
DNA Methylome and Transcriptome Maps of Primary Colorectal Cancer and Matched Liver Metastasis

 , , , , and
, , , , and
Abstract
:1. Introduction
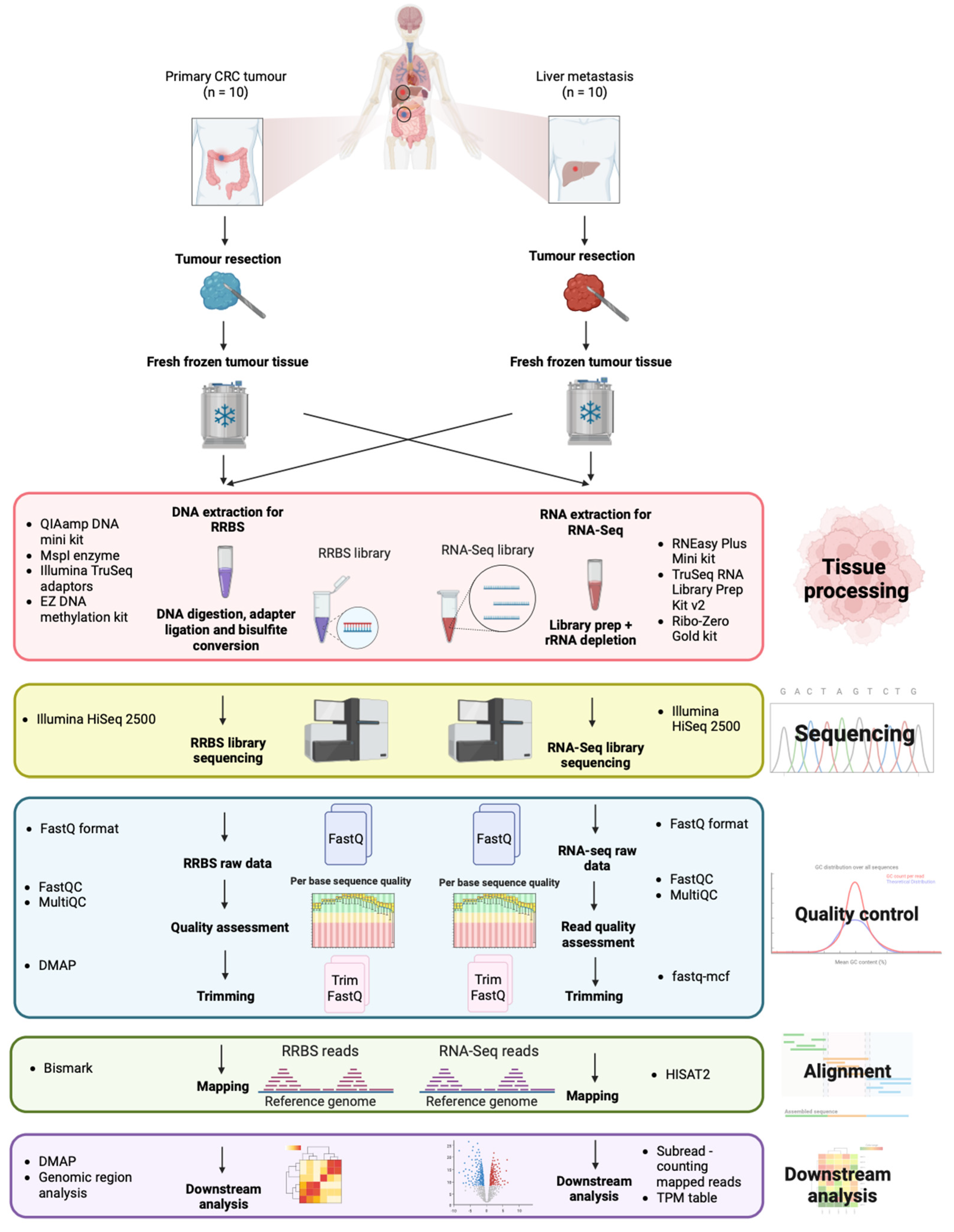
2. Data Description
3. Results
3.1. DNA and RNA Quality
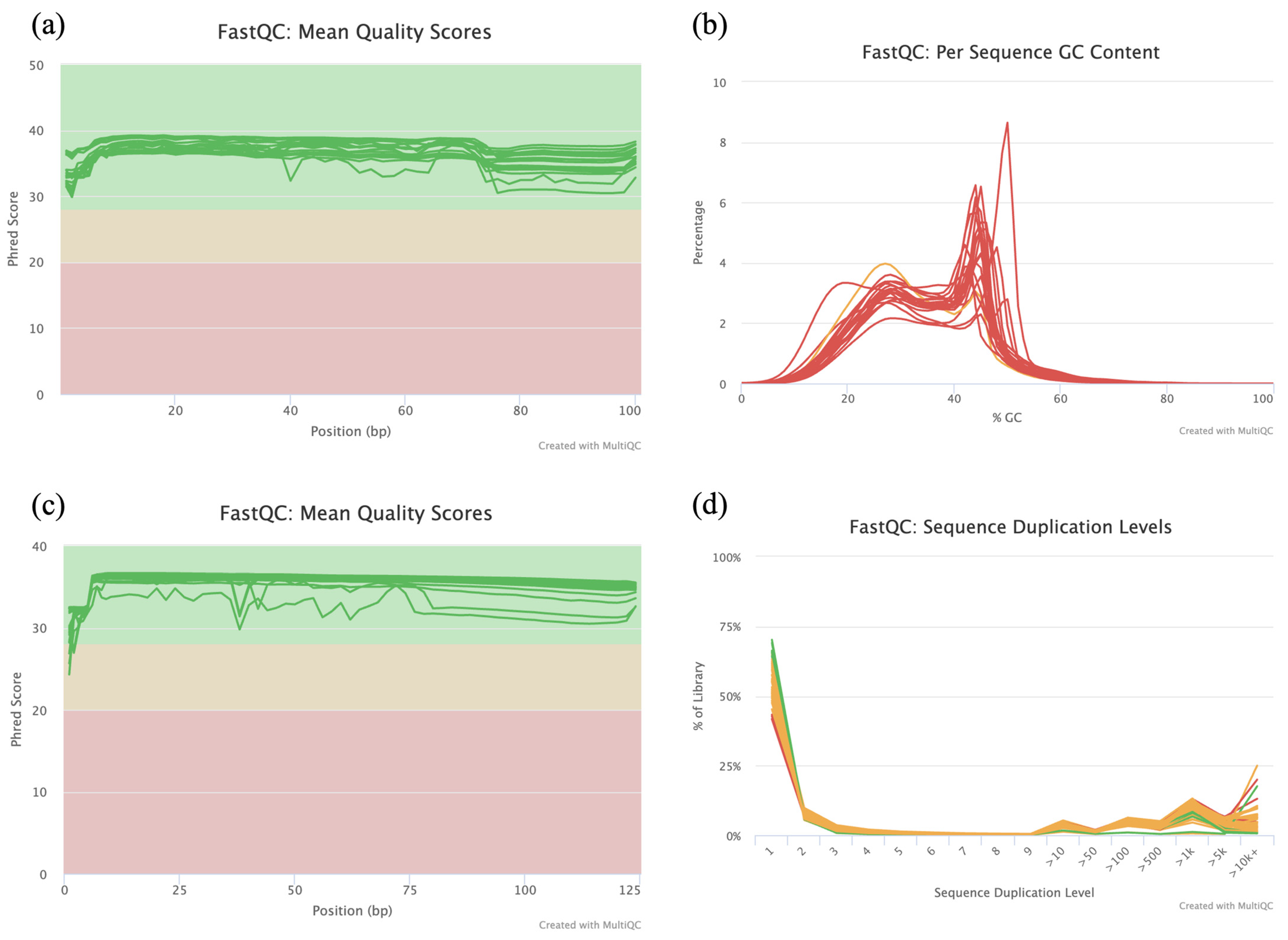
3.2. RRBS and RNA-Seq Data Quality
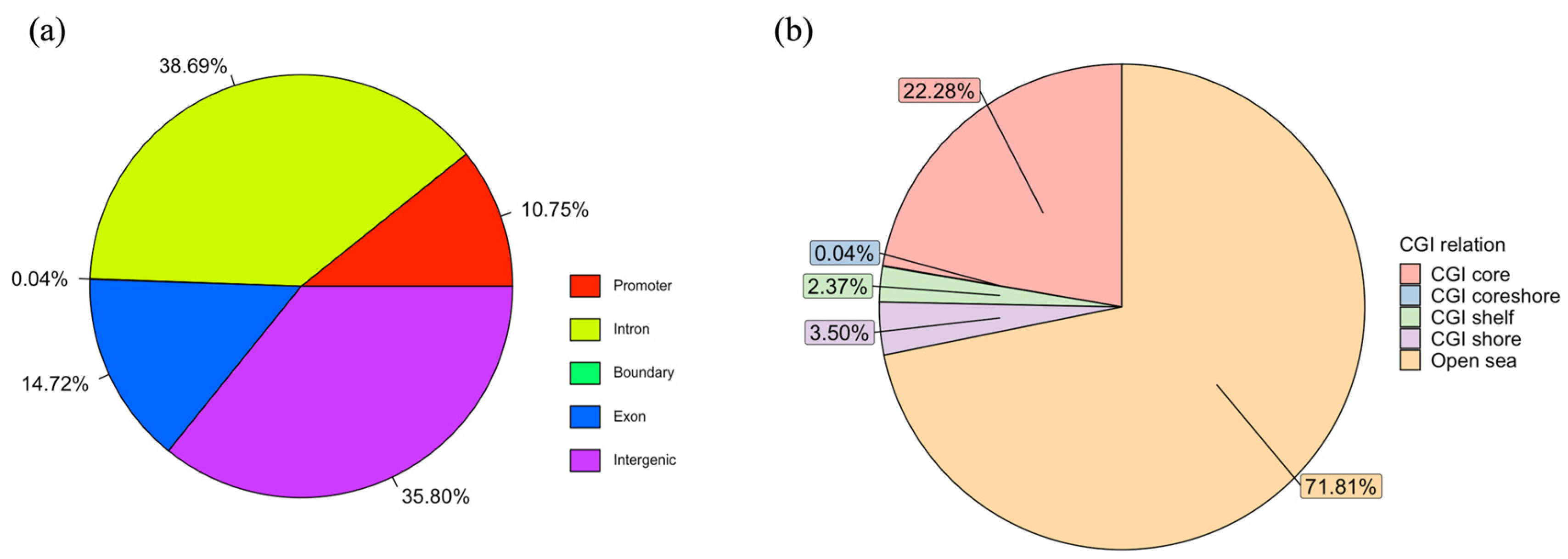
3.3. RRBS Alignment Quality and Characteristics of Methylome
3.4. RRBS Technical Replicates
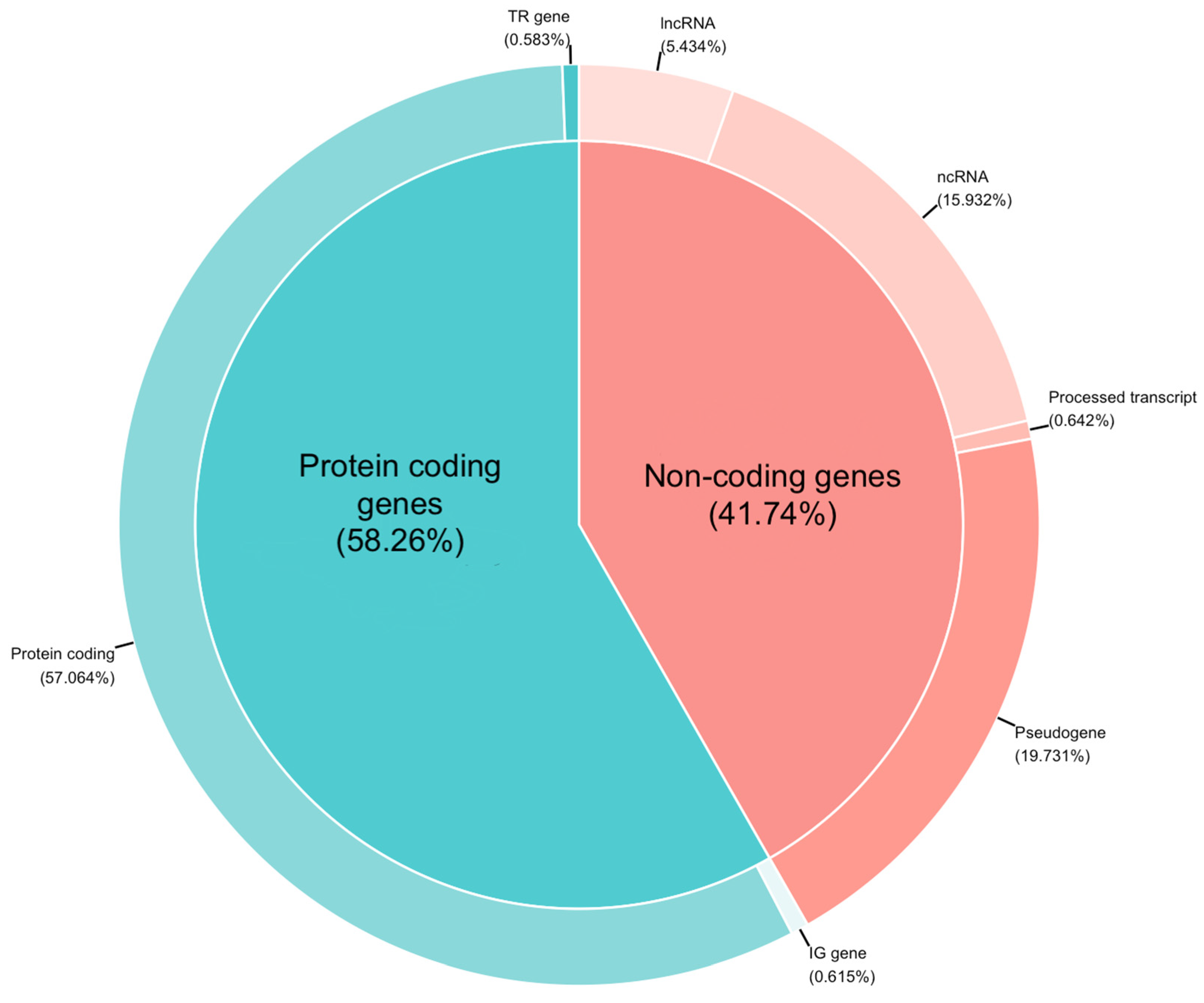
3.5. RNA-Seq Alignment Quality, Efficiency of rRNA Depletion and Transcriptome Analysis
4. Methods
4.1. Tumour Samples and Clinical Data Collection
4.2. Genomic DNA Extraction and RRBS Library Preparation
4.3. RRBS Data Analysis
4.4. RNA Extraction and Sequencing
4.5. RNA-Sequencing Data Analysis
5. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.J.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global burden of colorectal cancer in 2020 and 2040: Incidence and mortality estimates from GLOBOCAN. Gut 2023, 72, 338–344. [Google Scholar] [CrossRef]
- Cervantes, A.; Adam, R.; Roselló, S.; Arnold, D.; Normanno, N.; Taïeb, J.; Seligmann, J.; De Baere, T.; Osterlund, P.; Yoshino, T.; et al. Metastatic colorectal cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2022, 34, 10–32. [Google Scholar] [CrossRef]
- Lenos, K.J.; Bach, S.; Moreno, L.F.; Hoorn, S.T.; Sluiter, N.R.; Bootsma, S.; Braga, F.A.V.; Nijman, L.E.; Bosch, T.v.D.; Miedema, D.M.; et al. Molecular characterization of colorectal cancer related peritoneal metastatic disease. Nat. Commun. 2022, 13, 4443. [Google Scholar] [CrossRef]
- Pretzsch, E.; Nieß, H.; Bösch, F.; Westphalen, C.B.; Jacob, S.; Neumann, J.; Werner, J.; Heinemann, V.; Angele, M.K. Age and metastasis–how age influences metastatic spread in cancer. Colorectal cancer as a model. Cancer Epidemiol. 2022, 77, 102112. [Google Scholar] [CrossRef]
