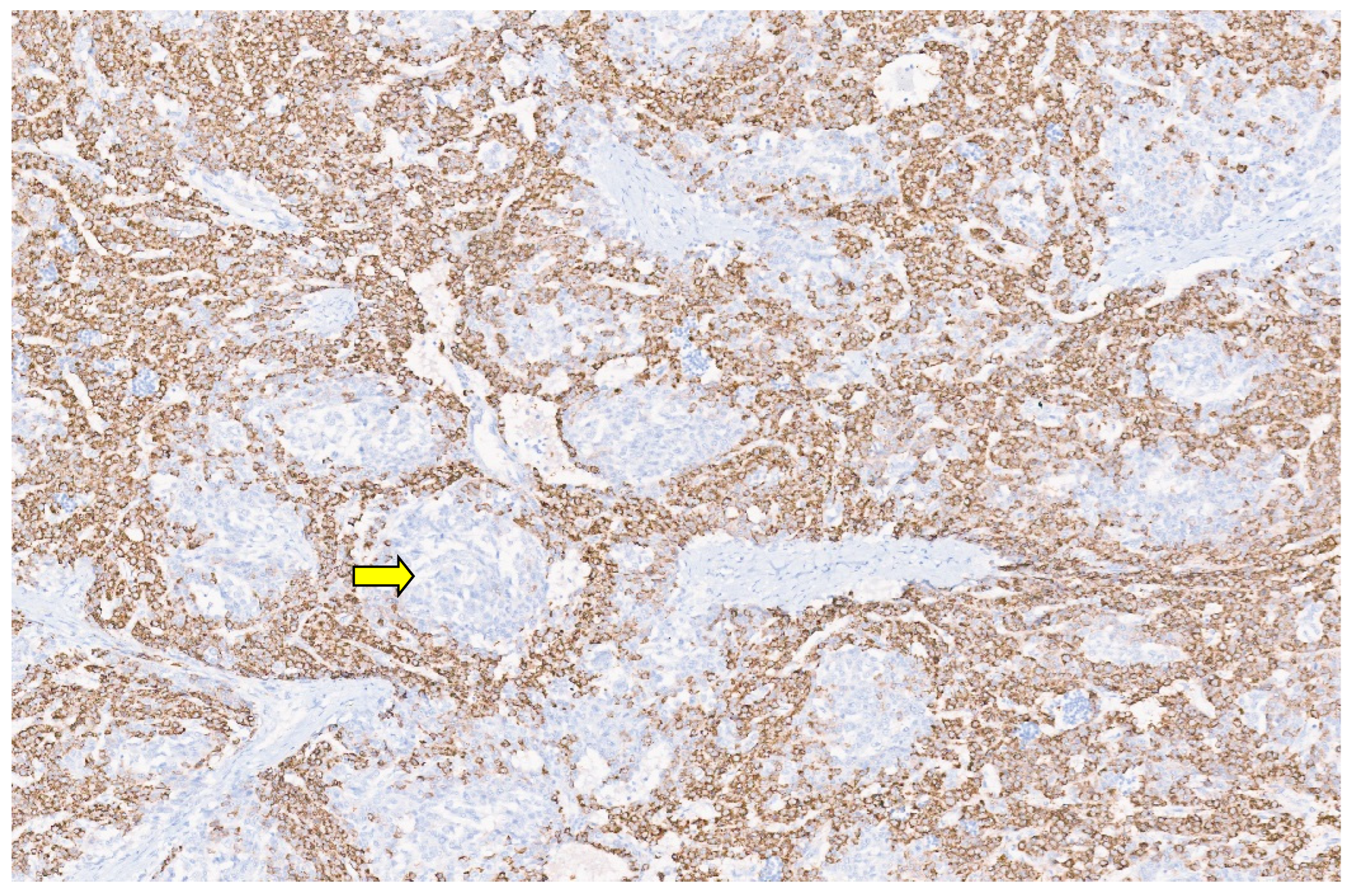
10. Embryonal Sarcoma
10.1. Clinical Presentation
10.2. Imaging Findings
10.3. Pathology
11. Hepatobiliary Rhabdomyosarcoma
11.1. Clinical Presentation
11.2. Imaging Findings
11.3. Pathology
12. Epstein–Barr-Virus-Associated Smooth Muscle Tumor
12.1. Clinical Features
12.2. Imaging
12.3. Pathology
13. Epithelial Tumors
13.1. Hepatocellular Adenoma
13.2. Clinical Presentation
13.3. Imaging Findings
13.4. Pathology
