
Integrated Genomic, Transcriptomic and Proteomic Analysis for Identifying Markers of Alzheimer’s Disease

 , , and
, , and 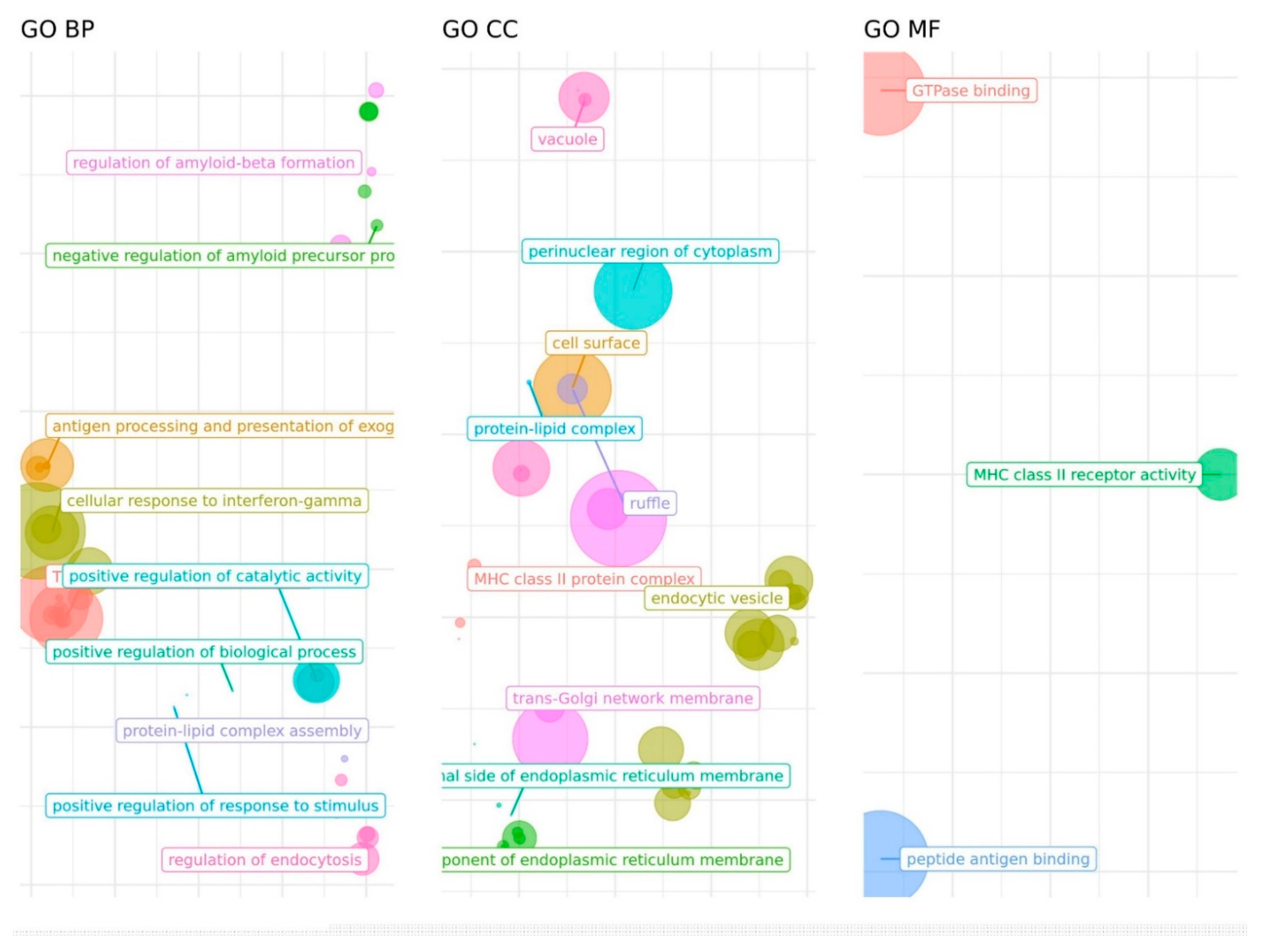{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome-Wide Data
2.2. Expression Quantitative Trait Loci (eQTL) Identification
2.3. Brain and Blood Transcriptomics
2.4. Proteomic Data Analysis
2.5. Enrichment Analysis
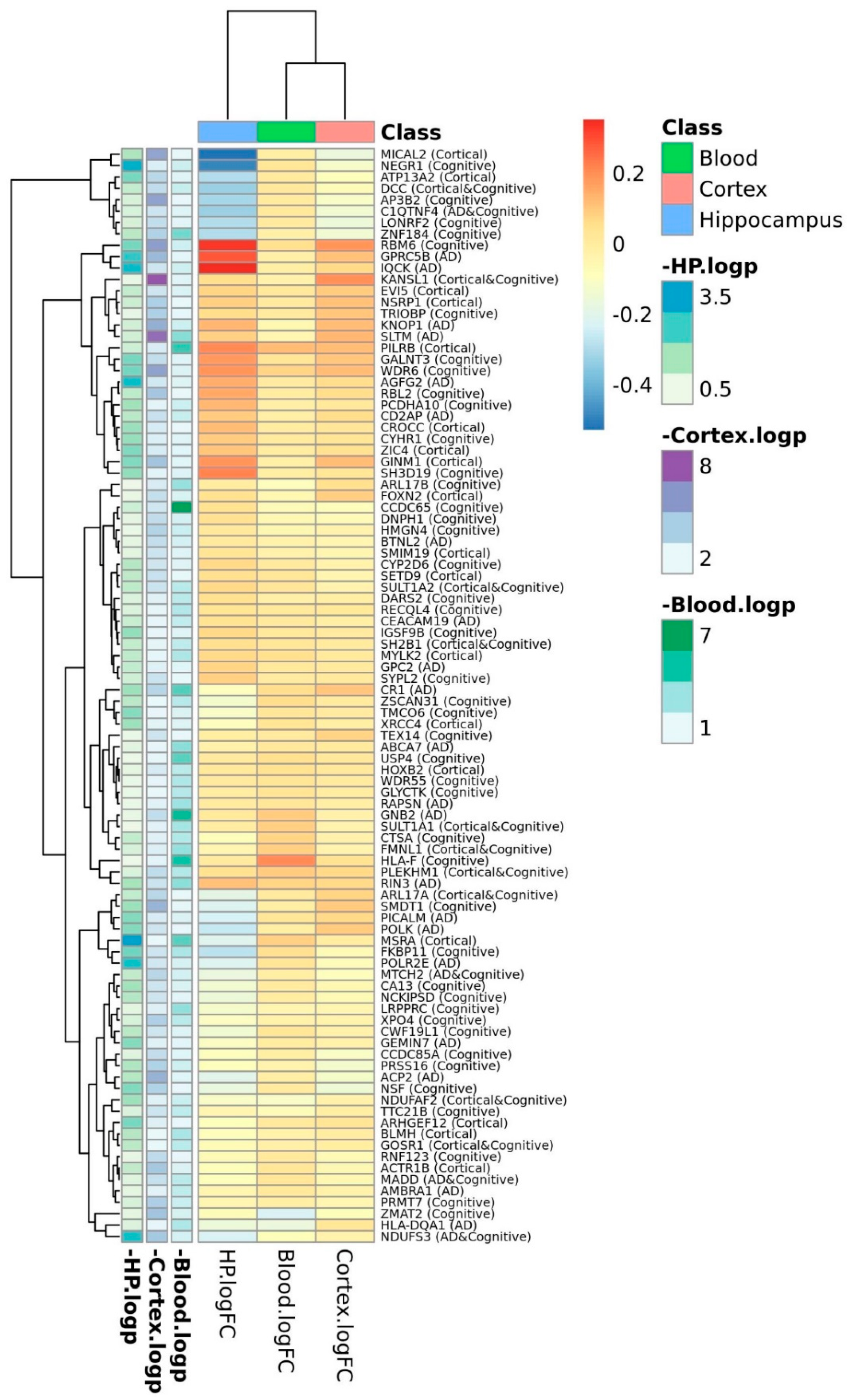
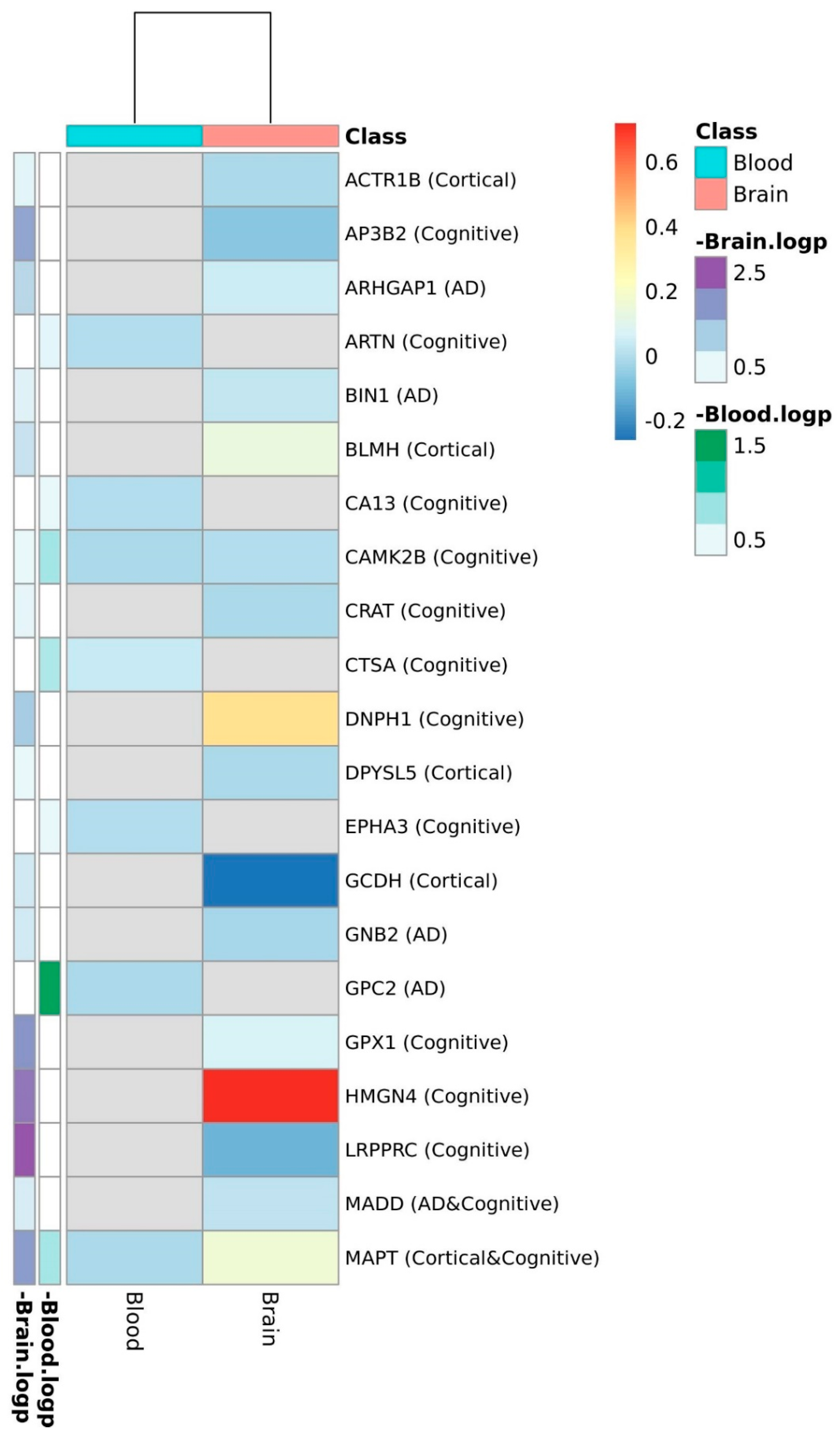
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lambert, J.-C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Moreno-Grau, S.; de Rojas, I.; Hernández, I.; Quintela, I.; Montrreal, L.; Alegret, M.; Hernández-Olasagarre, B.; Madrid, L.; González-Perez, A.; Maroñas, O.; et al. Genome-wide association analysis of dementia and its clinical endophenotypes reveal novel loci associated with Alzheimer disease and three causality networks of AD: The GR@ACE project. Alzheimers Dement. 2019, 15, 1331–1347. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; Van Der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. Available online: https://www.nature.com/articles/s41588-019-0358-2 (accessed on 9 June 2021). [CrossRef] [Green Version]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G.; DeStefano, A.L.; Bis, J.C.; Beecham, G.W.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. Available online: https://pubmed.ncbi.nlm.nih.gov/24162737/ (accessed on 12 July 2021). [CrossRef] [Green Version]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.; Steinberg, S.; Sealock, J.; Karlsson, I.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Sims, R.; Hill, M.; Williams, J. The multiplex model of the genetics of Alzheimer’s disease. Nat. Neurosci. 2020, 23, 311–322. [Google Scholar] [CrossRef]
- Bekris, L.M.; Yu, C.-E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213. Available online: /pmc/articles/PMC3044597/ (accessed on 11 July 2021). [CrossRef] [Green Version]