- Ciardiello, F.; Ciardiello, D.; Martini, G.; Napolitano, S.; Tabernero, J.; Cervantes, A. Clinical management of metastatic colorectal cancer in the era of precision medicine. CA A Cancer J. Clin. 2022, 72, 372–401. [Google Scholar] [CrossRef] [PubMed]
- Guidolin, K.; Choi, W.J.; Servidio-Italiano, F.; Quereshy, F.; Sapisochin, G. Attitudes of Canadian Colorectal Cancer Care Providers towards Liver Transplantation for Colorectal Liver Metastases: A National Survey. Curr. Oncol. 2022, 29, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.M.; Cardona, K.; Russell, M.C. Historical perspective: Two decades of progress in treating metastatic colorectal cancer. J. Surg. Oncol. 2019, 119, 549–563. [Google Scholar] [CrossRef]
- Viganò, L.; Branciforte, B.; Laurenti, V.; Costa, G.; Procopio, F.; Cimino, M.; Del Fabbro, D.; Di Tommaso, L.; Torzilli, G. The histopathological growth pattern of colorectal liver metastases impacts local recurrence risk and the adequate width of the surgical margin. Ann. Surg. Oncol. 2022, 29, 1–10. [Google Scholar] [CrossRef]
- Acciuffi, S.; Meyer, F.; Bauschke, A.; Croner, R.; Settmacher, U.; Altendorf-Hofmann, A. Solitary colorectal liver metastasis: Overview of treatment strategies and role of prognostic factors. J. Cancer Res. Clin. Oncol. 2022, 148, 657–665. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Lillemoe, H.A.; Vauthey, J. Dealing with an insufficient future liver remnant: Portal vein embolization and two-stage hepatectomy. J. Surg. Oncol. 2019, 119, 594–603. [Google Scholar] [CrossRef]
- Rodenhiser, D.I. Epigenetic contributions to cancer metastasis. Clin. Exp. Metastasis 2009, 26, 5–18. [Google Scholar] [CrossRef]