14. Focal Nodular Hyperplasia
14.1. Clinical Presentation
14.2. Imaging Findings
14.3. Pathology
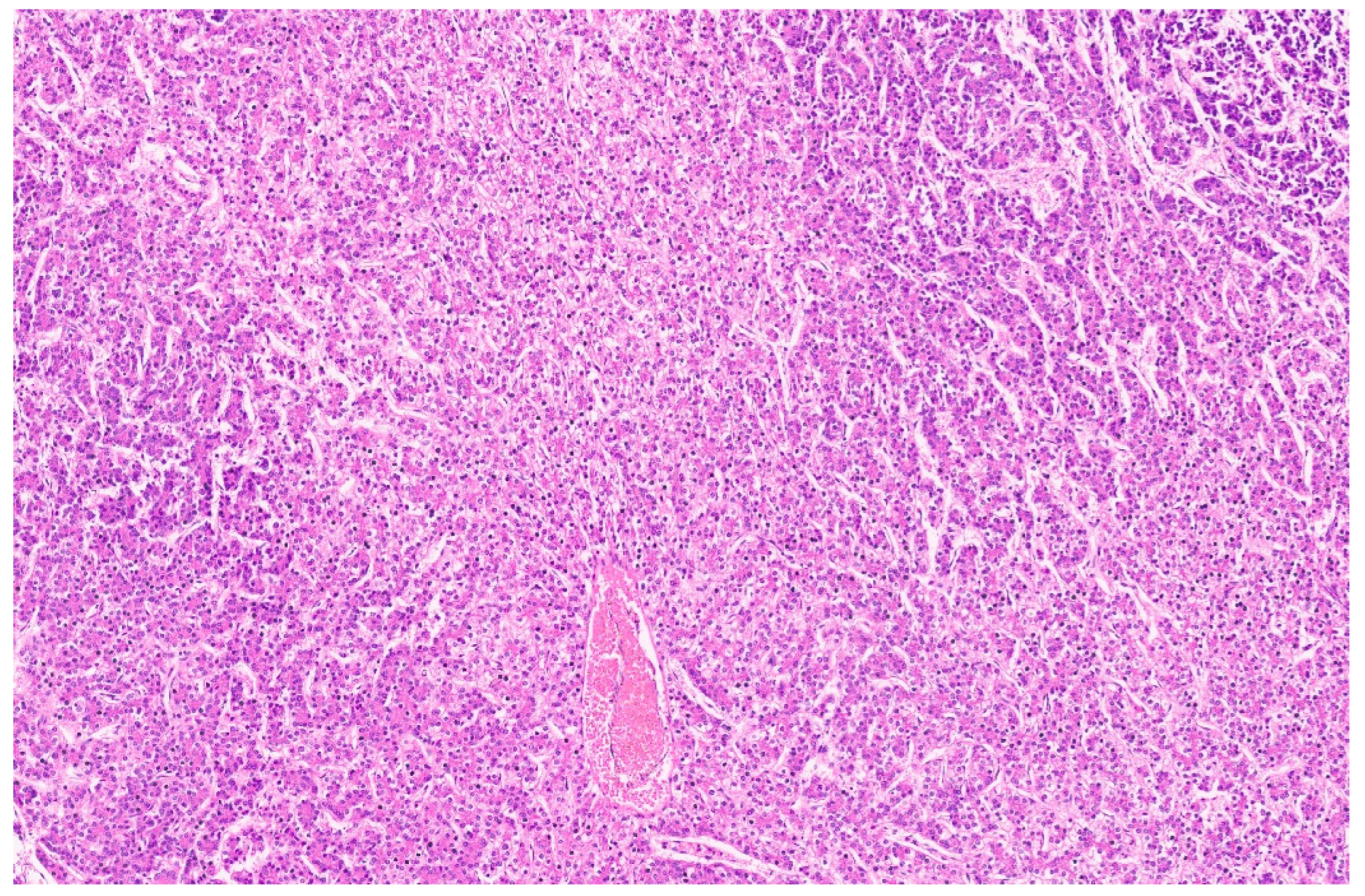
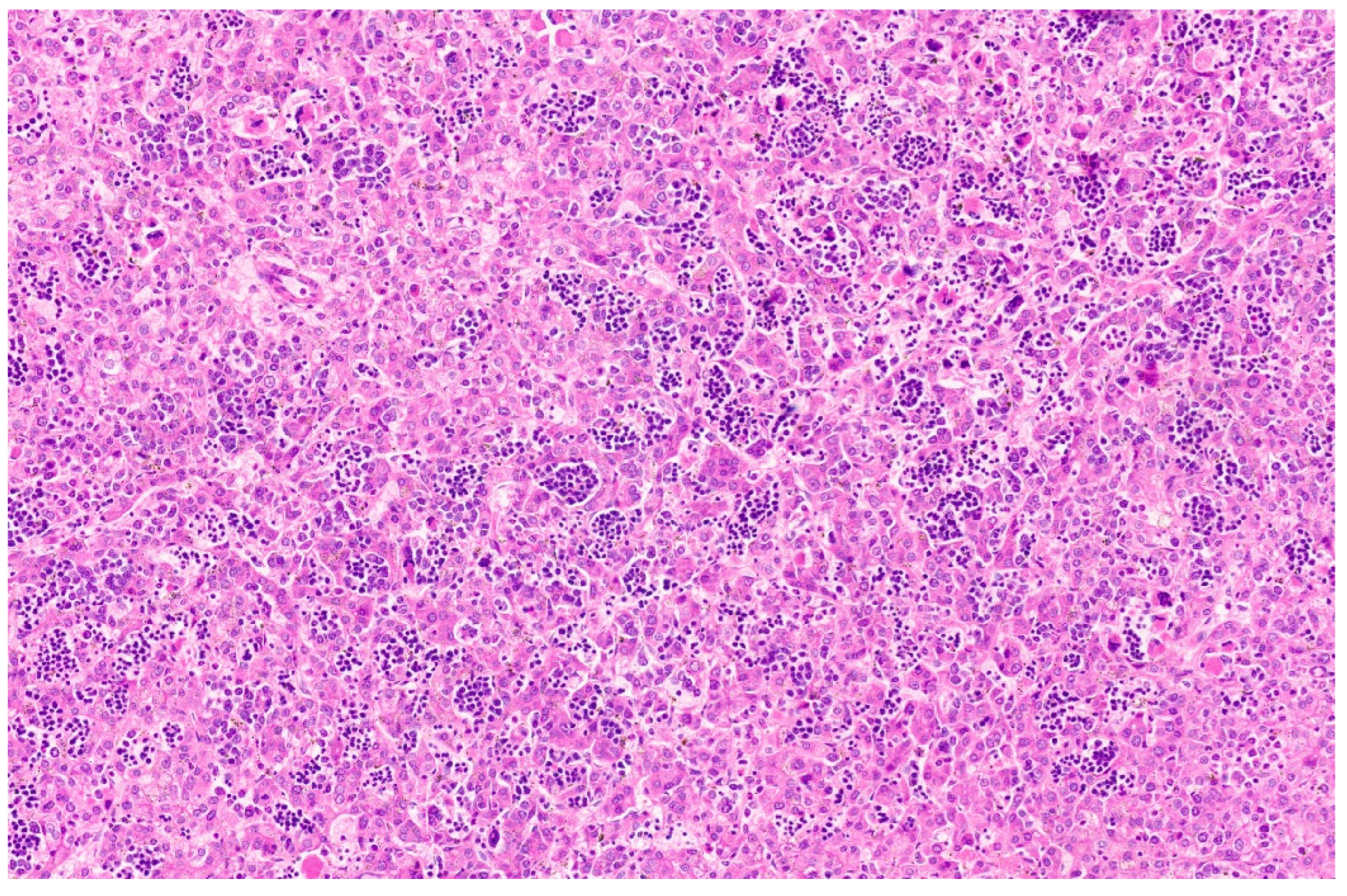
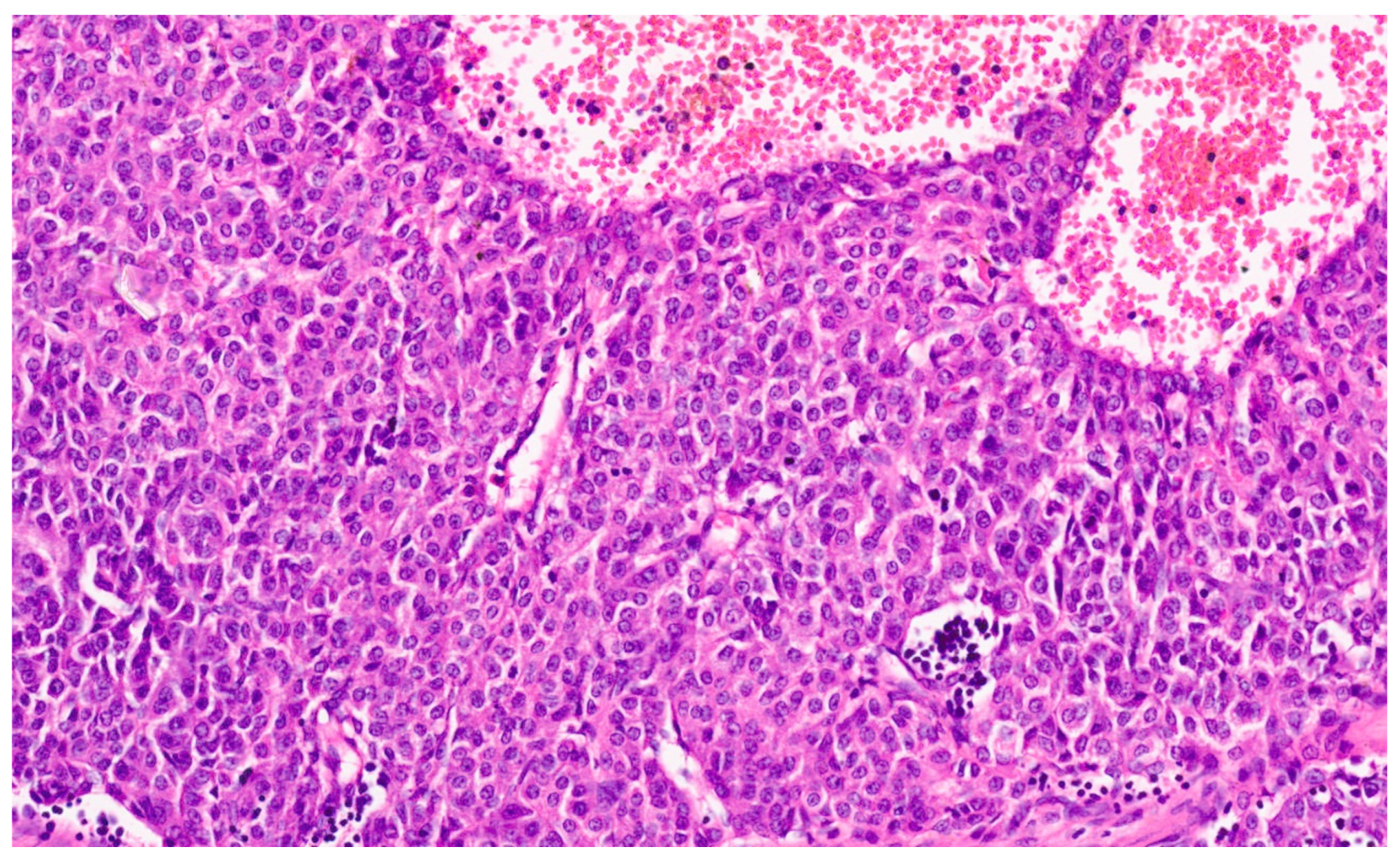
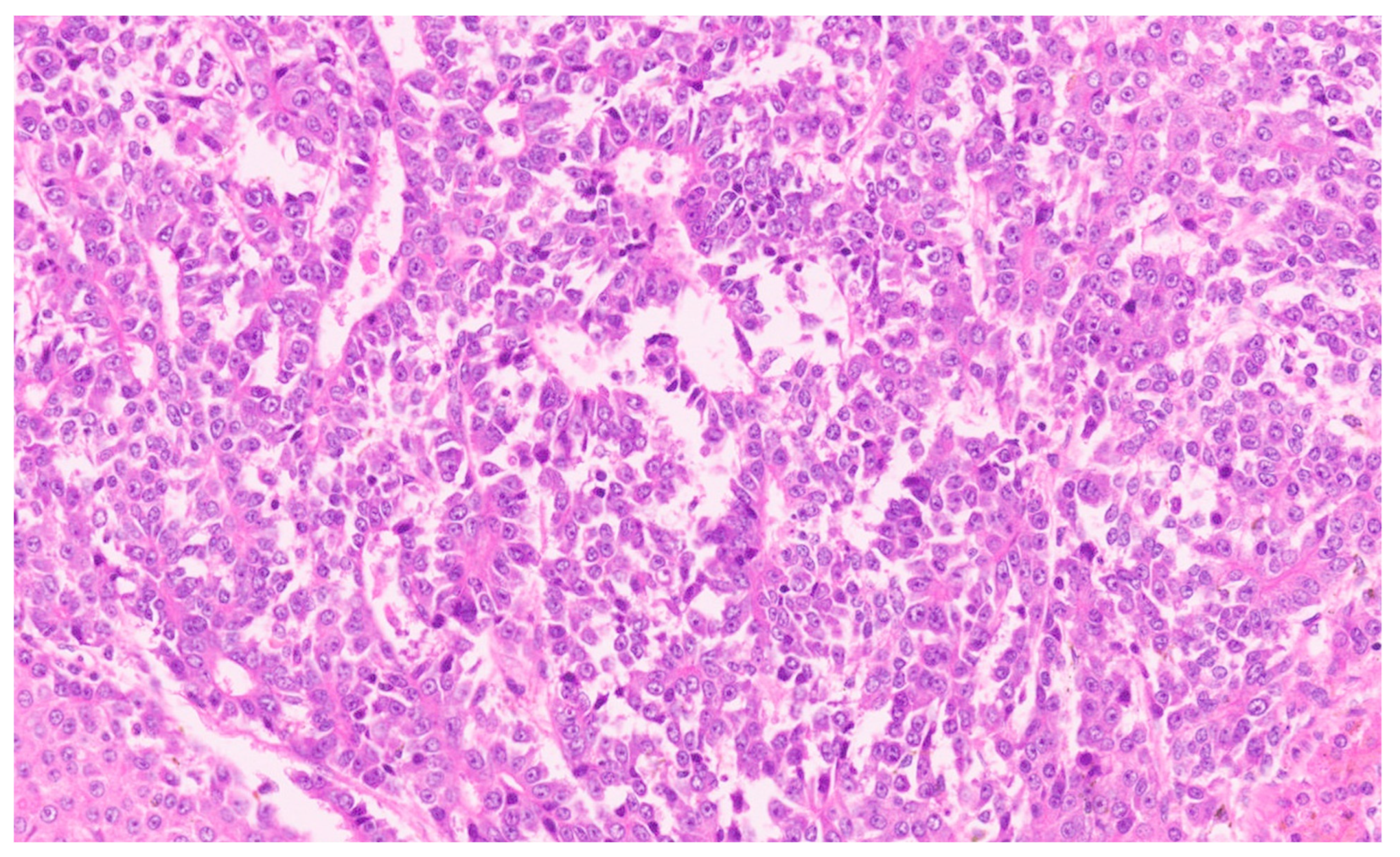
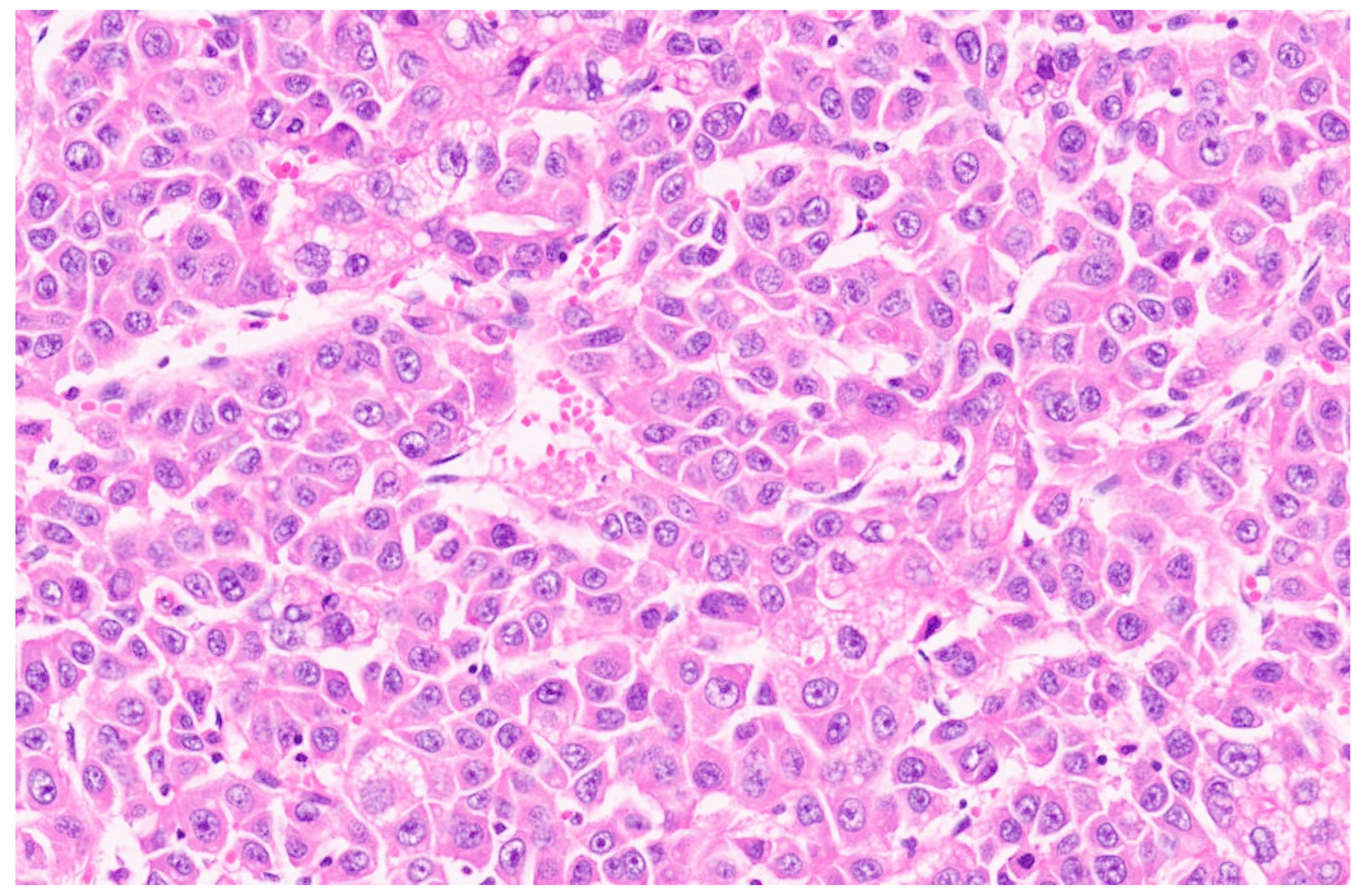
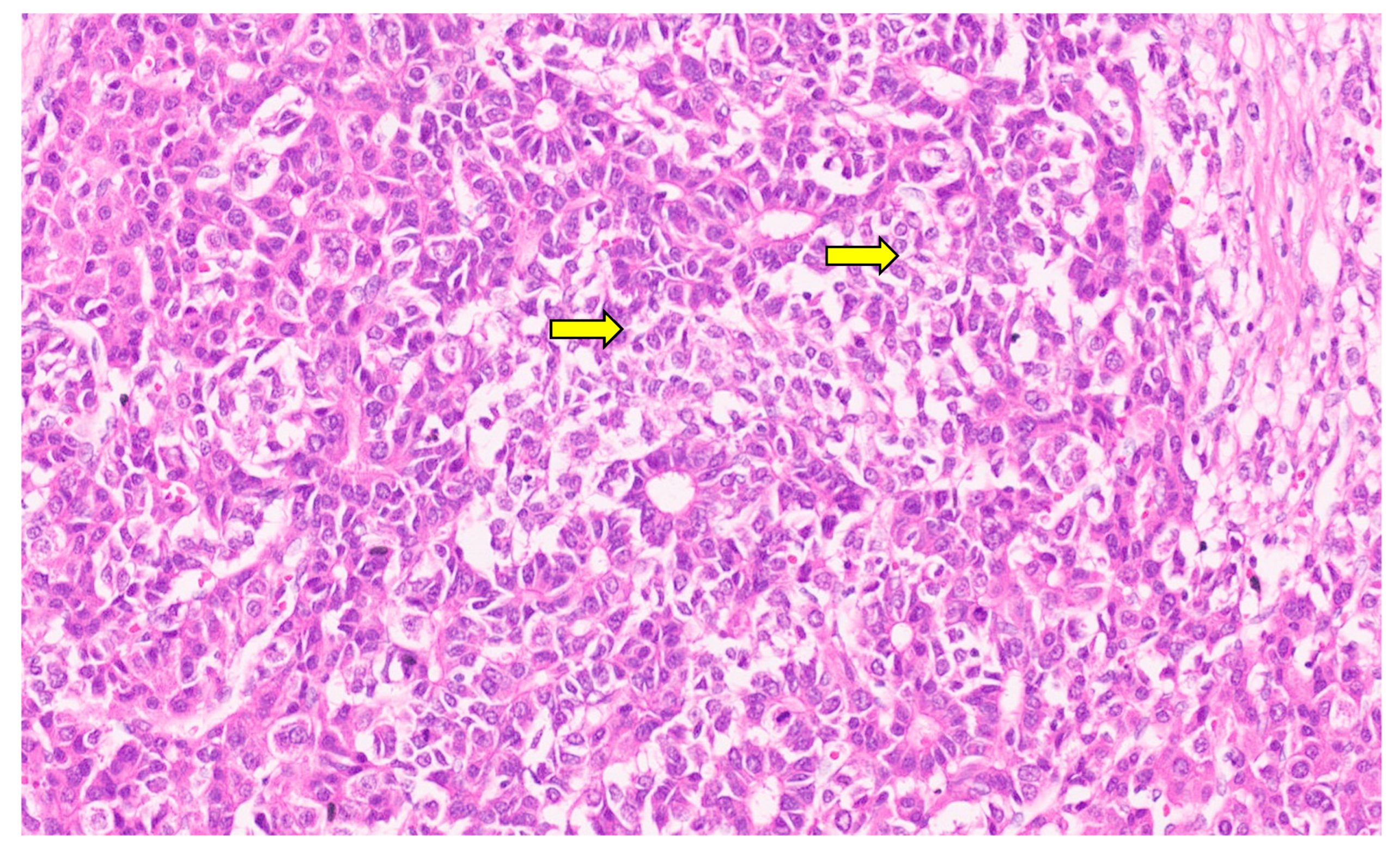
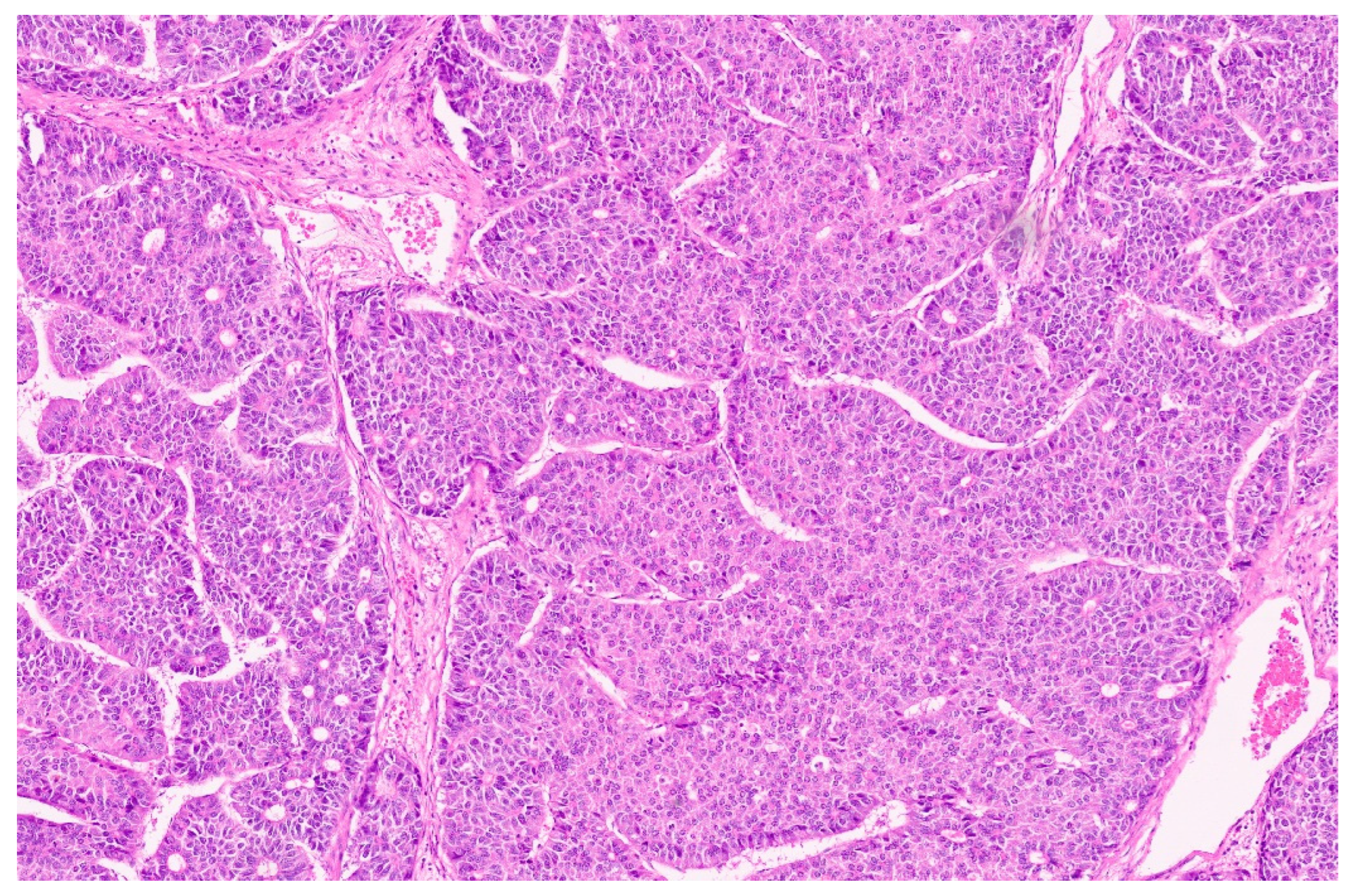
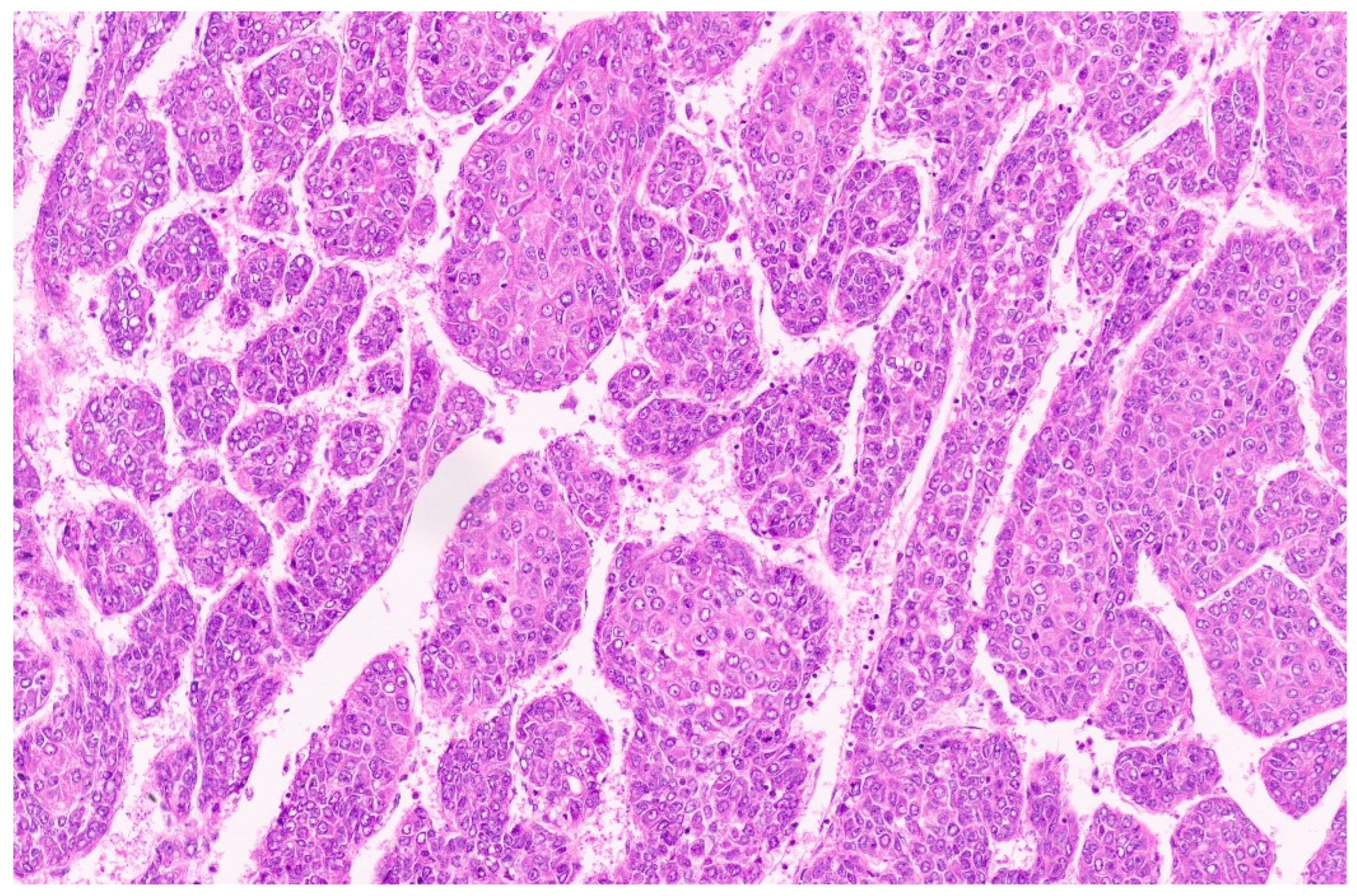
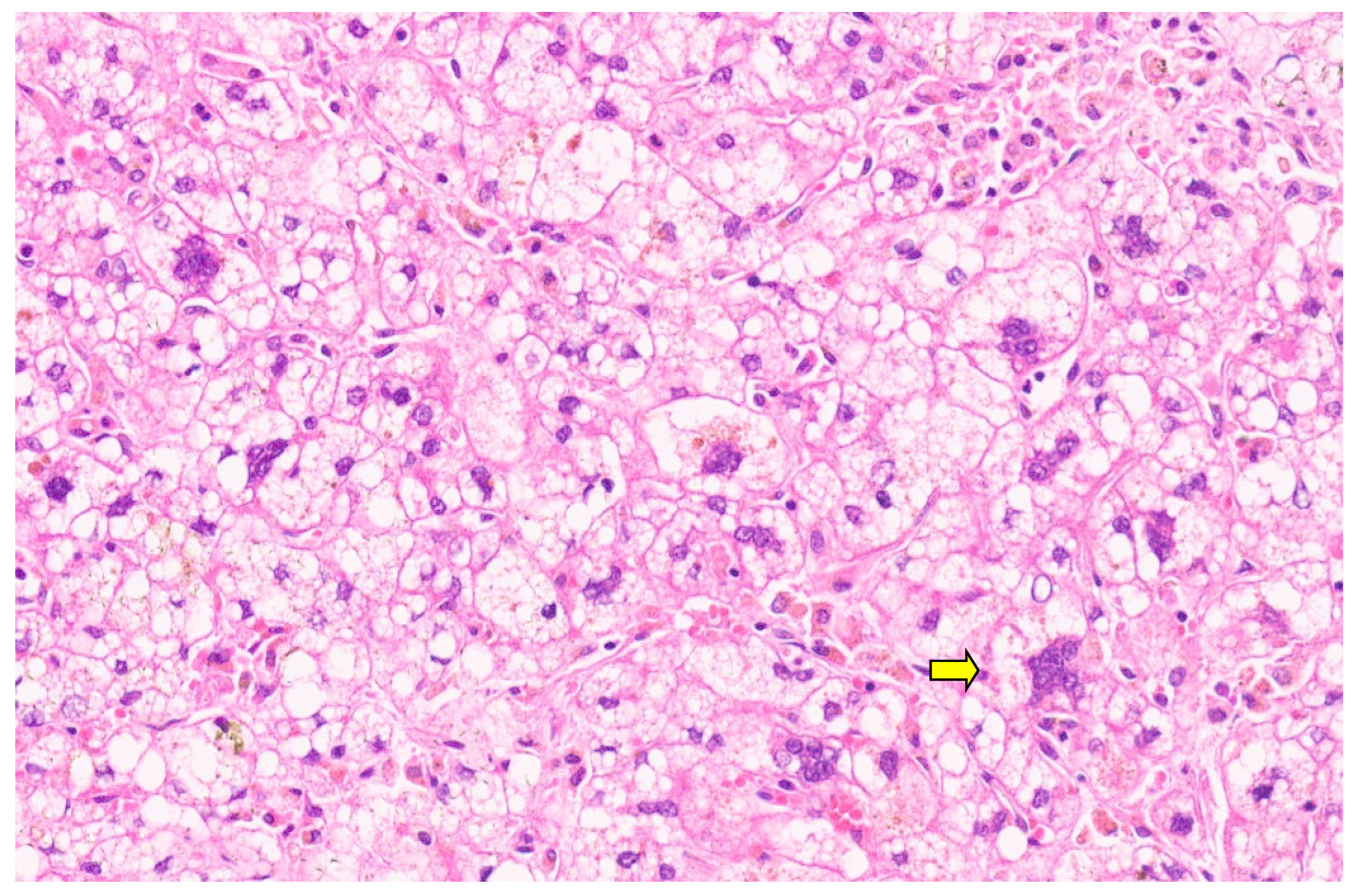
