
Transcriptome Analysis of Cambium Tissue of Paulownia Collected during Winter and Spring
by
 , and
, and
Zachary D. Perry
1, 
Thangasamy Saminathan
2,
Alok Arun
3,
Brajesh N. Vaidya
1,
Chhandak Basu
4,
Umesh K. Reddy
2 and
Nirmal Joshee
1,* 1
Agricultural Research Station, Fort Valley State University, Fort Valley, GA 31030, USA
2
Gus R. Douglass Institute and Department of Biology, West Virginia State University, Institute, WV 25112, USA
3
Institute of Sustainable Biotechnology, Inter American University of Puerto Rico, Barranquitas, PR 00794, USA
4
Department of Biology, California State University, Northridge, Los Angeles, CA 91330, USA
*
Author to whom correspondence should be addressed.
Diversity 2021, 13(9), 423; https://doi.org/10.3390/d13090423
Submission received: 14 July 2021
/
Revised: 27 August 2021
/
Accepted: 27 August 2021
/
Published: 1 September 2021
(This article belongs to the Section Plant Diversity)
Abstract
:Paulownia (Paulownia elongata) is a fast-growing, multipurpose deciduous hardwood species that grows in a wide range of temperatures from –30 °C to 45 °C. Seasonal cues influence the secondary growth of tree stems, including cambial activity, wood chemistry, and transition to latewood formation. In this study, a de novo transcriptome approach was conducted to identify the transcripts expressed in vascular cambial tissue from senescent winter and actively growing spring seasons. An Illumina paired-end sequenced cambial transcriptome generated 297,049,842 clean reads, which finally yielded 61,639 annotated unigenes. Based on non-redundant protein database analyses, Paulownia cambial unigenes shared the highest homology (64.8%) with Erythranthe guttata. KEGG annotation of 35,471 unigenes identified pathways enriched in metabolic activities. Transcriptome-wide DEG analysis showed that 2688 and 7411 genes were upregulated and downregulated, respectively, in spring tissues compared to winter. Interestingly, several transcripts encoding heat shock proteins were upregulated in the spring season. RT-qPCR expression results of fifteen wood-forming candidate genes involved in hemicellulose, cellulose, lignin, auxin, and cytokinin pathways showed that the hemicellulose genes (CSLC4, FUT1, AXY4, GATL1, and IRX19) were significantly upregulated in spring season tissues when compared to winter tissues. In contrast, lignin pathway genes CCR1 and CAD1 were upregulated in winter cambium. Finally, a transcriptome-wide marker analysis identified 11,338 Simple Sequence Repeat (SSRs). The AG/CT dinucleotide repeat predominately represented all SSRs. Altogether, the cambial transcriptomic analysis reported here highlights the molecular events of wood formation during winter and spring. The identification of candidate genes involved in the cambial growth provides a roadmap of wood formation in Paulownia and other trees for the seasonal growth variation.
1. Introduction
Paulownia (Paulownia elongata) is an extremely fast-growing woody plant growing up to 20 feet in one year when young. Some Paulownia spp., when in plantation, can be harvested for saw timber in as little as five years. The genus Paulownia consists of nine species of deciduous, fast-growing, multi-purpose, hardwood trees [1] that have long been shown to be extremely adaptive to wide environmental variations in both edaphic and climatic factors, as well as being capable of growing on marginal lands [2,3]. Species of Paulownia are native to Asia and are widely cultivated in China, Laos, Vietnam, Japan, and Korea. It has now been introduced and cultivated in Australia, Europe, and North and Central America. Ten-year-old Paulownia trees, in natural conditions, can attain 30–40 cm in diameter at breast height (DBH) and a timber volume of 0.3–0.5 m3 [1]. Craftworkers in Japan and other countries have used this valuable wood to create intricate carvings, surfboards, musical instruments, toys, and furniture. Paulownia wood has a high ignition point of 420–430 °C compared to other hardwoods, which generally range from 220–225 °C, thus making Paulownia wood fire retardant [4,5]. Paulownia plants bear abundant flowers that are highly nectariferous and yield premium honey [6], adding to the rural economy. By adding Paulownia wood flour (25–40%) to plastics, an attractive, equally strong, environmentally agreeable, and economically important biocomposite can be produced [7,8,9] to serve many industries. Additionally, the lightweight and strong nature of the wood makes it widely applicable in the music industry for preparing the finer components of instruments. Biochar produced from Paulownia is also a desirable organic soil amendment that allows the growth of beneficial microbes in the porous holes of the biochar [10]. Recently, researchers found its potential use as an animal feed resource [11].