- Nowak, E.; Bednarek, I. Aspects of the Epigenetic Regulation of EMT Related to Cancer Metastasis. Cells 2021, 10, 3435. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Yan, Q. The roles of epigenetics in cancer progression and metastasis. Biochem. J. 2021, 478, 3373–3393. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Vanharanta, S. Epigenetic determinants of metastasis. Mol. Oncol. 2017, 11, 79–96. [Google Scholar] [CrossRef]
- Liu, J.; Li, H.; Sun, L.; Shen, S.; Zhou, Q.; Yuan, Y.; Xing, C. Epigenetic Alternations of MicroRNAs and DNA Methylation Contribute to Liver Metastasis of Colorectal Cancer. Dig. Dis. Sci. 2019, 64, 1523–1534. [Google Scholar] [CrossRef]
- Chen, J.; Röcken, C.; Lofton-Day, C.; Schulz, H.-U.; Müller, O.; Kutzner, N.; Malfertheiner, P.; Ebert, M.P. Molecular analysis of APC promoter methylation and protein expression in colorectal cancer metastasis. Carcinogenesis 2005, 26, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-H.; Petko, Z.; Dzieciatkowski, S.; Lin, L.; Ghiassi, M.; Stain, S.; Chapman, W.C.; Washington, M.K.; Willis, J.; Markowitz, S.D.; et al. CpG island methylation of genes accumulates during the adenoma progression step of the multistep pathogenesis of colorectal cancer. Genes Chromosom. Cancer 2006, 45, 781–789. [Google Scholar] [CrossRef]
- Konishi, K.; Watanabe, Y.; Shen, L.; Guo, Y.; Castoro, R.J.; Kondo, K.; Chung, W.; Ahmed, S.; Jelinek, J.; Boumber, Y.A.; et al. DNA Methylation Profiles of Primary Colorectal Carcinoma and Matched Liver Metastasis. PLoS ONE 2011, 6, e27889. [Google Scholar] [CrossRef]
- Murata, A.; Baba, Y.; Watanabe, M.; Shigaki, H.; Miyake, K.; Ishimoto, T.; Iwatsuki, M.; Iwagami, S.; Sakamoto, Y.; Miyamoto, Y.; et al. Methylation levels of LINE-1 in primary lesion and matched metastatic lesions of colorectal cancer. Br. J. Cancer 2013, 109, 408–415. [Google Scholar] [CrossRef]
