
TP53-Mutated Circulating Tumor DNA for Disease Monitoring in Lymphoma Patients after CAR T Cell Therapy

Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Collection and Processing of Peripheral Blood (PB)
2.3. Cell-Free DNA Extraction
2.4. Probe Design
2.5. Droplet Digital PCR
2.6. QX200 Droplet Reader
2.7. Statistics
3. Results
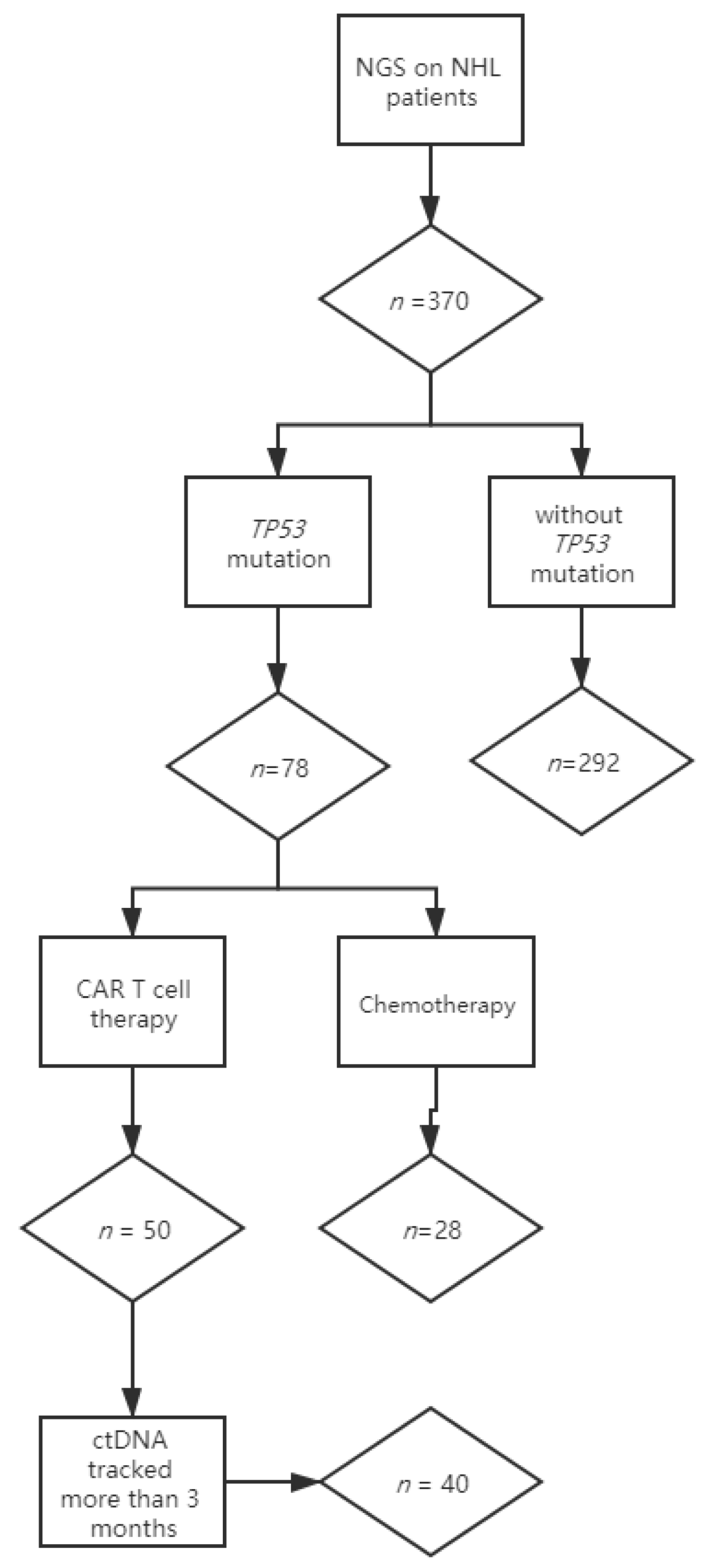
3.1. Monitoring Strategy
3.2. Distribution of TP53 Mutations in the Patient Cohort
3.3. Validation of ddPCR Assays
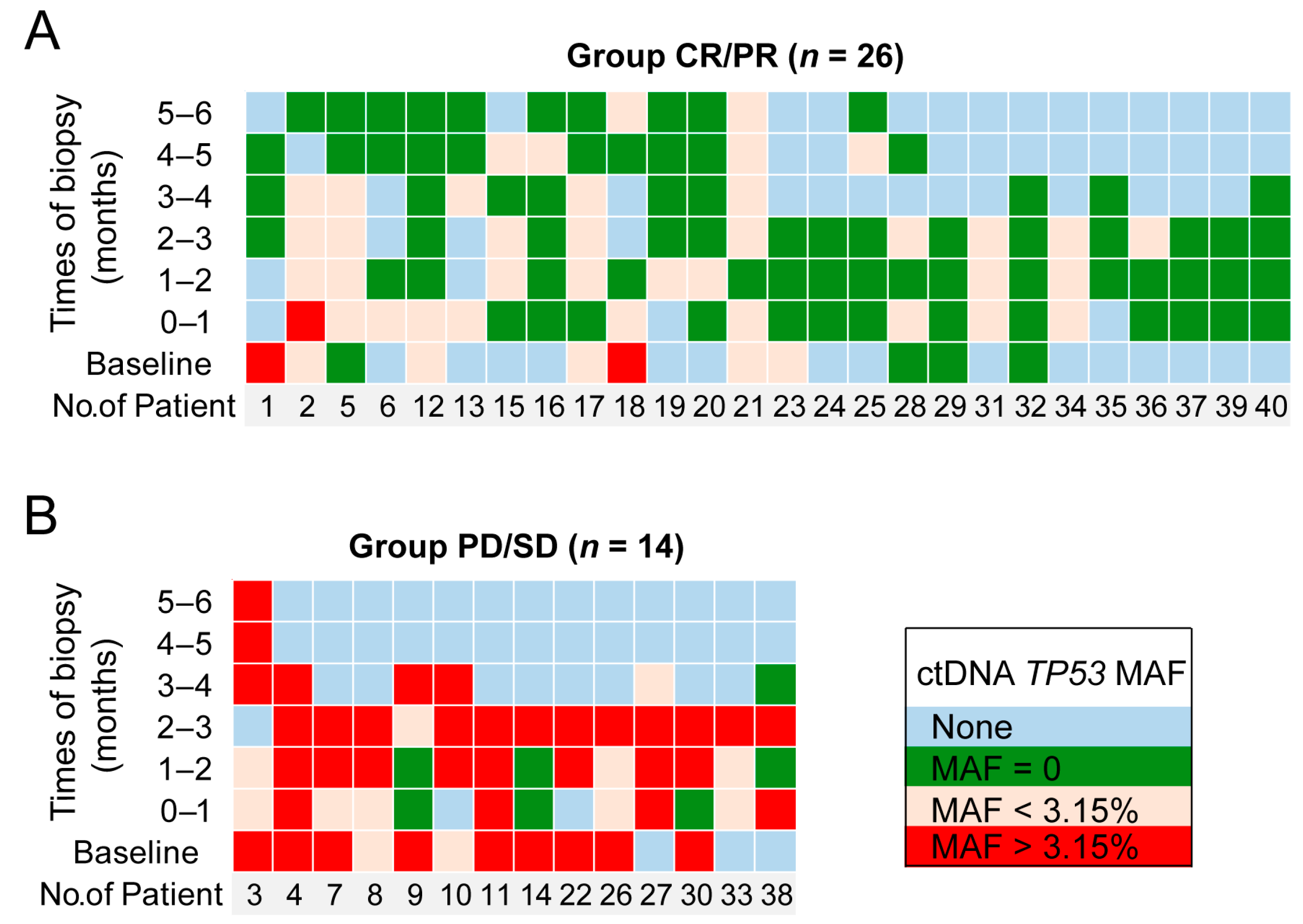
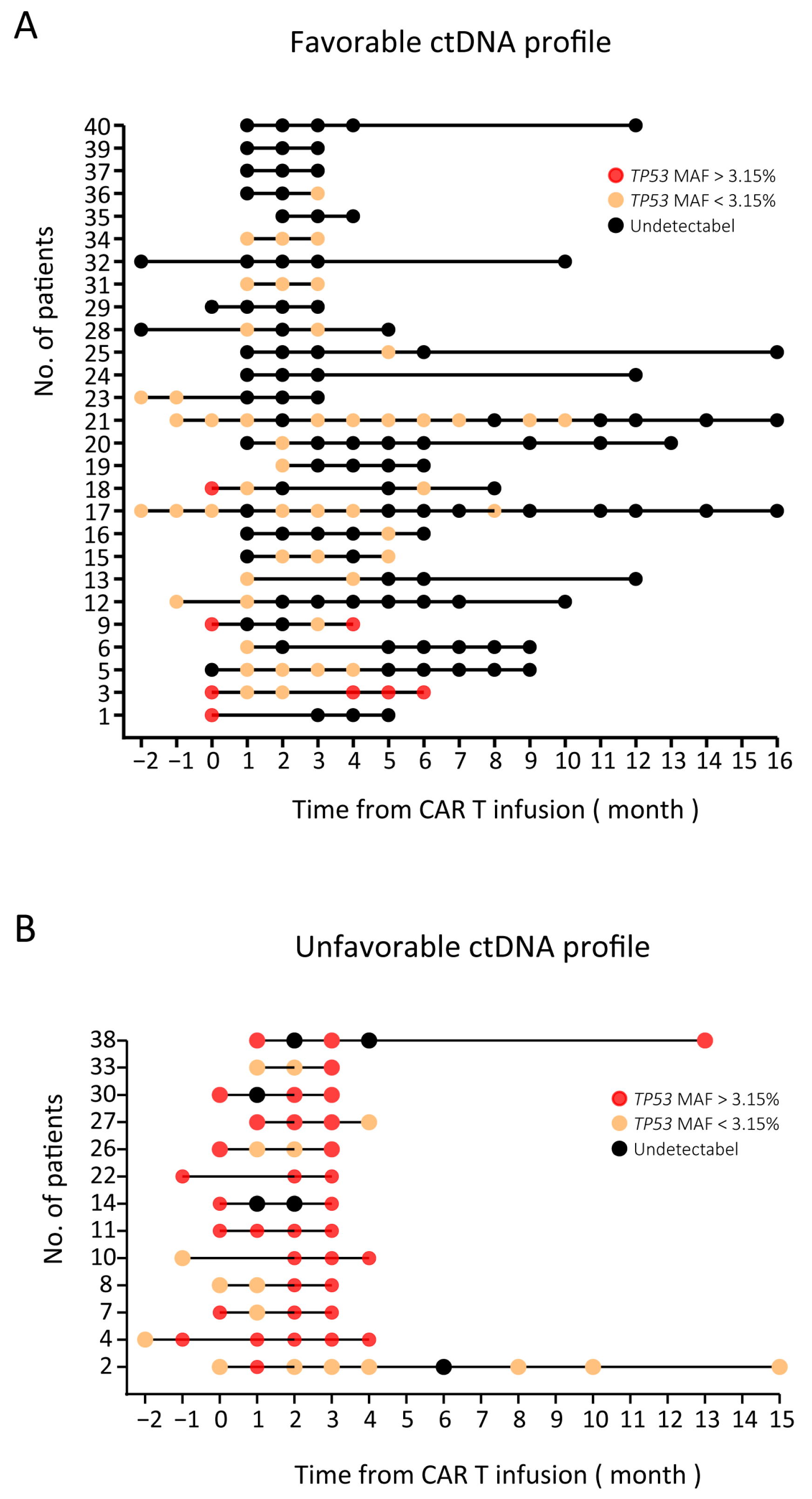
3.4. ctDNA Monitoring Results (TP53 MAF) in NHL Patients
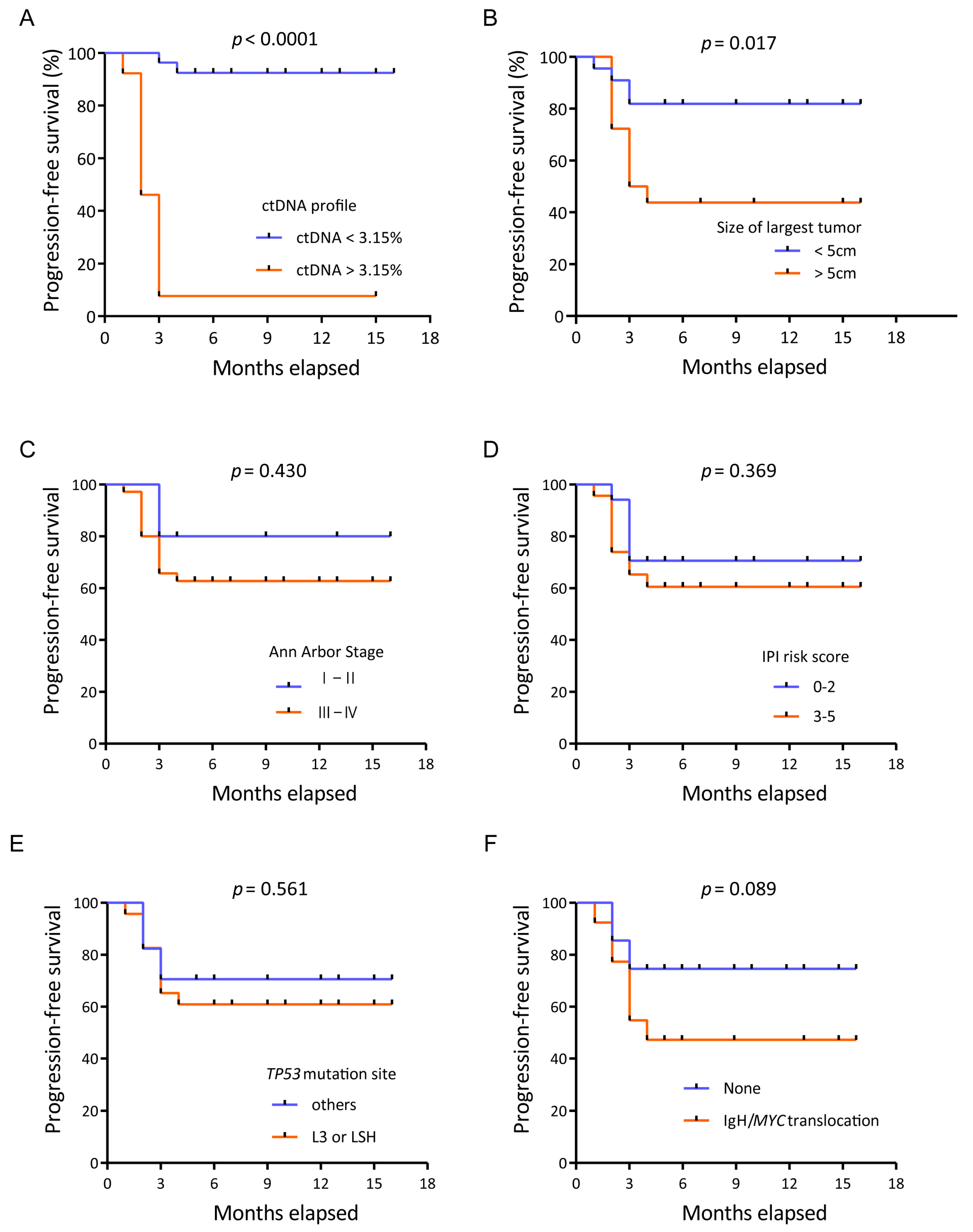
3.5. Prognostic Value of TP53 in ctDNA for Patients Receiving CAR T Cell Therapy
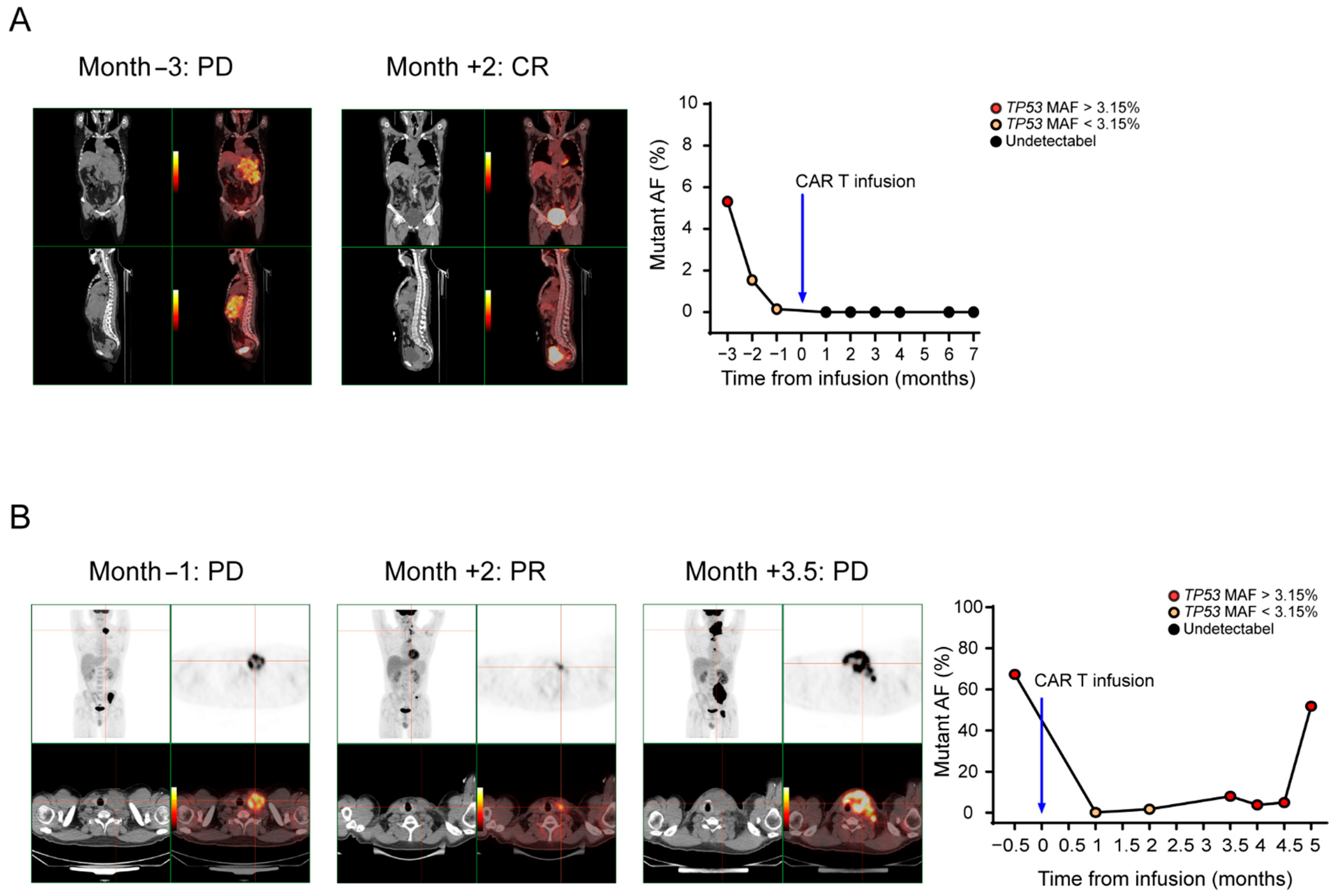
3.6. ctDNA Levels Can Reflect Tumor Burden
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CAR T | Chimeric antigen receptor T |
| NHL | Non-Hodgkin’s lymphoma |
| MRD | Minimal residual disease |
| ctDNA | Circulating tumor DNA |
| ddPCR | Droplet digital PCR |
| OS | Overall survival |
| PFS | Progression-free survival |
| PB | Peripheral blood |
| MAF | Mutant allele fraction |
| PROVEAN | Protein Variation Effect Analyzer |
| NGS | Next-generation sequencing |
| IWG | International Working Group |
| LSH | Loop-sheet-helix |
| HR | Hazard ratio |
| DLBCL | Diffuse large B cell lymphoma |
| FL | Follicular lymphoma |
| AUC | Area under the curve |
| ROC | Receiver operating characteristic |
References