Wood synthesis provides one of the most important sinks for atmospheric carbon dioxide [12]. Wood formation is a result of the regulated accumulation of secondary xylem cells (fibers, vessels, and rays in dicots) differentiated from the vascular cambium that involves wall thickening. This wall thickening is accompanied by the biosynthesis of wall components, lignin, cellulose, and hemicelluloses and is terminated by programmed cell death [13,14]. In order to survive multiple growing seasons, perennial plant species have adapted a dormancy regulation system that allows active growth during the desirable time of year and vegetative dormancy when climatic conditions are unfavorable for growth [15]. The Paulownia tree, due to its fast-growing nature, is capable of producing ~45 kg/tree in the first growing year and ~90 kg/tree at the end of the second year [16]. Being a perennial tree, Paulownia harvest is not limited to a small seasonal window but can be conducted year-round with proper management practices. Another beneficial property of Paulownia is coppicing, which is defined as the production of multiple sprouts from a stump after the removal of the tree or shrub. Harvest cycles of 2–3 years could be implemented to establish a fast-growing bioenergy crop. Since Paulownia is a short-rotation fast-growing perennial tree and serves as a good candidate to produce lignocellulosic biofuel, which can eliminate dependence on fossil fuel.
Transcriptome analyses from various tree species indicate that the putative role of gene families belonging to receptor kinases, transcription factors, and secondary wall biosynthesis are highly expressed in wood-forming cells [17,18,19,20,21]. At a molecular level, studies related to Paulownia gene expression profiling published in the last few years have primarily focused on drought tolerance and host-pathogen interactions [22,23,24,25]. Cambial development (the initiation and activity of the vascular cambium) leads to an accumulation of wood (secondary xylem tissue). Seasonal cues play a significant role in determining cambial growth as perennial plants growing in temperate and high-latitude regions show termination of cell division in the meristems [26] and reversal of growth arrest during long days [27]. Time-coursed RNA sequence studies identified downregulated phytohormone-related genes (IAA, ARF, and SAURs) and upregulated circadian genes (PIF3 and PRR5; [28,29]. A transcriptome-wide profiling study in P. elongata identified a subset of candidate genes that contribute to the production of wood [30] by investigating the differential expression of transcripts of the vascular cambium. Furthermore, there is evidence for the microRNAs controlling gene expression in Paulownia tomentosa cambial tissues in response to seasonal changes [31].
Transcriptomic analyses have been carried out to profile gene expression regulations for biotic and abiotic stresses and growth responses. However, to the best of our knowledge, no study has described how the gene expression profile changes in woody tissues under seasonal variations. In this study, we sequenced and analyzed the transcriptomes of cambium tissues collected during the winter and spring seasons to assess the impact of two seasons on biomass. A transcriptome-wide analysis identified 61,639 annotated unigenes, and 2688 and 7411 transcripts were up- and downregulated, respectively, in the spring season. Interestingly, among the selected wood-forming genes, hemicellulose-specific genes were upregulated in spring. Finally, 11,338 simple sequence repeats (SSRs) were identified from the transcriptome data. The identification of genes and pathways involved in cambial growth will be useful to further investigate the regulation of wood formation in Paulownia and other trees.