- Chen, S.; Liu, T.; Bu, D.; Zhu, J.; Wang, X.; Pan, Y.; Liu, Y.; Lu, Z.J.; Wang, P. Methylome profiling identifies TCHH methylation in CfDNA as a noninvasive marker of liver metastasis in colorectal cancer. FASEB J. 2021, 35, e21720. [Google Scholar] [CrossRef]
- Ebert, M.P.; Mooney, S.H.; Tonnes-Priddy, L.; Lograsso, J.; Hoffmann, J.; Chent, J.; Rocken, C.; Schulz, H.-U.; Malfertheiner, P.; Lofton-Day, C. Hypermethylation of the TPEF/HPP1 Gene in Primary, Metastatic Colorectal Cancers. Neoplasia 2005, 7, 771–778. [Google Scholar] [CrossRef]
- Ju, H.-X.; An, B.; Okamoto, Y.; Shinjo, K.; Kanemitsu, Y.; Komori, K.; Hirai, T.; Shimizu, Y.; Sano, T.; Sawaki, A.; et al. Distinct Profiles of Epigenetic Evolution between Colorectal Cancers with and without Metastasis. Am. J. Pathol. 2011, 178, 1835–1846. [Google Scholar] [CrossRef]
- Hur, K.; Toiyama, Y.; Takahashi, M.; Balaguer, F.; Nagasaka, T.; Koike, J.; Hemmi, H.; Koi, M.; Boland, C.R.; Goel, A. MicroRNA-200c modulates epithelial-to-mesenchymal transition (EMT) in human colorectal cancer metastasis. Gut 2013, 62, 1315–1326. [Google Scholar] [CrossRef]
- De Krijger, I.; Mekenkamp, L.J.; Punt, C.J.; Nagtegaal, I.D. MicroRNAs in colorectal cancer metastasis. J. Pathol. 2011, 224, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, M.; Mollevi, C.; Vezzio-Vie, N.; Bibeau, F.; Ychou, M.; Martineau, P. Specific Extracellular Matrix Remodeling Signature of Colon Hepatic Metastases. PLoS ONE 2013, 8, e74599. [Google Scholar] [CrossRef]
- Stevens, J.R.; Herrick, J.S.; Wolff, R.K.; Slattery, M.L. Power in pairs: Assessing the statistical value of paired samples in tests for differential expression. BMC Genom. 2018, 19, 953. [Google Scholar] [CrossRef]