- Andreasen, N.; Hesse, C.; Davidsson, P.; Minthon, L.; Wallin, A.; Winblad, B.; Vanderstichele, H.; Vanmechelen, E.; Blennow, K. Cerebrospinal fluid beta-amyloid(1-42) in Alzheimer disease: Differences between early- and late-onset Alzheimer disease and stability during the course of disease. Arch. Neurol. 1999, 56, 673–680. Available online: https://pubmed.ncbi.nlm.nih.gov/10369305/ (accessed on 4 November 2021). [CrossRef] [PubMed] [Green Version]
- Vanmechelen, E.; Vanderstichele, H.; Davidsson, P.; van Kerschaver, E.; van der Perre, B.; Sjögren, M. Quantification of tau phosphorylated at threonine 181 in human cerebrospinal fluid: A sandwich ELISA with a synthetic phosphopeptide for standardization. Neurosci. Lett. 2000, 285, 49–52. [Google Scholar] [CrossRef]
- Blennow, K.; Wallin, A.; Ågren, H.; Spenger, C.; Siegfried, J.; Vanmechelen, E. Tau protein in cerebrospinal fluid: A biochemical marker for axonal degeneration in Alzheimer disease? Mol. Chem. Neuropathol. 1995, 26, 231–245. Available online: https://pubmed.ncbi.nlm.nih.gov/8748926/ (accessed on 4 November 2021). [CrossRef]
- Rosengren, L.E.; Karlsson, J.E.; Sjögren, M.; Blennow, K.; Wallin, A. Neurofilament protein levels in CSF are increased in dementia. Neurology 1999, 52, 1090–1093. Available online: https://pubmed.ncbi.nlm.nih.gov/10102440/ (accessed on 4 November 2021). [CrossRef]
- Kvartsberg, H.; Duits, F.H.; Ingelsson, M.; Andreasen, N.; Öhrfelt, A.; Andersson, K.; Brinkmalm, G.; Lannfelt, L.; Minthon, L.; Hansson, O.; et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer’s disease. Alzheimer’s & Dementia. J. Alzheimer’s Assoc. 2015, 11, 1180–1190. Available online: https://pubmed.ncbi.nlm.nih.gov/25533203/ (accessed on 4 November 2021).
- Pametti, L.; Palumbo, B.; Cardinali, L.; Loreti, F.; Chionne, F.; Cecchetti, R.; Senin, U. Cerebrospinal fluid neuron-specific enolase in Alzheimer’s disease and vascular dementia. Neurosci. Lett. 1995, 183, 43–45. Available online: https://pubmed.ncbi.nlm.nih.gov/7746484/ (accessed on 4 November 2021). [CrossRef]