2. Materials and Methods
2.1. Tissue Sampling and RNA Isolation
Samples were selected from trees growing at the Fort Valley State University (FVSU) Paulownia Bioenergy Farm located at 32° 31′15.04″ N and 83° 52′12.95″ W. Paulownia trees bear flowers first, and after 3–4 weeks of flowering, leaves appear. Samples in replicates were collected from two seasonal points, each representing a different physiological state. The first sample (winter wood; hereafter referred to as WW) was collected before flowering on 1 March 2015 (min temp 33.1 °F and max temp 54 °F) and represented the senescent winter wood (Figure 1A). The second sample (spring wood; hereafter referred to as SW) was collected on 20 May 2015 (min temp 64 °F, max temp 91 °F) during spring and was representative of the actively growing spring wood (Figure 1B). Samples were harvested from twigs located at approximately 1.0–1.5 m from the ground with an average diameter of 2.5 cm. Since Paulownia has an opposite branching pattern, the WW and SW samples were taken from the mirrored axis of nodes. The samples were labeled, wrapped in aluminum foil, flash-frozen in liquid nitrogen, and subsequently stored in a −80 °C freezer until further use. Biological replicates were labeled as WW1, WW2, and WW3 for WW, and SW1, SW2, and SW3 for SW, respectively.
For high-quality, intact total RNA extraction, vascular cambium tissues were harvested from the frozen samples by first slicing a shallow, longitudinal cut into the outer bark with a sterile scalpel (Figure 1C). The bark was then removed using sterile forceps in a large, single piece. The frozen green vascular cambium was then gently scraped from the wood below into small strips using a sterile scalpel. A 100 mg vascular cambium tissue sample was powdered into microvials containing zirconia beads (BioSpec, Bartlesville, OK, USA) and 550 µL of TRIzol reagent (Invitrogen, Carlsbad, CA, USA) in a MagNA Lyser (Roche, Basel, Switzerland) as described earlier [32]. RNA was purified using Direct-Zol™ RNA mini-prep kit (Zymo Research, Irvine, CA, USA), and removal of genomic DNA contamination was carried out using DNase I treatment. RNA quality and quantity were analyzed using NanoDrop 1100 (NanoDrop, Wilmington, DE, USA) and Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA).
2.2. cDNA Synthesis and RNA Sequencing
RNA samples of each biological replicate from both treatments (a total of 6 samples) were sequenced at BGI International (http://bgi-international.com/). Briefly, magnetic beads coated with Oligo (dT) were used to isolate mRNA from the total RNA, and size selected mRNA were adapter-ligated and sequenced on the Illumina Hiseq 2000 platform following the manufacturer’s protocol.
2.3. Assembly and Annotation
Raw reads generated from pair-end sequencing were filtered for adapters and low-quality sequences by BGI proprietary software “filter_fq”. Clean reads were assembled into transcripts using Trinity (http://trinityrnaseq.sourceforge.net/) [33]. Three modules in Trinity (Inchworm, Chrysalis, and Butterfly) were applied sequentially to process raw RNA-seq reads to assemble them into contigs and full-length transcripts (unigenes). Butterfly also determined alternatively spliced isoforms of genes Unigenes were aligned with five databases, namely, KEGG (Kyoto Encyclopedia of Genes and Genomes), COG (Clusters of Orthologous Groups), NT (NCBI nucleotide database), NR (NCBI non-redundant protein database), and Swiss-Prot (Protein sequence database). The KEGG database [34] was used to perform a systematic analysis of metabolic pathways and the function of gene products within a cell. The COG database (http://www.ncbi.nlm.nih.gov/COG/) was used to classify orthologous gene products into clusters. COG clusters predicted the possible function of the transcripts. Both NCBI’s NR database and the Swiss-Prot (http://www.uniprot.org/uniprot/) annotated protein databases and added additional information about the possible function of the transcripts.