- Rodger, E.J.; Gimenez, G.; Ajithkumar, P.; Stockwell, P.A.; Almomani, S.; Bowden, S.A.; Leichter, A.L.; Ahn, A.; Pattison, S.; McCall, J.L.; et al. An epigenetic signature of advanced colorectal cancer metastasis. iScience 2023, 26, 106986. [Google Scholar] [CrossRef]
- Ashekyan, O.; Shahbazyan, N.; Bareghamyan, Y.; Kudryavzeva, A.; Mandel, D.; Schmidt, M.; Loeffler-Wirth, H.; Uduman, M.; Chand, D.; Underwood, D.; et al. Transcriptomic Maps of Colorectal Liver Metastasis: Machine Learning of Gene Activation Patterns and Epigenetic Trajectories in Support of Precision Medicine. Cancers 2023, 15, 3835. [Google Scholar] [CrossRef]
- Decruyenaere, P.; Verniers, K.; Poma-Soto, F.; Van Dorpe, J.; Offner, F.; Vandesompele, J. RNA Extraction Method Impacts Quality Metrics and Sequencing Results in Formalin-Fixed, Paraffin-Embedded Tissue Samples. Lab. Investig. 2023, 103, 100027. [Google Scholar] [CrossRef]
- Külahoglu, C.; Bräutigam, A. Quantitative Transcriptome Analysis Using RNA-seq. In Plant Circadian Networks: Methods and Protocols; Staiger, D., Ed.; Springer: New York, NY, USA, 2014; pp. 71–91. [Google Scholar]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Schillebeeckx, M.; Schrade, A.; Löbs, A.-K.; Pihlajoki, M.; Wilson, D.B.; Mitra, R.D. Laser capture microdissection–reduced representation bisulfite sequencing (LCM-RRBS) maps changes in DNA methylation associated with gonadectomy-induced adrenocortical neoplasia in the mouse. Nucleic Acids Res. 2013, 41, e116. [Google Scholar] [CrossRef]
- Gu, H.; Bock, C.; Mikkelsen, T.S.; Jäger, N.; Smith, Z.D.; Tomazou, E.; Gnirke, A.; Lander, E.S.; Meissner, A. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nat. Methods 2010, 7, 133–136. [Google Scholar] [CrossRef]
- Chatterjee, A.; Stockwell, P.A.; Rodger, E.J.; Morison, I.M. Genome-scale DNA methylome and transcriptome profiling of human neutrophils. Sci. Data 2016, 3, 160019. [Google Scholar] [CrossRef]