- Lee, J.M.; Blennow, K.; Andreasen, N.; Laterza, O.; Modur, V.; Olander, J.; Gao, F.; Ohlendorf, M.; Ladenson, J.H. The brain injury biomarker VLP-1 is increased in the cerebrospinal fluid of Alzheimer disease patients. Clin. Chem. 2008, 54, 1617–1623. Available online: https://pubmed.ncbi.nlm.nih.gov/18703769/ (accessed on 4 November 2021). [CrossRef] [PubMed] [Green Version]
- Ishizuka, K.; Kimura, T.; Igata-Yi, R.; Katsuragi, S.; Takamatsu, J.; Miyakawa, T. Identification of monocyte chemoattractant protein-1 in senile plaques and reactive microglia of Alzheimer’s disease. Psychiatry Clin. Neurosci. 1997, 51, 135–138. Available online: https://pubmed.ncbi.nlm.nih.gov/9225377/ (accessed on 4 November 2021). [CrossRef]
- Crols, R.; Saerens, J.; Noppe, M.; Lowenthal, A. Increased GFAp levels in CSF as a marker of organicity in patients with Alzheimer’s disease and other types of irreversible chronic organic brain syndrome. J. Neurol. 1986, 233, 157–160. Available online: https://pubmed.ncbi.nlm.nih.gov/3522811/ (accessed on 4 November 2021). [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. Available online: https://pubmed.ncbi.nlm.nih.gov/29377008/ (accessed on 4 November 2021). [CrossRef]
- Janelidze, S.; Stomrud, E.; Palmqvist, S.; Zetterberg, H.; van Westen, D.; Jeromin, A.; Song, L.; Hanlon, D.; Tan Hehir, C.A.; Baker, D.; et al. Plasma β-amyloid in Alzheimer’s disease and vascular disease. Sci. Rep. 2016, 6, 26801. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Hill, E.; Goodwill, A.; Gorelik, A.; Szoeke, C. Diet and biomarkers of Alzheimer’s disease: A systematic review and meta-analysis. Neurobiol. Aging 2019, 76, 45–52. [Google Scholar] [CrossRef]
- Rehiman, S.H.; Lim, S.M.; Neoh, C.F.; Majeed, A.B.; Chin, A.V.; Tan, M.P.; Kamaruzzaman, S.B.; Ramasamy, K. Proteomics as a reliable approach for discovery of blood-based Alzheimer’s disease biomarkers: A systematic review and meta-analysis. Ageing Res. Rev. 2020, 60, 101066. [Google Scholar] [CrossRef]