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Hay, K.A.; Turtle, C.J. Chimeric Antigen Receptor (CAR) T Cells: Lessons Learned from Targeting of CD19 in B-Cell Malignancies. Drugs 2017, 77, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, V.A.; Shadman, M.; Gopal, A.K. Translating anti-CD19 CAR T-cell therapy into clinical practice for relapsed/refractory diffuse large B-cell lymphoma. Blood 2018, 132, 777–781. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Hoon, D.S.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer Clin. Oncol. 2011, 1, 426. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Bhattacharya, S.; Kotsopoulos, S.; Olson, J.; Taly, V.; Griffiths, A.D.; Link, D.R.; Larson, J.W. Multiplex digital PCR: Breaking the one target per color barrier of quantitative PCR. Lab Chip 2011, 11, 2167–2174. [Google Scholar] [CrossRef] [Green Version]
- Beaver, J.A.; Jelovac, D.; Balukrishna, S.; Cochran, R.; Croessmann, S.; Zabransky, D.J.; Wong, H.Y.; Toro, P.V.; Cidado, J.; Blair, B.G.; et al. Detection of cancer DNA in plasma of patients with early-stage breast cancer. Clin. Cancer Res. 2014, 20, 2643–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joerger, A.C.; Fersht, A.R. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919. [Google Scholar] [CrossRef] [PubMed]
- Robles, A.I.; Harris, C.C. Clinical outcomes and correlates of TP53 mutations and cancer. Csh Perspect. Biol. 2010, 2, a001016. [Google Scholar] [CrossRef] [Green Version]
- Halldórsdóttir, A.M.; Lundin, A.; Murray, F.; Mansouri, L.; Knuutila, S.; Sundstr, M.C.; Laurell, A.; Ehrencrona, H.; Sander, B.; Rosenquist, R. Impact of TP53 mutation and 17p deletion in mantle cell lymphoma. Leukemia 2011, 25, 1904. [Google Scholar] [CrossRef] [Green Version]