- Chatterjee, A.; Macaulay, E.C.; Ahn, A.; Ludgate, J.L.; Stockwell, P.A.; Weeks, R.J.; Parry, M.F.; Foster, T.J.; Knarston, I.M.; Eccles, M.R.; et al. Comparative assessment of DNA methylation patterns between reduced representation bisulfite sequencing and Sequenom EpiTyper methylation analysis. Epigenomics 2017, 9, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Conley, A.; Zhang, H.; Kim, H.L. Whole-Transcriptome profiling of formalin-fixed, paraffin-embedded renal cell carcinoma by RNA-seq. BMC Genom. 2014, 15, 1087. [Google Scholar] [CrossRef] [PubMed]
- Shohdy, K.S.; Bareja, R.; Sigouros, M.; Wilkes, D.C.; Dorsaint, P.; Manohar, J.; Bockelman, D.; Xiang, J.Z.; Kim, R.; Ohara, K.; et al. Functional comparison of exome capture-based methods for transcriptomic profiling of formalin-fixed paraffin-embedded tumors. NPJ Genom. Med. 2021, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rodger, E.J.; Stockwell, P.A.; Almomani, S.; Eccles, M.R.; Chatterjee, A. Protocol for generating high-quality genome-scale DNA methylation sequencing data from human cancer biospecimens. STAR Protoc. 2023, 4, 102714. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Stockwell, P.A.; Chatterjee, A.; Rodger, E.J.; Morison, I. DMAP: Differential methylation analysis package for RRBS and WGBS data. Bioinformatics 2014, 30, 1814–1822. [Google Scholar] [CrossRef]
- Chatterjee, A.; Stockwell, P.A.; Rodger, E.J.; Morison, I.M. Comparison of alignment software for genome-wide bisulphite sequence data. Nucleic Acids Res. 2012, 40, e79. [Google Scholar] [CrossRef] [PubMed]
- Aronesty, E. ea-utils: Command-Line Tools for Processing Biological Sequencing Data. 2011. Available online: https://github.com/ExpressionAnalysis/ea-utils (accessed on 20 February 2023).