- Davies, G.; Lam, M.; Harris, S.E.; Trampush, J.W.; Luciano, M.; Hill, W.D.; Hagenaars, S.P.; Ritchie, S.J.; Marioni, R.E.; Fawns-Ritchie, C.; et al. Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Savage, J.E.; Jansen, P.R.; Stringer, S.; Watanabe, K.; Bryois, J.; de Leeuw, C.; Nagel, M.; Awasthi, S.; Barr, P.B.; Coleman, J.R.I.; et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat. Genet. 2018, 50, 912–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofer, E.; Roshchupkin, G.V.; Adams, H.H.H.; Knol, M.J.; Lin, H.; Li, S.; Zare, H.; Ahmad, S.; Armstrong, N.J.; Satizabal, C.L.; et al. Genetic correlations and genome-wide associations of cortical structure in general population samples of 22,824 adults. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Grasby, K.L.; Jahanshad, N.; Painter, J.N.; Colodro-Conde, L.; Bralten, J.; Hibar, D.P.; Lind, P.A.; Pizzagalli, F.; Ching, C.R.K.; McMahon, M.A.B.; et al. The genetic architecture of the human cerebral cortex. Science 2020, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satizabal, C.L.; Adams, H.H.H.; Hibar, D.P.; White, C.C.; Knol, M.J.; Stein, J.L.; Scholz, M.; Sargurupremraj, M.; Jahanshad, N.; Roshchupkin, G.V.; et al. Genetic architecture of subcortical brain structures in 38,851 individuals. Nat. Genet. 2019, 51, 1624–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, J.A.; Gibbs, J.R.; Clarke, J.; Ray, M.; Zhang, W.; Holmans, P.; Rohrer, K.; Zhao, A.; Marlowe, L.; Kaleem, M.; et al. Genetic Control of Human Brain Transcript Expression in Alzheimer Disease. Am. J. Hum. Genet. 2009, 84, 445–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berchtold, N.C.; Cribbs, D.H.; Coleman, P.D.; Rogers, J.; Head, E.; Kim, R.; Beach, T.; Miller, C.; Troncoso, J.; Trojanowski, J.Q.; et al. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc. Natl. Acad. Sci. USA 2008, 105, 15605–15610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamimura, K.; Maeda, N. Glypicans and Heparan Sulfate in Synaptic Development, Neural Plasticity, and Neurological Disorders. Front. Neural Circuits 2021, 15, 15. [Google Scholar] [CrossRef]