- Young, K.H.; Leroy, K.; Moller, M.B.; Colleoni, G.W.; Sanchez-Beato, M.; Kerbauy, F.R.; Haioun, C.; Eickhoff, J.C.; Young, A.H.; Gaulard, P.; et al. Structural profiles of TP53 gene mutations predict clinical outcome in diffuse large B-cell lymphoma: An international collaborative study. Blood 2008, 112, 3088–3098. [Google Scholar] [CrossRef] [Green Version]
- Xu-Monette, Z.Y.; Wu, L.; Visco, C.; Tai, Y.C.; Tzankov, A.; Liu, W.M.; Montes-Moreno, S.; Dybkaer, K.; Chiu, A.; Orazi, A.; et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: Report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood 2012, 120, 3986–3996. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Hilden, P.; Coiffier, B.; Hagenbeek, A.; Salles, G.; Wilson, W.; Seymour, J.F.; Kelly, K.; Gribben, J.; Pfreunschuh, M. International Working Group consensus response evaluation criteria in lymphoma (RECIL 2017). Ann. Oncol. 2017, 28, 1436. [Google Scholar] [CrossRef] [PubMed]
- Voropaeva, E.N.; Pospelova, T.I.; Voevoda, M.I.; Maksimov, V.N. Frequency, spectrum, and functional significance of TP53 mutations in patients with diffuse large B-cell lymphoma. Mol. Biol. 2017, 51, 53–60. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baugh, E.H.; Hua, K.; Levine, A.J.; Bonneau, R.A.; Chang, S.C. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2017, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Hagn, F.; Lagleder, S.; Retzlaff, M.; Rohrberg, J.; Demmer, O.; Richter, K.; Buchner, J.; Kessler, H. Structural analysis of the interaction between Hsp90 and the tumor suppressor protein p53. Nat. Struct. Mol. Biol. 2011, 18, 1086–1093. [Google Scholar] [CrossRef]