- Bahrami, A. Which Aligner Software is the Best for Our Study. J. Genet. Genome Res. 2020, 7, 048. [Google Scholar]
- Liao, Y.; Smyth, G.K.; Shi, W. feature Counts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cooper, N.G.F.; O’Toole, T.E.; Rouchka, E.C. Choice of library size normalization and statistical methods for differential gene expression analysis in balanced two-group comparisons for RNA-seq studies. BMC Genom. 2020, 21, 75. [Google Scholar] [CrossRef]
- Yassi, M.; Chatterjee, A.; Parry, M. Application of deep learning in cancer epigenetics through DNA methylation analysis. Briefings Bioinform. 2023, 24, bbad411. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient No. | Clinical Details | CRC Primary Tumour Accession Number | Liver Metastasis Accession Number | ||||
|---|---|---|---|---|---|---|---|
| Sex | Age | Primary Tumour Site | RRBS Data | RNA-Seq Data | RRBS Data | RNA-Seq Data | |
| 8 | M | 74 | Colon | GSM6585685 | GSM6585705 | GSM6585695 | GSM6585715 |
| 87 | M | 74 | Colon | GSM6585686 | GSM6585706 | GSM6585696 | GSM6585716 |
| 208 | M | 57 | Colon | GSM6585687 | GSM6585707 | GSM6585697 | GSM6585717 |
| 241 | F | 60 | Colon | GSM6585688 | GSM6585708 | GSM6585698 | GSM6585718 |
| 269 | F | 61 | Colon | GSM6585689 | GSM6585709 | GSM6585699 | GSM6585719 |
| 270 | M | 64 | Colon | GSM6585690 | GSM6585710 | GSM6585700 | GSM6585720 |
| 275 | F | 60 | Rectum | GSM6585691 | GSM6585711 | GSM6585701 | GSM6585721 |
| 311 | M | 57 | Colon | GSM6585692 | GSM6585712 | GSM6585702 | GSM6585722 |
| 314 | M | 65 | Colon | GSM6585693 | GSM6585713 | GSM6585703 | GSM6585723 |
| 316 | M | 75 | Colon | GSM6585694 | GSM6585714 | GSM6585704 | GSM6585724 |
| Sample ID | Sequenced Reads | Unique Reads | Mapping Efficiency (%) | Total CpG Meth (%) | Total Non-CpG Meth (%) |
|---|---|---|---|---|---|
| CRC8 | 67,293,600 | 342,740,70 | 50.90 | 37.00 | 0.35 |
| CRC87 | 79,942,689 | 42,651,395 | 53.40 | 56.20 | 0.49 |
| CRC208 | 15,865,582 | 9,962,519 | 62.80 | 56.60 | 0.40 |
| CRC241 | 49,656,769 | 30,745,536 | 61.90 | 50.40 | 0.42 |
| CRC269 | 54,605,569 | 27,044,502 | 49.50 | 47.90 | 0.37 |
| CRC270 | 30,066,202 | 16,094,322 | 53.50 | 42.50 | 0.38 |
| CRC275 | 90,499,303 | 55,671,910 | 61.50 | 42.20 | 0.54 |
| CRC311 | 85,268,731 | 46,371,408 | 54.40 | 48.50 | 0.45 |
| CRC314 | 94,196,062 | 51,990,759 | 55.20 | 41.10 | 0.50 |
| CRC316 | 89,855,788 | 50,133,332 | 55.80 | 48.40 | 0.53 |
| LM8 | 73,033,866 | 44,328,416 | 60.70 | 61.60 | 0.33 |
| LM87 | 98,621,906 | 44,354,592 | 45.00 | 39.70 | 0.50 |
| LM208 | 74,482,754 | 18,899,940 | 25.40 | 50.50 | 0.39 |
| LM241 | 90,013,168 | 51,489,691 | 57.20 | 47.60 | 0.48 |
| LM269 | 78,414,515 | 41,592,069 | 53.00 | 54.30 | 0.36 |