- Lugert, S.; Kremer, T.; Jagasia, R.; Herrmann, A.; Aigner, S.; Giachino, C.; Mendez-David, I.; Gardier, A.M.; Carralot, J.P.; Meistermann, H.; et al. Glypican-2 levels in cerebrospinal fluid predict the status of adult hippocampal neurogenesis. Sci. Rep. 2017, 7, 46543. [Google Scholar] [CrossRef] [Green Version]
- de Rojas, I.; Moreno-Grau, S.; Tesi, N.; Grenier-Boley, B.; Andrade, V.; Jansen, I.E.; Pedersen, N.L.; Stringa, N.; Zettergren, A.; Hernández, I.; et al. Common variants in Alzheimer’s disease and risk stratification by polygenic risk scores. Nat. Commun. 2021, 12, 3417. [Google Scholar] [CrossRef]
- Shen, L.; Yao, X. Biomarkers. Poster presentations Biomarkers (non-neuroimaging)/Differential diagnosis Integrative analysis of summary data from GWAS and eQTL studies predicts tissue-specific gene targets for Alzheimer’s disease. Alzheimer’s Dement. 2020, 16. [Google Scholar] [CrossRef]
- Whelan, C.D.; Mattsson, N.; Nagle, M.W.; Vijayaraghavan, S.; Hyde, C.; Janelidze, S.; Stomrud, E.; Lee, J.; Fitz, L.; Samad, T.A.; et al. Multiplex proteomics identifies novel CSF and plasma biomarkers of early Alzheimer’s disease. Acta Neuropathol. Commun. 2019, 7, 169. [Google Scholar] [CrossRef]
- Van Horssen, J.; Wesseling, P.; Van Den Heuvel, L.P.W.J.; De Waal, R.M.W.; Verbeek, M.M. Heparan sulphate proteoglycans in Alzheimer’s disease and amyloid-related disorders. Lancet Neurol. 2003, 2, 482–492. [Google Scholar] [CrossRef]
- Sierksma, A.; Lu, A.; Mancuso, R.; Fattorelli, N.; Thrupp, N.; Salta, E.; Zoco, J.; Blum, D.; Buée, L.; De Strooper, B.; et al. Novel Alzheimer risk genes determine the microglia response to amyloid-β but not to TAU pathology. EMBO Mol. Med. 2020, 12, e10606. [Google Scholar] [CrossRef]
- O’Callaghan, P.; Sandwall, E.; Li, J.-P.; Yu, H.; Ravid, R.; Guan, Z.-Z.; van Kuppevelt, T.H.; Nilsson, L.N.G.; Ingelsson, M.; Hyman, B.T.; et al. Heparan Sulfate Accumulation with Aβ Deposits in Alzheimer’s Disease and Tg2576 Mice is Contributed by Glial Cells. Brain Pathol. 2008, 18, 548–561. [Google Scholar] [CrossRef] [Green Version]