- Fesnak, A.D.; June, C.H.; Levine, B.L. Engineered T cells: The promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Hu, X.; Cao, W.; Li, C.; Xiao, Y.; Cao, Y.; Gu, C.; Zhang, S.; Chen, L.; Cheng, J.; et al. Efficacy and Safety of CAR19/22 T-cell “Cocktail” Therapy in Patients with Refractory/ Relapsed B-Cell Malignancies. Blood 2019, 135, 17–27. [Google Scholar] [CrossRef]
- Huntington, S.F.; Jakub, S.; Doshi, J.A. Cost-effectiveness analysis of routine surveillance imaging of patients with diffuse large B-cell lymphoma in first remission. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 1467–1474. [Google Scholar] [CrossRef]
- Christopher, M.; Wilson, W.H.; Mark, R. Monitoring Clinical Outcomes in Aggressive B-cell Lymphoma: From Imaging Studies to Circulating Tumor DNA. Best Pract. Res. Clin. Haematol. 2018, 31, 285–292. [Google Scholar]
- Rivas-Delgado, A.; Nadeu, F.; Enjuanes, A.; Casanueva-Eliceiry, S.; Mozas, P.; Magnano, L.; Castrejon, D.A.N.; Rovira, J.; Dlouhy, I.; Martin, S.; et al. Mutational Landscape and Tumor Burden Assessed by Cell-free DNA in Diffuse Large B-Cell Lymphoma in a Population-Based Study. Clin. Cancer Res. 2021, 27, 513–521. [Google Scholar] [CrossRef]
- Roschewski, M.; Staudt, L.M.; Wilson, W.H. Dynamic monitoring of circulating tumor DNA in non-Hodgkin lymphoma. Blood 2016, 127, 3127–3132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, D.M.; Green, M.R.; Bratman, S.V.; Florian, S.; Chih Long, L.; Kunder, C.A.; Kazuhiro, T.; Cynthia, G.; Colm, K.; Shingo, K. Noninvasive monitoring of diffuse large B-cell lymphoma by immunoglobulin high-throughput sequencing. Blood 2015, 125, 3679–3687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, D.; Diop, F.; Spaccarotella, E.; Monti, S.; Zanni, M.; Rasi, S.; Deambrogi, C.; Spina, V.; Bruscaggin, A.; Favini, C. Diffuse large B-cell lymphoma genotyping on the liquid biopsy. Blood 2017, 129, 1947. [Google Scholar] [CrossRef]