15. Hepatoblastoma
15.1. Clinical Presentation
15.2. Imaging Findings
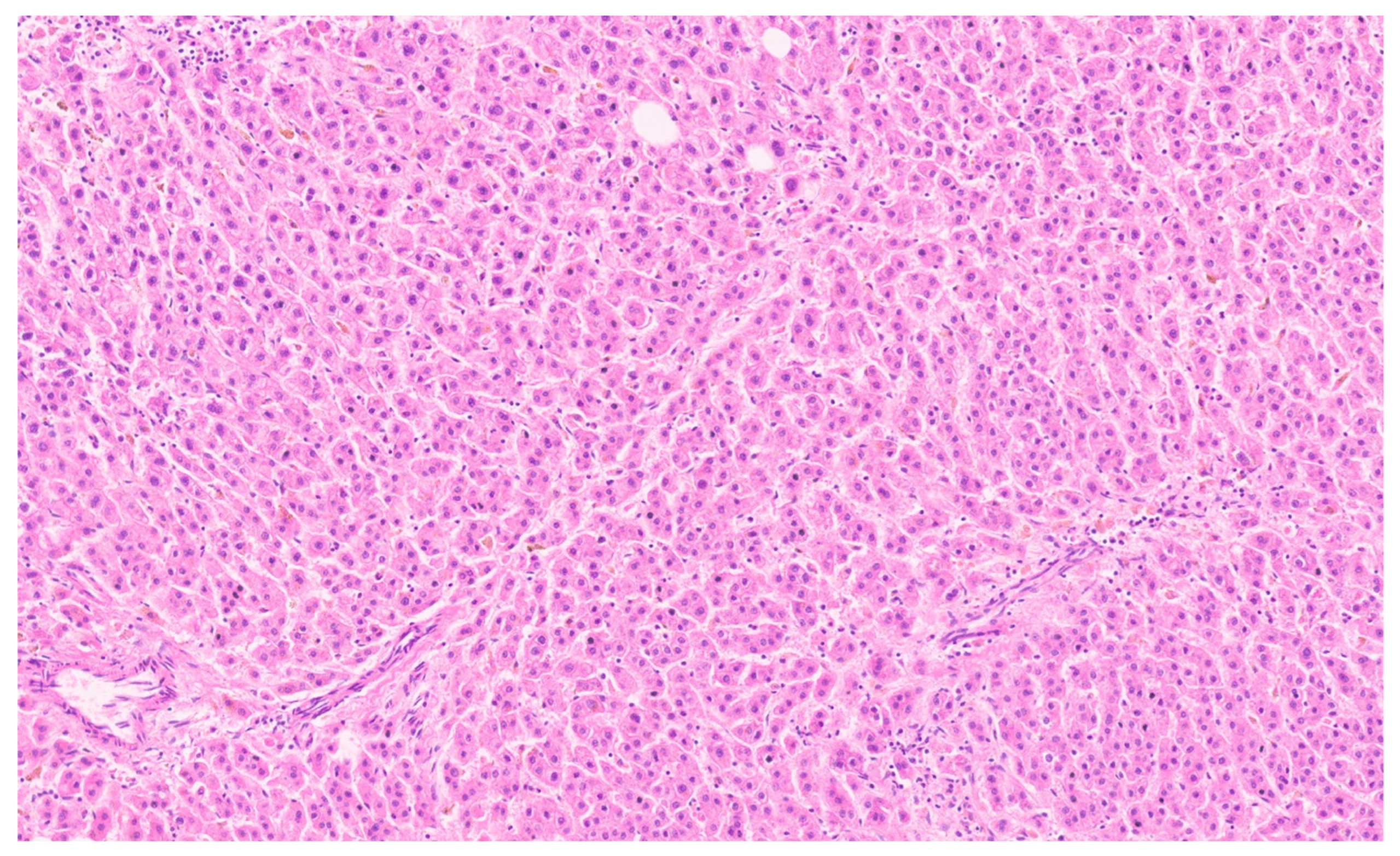
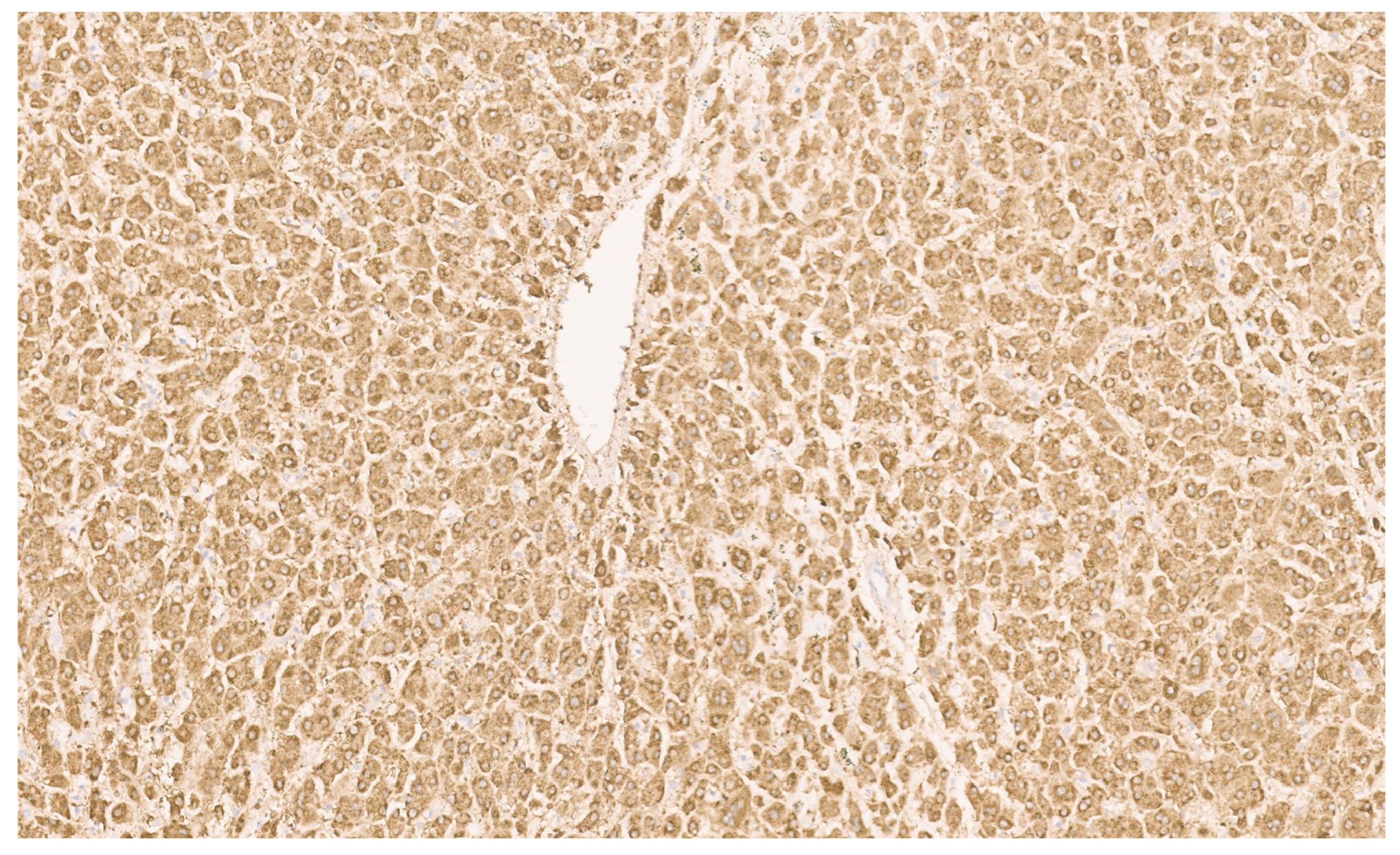
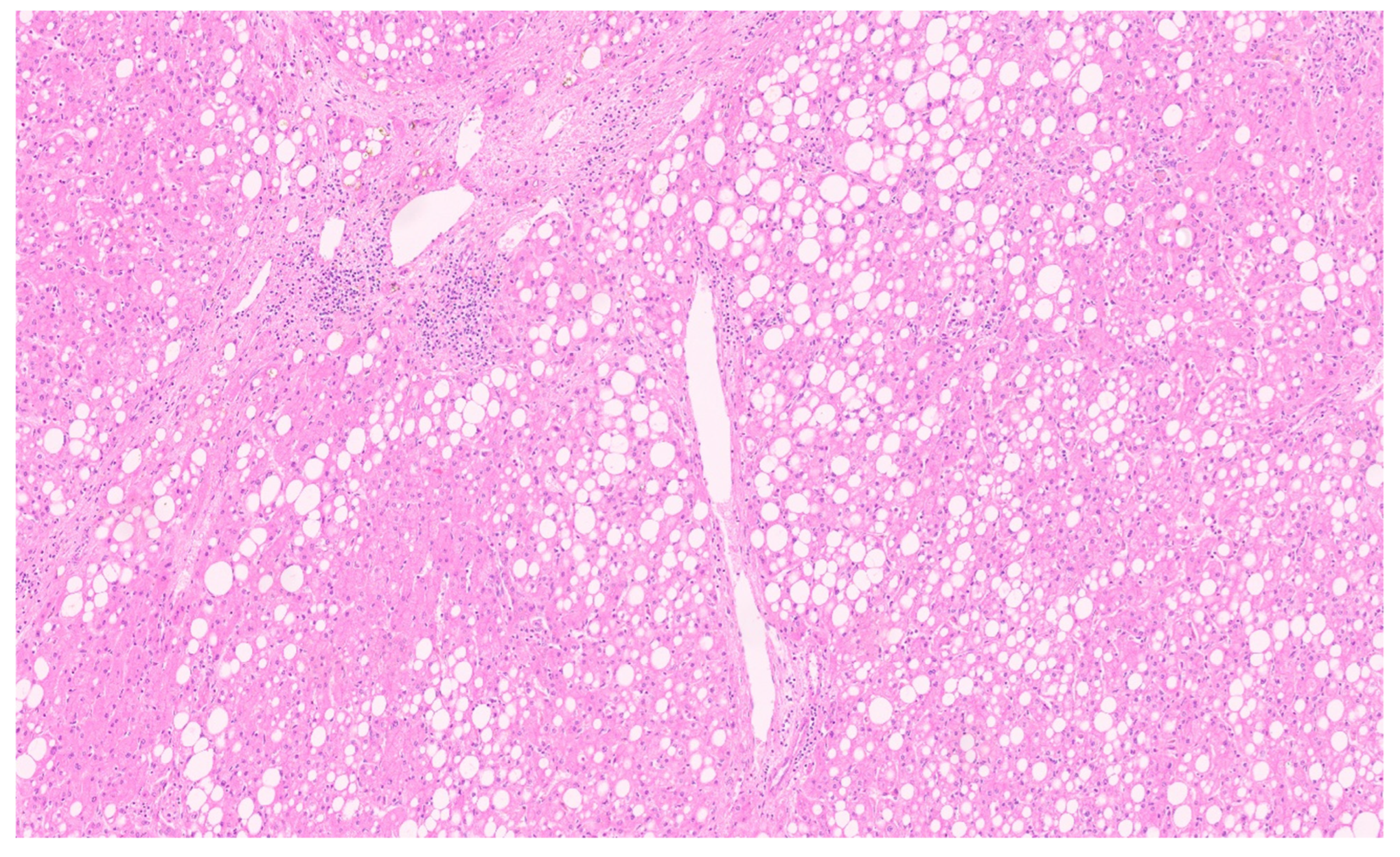
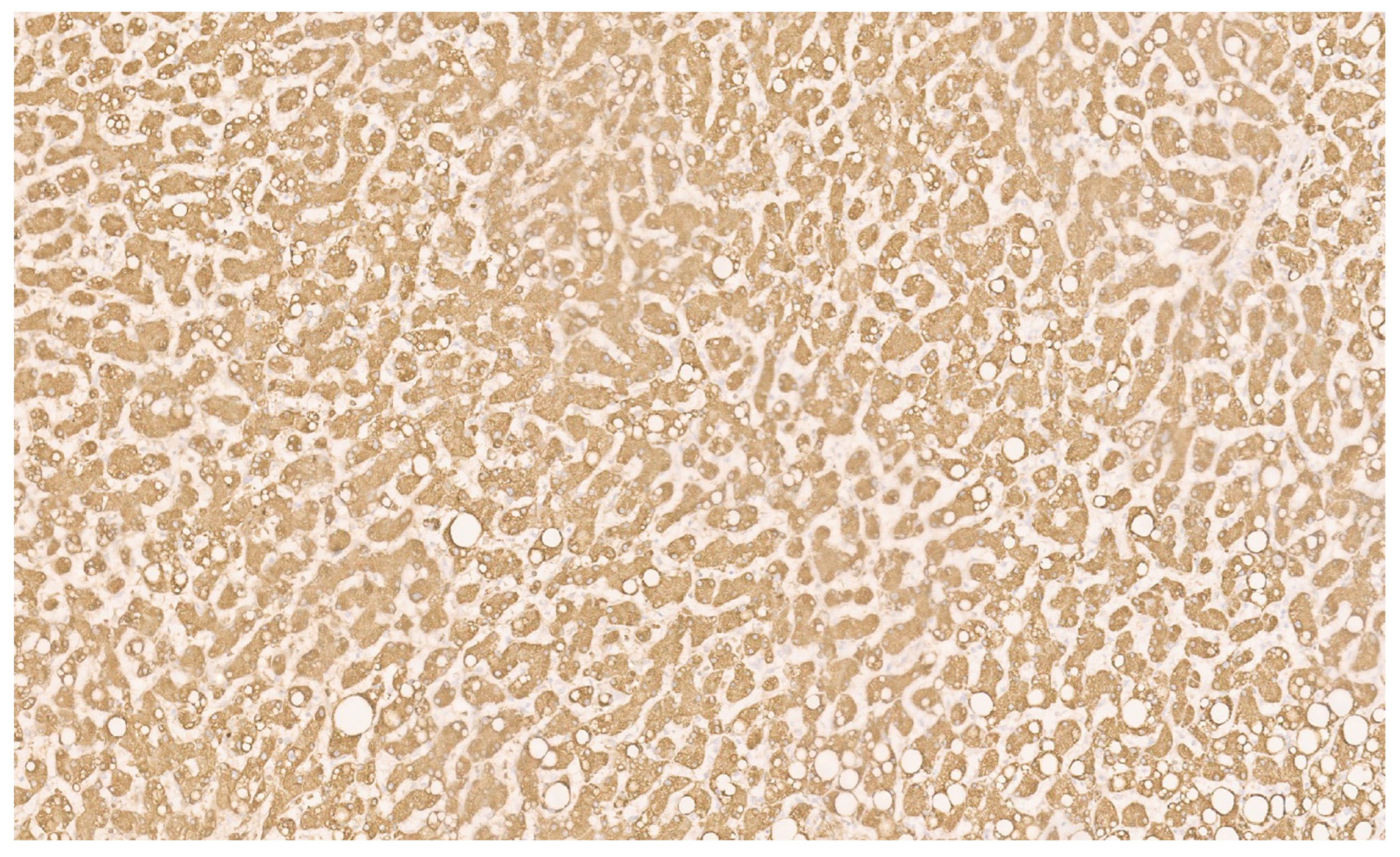
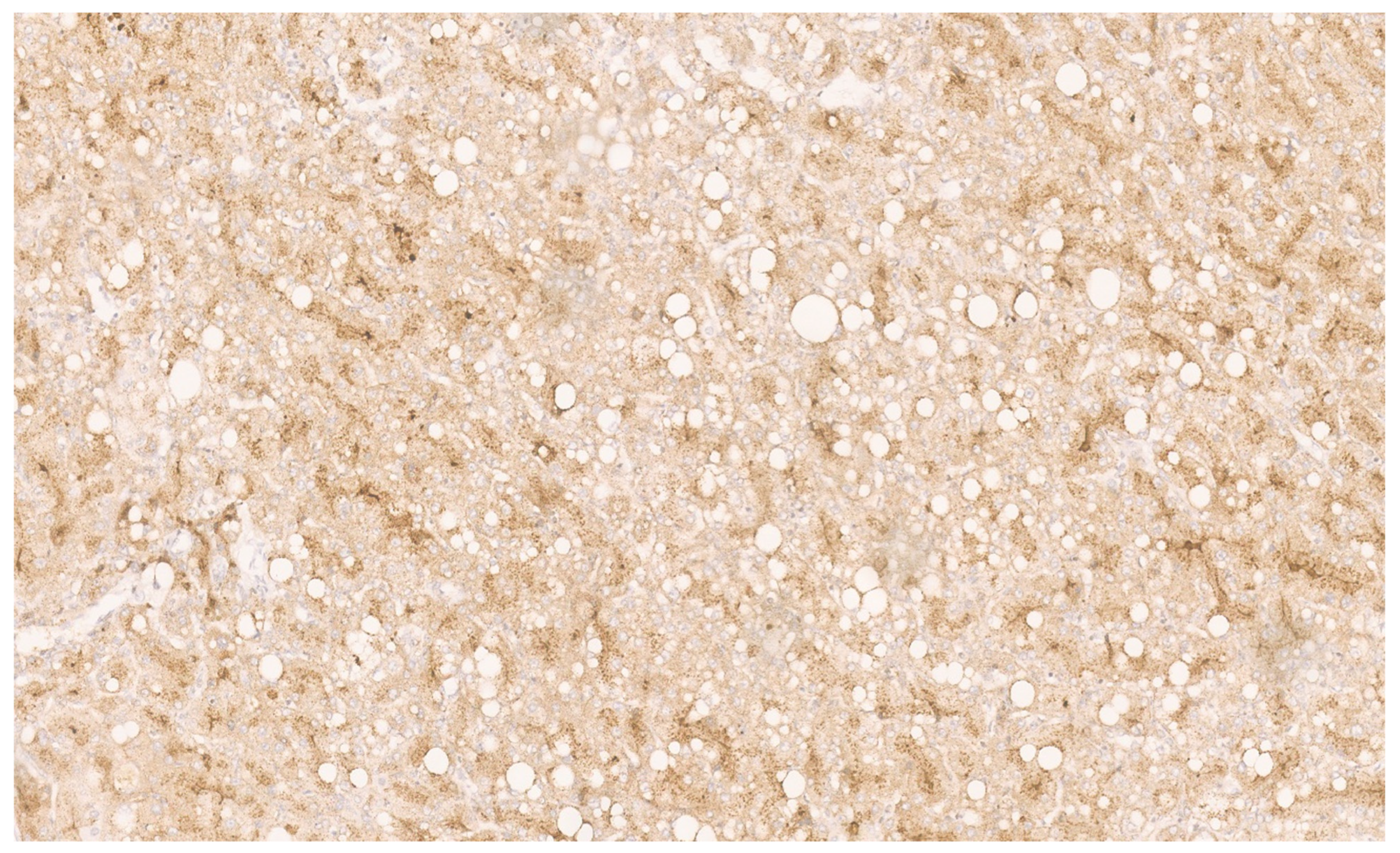
15.3. Pathology
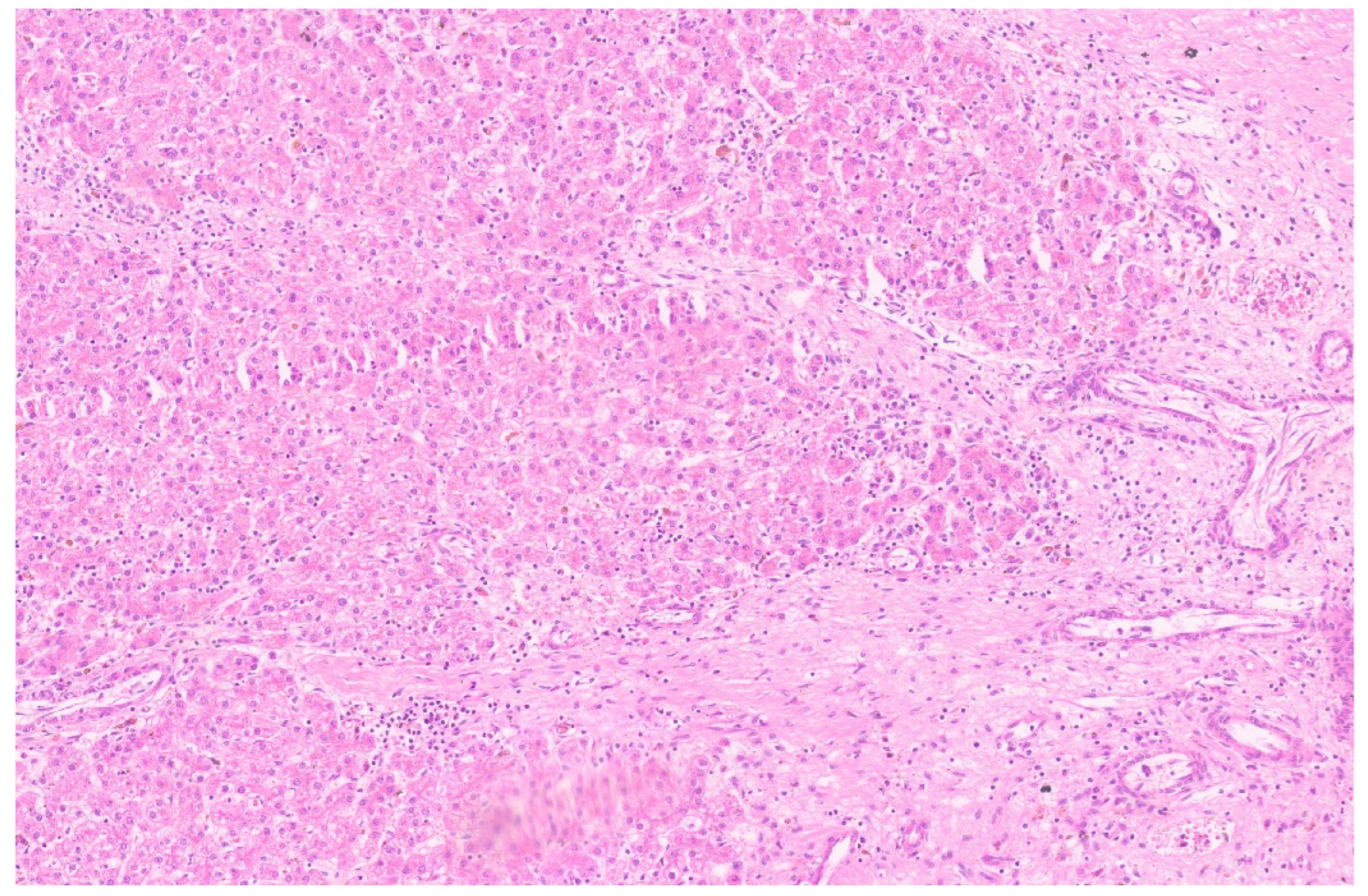
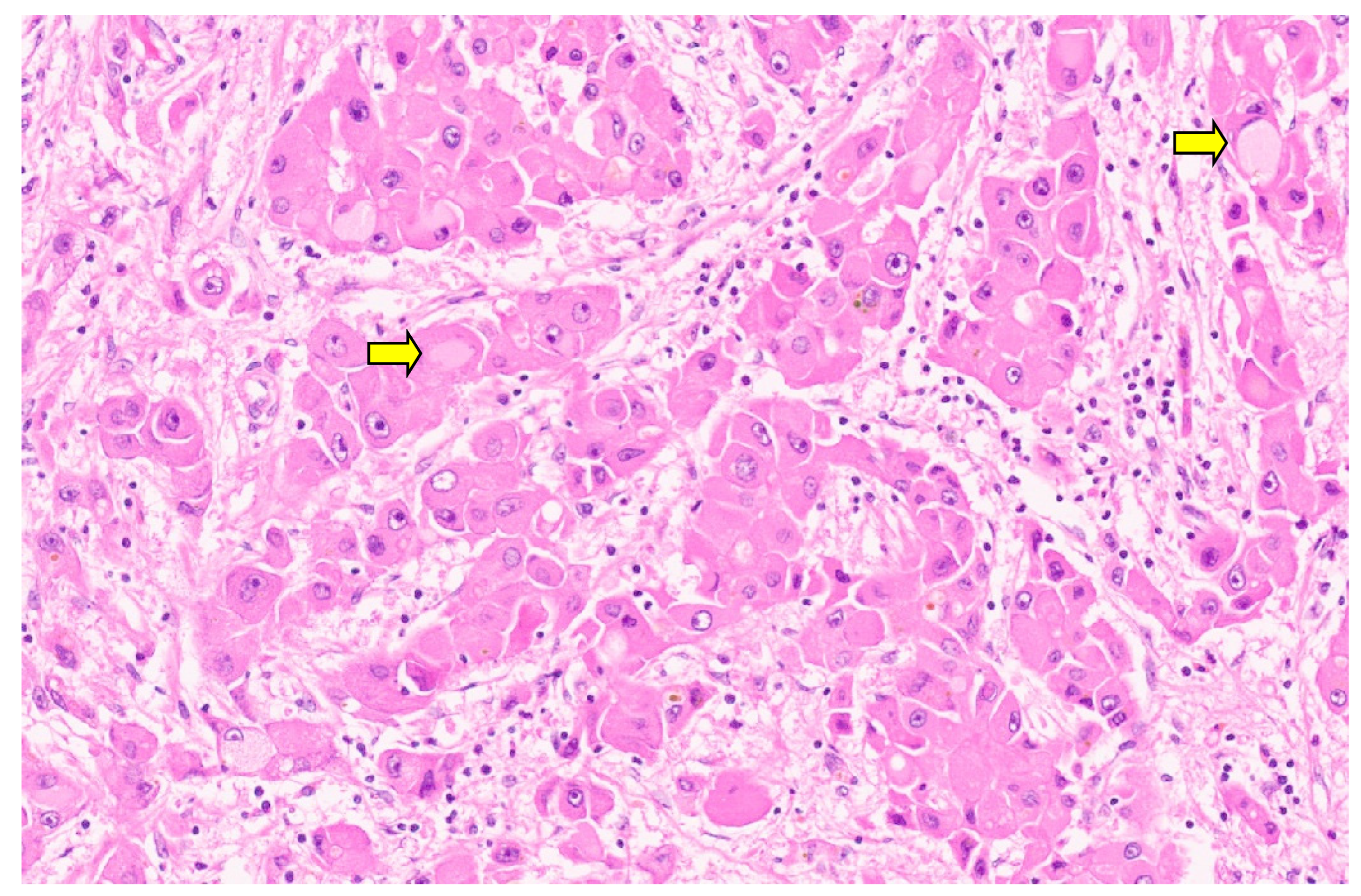
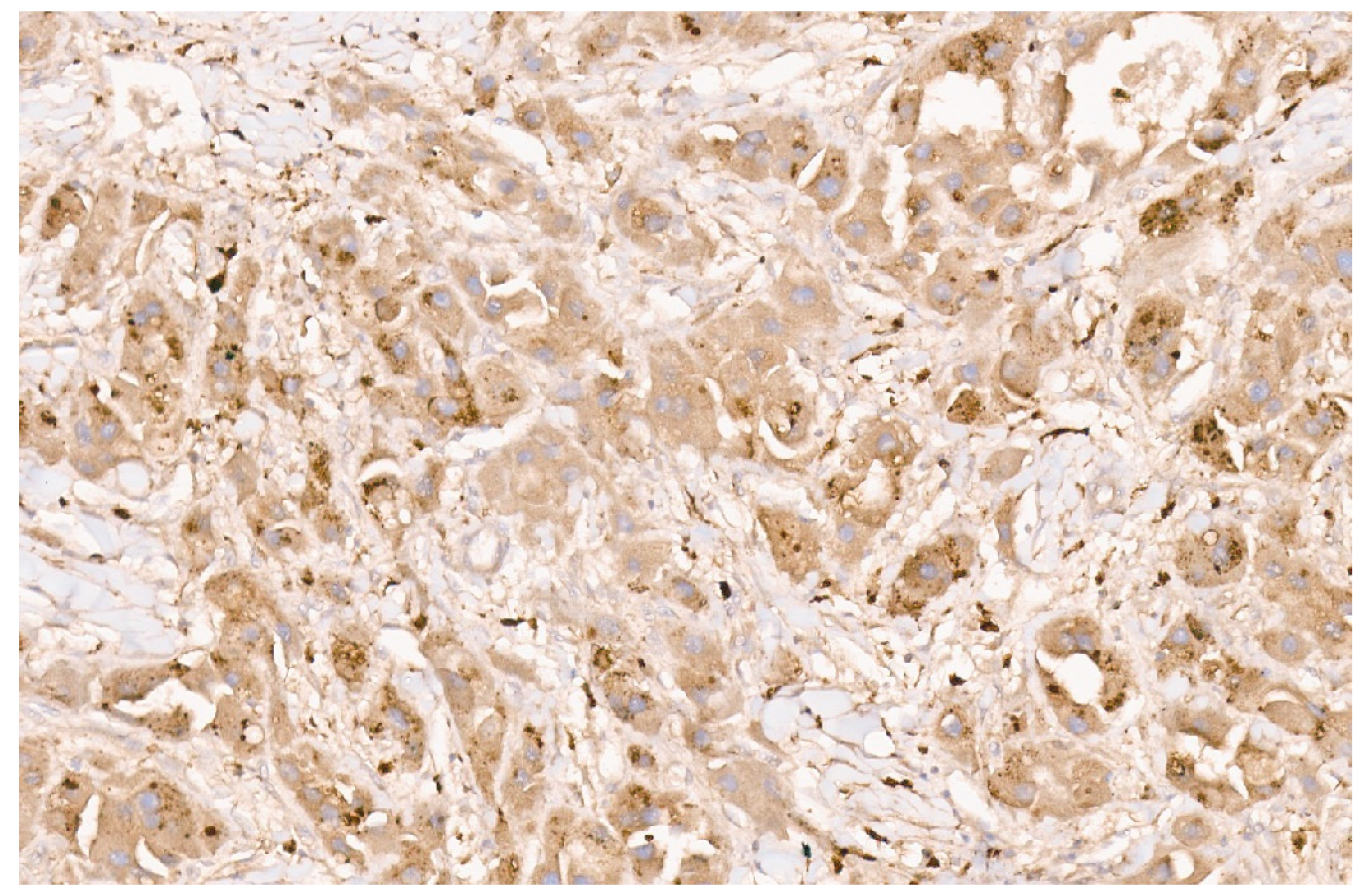
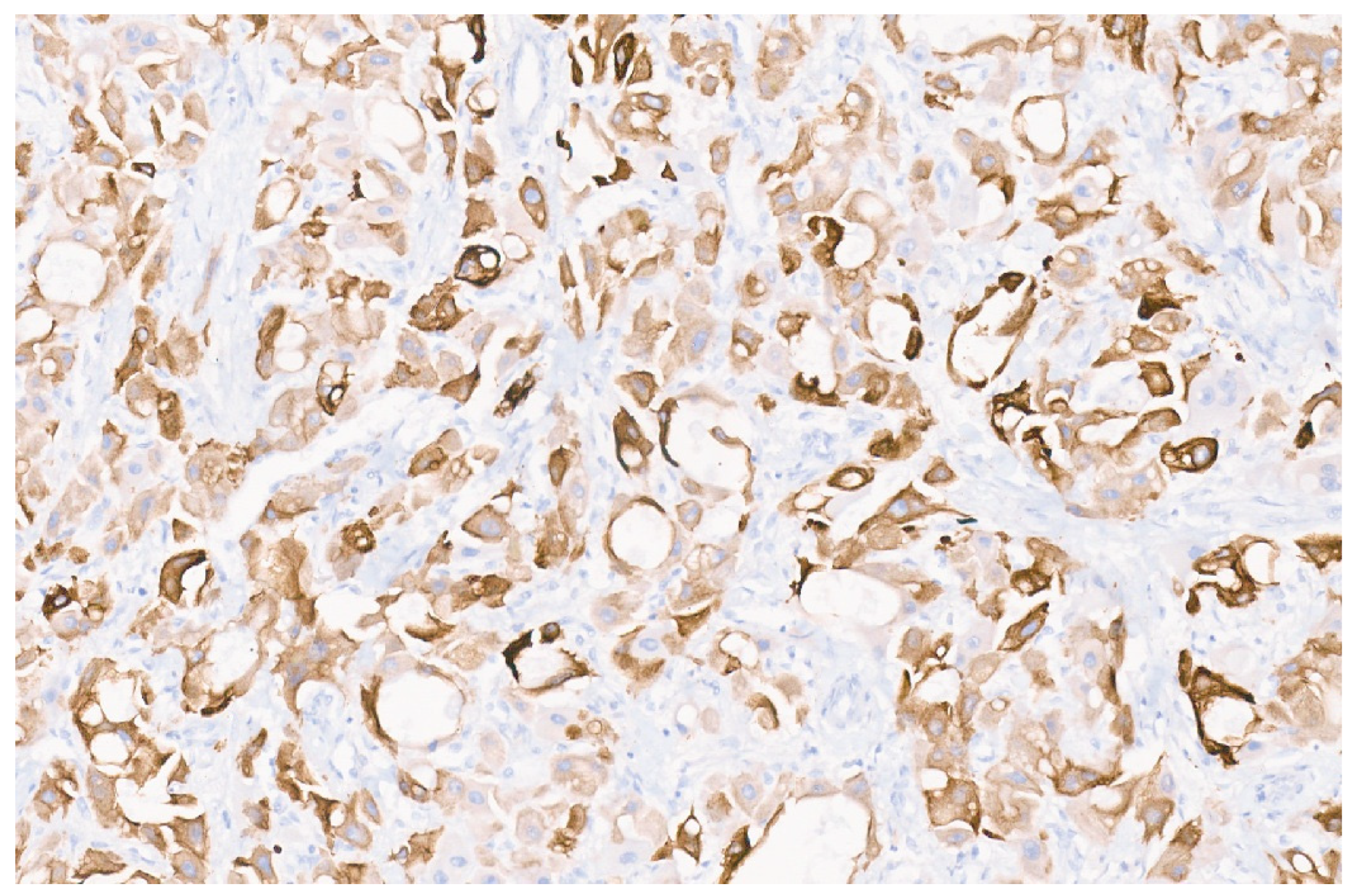