| LM270 | 89,824,235 | 38,806,877 | 43.20 | 45.30 | 0.46 |
| LM275 | 71,135,244 | 41,994,300 | 59.00 | 54.20 | 0.36 |
| LM311 | 62,045,345 | 31,262,905 | 50.40 | 56.00 | 0.31 |
| LM314 | 69,843,997 | 38,398,803 | 55.00 | 47.60 | 0.34 |
| LM316 | 33,679,596 | 20,021,531 | 55.75 | 59.40 | 0.52 |
| Average | 69,917,246.1 | 36,804,443.9 | 53.18 | 49.35 | 0.424 |
| Sample ID | Total Number of Sequenced Reads | Total Number of Uniquely Mapped Reads (GRCh37) | Uniquely Mapped Reads (%) | Percentage of Reads Aligned to rRNA Regions (%) |
|---|---|---|---|---|
| CRC8 | 23,384,046 | 19,026,422 | 81.36 | 0.0003 |
| CRC87 | 24,339,816 | 22,145,940 | 90.99 | 0.0003 |
| CRC208 | 11,577,334 | 7,738,152 | 66.83 | 0.0002 |
| CRC241 | 28,268,750 | 23,299,700 | 82.42 | 0.0002 |
| CRC269 | 21,423,020 | 18,013,129 | 84.08 | 0.0002 |
| CRC270 | 19,305,816 | 14,088,264 | 72.98 | 0.0002 |
| CRC275 | 27,242,458 | 23,347,216 | 85.70 | 0.0002 |
| CRC311 | 14,002,078 | 7,888,497 | 56.34 | 0.0001 |
| CRC314 | 20,987,192 | 18,011,052 | 85.82 | 0.0004 |
| CRC316 | 20,464,570 | 16,317,115 | 79.73 | 0.0005 |
| LM8 | 31,508,190 | 26,257,882 | 83.34 | 0.0001 |
| LM87 | 20,470,182 | 14,708,258 | 71.85 | 0.0003 |
| LM208 | 18,987,032 | 15,298,388 | 80.57 | 0.0002 |
| LM241 | 23,978,748 | 20,965,130 | 87.43 | 0.0003 |
| LM269 | 20,095,084 | 14,373,695 | 71.53 | 0.0004 |
| LM270 | 20,743,994 | 18,185,861 | 87.67 | 0.0002 |
| LM275 | 19,603,228 | 16,678,527 | 85.08 | 0.0005 |
| LM311 | 27,400,080 | 22,939,465 | 83.72 | 0.0002 |
| LM314 | 21,182,554 | 17,709,592 | 83.60 | 0.0002 |
| LM316 | 22,810,830 | 19,212,321 | 84.22 | 0.0003 |
| Average | 21,888,750.1 | 17,810,230.3 | 80.26 | 0.0005 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ajithkumar, P.; Gimenez, G.; Stockwell, P.A.; Almomani, S.; Bowden, S.A.; Leichter, A.L.; Ahn, A.; Pattison, S.; Schmeier, S.; Frizelle, F.A.; et al. DNA Methylome and Transcriptome Maps of Primary Colorectal Cancer and Matched Liver Metastasis. Data 2024, 9, 8. https://doi.org/10.3390/data9010008
Ajithkumar P, Gimenez G, Stockwell PA, Almomani S, Bowden SA, Leichter AL, Ahn A, Pattison S, Schmeier S, Frizelle FA, et al. DNA Methylome and Transcriptome Maps of Primary Colorectal Cancer and Matched Liver Metastasis. Data. 2024; 9(1):8. https://doi.org/10.3390/data9010008
Chicago/Turabian StyleAjithkumar, Priyadarshana, Gregory Gimenez, Peter A. Stockwell, Suzan Almomani, Sarah A. Bowden, Anna L. Leichter, Antonio Ahn, Sharon Pattison, Sebastian Schmeier, Frank A. Frizelle, and et al. 2024. "DNA Methylome and Transcriptome Maps of Primary Colorectal Cancer and Matched Liver Metastasis" Data 9, no. 1: 8. https://doi.org/10.3390/data9010008