- Postel, M.; Roosen, A.; Laurentpuig, P.; Taly, V.; Wangrenault, S.F. Droplet-based digital PCR and next generation sequencing for monitoring circulating tumor DNA: A cancer diagnostic perspective. Expert Rev. Mol. Diagn. 2017, 18, 14737159. [Google Scholar] [CrossRef]
- Cheson, B.D. Hematologic malignancies: New developments and future treatments. Semin Oncol. 2002, 29, 33–45. [Google Scholar] [CrossRef]
- Hainaut, P.; Hollstein, M. p 53 and Human Cancer: The First Ten Thousand Mutations. Adv. Cancer Res. 1999, 77, 81–86. [Google Scholar]
- Levine, A.J.; Hu, W.; Feng, Z. The P53 pathway: What questions remain to be explored? Cell Death Differ. 2006, 13, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Chasov, V.; Mirgayazova, R.; Zmievskaya, E.; Khadiullina, R.; Valiullina, A.; Stephenson, C.J.; Rizvanov, A.; Baud, M.; Bulatov, E. Key Players in the Mutant p53 Team: Small Molecules, Gene Editing, Immunotherapy. Front. Oncol. 2020, 10, 1460. [Google Scholar] [CrossRef] [PubMed]
- Xu-Monette, Z.Y.; Young, K.H. The TP53 tumor suppressor and autophagy in malignant lymphoma. Autophagy 2015, 8, 842–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. of Patient | Protein Change | Nucleotide Change | Mutant Probe | Forward Primer | Reverse Primer |
|---|---|---|---|---|---|
| 28 | p.E336fs | c.1007_1020del | TGGGCGTGTTCCGAGAG | CCTCCTCTGTTGCTGCAGATC | TGAGTTCCAAGGCCTCATTCA |
| 40 | p.S106R | c.318C > G | CAGGGCAGGTACGGT | CTGTCCCTTCCCAGAAAACCT | GGCTGTCCCAGAATGCAAGA |
| 6 | - | c.376-1G > A | TTCCTACAATACTCCCCT | TGACTTTCAACTCTGTCTCCTTCCT | GCTGCACAGGGCAGGTCTT |