16. Pediatric Hepatocellular Carcinoma
16.1. Clinical Presentation
16.2. Imaging Findings
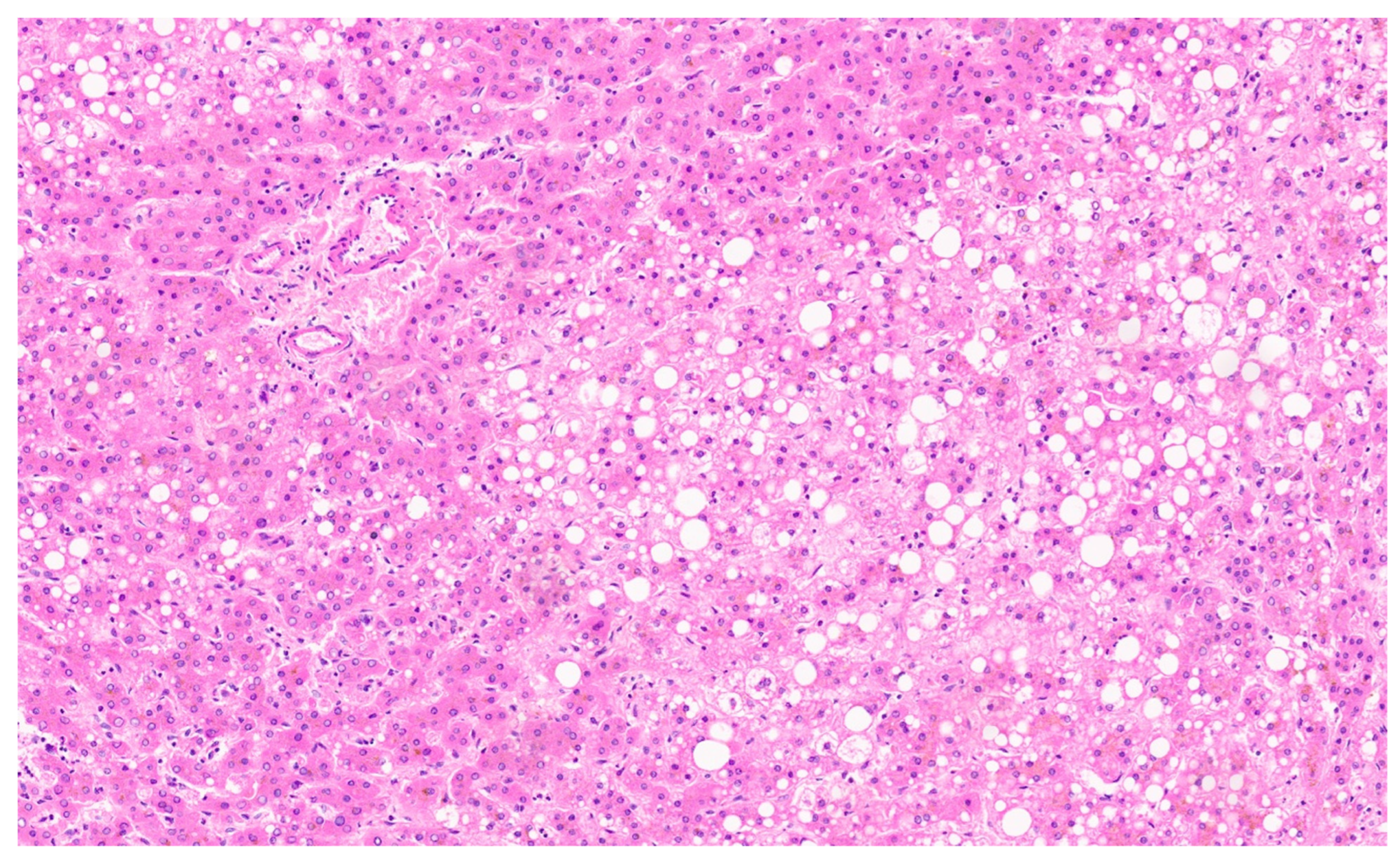
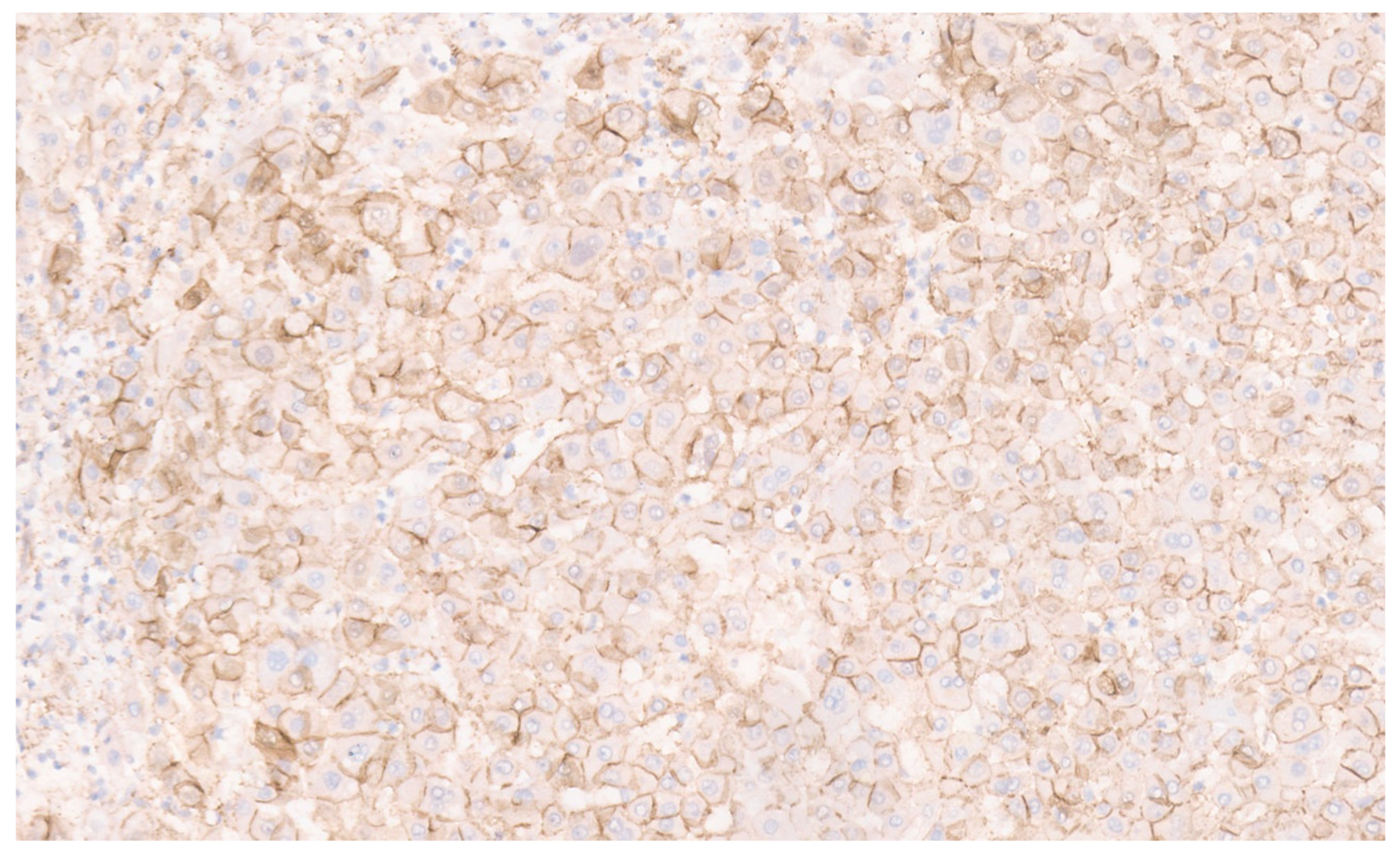
16.3. Pathology
17. Hepatoblastoma with Carcinoma Features
- Biphasic HCN NOS, which shows distinct areas with HB- and HCC-like features;
- Equivocal HCN NOS, with features intermediate between HB and HCC;
- Hepatoblastoma with a focal macrotrabecular pattern, pleomorphism, and anaplasia (HBFPA).
18. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. (Eds.) SEER Cancer Statistics Review, 1975–2017; National Cancer Institute: Bethesda, MD, USA, 2020. Available online: https://seer.cancer.gov/csr/1975_2017/ (accessed on 7 July 2023).
- Ng, K.; Mogul, D.B. Pediatric Liver Tumors. Clin. Liver Dis. 2018, 22, 753–772. [Google Scholar] [CrossRef]
- Cho, S.J. Pediatric Liver Tumors Updates in Classification. Surg. Pathol. 2020, 13, 601–623. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, I.; Phung, T.L.; Adams, D.M.; Trenor, C.C., 3rd; Blei, F.; Fishman, D.S.; Hammill, A.; Masand, P.M.; Fishman, S.J. Guidance Document for Hepatic Hemangioma (Infantile and Congenital) Evaluation and Monitoring. J. Pediatr. 2018, 203, 294–300.e2. [Google Scholar] [CrossRef]
- Gupta, A.; Kozakewich, H.P. Congenital Hepatic Hemangioma. In WHO Classification of Tumours Editorial Board, Pediatric Tumours, 5th ed.; WHO Classification of Tumours Series; International Agency for Research on Cancer: Lyon, France, 2022; Volume 7, Available online: https://tumourclassification.iarc.who.int/chapters/44 (accessed on 7 July 2023).
- Cordier, F.; Hoorens, A.; Van Dorpe, J.; Creytens, D. Pediatric vascular tumors of the liver: Review from the pathologist’s point of view. World J. Hepatol. 2021, 13, 1316–1327. [Google Scholar] [CrossRef] [PubMed]
- Dickie, B.; Dasgupta, R.; Nair, R.; Alonso, M.H.; Ryckman, F.C.; Tiao, G.M.; Adams, D.M.; Azizkhan, R.G. Spectrum of hepatic hemangiomas: Management and outcome. J. Pediatr. Surg. 2009, 44, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Berklite, L.; Malik, F.; Ranganathan, S.; Gupta, A. Pediatric hepatic vascular tumors: Clinicopathologic characteristics of 33 cases and proposed updates tocurrent classification schemes. Hum. Pathol. 2023, in press. [CrossRef]
- El Zein, S.; Boccara, O.; Soupre, V.; Vieira, A.F.; Bodemer, C.; Coulomb, A.; Wassef, M.; Fraitag, S. The histopathology of congenital hemangioma and its clinical correlations: A long-term follow-up study of 55 cases. Histopathology 2020, 77, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Trindade, F.; Tellechea, O.; Torrelo, A.; Requena, L.; Colmenero, I. Wilms tumor 1 expression in vascular neoplasms and vascular malformations. Am. J. Dermatopathol. 2011, 33, 569–572. [Google Scholar] [CrossRef]
- Triana, P.; Rodríguez-Laguna, L.; Giacaman, A.; Salinas-Sanz, J.A.; Martín-Santiago, A.; López-Santamaría, M.; Palacios, E.; Beato, M.J.; Martinez-González, V.; López-Gutierrez, J.C. Congenital hepatic hemangiomas: Clinical, histologic, and geneticcorrelation. J. Pediatr. Surg. 2020, 55, 2170–2176. [Google Scholar] [CrossRef]
- Zavras, N.; Dimopoulou, A.; Machairas, N.; Paspala, A.; Vaos, G. Infantile hepatic hemangioma: Current state of the art, controversies, and perspectives. Eur. J. Pediatr. 2020, 179, 1–8. [Google Scholar] [CrossRef]
- Gupta, A.; Kozakewich, H.P. Infantile Hepatic Hemangioma. In WHO Classification of Tumours Editorial Board. Pediatric Tumours, 5th ed.; WHO Classification of Tumours Series; International Agency for Research on Cancer: Lyon, France, 2022; Volume 7, Available online: https://tumourclassification.iarc.who.int/chapters/44 (accessed on 7 July 2023).
- Kulungowski, A.M.; Alomari, A.I.; Chawla, A.; Christison-Lagay, E.R.; Fishman, S.J. Lessons from a liver hemangioma registry: Subtype classification. J. Pediatr. Surg. 2012, 47, 165–170. [Google Scholar] [CrossRef]
- Siano, M.A.; Ametrano, O.; Barbato, F.; Sammarco, E.; Ranucci, G.; Pietrobattista, A.; Rossomando, A.; Mandato, C. Consumptive Hypothyroidism due to Hepatic Hemangiomas: A Case Series and Review of the Literature. JPGN Rep. 2022, 3, e270. [Google Scholar] [CrossRef]
- Smith, C.J.F.; Friedlander, S.F.; Guma, M.; Kavanaugh, A.; Chambers, C.D. Infantile Hemangiomas: An Updated Review on Risk Factors, Pathogenesis, and Treatment. Birth Defects Res. 2017, 109, 809–815. [Google Scholar] [CrossRef]
- Cournoyer, E.; Al-Ibraheemi, A.; Engel, E.; Chaudry, G.; Stapleton, S.; Adams, D.M. Clinical characterization and long-term outcomes in pediatric epithelioidhemangioendothelioma. Pediatr. Blood Cancer 2020, 67, e28045. [Google Scholar] [CrossRef]
- Bannoura, S.; Putra, J. Primary malignant vascular tumors of the liver in children: Angiosarcoma and epithelioid hemangioendothelioma. World J. Gastrointest. Oncol. 2021, 13, 223–230. [Google Scholar] [CrossRef]
- Hettmer, S.; Andrieux, G.; Hochrein, J.; Kurz, P.; Rössler, J.; Lassmann, S.; Werner, M.; von Bubnoff, N.; Peters, C.; Koscielniak, E.; et al. Epithelioid hemangioendotheliomas of the liver and lung inchildren and adolescents. Pediatr. Blood Cancer 2017, 64, e26675. [Google Scholar] [CrossRef]
- Chavhan, G.B.; Siddiqui, I.; Ingley, K.M.; Gupta, A.A. Rare malignant liver tumors in children. Pediatr. Radiol. 2019, 49, 1404–1421. [Google Scholar] [CrossRef]
- Lee, H.E.; Torbenson, M.S.; Wu, T.T.; Chandan, V.S. Aberrant keratin expression is common in primary hepatic malignant vascular tumors: A potential diagnosticpitfall. Ann Diagn Pathol. 2020, 49, 151589. [Google Scholar] [CrossRef] [PubMed]
- Mendlick, M.R.; Nelson, M.; Pickering, D.; Johansson, S.L.; Seemayer, T.A.; Neff, J.R.; Vergara, G.; Rosenthal, H.; Bridge, J.A. Translocation t(1;3)(p36.3;q25) is a nonrandom aberration in epithelioid hemangioendothelioma. Am. J. Surg. Pathol. 2001, 25, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Tanas, M.R.; Sboner, A.; Oliveira, A.M.; Erickson-Johnson, M.R.; Hespelt, J.; Hanwright, P.J.; Flanagan, J.; Luo, Y.; Fenwick, K.; Natrajan, R.; et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci. Transl. Med. 2011, 3, 98ra82. [Google Scholar] [CrossRef] [PubMed]
- Flucke, U.; Vogels, R.J.C.; de Saint Aubain Somerhausen, N.; Creytens, D.H.; Riedl, R.G.; van Gorp, J.M.; Milne, A.N.; Huysentruyt, C.J.; Verdijk, M.A.; van Asseldonk, M.M.; et al. Epithelioid Hemangioendothelioma: Clinicopathologic, immunohistochemical, and molecular genetic analysis of 39 cases. Diagn. Pathol. 2014, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C.; Le Loarer, F.; Mosquera, J.M.; Sboner, A.; Zhang, L.; Chen, C.L.; Chen, H.W.; Pathan, N.; Krausz, T.; Dickson, B.C.; et al. Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer 2013, 52, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, R.; Matsuyama, A.; Shiba, E.; Harada, H.; Yabuki, K.; Hisaoka, M. CAMTA1 is a useful immunohistochemical marker for diagnosing epithelioid haemangioendothelioma. Histopathology. 2015, 67, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, E.; Jadeja, B.; Xu, B.; Zhang, L.; Agaram, N.P.; Travis, W.; Singer, S.; Tap, W.D.; Antonescu, C.R. Prognostic stratification of clinical and molecular epithelioid hemangioendothelioma subsets. Mod. Pathol. 2020, 33, 591–602. [Google Scholar] [CrossRef]
- Dermawan, J.K.; Azzato, E.M.; Billings, S.D.; Fritchie, K.J.; Aubert, S.; Bahrami, A.; Barisella, M.; Baumhoer, D.; Blum, V.; Bode, B.; et al. YAP1-TFE3-fused hemangioendothelioma: A multi-institutionalclinicopathologic study of 24 genetically-confirmed cases. Mod. Pathol. 2021, 34, 2211–2221. [Google Scholar] [CrossRef]
- Shibayama, T.; Makise, N.; Motoi, T.; Mori, T.; Hiraoka, N.; Yonemori, K.; Watanabe, S.I.; Esaki, M.; Morizane, C.; Okuma, T.; et al. Clinicopathologic Characterization of Epithelioid Hemangioendothelioma in a Series of 62 Cases: A Proposal of Risk Stratification and Identification of a Synaptophysin-positive Aggressive Subset. Am. J. Surg. Pathol. 2021, 45, 616–626. [Google Scholar] [CrossRef]
- Geramizadeh, B.; Safari, A.; Bahador, A.; Nikeghbalian, S.; Salahi, H.; Kazemi, K.; Dehghani, S.-M.; Malek-Hosseini, S.-A. Hepatic Angiosarcoma of Childhood: A Case Report and Review of Literature. J. Pediatric. Surg. 2011, 46, e9–e11. [Google Scholar] [CrossRef]
- McLean, C.K.; Squires, J.H.; Reyes-Múgica, M.; McCormick, A.; Mahmood, B. Hepatic Vascular Tumors in the Neonate: Angiosarcoma. J. Pediatr. 2018, 193, 245–248.e1. [Google Scholar] [CrossRef]
- Peterson, M.S.; Baron, R.L.; Rankin, S.C. Hepatic angiosarcoma: Findings on multiphasic contrast-enhanced helical CT do not mimic hepatic hemangioma. AJR Am. J. Roentgenol. 2000, 175, 165–170. [Google Scholar] [CrossRef]
- Pickhardt, P.J.; Kitchin, D.; Lubner, M.G.; Ganeshan, D.M.; Bhalla, S.; Covey, A.M. Primary hepatic angiosarcoma: Multi-institutional comprehensive cancer centre review of multiphasic CT and MR imaging in 35 patients. Eur. Radiol. 2015, 25, 315–322. [Google Scholar] [CrossRef]
- Dimashkieh, H.H.; Mo, J.Q.; Wyatt-Ashmead, J.; Collins, M.H. Pediatric hepaticangiosarcoma: Case report and review of the literature. Pediatr. Dev. Pathol. 2004, 7, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Awan, S.; Davenport, M.; Portmann, B.; Howard, E.R. Angiosarcoma of the liver in children. J. Pediatr. Surg. 1996, 31, 1729–1732. [Google Scholar] [CrossRef] [PubMed]
- Murali, R.; Chandramohan, R.; Möller, I.; Scholz, S.L.; Berger, M.; Huberman, K.; Viale, A.; Pirun, M.; Socci, N.D.; Bouvier, N.; et al. Targeted massively parallel sequencing of angiosarcomas reveals frequent activation of the mitogen activated protein kinase pathway. Oncotarget. 2015, 6, 36041–36052. [Google Scholar] [CrossRef] [PubMed]
- Stringer, M.D.; Alizai, N.K. Mesenchymal Hamartoma of the Liver: A Systematic Review. J. Pediatr. Surg. 2005, 40, 1681–1690. [Google Scholar] [CrossRef]
- Cignini, P.; Coco, C.; Giorlandino, M.; Bagolan, P.; Morini, F.; Giorlandino, C. Fetal Hepatic Mesenchymal Hamartoma: A Case Report. J. Prenat. Med. 2007, 1, 45–46. [Google Scholar]
- Armanian, A.-M.; Nazem, M. Mesenchymal Hamartoma of Liver in a Neonate. J. Neonatol. Biol. 2016, 5, 1000223. [Google Scholar] [CrossRef]
- Gray, S.C.; Pienaar, J.A.; Sofianos, Z.; Varghese, J.; Warnich, I. Hepatic Mesenchymal Hamartoma: An Uncommon but Important Paediatric Diagnosis. SA J. Radiol. 2020, 24, 1891. [Google Scholar] [CrossRef]
- Wu, H.; Ferguson, W.; Castro, E.; Finegold, M.; Patel, K. Pediatric Mesenchymal Hamartomas of the Liver Can Show Both Foregut and Hindgut Phenotype. Pediatr. Dev. Pathol. 2017, 20, 490–497. [Google Scholar] [CrossRef]
- Hassan, M.A. Giant Solid Mesenchymal Hamartoma of the Liver in a Neonate: Case Report. Ann. Pediatr. Surg. 2020, 16, 29. [Google Scholar] [CrossRef]
- Chung, E.M.; Cube, R.; Lewis, R.B.; Conran, R.M. From the archives of the AFIP: Pediatric liver masses: Radiologic-pathologic correlation part 1. Benign tumors. Radiographics 2010, 30, 801–826. [Google Scholar] [CrossRef]
- Gow, K.W.; Lee, L.; Pruthi, S.; Patterson, K.; Healey, P.J. Mesenchymal hamartoma of the liver. J. Pediatr. Surg. 2009, 44, 468–470. [Google Scholar] [CrossRef]
- Anil, G.; Fortier, M.; Low, Y. Cystic hepatic mesenchymal hamartoma: The role of radiology in diagnosis and perioperative management. Br. J. Radiol. 2011, 84, e91–e94. [Google Scholar] [CrossRef] [PubMed]
- Koumanidou, C.; Vakaki, M.; Papadaki, M.; Pitsoulakis, G.; Savvidou, D.; Kakavakis, K. New sonographic appearance of hepatic mesenchymal hamartoma in childhood. J. Clin. Ultrasound 1999, 27, 164–167. [Google Scholar] [CrossRef]
- Horton, K.M.; Bluemke, D.A.; Hruban, R.H.; Soyer, P.; Fishman, E.K. CT and MR imaging of benign hepatic and biliary tumors. Radiographics 1999, 19, 431–451. [Google Scholar] [CrossRef] [PubMed]
- Ros, P.R.; Goodman, Z.D.; Ishak, K.G.; Dachman, A.H.; Olmsted, W.W.; Hartman, D.S.; Lichtenstein, J.E. Mesenchymal hamartoma of the liver: Radiologic-pathologic correlation. Radiology 1986, 158, 619–624. [Google Scholar] [CrossRef]
- Mortele, K.J.; Ros, P.R. Benign liver neoplasms. Clin. Liver Dis. 2002, 6, 119–145. [Google Scholar] [CrossRef]
- Powers, C.; Ros, P.R.; Stoupis, C.; Johnson, W.K.; Segel, K.H. Primary liver neoplasms: MR imaging with pathologic correlation. Radiographics 1994, 14, 459–482. [Google Scholar] [CrossRef]
- Martins-Filho, S.N.; Putra, J. Hepatic mesenchymal hamartoma and undifferentiated embryonal sarcoma of the liver: A pathologic review. Hepat. Oncol. 2020, 7, HEP19. [Google Scholar] [CrossRef]
- Stocker, J.T.; Ishak, K.G. Mesenchymal hamartoma of the liver: Report of 30 cases and review of the literature. Pediatr. Pathol. 1983, 1, 245–267. [Google Scholar] [CrossRef]
- Papke, D.J., Jr. Mesenchymal Neoplasms of the Liver. Surg. Pathol. Clin. 2023, 16, 609–634. [Google Scholar] [CrossRef]
- Rao, S.; Shetty, D.; Sharan, A.; Mukharji, S. Pediatric Mesenchymal Hamartoma of Liver: A Case Report with Histomorphological Differential Diagnosis and Review of Literature. Curr. Health Sci. J. 2022, 48, 230–234. [Google Scholar] [PubMed]
- Speleman, F.; de Telder, V.; de Potter, K.R.; Cin, P.D.; Van Daele, S.; Benoit, Y.; Leroy, J.G.; Van den Berghe, H. Cytogenetic analysis of a mesenchymal hamartoma of the liver. Cancer Genet. Cytogenet. 1989, 40, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Mascarello, J.T.; Krous, H.F. Second report of a translocation involving 19q13.4 in a mesenchymal hamartoma of the liver. Cancer Genet. Cytogenet. 1992, 58, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Mathews, J.; Duncavage, E.J.; Pfeifer, J.D. Characterization of translocations in mesenchymal hamartoma and undifferentiated embryonal sarcoma of the liver. Exp. Mol. Pathol. 2013, 95, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Bentwich, I.; Avniel, A.; Karov, Y.; Aharonov, R.; Gilad, S.; Barad, O.; Barzilai, A.; Einat, P.; Einav, U.; Meiri, E.; et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet. 2005, 37, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Noguer-Dance, M.; Abu-Amero, S.; Al-Khtib, M.; Lefèvre, A.; Coullin, P.; Moore, G.E.; Cavaillé, J. The primate-specific microRNA gene cluster (C19MC) is imprinted in the placenta. Hum. Mol. Genet. 2010, 19, 3566–3582. [Google Scholar] [CrossRef] [PubMed]
- Keller, R.B.; Demellawy, D.E.; Quaglia, A.; Finegold, M.; Kapur, R.P. Methylation status of the chromosome arm 19q microRNA cluster in sporadic and androgenetic-biparental mosaicism-associated hepatic mesenchymal hamartoma. Pediatr. Dev. Pathol. 2015, 18, 218–227. [Google Scholar] [CrossRef]
- Nagarajan, S.; Jayabose, S.; McBride, W.; Prasadh, I.; Tanjavur, V.; Marvin, M.R.; Rodriguez-Davalos, M.I. Inflammatory myofibroblastic tumor of the liver in children. J. Pediatr. Gastroenterol. Nutr. 2013, 57, 277–280. [Google Scholar] [CrossRef]
- Tong, M.; Zhang, B.C.; Jia, F.Y.; Wang, J.; Liu, J.H. Hepatic inflammatory myofibroblastic tumor: A case report. World J. Clin. Cases 2023, 11, 4318–4325. [Google Scholar] [CrossRef]