- Madrid, L.; Moreno-Grau, S.; Ahmad, S.; González-Pérez, A.; de Rojas, I.; Xia, R.; Adami, P.V.M.; García-González, P.; Kleineidam, L.; Yang, Q.; et al. Multiomics integrative analysis identifies APOE allele-specific blood biomarkers associated to Alzheimer’s disease etiopathogenesis. Aging 2021, 13, 9277–9329. [Google Scholar] [CrossRef]
- Chen, J.-F.; Liu, K.; Hu, B.; Li, R.-R.; Xin, W.; Chen, H.; Wang, F.; Chen, L.; Li, R.-X.; Ren, S.-Y.; et al. Enhancing myelin renewal reverses cognitive dysfunction in a murine model of Alzheimer’s disease. Neuron 2021, 109, 2292–2307.e5. [Google Scholar] [CrossRef] [PubMed]
- Hudák, A.; Jósvay, K.; Domonkos, I.; Letoha, A.; Szilák, L.; Letoha, T. The Interplay of Apoes with Syndecans in Influencing Key Cellular Events of Amyloid Pathology. Int. J. Mol. Sci. 2021, 22, 7070. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Fransson, L.Å.; Mani, K. Reversal of apolipoprotein E4-dependent or chemical-induced accumulation of APP degradation products by vitamin C-induced release of heparan sulfate from glypican-1. Glycobiology 2021, 31, 800–811. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madrid, L.; Labrador, S.C.; González-Pérez, A.; Sáez, M.E.; The Alzheimer’s Disease Neuroimaging Initiative (ADNI). Integrated Genomic, Transcriptomic and Proteomic Analysis for Identifying Markers of Alzheimer’s Disease. Diagnostics 2021, 11, 2303. https://doi.org/10.3390/diagnostics11122303
Madrid L, Labrador SC, González-Pérez A, Sáez ME, The Alzheimer’s Disease Neuroimaging Initiative (ADNI). Integrated Genomic, Transcriptomic and Proteomic Analysis for Identifying Markers of Alzheimer’s Disease. Diagnostics. 2021; 11(12):2303. https://doi.org/10.3390/diagnostics11122303
Chicago/Turabian StyleMadrid, Laura, Sandra C. Labrador, Antonio González-Pérez, María E. Sáez, and The Alzheimer’s Disease Neuroimaging Initiative (ADNI). 2021. "Integrated Genomic, Transcriptomic and Proteomic Analysis for Identifying Markers of Alzheimer’s Disease" Diagnostics 11, no. 12: 2303. https://doi.org/10.3390/diagnostics11122303