| 3 | p.Y126N | c.376T > A | CAGGACTCCCCTGCC | CAACTCTGTCTCCTTCCTCTTCCT | GGCCAGTTGGCAAAACATCTT |
| 24 | p.A138V | c.413C > T | CAGGTCTTGACCAGTT | ATGTGCTGTGACTGCTTGTAGATG | GCCCTCAACAAGATGTTTTGC |
| 1 32 | p.W146 * | c.437G > A | TGCAGCTGTAGGTTGAT | TCAACAAGATGTTTTGCCAACTG | ATGTGCTGTGACTGCTTGTAGATG |
| 15 | p.Y163C | c.488A > G | CCATCTGCAAGCAG | CTGCCCTCAACAAGATGTTTTG | CCTCACAACCTCCGTCATGTG |
| 20 23 | p.R175H | c.524G > A | ATGGGCCTCCAGTTC | TGGAGTCTTCCAGTGTGATGATG | CAACTACATGTGTAACAGTTCCTGCAT |
| 19 | p.D208Y | c.622G > T | ATGGGTTTAACTATTTCACAG | ACCCCATGAGATGTGCAAAGTA | GCCTCTGTAAGCTTCAGTTTTTTCA |
| 25 | p.R213 * | c.637C > T | CACTTTTTGACATAGTGTG | CGTGTGGAGTATTTGGATGACAGA | AGACCTCAGGCGGCTCATAG |
| 26 | p.R213Q | c.638G > A | CACTTTTCAACATAGTG | CGTGTGGAGTATTTGGATGACAGA | AGACCTCAGGCGGCTCATAG |
| 5 | p.I232N | c.695T > A | TACCACCAACCACTACA | TGGGCCTGTGTTATCTCCTAGGT | CCATGCAGGAACTGTTACACATG |
| 30 | p.Y234D | c.700T > G | ACCATCCACGACAAC | TCTCCTAGGTTGGCTCTGACTGT | CCATGCAGGAACTGTTACACATG |
| 33 | p.Y236D | c.706T > G | CACTACAACGACATGTGTAA | TTGGCTCTGACTGTACCACCAT | TGGGCCTCCGGTTCATG |
| 9 | p.C238W | c.714T > G | CATGTGGAACAGTTCC | TCTGACTGTACCACCATCCACTACA | TGGGCCTCCGGTTCATG |
| 14 36 | p.N239D | c.715A > G | TGTGTGACAGTTCC | TGACTGTACCACCATCCACTACAA | GATGGGCCTCCGGTTCAT |
| 11 | p.C242Y | c.725G > A | TGTAACAGTTCCTACATG | CTGTACCACCATCCACTACAACTACA | TGGGCCTCCGGTTCATG |
| 7 13 31 | p.G245S | c.733G > A | CCGGTTCATGCTGCC | TGGAGTCTTCCAGTGTGATGATG | AACTACATGTGTAACAGTTCCTGCAT |
| 35 | p.R248W | c.742C > T | ATGGGCCTCCAGTTC | TGGAGTCTTCCAGTGTGATGATG | CAACTACATGTGTAACAGTTCCTGCAT |