- Kato, H.; Mitani, Y.; Goda, T.; Ueno, M.; Hayami, S.; Tsujimoto, H.; Kounami, S.; Ichikawa, T.; Yamaue, H. Hepatic Inflammatory Myofibroblastic Tumor Detected in the Fetal Period That Caused an Oncologic Emergency. Case Rep. Oncol. 2021, 13, 1513–1519. [Google Scholar] [CrossRef]
- Dasgupta, D.; Guthrie, A.; McClean, P.; Davison, S.; Luntley, J.; Rajwal, S.; Lodge, J.P.; Prasad, K.R.; Wyatt, J.I.; Stringer, M.D. Liver transplantation for a hilar inflammatory myofibroblastic tumor. Pediatr. Transplant. 2004, 8, 517–521. [Google Scholar] [CrossRef]
- Park, J.Y.; Choi, M.S.; Lim, Y.S.; Park, J.W.; Kim, S.U.; Min, Y.W.; Gwak, G.Y.; Paik, Y.H.; Lee, J.H.; Koh, K.C.; et al. Clinical features, image findings, and prognosis of inflammatory pseudotumor of the liver: A multicenter experience of 45 cases. Gut Liver 2014, 8, 58–63. [Google Scholar] [CrossRef]
- Patnana, M.; Sevrukov, A.B.; Elsayes, K.M.; Viswanathan, C.; Lubner, M.; Menias, C.O. Inflammatory pseudotumor: The great mimicker. Am. J. Roentgenol. 2012, 198, W217–W227. [Google Scholar] [CrossRef] [PubMed]
- Fukuya, T.; Honda, H.; Matsumata, T.; Kawanami, T.; Shimoda, Y.; Muranaka, T.; Hayashi, T.; Maeda, T.; Sakai, H.; Masuda, K. Diagnosis of inflammatory pseudotumor of the liver: Value of CT. Am. J. Radiol. 1994, 163, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Ikeda, H.; Suzuki, N.; Takahashi, A.; Kuroiwa, M.; Hirato, J.; Hatakeyama Si Tsuchida, Y. Inflammatory pseudotumor of the liver: Case report and review of the literature. J. Pediatr. Surg. 2001, 36, 663–666. [Google Scholar] [CrossRef]
- Qiu, X.; Montgomery, E.; Sun, B. Inflammatory myofibroblastic tumor and low-grade myofibroblastic sarcoma: A comparative study of clinicopathologic features and further observations on the immunohistochemical profile of myofibroblasts. Hum. Pathol. 2008, 39, 846–856. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Suurmeijer, A.J.; Zhang, L.; Sung, Y.S.; Jungbluth, A.A.; Travis, W.D.; Al-Ahmadie, H.; Fletcher, C.D.; Alaggio, R. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 fusions and rare novel RET gene rearrangement. Am. J. Surg. Pathol. 2015, 39, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Coffin, C.M.; Hornick, J.L.; Fletcher, C.D. Inflammatory myofibroblastic tumor: Comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am. J. Surg. Pathol. 2007, 31, 509–520. [Google Scholar] [CrossRef]
- Yamamoto, H.; Yoshida, A.; Taguchi, K.; Kohashi, K.; Hatanaka, Y.; Yamashita, A.; Mori, D.; Oda, Y. ALK, ROS1 and NTRK3 gene rearrangements in inflammatory myofibroblastic tumours. Histopathology 2016, 69, 72–83. [Google Scholar] [CrossRef]
- Coffin, C.M.; Fletcher, J.A. Inflammatory myofibroblastic tumor. In World Health Organization Classification of Tumours of Soft Tissue and Bone; Fletcher, C.D.M., Bridge, J.A., Hogendoorn, P.C.W., Mertens, F., Eds.; IARC Press: Lyon, France, 2013; pp. 83–84. [Google Scholar]
- Fazlollahi, L.; Hsiao, S.J.; Kochhar, M.; Mansukhani, M.M.; Yamashiro, D.J.; Remotti, H.E. Malignant rhabdoid tumor, an aggressive tumor often misclassified as small cell variant of hepatoblastoma. Cancers 2019, 11, 1992. [Google Scholar] [CrossRef]
- Trobaugh-Lotrario, A.D.; Finegold, M.J.; Feusner, J.H. Rhabdoid tumors of the liver: Rare, aggressive, and poorly responsive to standard cytotoxic chemotherapy. Pediatr. Blood Cancer 2011, 57, 423–428. [Google Scholar] [CrossRef]
- Oita, S.; Terui, K.; Komatsu, S.; Hishiki, T.; Saito, T.; Mitsunaga, T.; Nakata, M.; Yoshida, H. Malignant Rhabdoid Tumor of the Liver: A Case Report and Literature Review. Pediatr. Rep. 2015, 7, 5578. [Google Scholar] [CrossRef] [PubMed]
- Kupeli, S.; Yöntem, A.; Narli, N.; Doran, F.; Soyupak, S. Infantile Rhabdoid Tumor Mimicking Hepatic Hemangioendothelioma. J. Cancer Res. Ther. 2016, 12, 430–431. [Google Scholar] [CrossRef] [PubMed]
- Kapral, N.; Melmer, P.; Druzgal, C.H.; Lancaster, L. Pediatric Hepatic Rhabdoid Tumor: A Rare Cause of Abdominal Mass in Children. Radiol. Case Rep. 2018, 13, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Cornet, M.; De Lambert, G.; Pariente, D.; Planchon, J.M.; Guettier, C.; Martelli, H.; Guérin, F.; Branchereau, S. Rhabdoid Tumor of the Liver: Report of 6 Pediatric Cases Treated at a Single Institute. J. Pediatr. Surg. 2018, 53, 567–571. [Google Scholar] [CrossRef]
- Agarwala, S.; Jindal, B.; Jana, M.; Bhatnagar, V.; Gupta, A.K.; Iyer, V.K. Malignant Rhabdoid Tumor of Liver. J. Indian Assoc. Pediatr. Surg. 2014, 19, 38–40. [Google Scholar] [CrossRef] [PubMed]
- Ravindra, K.V.; Cullinane, C.; Lewis, I.J.; Squire, B.R.; Stringer, M.D. Long-term survival after spontaneous rupture of a malignant rhabdoid tumor of the liver. J. Pediatr. Surg. 2002, 37, 1488–1490. [Google Scholar] [CrossRef]
- Kachanov, D.; Teleshova, M.; Kim, E.; Dobrenkov, K.; Moiseenko, R.; Usychkina, A.; Filin, A.; Semenkov, A.; Mitrofanova, A.; Konovalov, D.; et al. Malignant Rhabdoid Tumor of the Liver Presented with Initial Tumor Rupture. Cancer Genet. 2014, 207, 412–414. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Stelling, A.; Kuramoto, A.; Patel, C.; Keller, J. Malignant Rhabdoid Tumor of the Liver: Findings at US, CT, and MRI, with Histopathologic Correlation. Radiol. Case Rep. 2014, 9, 854. [Google Scholar] [CrossRef]
- Garcés-Iñigo, E.F.; Leung, R.; Sebire, J.N.; McHugh, K. Extrarenal rhabdoid tumours outside the central nervous system in infancy. Pediatr. Radiol. 2009, 39, 817–822. [Google Scholar] [CrossRef]
- Hoot, A.C.; Russo, P.; Judkins, A.R.; Perlman, E.J.; Biegel, J.A. Immunohistochemical analysis of hSNF5/INI1 distinguishes renal and extra-renal malignant rhabdoid tumors from other pediatric soft tissue tumors. Am. J. Surg. Pathol. 2004, 28, 1485–1491. [Google Scholar] [CrossRef]
- Versteege, I.; Sévenet, N.; Lange, J.; Rousseau-Merck, M.F.; Ambros, P.; Handgretinger, R.; Aurias, A.; Delattre, O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998, 394, 203–206. [Google Scholar] [CrossRef]
- Biegel, J.A.; Zhou, J.Y.; Rorke, L.B.; Stenstrom, C.; Wainwright, L.M.; Fogelgren, B. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999, 59, 74–79. [Google Scholar]
- Fanburg-Smith, J.C.; Hengge, M.; Hengge, U.R.; Smith, J.S., Jr.; Miettinen, M. Extrarenal rhabdoid tumors of soft tissue: A clinicopathologic and immunohistochemical study of 18 cases. Ann. Diagn. Pathol. 1998, 2, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Fei, L.; LI, S.; Cui, K.; Zhang, J.; YU, F.; Zhang, B. Undifferentiated Embryonal Sarcoma of the Liver in a Child: A Case Report and Review of the Literature. Oncol. Lett. 2013, 5, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Putra, J.; Ornvold, K. Undifferentiated Embryonal Sarcoma of the Liver: A Concise Review. Arch. Pathol. Lab. Med. 2015, 139, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Mathias, M.D.; Ambati, S.R.; Chou, A.J.; Slotkin, E.K.; Wexler, L.H.; Meyers, P.A.; Magnan, H. A Single-Center Experience with Undifferentiated Embryonal Sarcoma of the Liver. Pediatr. Blood Cancer 2016, 63, 2246–2248. [Google Scholar] [CrossRef]
- Shi, Y.; Rojas, Y.; Zhang, W.; Beierle, E.A.; Doski, J.J.; Goldfarb, M.; Goldin, A.B.; Gow, K.W.; Langer, M.; Meyers, R.L.; et al. Characteristics and Outcomes in Children with Undifferentiated Embryonal Sarcoma of the Liver: A Report from the National Cancer Database. Pediatr. Blood Cancer 2017, 64, e26272. [Google Scholar] [CrossRef]
- Shi, M.; Xu, H.; Sangster, G.P.; Gu, X. Pulmonary Metastases from an Undifferentiated Embryonal Sarcoma of the Liver: A Case Report and Review. Case Rep. Oncol. Med. 2018, 2018, 7840865. [Google Scholar] [CrossRef]
- Hu, H.-M.; Zhang, W.-L.; Li, J.; Wen, Y.; Li, F.; Zhi, T.; Huang, D.-S. Report of Seven Children with Undifferentiated Embryonal Sarcoma of the Liver. Chin. Med. J. 2019, 132, 2244–2245. [Google Scholar] [CrossRef]
- Stocker, J.T.; Ishak, K.G. Undifferentiated (Embryonal) Sarcoma of the Liver: Report of 31 Cases. Cancer 1978, 42, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Shehata, B.M.; Gupta, N.A.; Katzenstein, H.M.; Steelman, C.K.; Wulkan, M.L.; Gow, K.W.; Bridge, J.A.; Kenney, B.D.; Thompson, K.; de Chadarévian, J.P.; et al. Undifferentiated embryonal sarcoma of the liver is associated with mesenchymal hamartoma and multiple chromosomal abnormalities: A review of eleven cases. Pediatr. Dev. Pathol. 2011, 14, 111–116. [Google Scholar] [CrossRef]
- Setty, B.A.; Jinesh, G.G.; Arnold, M.; Pettersson, F.; Cheng, C.-H.; Cen, L.; Yoder, S.J.; Teer, J.K.; Flores, E.R.; Reed, D.R.; et al. The Genomic Landscape of Undifferentiated Embryonal Sarcoma of the Liver Is Typified by C19MC Structural Rearrangement and Overexpression Combined with TP53 Mutation or Loss. PLoS Genet. 2020, 16, e1008642. [Google Scholar] [CrossRef]
- Zhang, C.; Jia, C.-J.; Xu, C.; Sheng, Q.-J.; Dou, X.-G.; Ding, Y. Undifferentiated Embryonal Sarcoma of the Liver: Clinical Characteristics and Outcomes. World J. Clin. Cases 2020, 8, 4763–4772. [Google Scholar] [CrossRef]
- Letherer, A.; Mastenbrook, J.; VanEnk, R.A.; Bauler, L.D. Undifferentiated Embryonal Sarcoma of the Liver Presents as a Molecular Mimic of Parasitic Infection. Cureus 2020, 12, e6800. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.L.; Yu, R.S.; Chen, Y.; Zhang, Q. Sarcomas of abdominal organs: Computed tomography and magnetic resonance imaging findings. Semin. Ultrasound CT MR 2011, 32, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Thombare, P.; Verma, M.; Shah, K.; Doshi, H.; Verma, G.; Patkar, D. Undifferentiated Embryonal Sarcoma of Liver: Paradoxical Imaging Appearance. Radiol. Case Rep. 2020, 15, 1095–1098. [Google Scholar] [CrossRef]
- Zheng, J.M.; Tao, X.; Xu, A.M.; Chen, X.F.; Wu, M.C.; Zhang, S.H. Primary and recurrent embryonal sarcoma of the liver: Clinicopathological and immunohistochemical analysis. Histopathology 2007, 51, 195–203. [Google Scholar] [CrossRef]
- Fornari, F.; Milazzo, M.; Chieco, P.; Negrini, M.; Marasco, E.; Capranico, G.; Mantovani, V.; Marinello, J.; Sabbioni, S.; Callegari, E.; et al. In hepatocellular carcinoma miR-519d is up-regulated by p53 and DNA hypomethylation and targets CDKN1A/p21, PTEN, AKT3 and TIMP2. J. Pathol. 2012, 227, 275–285. [Google Scholar] [CrossRef]
- Flor, I.; Bullerdiek, J. The dark side of a success story: microRNAs of the C19MC cluster in human tumours. J. Pathol. 2012, 227, 270–274. [Google Scholar] [CrossRef]
- Fuchs, J.; Murtha-Lemekhova, A.; Kessler, M.; Günther, P.; Fichtner, A.; Pfeiffenberger, J.; Probst, P.; Hoffmann, K. Biliary Rhabdomyosarcoma in Pediatric Patients: A Systematic Review and Meta-Analysis of Individual Patient Data. Front. Oncol. 2021, 11, 701400. [Google Scholar] [CrossRef]
- Malkan, A.D.; Fernandez-Pineda, I. The Evolution of Diagnosis and Management of Pediatric Biliary Tract Rhabdomyosarcoma. Curr. Pediatr. Rev. 2016. ahead of print. [Google Scholar] [CrossRef]
- Meyers, R.; Hiyama, E.; Czauderna, P.; Tiao, G.M. Liver Tumors in Pediatric Patients. Surg. Oncol. Clin. N. Am. 2021, 30, 253–274. [Google Scholar] [CrossRef] [PubMed]
- Saeed, O.; Saxena, R. Primary mesenchymal liver tumors of childhood. Semin. Diagn Pathol. 2017, 34, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Vij, M.; Sivasankaran, M.; Jayaraman, D.; Sankaranarayanan, S.; Kumar, V.; Munirathnam, D.; Scott, J. CARMIL2 Immunodeficiency with Epstein Barr Virus Associated Smooth Muscle Tumor (EBV-SMT). Report of a Case with Comprehensive Review of Literature. Fetal Pediatr. Pathol. 2022, 41, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Arva, N.C.; Schafernak, K.T. Rare Presentations of Epstein-Barr Virus—Associated Smooth Muscle Tumor in Children. Pediatr. Dev. Pathol. 2016, 19, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Magg, T.; Schober, T.; Walz, C.; Ley-Zaporozhan, J.; Facchetti, F.; Klein, C.; Hauck, F. Epstein-Barr Virus (+) Smooth Muscle Tumors as Manifestation of Primary Immunodeficiency Disorders. Front Immunol. 2018, 9, 368. [Google Scholar] [CrossRef]
- Deyrup, A.T. Epstein-Barr virus-associated epithelial and mesenchymal neoplasms. Hum. Pathol. 2008, 39, 473–483. [Google Scholar] [CrossRef]
- Hussein, K.; Rath, B.; Ludewig, B.; Kreipe, H.; Jonigk, D. Clinico-pathological characteristics of different types of immunodeficiency-associated smooth muscle tumours. Eur. J. Cancer 2014, 50, 2417–2424. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, M.C.; Torbenson, M.S.; Wu, T.T.; Kakar, S.; Jain, D.; Yeh, M.M. Pediatric Hepatocellular Adenomas: The Influence of Age and Syndrome on Subtype. Am. J. Surg. Pathol. 2021, 45, 1641–1647. [Google Scholar] [CrossRef]
- Hahn, E.; Putra, J. Hepatocellular adenoma in the paediatric population: Molecular classification and clinical associations. World J. Gastroenterol. 2020, 26, 2294–2304. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Jeannot, E.; Nhieu, J.T.; Scoazec, J.Y.; Guettier, C.; Rebouissou, S.; Bacq, Y.; Leteurtre, E.; Paradis, V.; Michalak, S.; et al. Genotype-phenotype correlation in hepatocellularadenoma: New classification and relationship with HCC. Hepatology 2006, 43, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Couchy, G.; Balabaud, C.; Morcrette, G.; Caruso, S.; Blanc, J.F.; Bacq, Y.; Calderaro, J.; Paradis, V.; Ramos, J.; et al. Molecular Classification of Hepatocellular Adenoma Associates with Risk Factors, Bleeding, and Malignant Transformation. Gastroenterology 2017, 152, 880–894.e6. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Agarwal, S.; Arnason, T.; Saini, S.; Belghiti, J. Management of Hepatocellular Adenoma: Recent Advances. Clin. Gastroenterol. Hepatol. 2015, 13, 1221–1230. [Google Scholar] [CrossRef]
- Sempoux, C.; Balabaud, C.; Bioulac-Sage, P. Malignant Transformation of Hepatocellular Adenoma. Hepatic Oncol. 2014, 1, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Haring, M.P.D.; Gouw, A.S.H.; de Haas, R.J.; Cuperus, F.J.C.; de Jong, K.P.; de Meijer, V.E. The Effect of Oral Contraceptive Pill Cessation on Hepatocellular Adenoma Diameter: A Retrospective Cohort Study. Liver Int. 2019, 39, 905–913. [Google Scholar] [CrossRef]
- Crosnier, H.; Thibaud, E.; Duflos, C.; Polak, M. Norethisterone-Induced Hepatic Adenomas Can Cause Life-Threatening Bleeding in Girls with Inherited Platelet Disorders. Fertil. Steril. 2010, 94, 2329.e1–2329.e3. [Google Scholar] [CrossRef]
- Brady, P.C.; Missmer, S.A.; Laufer, M.R. Hepatic Adenomas in Adolescents and Young Women with Endometriosis Treated with Norethindrone Acetate. J. Pediatr. Adolesc. Gynecol. 2017, 30, 422–424. [Google Scholar] [CrossRef]