| 8 18 21 27 | p.R248Q | c.743G > A | ATGGGCCTCTGGTTCA | TGGAGTCTTCCAGTGTGATGATG | CAACTACATGTGTAACAGTTCCTGCAT |
| 39 | p.R249S | c.747G > C | AACCGGAGCCCCAT | GTACCACCATCCACTACAACTACATGT | GGCTCCTGACCTGGAGTCTTC |
| 22 | p.T253Pfs * 92 | c.756delC | CCATCCTACCATCATC | GCATGGGCGGCATGAA | GGCTCCTGACCTGGAGTCTTC |
| 37 | p.E258D | c.774A > C | CACTGGACGACTCC | TTGGCTCTGACTGTACCACCAT | TGTGCAGGGTGGCAAGTG |
| 10 | p.R267W | c.799C > T | CTACTGGGATGGAACAG | GCTTCTCTTTTCCTATCCTGAGTAGTG | GCACAAACACGCACCTCAAA |
| 17 | p.R273C | c.817C > T | CTTTGAGGTGTGTGTTT | TGGTAATCTACTGGGACGGAACA | CTCTGTGCGCCGGTCTCT |
| 2 4 34 38 | p.R273H | c.818G > A | AGGTGCATGTTTGTGCC | TCTACTGGGACGGAACAGCTTT | CTCTGTGCGCCGGTCTCT |
| 16 | p.D281V | c.842A > T | TGGGAGAGTCCGGCG | AGCTTTGAGGTGCGTGTTTGT | TGCGGAGATTCTCTTCCTCTGT |
| 29 | p.R282W | c.844C > T | TGTGCGCCAGTCT | CCTTTCTTGCGGAGATTCTCTTC | AGCTTTGAGGTGCGTGTTTGT |
| 12 | p.R282-R283insHR | c.847-848insATCGGG | CCGGTTCATGCTGCC | TGGAGTCTTCCAGTGTGATGATG | AACTACATGTGTAACAGTTCCTGCAT |
| Characteristic | Favorable | Unfavorable | Total | p Value |
|---|---|---|---|---|
| Age, median (range) | 43.5 (27–64) | |||
| Age group, years | ||||
| Younger than 60 y | 26 | 12 | 38 (95%) | 0.59 |
| At least 60 y | 1 | 1 | 2 (5%) | |
| Gender | ||||
| Male | 21 | 11 | 32 (80%) | 0.61 |
| Female | 6 | 2 | 8 (20%) | |
| IPI risk score | ||||
| 0–2 | 13 | 4 | 17 (42.5%) | 0.30 |
| 3–5 | 14 | 9 | 23 (57.5%) | |
| Ann Arbor stage | ||||
| I–II | 4 | 1 | 5 (12.5%) | 0.95 |
| III–IV | 23 | 12 | 35 (87.5%) | |
| Volume of disease | ||||
| <5 cm | 19 | 3 | 22 (55%) | 0.01 |
| >5 cm | 8 | 10 | 18 (45%) | |
| Disease status | ||||
| Primary refractory | 12 | 7 | 19 (47.5%) | |
| First relapse | 8 | 4 | 12 (30%) | 0.74 |