- Triantafyllopoulou, M.; Whitington, P.F.; Melin-Aldana, H.; Benya, E.C.; Brickman, W. Hepatic Adenoma in an Adolescent with Elevated Androgen Levels. J. Pediatr. Gastroenterol. Nutr. 2007, 44, 640–642. [Google Scholar] [CrossRef]
- Wang, L.; Wang, C.; Li, W.; Meng, F.; Li, Y.; Fan, H.; Zhou, Y.; Bharathi, G.; Gao, S.; Yang, Y. Multiple Hepatocellular Adenomas Associated with Long-Term Administration of Androgenic Steroids for Aplastic Anemia: A Case Report and Literature Review. Medicine 2020, 99, e20829. [Google Scholar] [CrossRef]
- Daga, B.V.; Shah, V.R.; More, R.B. CT scan diagnosis of hepatic adenoma in a case of von Gierke disease. Indian J. Radiol. Imaging 2012, 22, 54–57. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Chuang, T.-P.; Bali, D.; Koeberl, D.; Austin, S.; Weinstein, D.A.; Murphy, E.; Chen, Y.-T.; Boyette, K.; Liu, C.-H.; et al. Chromosomal and Genetic Alterations in Human Hepatocellular Adenomas Associated with Type Ia Glycogen Storage Disease. Hum. Mol. Genet. 2009, 18, 4781–4790. [Google Scholar] [CrossRef]
- Calderaro, J.; Labrune, P.; Morcrette, G.; Rebouissou, S.; Franco, D.; Prévot, S.; Quaglia, A.; Bedossa, P.; Libbrecht, L.; Terracciano, L.; et al. Molecular characterization of hepatocellular adenomas developed in patients with glycogen storage disease type I. J. Hepatol. 2013, 58, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Glonnegger, H.; Schulze, M.; Kathemann, S.; Berg, S.; Füllgraf, H.; Tannapfel, A.; Gerner, P.; Grohmann, J.; Niemeyer, C.; Hettmer, S. Case Report: Hepatic Adenoma in a Child with a Congenital Extrahepatic Portosystemic Shunt. Front. Pediatr. 2020, 8, 501. [Google Scholar] [CrossRef]
- Yoon, H.J.; Jeon, T.Y.; Yoo, S.Y.; Kim, J.H.; Eo, H.; Lee, S.K.; Kim, J.S. Hepatic tumours in children with biliary atresia: Single-centre experience in 13 cases and reviewof the literature. Clin. Radiol. 2014, 69, e113–e119. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, D.; Bruneton, J.N.; Drouillard, J.; Pointreau, C.C.; Vasile, N. Hepatic adenomas and focal nodular hyperplasia: Dynamic CT study. Radiology 1986, 160, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Grazioli, L.; Federle, M.P.; Brancatelli, G.; Ichikawa, T.; Olivetti, L.; Blachar, A. Hepatic adenomas: Imaging and pathologic findings. Radiographics 2001, 21, 877–892, discussion 892–894. [Google Scholar] [CrossRef]
- Ichikawa, T.; Federle, M.P.; Grazioli, L.; Nalesnik, M. Hepatocellular adenoma: Multiphasic CT and histopathologic findings in 25 patients. Radiology 2000, 214, 861–868. [Google Scholar] [CrossRef]
- Dong, Q.; Chen, J.J. CT Scan of Pediatric Liver Tumors. In CT Scanning-Techniques and Applications; Subburaj, K., Ed.; InTech: Rijeka, Croatia, 2011. [Google Scholar]
- Bioulac-Sage, P.; Balabaud, C.; Zucman-Rossi, J. Subtype Classification of Hepatocellular Adenoma. Dig. Surg. 2010, 27, 39–45. [Google Scholar] [CrossRef]
- Espinoza, A.F.; Vasudevan, S.A.; Masand, P.M.; Lòpez-Terrada, D.H.; Patel, K.R. Pediatric Hepatocellular Adenomas: What Is Known and What Is New? Cancers 2023, 15, 4790. [Google Scholar] [CrossRef]
- Franchi-Abella, S.; Branchereau, S. Benign hepatocellular tumors in children: Focal nodular hyperplasia and hepatocellular adenoma. Int. J. Hepatol. 2013, 2013, 215064. [Google Scholar] [CrossRef]
- Zhuang, L.; Ni, C.; Din, W.; Zhang, F.; Zhuang, Y.; Sun, Y.; Xi, D. Huge Focal Nodular Hyperplasia Presenting in a 6-Year-Old Child: A Case Presentation. Int. J. Surg. Case Rep. 2016, 29, 76–79. [Google Scholar] [CrossRef]
- Gürses, C.; Ok¸sar, F.S.; Erol, B.; Yalçın, M.; Kahvecioğlu, N.; Alparslan, A.Ş. Natural Course of Hepatic Focal Nodular Hyperplasia from Childhood to Adulthood and Review of the Literature. Turk. J. Gastroenterol. 2017, 28, 492–497. [Google Scholar] [CrossRef]
- Lautz, T.; Tantemsapya, N.; Dzakovic, A.; Superina, R. Focal nodular hyperplasia in children: Clinical features and current management practice. J. Pediatr. Surg. 2010, 45, 1797–1803. [Google Scholar] [CrossRef]
- Lucas, B.; Ravishankar, S.; Pateva, I. Pediatric Primary Hepatic Tumors: Diagnostic Considerations. Diagnostics 2021, 11, 333. [Google Scholar] [CrossRef]
- Ji, Y.; Chen, S.; Xiang, B.; Wen, T.; Yang, J.; Zhong, L.; Li, F.; Li, L. Clinical Features of Focal Nodular Hyperplasia of the Liver in Children. J. Pediatr. Gastroenterol. Nutr. 2016, 62, 813–818. [Google Scholar] [CrossRef]
- Cattoni, A.; Rovelli, A.; Prunotto, G.; Bonanomi, S.; Invernizzi, P.; Perego, R.; Mariani, A.M.; Balduzzi, A. Hepatic Focal Nodular Hyperplasia after Pediatric Hematopoietic Stem Cell Transplantation: The Impact of Hormonal Replacement Therapy and Iron Overload. Pediatr. Blood Cancer 2020, 67, e28137. [Google Scholar] [CrossRef] [PubMed]
- Chiorean, L.; Cui, X.W.; Tannapfel, A.; Franke, D.; Stenzel, M.; Kosiak, W.; Schreiber- Dietrich, D.; Jüngert, J.; Chang, J.M.; Dietrich, C.F. Benign liver tumors in pediatric patients—Review with emphasis on imaging features. World J. Gastroenterol. 2015, 21, 8541–8561. [Google Scholar] [CrossRef]
- Denk, H.; Pabst, D.; Abuja, P.M.; Reihs, R.; Tessaro, B.; Zatloukal, K.; Lackner, C. Senescence markers in focal nodular hyperplasia of the liver: Pathogenic considerations on the basis of immunohistochemical results. Mod. Pathol. 2022, 35, 87–95. [Google Scholar] [CrossRef]
- Umetsu, S.E.; Joseph, N.M.; Cho, S.J.; Morotti, R.; Deshpande, V.; Jain, D.; Kakar, S. FocalNodular Hyperplasia-like Nodules Arising in the Setting of Hepatic Vascular Disorders with Portosystemic Shunting Show β-catenin Activation. Hum. Pathol. 2023, 142, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Torbenson, M.S. Hamartomas and malformations of the liver. Semin. Diagn. Pathol. 2019, 36, 39–47. [Google Scholar] [CrossRef]
- Shamsi, K.; De Schepper, A.; Degryse, H.; Deckers, F. Focal nodular hyperplasia of the liver: Radiologic findings. Abdom. Imaging 1993, 18, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Kehagias, D.; Moulopoulos, L.; Antoniou, A.; Hatziioannou, A.; Smyrniotis, V.; Trakadas, S.; Lahanis, S.; Vlahos, L. Focal nodular hyperplasia: Imaging findings. Eur. Radiol. 2001, 11, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Carlson, S.K.; Johnson, C.D.; Bender, C.E.; Welch, T.J. CT of focal nodular hyperplasia of the liver. AJR Am. J. Roentgenol. 2000, 174, 705–712. [Google Scholar] [CrossRef]
- Martí-Bonmatí, L.; Casillas, C.; Dosdá, R. Enhancement characteristics of hepatic focal nodular hyperplasia and its scar by dynamic magnetic resonance imaging. MAGMA 2000, 10, 200–204. [Google Scholar] [CrossRef]
- Hussain, S.M.; Terkivatan, T.; Zondervan, P.E.; Lanjouw, E.; de Rave, S.; Ijzermans, J.N.; de Man, R.A. Focal nodular hyperplasia: Findings at state-of-the-art MR imaging, US, CT, and pathologic analysis. Radiographics 2004, 24, 3–17, discussion 18–19. [Google Scholar] [CrossRef] [PubMed]
- Courtier, J.L.; Perito, E.R.; Rhee, S.; Tsai, P.; Heyman, M.B.; MacKenzie, J.D. Targeted MRI contrast agents for pediatric hepatobiliary disease. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.S.; Leary, C.; Bluemke, D.A.; Amendola, M.; Sahani, D.; McTavish, J.D.; Brody, J.; Outwater, E.; Mitchell, D.; Sheafor, D.H.; et al. Improved characterization of focal liver lesions with liver-specific gadoxetic acid disodium-enhanced magnetic resonance imaging: A multicenter phase 3 clinical trial. J. Comput. Assist. Tomogr. 2010, 34, 163–172. [Google Scholar] [CrossRef]
- Geller, S.A.; Petrovic, L.M. Biopsy Interpretation of the Liver, 2nd ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2009; pp. 324–326. [Google Scholar]
- Ma, I.T.; Rojas, Y.; Masand, P.M.; Castro, E.C.; Himes, R.W.; Kim, E.S.; Goss, J.A.; Nuchtern, J.G.; Finegold, M.J.; Thompson, P.A.; et al. Focal Nodular Hyperplasia in Children: An Institutional Experience with Review of the Literature. J. Pediatr. Surg. 2015, 50, 382–387. [Google Scholar] [CrossRef]
- Czauderna, P.; Lopez-Terrada, D.; Hiyama, E.; Häberle, B.; Malogolowkin, M.H.; Meyers, R.L. Hepatoblastoma state of the art: Pathology, genetics, risk stratification, and chemotherapy. Curr. Opin. Pediatr. 2014, 26, 19–28. [Google Scholar] [CrossRef]
- Spector, L.G.; Birch, J. The epidemiology of hepatoblastoma. Pediatr. Blood Cancer. 2012, 59, 776–779. [Google Scholar] [CrossRef]
- Devi, L.P.; Kumar, R.; Handique, A.; Kumar, M. Hepatoblastoma--a rare liver tumor with review of literature. J. Gastrointest. Cancer 2014, 45 (Suppl. 1), 261–264. [Google Scholar] [CrossRef] [PubMed]
- Yoo, G.H.Y.; Mugarab-Samedi, V.; Hansen, G.; Miller, G.; Givelichian, L.; Kalaniti, K.; Daspal, S. Rare Cause of Emergency in the First Week of Life: Congenital Hepatoblastoma (Case Report). Oxf. Med. Case Rep. 2020, 2020, omaa002. [Google Scholar] [CrossRef]
- Feng, J.; Polychronidis, G.; Heger, U.; Frongia, G.; Mehrabi, A.; Hoffmann, K. Incidence trends and survival prediction of hepatoblastoma in children: A population-based study. Cancer Commun. 2019, 39, 62. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, L.M.; Georgieff, M.K.; Ross, J.A.; Feusner, J.H.; Tomlinson, G.E.; Malogolowkin, M.H.; Krailo, M.D.; Miller, N.; Fonstad, R.; Spector, L.G. Neonatal Medical Exposures and Characteristics of Low Birth Weight Hepatoblastoma Cases: A Report from the Children’s Oncology Group. Pediatr. Blood Cancer 2014, 61, 2018–2023. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Subbarao, G.; Saxena, R. Hepatoblastoma. Semin. Diagn. Pathol. 2017, 34, 192–200. [Google Scholar] [CrossRef]
- Lin, J.; Guo, J.; Chen, C.; Jiang, L.; Lai, C.; Wang, C. Congenital hepatoblastoma presenting with hepatic arteriovenous fistulas: A case report. Front. Pediatr. 2023, 11, 1199224. [Google Scholar] [CrossRef]
- Lai, M.; Burjonrappa, S. Perinatal Hemorrhage Complicating Neonatal Hepatoblastoma: Case Report. J. Pediatr. Surg. 2012, 47, e29–e32. [Google Scholar] [CrossRef]
- Towbin, A.J.; Braga, F.D.C.B.; Zhang, B.; Geller, J.I.; Tiao, G.M.; Podberesky, D.J. Fractures in Children with Newly Diagnosed Hepatoblastoma. Pediatr. Radiol. 2018, 48, 581–585. [Google Scholar] [CrossRef]
- Al-Jumaily, U.; Sammour, I.; Al-Muhaisen, F.; Ajlouni, F.; Sultan, I. Precocious Puberty in an Infant with Hepatoblastoma: A Case Report. J. Med. Case Rep. 2011, 5, 422. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Ishii, E.; Hayashida, Y.; Hirata, Y.; Sakai, R.; Miyazaki, S. Mechanism of Thrombocytosis in Hepatoblastoma: A Case Report. Pediatr. Hematol. Oncol. 1996, 13, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, N.; Scott, J.X.; Kumar, V.; Vij, M.; Ramachandran, P.; Narasimhan, G.; Reddy, M.S.; Kota, V.; Munirathnam, D.; Kelgeri, C.; et al. Multidisciplinary management of hepatoblastoma in children: Experience from a developing country. Pediatr. Blood Cancer 2017, 64, e26249. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Zheng, Q.-Z.; Cao, D.-Y.; Xie, Y.-N.; Song, T.; Jiang, Q.-P.; Chen, M. Prenatal Diagnosis of Congenital Hepatoblastoma. Matern. Fet. Med. 2020, 2, 115–118. [Google Scholar] [CrossRef]
- Chung, E.M.; Lattin GEJr Cube, R.; Lewis, R.B.; Marichal-Hernández, C.; Shawhan, R.; Conran, R.M. From the archives of the AFIP: Pediatric liver masses: Radiologic-pathologic correlation. Part 2. Malignant tumors. Radiographics 2011, 31, 483–507. [Google Scholar] [CrossRef] [PubMed]
- Meyers, A.B.; Towbin, A.J.; Geller, J.I.; Podberesky, D.J. Hepatoblastoma imaging with gadoxetate disodium-enhanced MRI-typical, atypical, pre- and post-treatment evaluation. Pediatr. Radiol. 2012, 42, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Baheti, A.D.; Chapman, T.; Rudzinski, E.; Albert, C.M.; Stanescu, A.L. Diagnosis, histopathologic correlation and management of hepatoblastoma: What the radiologist needs to know. Clin. Imaging 2018, 52, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Ferrell, L.D.; Kakar, S.; Terracciano, L.M.; Wee, A. Tumors and tumor like lesions of the liver. In MacSween’s Pathology of the Liver, 7th ed.; Burt, A.D., Ferrell, L.D., Hubscher, S.G., Eds.; Elsevier: Philadelphia, PA, USA, 2018; pp. 780–879. [Google Scholar]
- Wang, L.L.; Filippi, R.Z.; Zurakowski, D.; Archibald, T.; Vargas, S.O.; Voss, S.D.; Shamberger, R.C.; Davies, K.; Kozakewich, H.; Perez-Atayde, A.R. Effects of neoadjuvant chemotherapy on hepatoblastoma: A morphologic and immunohistochemical study. Am. J. Surg. Pathol. 2010, 34, 287–299. [Google Scholar] [CrossRef]
- López-Terrada, D.; Alaggio, R.; de Dávila, M.T.; Czauderna, P.; Hiyama, E.; Katzenstein, H.; Leuschner, I.; Malogolowkin, M.; Meyers, R.; Ranganathan, S.; et al. Towards an international pediatric liver tumor consensus classification: Proceedings of the Los Angeles COG liver tumors symposium. Mod. Pathol. 2014, 27, 472–491. [Google Scholar] [CrossRef]
- Ranganathan, S.; Lopez-Terrada, D.; Alaggio, R. Hepatoblastoma and Pediatric Hepatocellular Carcinoma: An Update. Pediatr. Dev. Pathol. 2020, 23, 79–95. [Google Scholar] [CrossRef]
- Trobaugh-Lotrario, A.D.; Tomlinson, G.E.; Finegold, M.J.; Gore, L.; Feusner, J.H. Small cell undifferentiated variant of hepatoblastoma: Adverse clinical and molecular features similar to rhabdoid tumors. Pediatr. Blood Cancer 2009, 52, 328–334. [Google Scholar] [CrossRef]
- Haas, J.E.; Feusner, J.H.; Finegold, M.J. Small cell undifferentiated histology in hepatoblastoma may be unfavorable. Cancer 2001, 92, 3130–3134. [Google Scholar] [CrossRef]
- Meyers, R.L.; Rowland, J.R.; Krailo, M.; Chen, Z.; Katzenstein, H.M.; Malogolowkin, M.H. Predictive power of pretreatment prognostic factors in children with hepatoblastoma: A report from the Children's Oncology Group. Pediatr. Blood Cancer 2009, 53, 1016–1022. [Google Scholar] [CrossRef]
- Trobaugh-Lotrario, A.; Katzenstein, H.M.; Ranganathan, S.; Lopez-Terrada, D.; Krailo, M.D.; Piao, J.; Chung, N.; Randazzo, J.; Malogolowkin, M.H.; Furman, W.L.; et al. Small Cell UndifferentiatedHistology Does Not Adversely Affect Outcome in Hepatoblastoma: A Report From the Children’s Oncology Group (COG) AHEP0731 Study Committee. J. Clin. Oncol. 2022, 40, 459–467. [Google Scholar] [CrossRef]
- Sumazin, P.; Chen, Y.; Treviño, L.R.; Sarabia, S.F.; Hampton, O.A.; Patel, K.; Mistretta, T.A.; Zorman, B.; Thompson, P.; Heczey, A.; et al. Genomic analysis of hepatoblastoma identifies distinct molecular and prognostic subgroups. Hepatology 2017, 65, 104–121. [Google Scholar] [CrossRef] [PubMed]
- Hadzic, N.; Finegold, M.J. Liver neoplasia in children. Clin. Liver Dis. 2011, 15, 443–462. [Google Scholar] [CrossRef]