| ≥Second relapse | 7 | 2 | 9 (22.5%) | |
| Genetics | ||||
| IgH/MYC translocation | 7 | 3 | 13 (32.5%) | 0.72 |
| TP53 mutation with del (17p) | 5 | 2 | 7 (17.5%) | 0.81 |
| All TP53 mutations | ||||
| Outside L3 and LSH | 12 | 5 | 17 (42.5%) | 0.72 |
| Loop-L3 and LSH motifs | 15 | 8 | 23 (57.5%) |
| Characteristic | Variable | Progression-Free Survival | |
|---|---|---|---|
| Hazard Ratio (95% CI) | p Value | ||
| ctDNA | Unfavorable/favorable | 19.45 (5.62–66.82) | <0.0001 |
| Volume of disease | >5 cm/<5 cm | 3.44 (1.19–9.95) | 0.017 |
| Ann Arbor stage | Ⅲ–Ⅳ/I–II | 2.11 (0.47–9.57) | 0.430 |
| IPI risk score | 3–5/0–2 | 1.52 (0.53–4.35) | 0.369 |
| TP53 mutation site | LSH L3/others | 1.35 (0.47–3.90) | 0.561 |
| IgH/MYC translocation | Translocation/none | 2.29 (0.73–7.14) | 0.089 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Mu, W.; Gu, J.; Xiao, M.; Huang, L.; Zheng, M.; Li, C.; Xiao, Y.; Zhou, J.; Long, X. TP53-Mutated Circulating Tumor DNA for Disease Monitoring in Lymphoma Patients after CAR T Cell Therapy. Diagnostics 2021, 11, 844. https://doi.org/10.3390/diagnostics11050844
Chen L, Mu W, Gu J, Xiao M, Huang L, Zheng M, Li C, Xiao Y, Zhou J, Long X. TP53-Mutated Circulating Tumor DNA for Disease Monitoring in Lymphoma Patients after CAR T Cell Therapy. Diagnostics. 2021; 11(5):844. https://doi.org/10.3390/diagnostics11050844
Chicago/Turabian StyleChen, Liting, Wei Mu, Jia Gu, Min Xiao, Liang Huang, Miao Zheng, Chunrui Li, Yi Xiao, Jianfeng Zhou, and Xiaolu Long. 2021. "TP53-Mutated Circulating Tumor DNA for Disease Monitoring in Lymphoma Patients after CAR T Cell Therapy" Diagnostics 11, no. 5: 844. https://doi.org/10.3390/diagnostics11050844