- Emre, S.; Umman, V.; Rodriguez-Davalos, M. Current concepts in pediatric liver tumors. Pediatr. Transplant 2012, 16, 549–563. [Google Scholar] [CrossRef]
- Palaniappan, K.; Borkar, V.V.; Safwan, M.; Vij, M.; Govil, S.; Shanmugam, N.; Rela, M. Pediatric hepatocellular carcinoma in a developing country: Is the etiology changing? Pediatr. Transplant. 2016, 20, 898903. [Google Scholar] [CrossRef]
- Valamparampil, J.J.; Shanmugam, N.; Vij, M.; Reddy, M.S.; Rela, M. Hepatocellular Carcinoma in Paediatric Patients with Alagille Syndrome: Case Series and Review of Literature. J. Gastrointest. Cancer 2020, 51, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Menon, J.; Shanmugam, N.; Valamparampil, J.J.; Hakeem, A.; Vij, M.; Jalan, A.; Reddy, M.S.; Rela, M. Liver Transplantation: A Safe and Definitive Alternative to Lifelong Nitisinone for Tyrosinemia Type 1. Indian J. Pediatr. 2022, 89, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Vij, M.; Shanmugam, N.P.; Reddy, M.S.; Govil, S.; Rela, M. Hepatocarcinogenesis in multidrug-resistant P-glycoprotein 3 deficiency. Pediatr. Transplant. 2017, 21, e12889. [Google Scholar] [CrossRef]
- Vij, M.; Safwan, M.; Shanmugam, N.P.; Rela, M. Liver pathology in severe multidrug resistant 3 protein deficiency: A series of 10 pediatric cases. Ann. Diagn. Pathol. 2015, 19, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Menon, J.; Shanmugam, N.; Vij, M.; Rammohan, A.; Rela, M. Liver transplantation in tight junction protein-2 deficiency associated liver disease. Liver Transpl. 2022, 28, 1796–1799. [Google Scholar] [CrossRef] [PubMed]
- Vij, M.; Menon, J.; Subbiah, K.; Raju, L.P.; Gowrisankar, G.; Shanmugum, N.; Kaliamoorthy, I.; Rammohan, A.; Rela, M. Pathologic and Immunophenotypic Characterization of Syncytial Giant Cell Variant of Pediatric Hepatocellular Carcinoma. A Distinct Subtype. Fetal Pediatr. Pathol. 2023, 42, 709–718. [Google Scholar] [CrossRef]
- Hadžić, N.; Quaglia, A.; Portmann, B.; Paramalingam, S.; Heaton, N.D.; Rela, M.; Mieli-Vergani, G.; Davenport, M. Hepatocellular Carcinoma in Biliary Atresia: King’s College Hospital Experience. J. Pediatr. 2011, 159, 617–622.e1. [Google Scholar] [CrossRef] [PubMed]
- Walther, A.; Tiao, G. Approach to Pediatric Hepatocellular Carcinoma. Clin. Liver Dis. 2013, 2, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Cowell, E.; Patel, K.; Heczey, A.; Finegold, M.; Venkatramani, R.; Wu, H.; López-Terrada, D.; Miloh, T. Predisposing Conditions to Pediatric Hepatocellular Carcinoma and Association with Outcomes: Single-Center Experience. J. Pediatr. Gastroenterol. Nutr. 2019, 68, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Thakral, N.; Simonetto, D.A. Hyperammonemic Encephalopathy: An Unusual Presentation of Fibrolamellar Hepatocellular Carcinoma. Clin. Mol. Hepatol. 2020, 26, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Tajiri, H.; Takano, T.; Tanaka, H.; Ushijima, K.; Inui, A.; Miyoshi, Y.; Ozono, K.; Abukawa, D.; Endo, T.; Brooks, S.; et al. Hepatocellular Carcinoma in Children and Young Patients with Chronic HBV Infection and the Usefulness of Alpha-Fetoprotein Assessment. Cancer Med. 2016, 5, 3102–3110. [Google Scholar] [CrossRef]
- Kelly, D.; Sharif, K.; Brown, R.M.; Morland, B. Hepatocellular carcinoma in children. Clin. Liver Dis. 2015, 19, 433–447. [Google Scholar] [CrossRef]
- Choi, J.Y.; Lee, J.M.; Sirlin, C.B. CT and MR imaging diagnosis and staging of hepatocellular carcinoma: Part Ⅱ. Extracellular agents, hepatobiliary agents, and ancillary imaging features. Radiology 2014, 273, 30–50. [Google Scholar] [CrossRef]
- Saar, B.; Kellner-Weldon, F. Radiological Diagnosis of Hepatocellular Carcinoma. Liver Int. 2008, 28, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Heimbach, J.K.; Kulik, L.M.; Finn, R.S.; Sirlin, C.B.; Abecassis, M.M.; Roberts, L.R.; Zhu, A.X.; Murad, M.H.; Marrero, J.A. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 2018, 67, 358–380. [Google Scholar] [CrossRef] [PubMed]
- Ganeshan, D.; Szklaruk, J.; Kundra, V.; Kaseb, A.; Rashid, A.; Elsayes, K.M. Imaging Features of Fibrolamellar Hepatocellular Carcinoma. Am. J. Roentgenol. 2014, 202, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Vij, M.; Calderaro, J. Pathologic and molecular features of hepatocellular carcinoma: An update. World J. Hepatol. 2021, 13, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.J.; Makhlouf, H.R.; Ranganathan, S. Fibrolamellar variant of hepatocellular carcinoma. In WHO Classification of Tumours Editorial Board, Digestive System Tumours, 5th ed.; WHO Classification of Tumours Series; International Agency for Research on Cancer: Lyon, France, 2019. [Google Scholar]
- Cho, S.J.; Guettier, C.; Hiyama, E.; Hala, R.; Makhlouf, H.R.; Ranganathan, S.; Rangaswami, A. Pediatric hepatocellular carcinoma. In WHO Classification of Tumours Editorial Board, Digestive System Tumours, 5th ed.; WHO Classification of Tumours Series; International Agency for Research on Cancer: Lyon, France, 2019. [Google Scholar]
- Allan, B.J.; Wang, B.; Davis, J.S.; Parikh, P.P.; Perez, E.A.; Neville, H.L.; Sola, J.E. A review of 218 pediatric cases of hepatocellular carcinoma. J. Pediatr. Surg. 2014, 49, 166–171, discussion 171. [Google Scholar] [CrossRef]
- Zen, Y.; Vara, R.; Portmann, B.; Hadzic, N. Childhood hepatocellular carcinoma: A clinicopathological study of 12 cases with special reference to EpCAM. Histopathology 2014, 64, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Yang, H.M. Fibrolamellar Carcinoma: A Concise Review. Arch. Pathol. Lab. Med. 2018, 142, 1141–1145. [Google Scholar] [CrossRef]
- Safwan, M.; Vij, M.; Narasimhan, G.; Shanmugam, N.; Rela, M. Caroli’s Syndrome with Incidental Fibrolamellar Carcinoma on Liver Explant. Indian J. Pediatr. 2016, 83, 85–86. [Google Scholar] [CrossRef]
- Graham, R.P. Fibrolamellar Carcinoma: What Is New and Why It Matters. Surg. Pathol. Clin. 2018, 11, 377–387. [Google Scholar] [CrossRef]
- Graham, R.P.; Terracciano, L.M.; Meves, A.; Vanderboom, P.M.; Dasari, S.; Yeh, M.M.; Torbenson, M.S.; Cruise, M.W. Hepatic adenomas with synchronous or metachronous fibrolamellar carcinomas: Both are characterized by LFABP loss. Mod. Pathol. 2016, 29, 607–615. [Google Scholar] [CrossRef]
- Honeyman, J.N.; Simon, E.P.; Robine, N.; Chiaroni-Clarke, R.; Darcy, D.G.; Lim, I.I.; Gleason, C.E.; Murphy, J.M.; Rosenberg, B.R.; Teegan, L.; et al. Detection of a recurrent DNAJB1-PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science 2014, 343, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.P.; Lackner, C.; Terracciano, L.; González-Cantú, Y.; Maleszewski, J.J.; Greipp, P.T.; Simon, S.M.; Torbenson, M.S. Fibrolamellar carcinoma in the Carney complex: PRKAR1A loss instead of the classic DNAJB1-PRKACA fusion. Hepatology 2018, 68, 1441–1447. [Google Scholar] [CrossRef] [PubMed]
- Malouf, G.G.; Job, S.; Paradis, V.; Fabre, M.; Brugières, L.; Saintigny, P.; Vescovo, L.; Belghiti, J.; Branchereau, S.; Faivre, S.; et al. Transcriptional profiling of pure fibrolamellar hepatocellular carcinoma reveals an endocrine signature. Hepatology 2014, 596, 2228–2237. [Google Scholar] [CrossRef]
- Sumazin, P.; Peters, T.L.; Sarabia, S.F.; Kim, H.R.; Urbicain, M.; Hollingsworth, E.F.; Alvarez, K.R.; Perez, C.R.; Pozza, A.; Najaf Panah, M.J.; et al. Hepatoblastomas with carcinoma features represent a biological spectrum of aggressive neoplasms in children and young adults. J. Hepatol. 2022, 77, 1026–1037. [Google Scholar] [CrossRef]



























































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mesenchymal Tumors |
| Hepatic congenital hemangioma |
| Hepatic infantile hemangioma |
| Hepatic epithelioid hemangioendothelioma |
| Hepatic angiosarcoma |
| Mesenchymal hamartoma |
| Inflammatory myofibrobalstic tumor |
| Embryonal sarcoma of the liver |
| Hepatobiliary Rhabdomyosarcoma |
| Epstein Barr Virus-associated smooth muscle tumor |
| Tumors of uncertain origin |
| Malignant rhabdoid tumor of the liver (INI-1 negative) |
| Epithelial tumors |
| Hepatic adenoma |
| Focal nodular hyperplasia (FNH) |
| Hepatoblastoma |
| Pediatric hepatocellular carcinoma |
| Fibrolamellar hepatocellular carcinoma |
| Hepatoblastoma with carcinoma features |
| Classification | Architecture, Cell Size and Shape, Mitosis | Nucleus | Cytoplasm | IHC |
|---|---|---|---|---|
| Epithelial type | ||||
| Fetal | ||||
| 1–2-cell-thick cords of polygonal cells measuring 10–20 µ with central nuclei. Mitosis <2/10 HPF. Closely packed cells with high N/C ratio, well-delineated plasma membranes, mitosis ≥2/10HPF. | Round nucleus with finely stippled chromatin, well-delineated nuclear membrane, inconspicuous nucleoli. Round nuclei with fine chromatin. | Clear or finely granular and eosinophilic due to variable amounts of glycogen and lipids. Amphophilic cytoplasm due to decreased glycogen | Glypican 3 with finely granular cytoplasmic positivity, strong glutamine synthetase positivity, Hep par-1-positive, b-catenin membranous, cytoplasmic, with rare nuclear staining Strong coarse glypican 3 positivity, glutamine synthetase variable positivity, Hep par-1 positivity, uniform nuclear b-catenin positivity |
| Embryonal | Solid nests, glands, acini, pseudorosettes, or papillae. High mitotic activity level, indistinct cell borders | Enlarged angulated to oval nuclei with coarse chromatin and prominent nucleoli | Scanty dark granular cytoplasm, devoid of lipids and glycogen. | Strong coarse glypican 3 positivity, week glutamine synthetase variable b-catenin nuclear positivity, Hep par-1 positivity |
| Pleomorphic | Bizarre pleomorphic cells. May resemble fetal or embryonal cell types | Anisomorphic irregular large nuclei with coarse chromatin and conspicuous to prominent nucleoli | Eosinophilic to amphophilic cytoplasm | Strong coarse glypican 3 positivity, glutamine synthetase and Hep par-1 variable, strong nuclear b-catenin positivity |
| Small-cell undifferentiated | ||||
| Diffuse pattern, nests, round to oval cells measuring 7–8 µ. Variable mitotis with apoptosis and necrosis. Discohesive cells, resembles rhabdoid tumor | Fine nuclear chromatin with inconspicuous nucleoli. Eccentric irregular nuclei, prominent nucleoli. | Scanty cytoplasm. Abundant cytoplasmic filaments | Co expression of pan-cytokeratin and vimentin. Glypican 3, glutamine synthetase and Hep par-1 are negative |
| Cholangioblastic | Ductular differentiation | Large nuclei with coarse chromatin and prominent nucleoli. | Scanty dark granular cytoplasm | Nuclear b-catenin, CK7, and CK 19 are positive; glypican 3, glutamine synthetase, and Hep par-1 are negative |
| Macrotrabecular | The trabeculae are >5 cells thick. Variable and may resemble fetal or embryonal cell types | Strong nuclear b-cat expression | ||
| Mixed epithelial and Mesenchymal type | ||||
| Mesenchymal components comprise mature and immature fibrous tissue, osteoid and hyaline cartilage. Neuroectodermal-tissue-like glial tissue, neuronal cells, primitive neuroepithelium forming tubules and rosettes, retinal pigment, and squamous epithelium. Mesenchymal-tissue-like striated muscle cartilage and bone. | |||
| Histopathology | Well Differentiated | Moderately Differentiated | Poorly Differentiated |
|---|---|---|---|
| Arrangement | Trabeculae that are at least 3 cells thick and lined by sinusoids, pseudoacinar arrangement | Trabeculae that are 15–20 cells thick lined by sinusoids, pseudoacinar arrangement | Sheets or nests of cells not resembling hepatocytes. |
| Cells and nuclei | Resemble hepatocytes with mild nuclear pleomorphism, centrally placed round nuclei, and abundant cytoplasm | Moderate nuclear pleomorphism, large nuclei with prominent nucleoli and intranuclear inclusions, abundant cytoplasm. | High N/C ratio, nuclear pleomorphism, prominent nucleoli and intranuclear inclusions, numerous mitosis |
| Necrosis | Usually absent | May be seen | Present |
| Immunohistochemistry | Variable glypican 3 and glutamine synthetase levels. Negative for β-catenin nuclear staining | Glypican 3 and glutamine synthetase are positive in 50% of cases, β-catenin is variable | Glypican 3 and glutamine synthetase |
| Tumor | Age at Diagnosis | Gender Preponderance | Risk Factors | Prognosis |
|---|---|---|---|---|
| Hepatic congenital hemangioma | Inutero | Female predominance | - | Good |
| Hepatic infantile hemangioma | <12 months | Female predominance | Multigestational pregnancy, low birth weight, prematurity | Good |
| Epithelioid hemangioendothelioma | 12 years | Female predominance | - | Variable/Uncertain |
| Hepatic angiosarcoma | 2–7 years | Slight female predominance | Exposure to chemical carcinogens, radiation | Poor |
| Mesenchymal mamartoma | <2 years | Slight male predominance | - | Excellent |
| Inflammatory Myofibroblastic tumor | <15 years | No gender predominance | - | Good |
| Malignant rhabdoid tumor | <2 years | Slight male predominance | - | Poor |
| Embryonal sarcoma | 6–10 years | No gender predominance | - | Poor |
| Hepatobiliary rhabdomyosarcoma | 3–4 years | Male predominance | - | Poor |
| Epstein–Barr-virus-associated smooth muscle tumor | 4 years | Female predominance | Immunodeficiency | Poor |
| Hepatocellular adenoma | 14 years | No gender predominance | Genetic disorders, hepatic parenchymal diseases, obesity | Good |
| Focal nodular hyperplasia | 8–11 years | Female predominance | Portosystemic shunts, chemotherapy, radiation therapy | Good |
| Hepatoblastoma | 6 months–3 years | Slight Male predominance | Premature delivery, low birth weight | Good |
| Hepatocellular carcinoma | 10–14 years | Slight male predominance | Hepatitis B, inherited liver diseases, biliary atresia | Poor |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhagat, P.; Vij, M.; Raju, L.P.; Gowrishankar, G.; Menon, J.; Shanmugam, N.; Kaliamoorthy, I.; Rammohan, A.; Rela, M. Update on the Pathology of Pediatric Liver Tumors: A Pictorial Review. Diagnostics 2023, 13, 3524. https://doi.org/10.3390/diagnostics13233524
Bhagat P, Vij M, Raju LP, Gowrishankar G, Menon J, Shanmugam N, Kaliamoorthy I, Rammohan A, Rela M. Update on the Pathology of Pediatric Liver Tumors: A Pictorial Review. Diagnostics. 2023; 13(23):3524. https://doi.org/10.3390/diagnostics13233524
Chicago/Turabian StyleBhagat, Priyanka, Mukul Vij, Lexmi Priya Raju, Gowripriya Gowrishankar, Jagadeesh Menon, Naresh Shanmugam, Ilankumaran Kaliamoorthy, Ashwin Rammohan, and Mohamed Rela. 2023. "Update on the Pathology of Pediatric Liver Tumors: A Pictorial Review" Diagnostics 13, no. 23: 3524. https://doi.org/10.3390/diagnostics13233524