2.4. Gene Ontology and Coding Sequences
Gene ontology (GO) was employed to standardize functional gene classification, including molecular function, cellular components, and biological processes. The Blast2GO program (http://www.blast2go.com/b2ghome) was used to retrieve GO functional classification for all transcripts [35]. In order to determine the CDS for the transcripts, unigenes were first aligned to the protein databases, listed in order of priority of NR, Swiss-Prot, KEGG, and COG by using a local blastx (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/release/LATEST/), with a significant cut-off value of e<0.00001, of the unigene sequences. Unigenes with alignments to higher priority databases, for example, the NR database, were not aligned to lower priority databases. The highest-ranking proteins in the blastx results were used to decide the coding region sequences of unigenes. Results of the blastx alignment used a standard codon table to translate the nucleotide query sequence into a translated amino acid sequence. Unigenes that could not be aligned to any database were further scanned by ESTScan [36], producing nucleotide sequence (5′→3′) direction and amino sequence of the predicted coding region.
2.5. Gene Expression Analysis
In order to determine the expression pattern of the unigenes, clean reads were first mapped to unigenes using the program Bowtie2 (v. 2.2.5) [37]. SAM files generated through Bowtie were used with the RSEM (RNA-Seq by Expectation-Maximization) software package (http://deweylab.github.io/RSEM/; v1.2.12) in R (v1.03; http://www.r-project.org/) to measure the expression level of unigenes. RSEM software was used to estimate gene expression levels from RNA-seq data [38] in FPKM (fragments per kilobase of transcript per million mapped reads) format, which was subsequently used to perform differential gene expression analysis in this study. To detect differentially expressed genes (DEGs), the program NOIseq was utilized [39]. In this study, WW and SW unigenes with a fold change of ≥2 and a probability ≥0.8 were considered to be significantly differentially expressed. Principal component analysis (PCA) was performed using the R statistical package.
2.6. Simple Sequence Repeats Analyses
Simple sequence repeat (SSR) identification was accomplished with MIcroSAtellite (MISA) software (http://pgrc.ipk-gatersleben.de/misa/misa.html), using unigenes as input sequences. The identified SSRs with nucleotide size > 150 bps on both ends of the unigenes were used for oligonucleotide design using Primer3 (v2.3.4; http://www.onlinedown.net/soft/51549.htm).
2.7. Validation of Wood-Forming Candidate Genes with RT-qPCR
Based on existing research information, fifteen wood-forming candidate genes corresponding to the biosynthesis of cellulose, hemicellulose, or lignin [18,40,41,42,43], were selected for validation of expression level. Using the local blast utility (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/release/LATEST/), a database of all Paulownia unigenes was created. The mRNA sequences acquired for each of the selected genes were then aligned to the database of all unigenes using the local blastx utility. The unigenes showing maximum homology for each of the genes were selected for a two-step RT-qPCR primer design. The software Primer Express v3.01 (Applied Biosystems, Foster City, CA) was used to design primers for the unigenes corresponding to the selected wood forming genes. cDNA was synthesized from Paulownia vascular cambium total RNA using SuperScript™ II Reverse Transcriptase (Invitrogen, Waltham, Massachusetts, USA) with the suggested protocol. FastStart SYBR Green Master Mix (Roche, Grenzacherstrasse, Basel, Switzerland) reagent was used in combination with primers and cDNA. RT-qPCR of three biological replications with no-template control (NTC) involved StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) and FastStart SYBR Green (Roche). The expression of selected genes was normalized with reference gene 18S rRNA. Finally, the relative gene expression was measured using the 2–ΔΔCt method [44].
All raw files of sequencing data have been submitted to the NCBI Sequenced Read Archive (SRA) database with accession numbers SRX9298494—SRX9298499.
3. Results and Discussion
3.1. RNA-Seq and Transcriptome Assembly of Paulownia Cambial Tissue
To obtain the candidate genes associated with cambium development of the empress tree (common name for all Paulownia species) during seasonal growth, transcriptome sequencing analysis for tissues representing winter and spring seasons (Figure 1A,B) was carried out by collecting cambial tissues from tree twigs (Figure 1C). A total of 305,882,370 (~29 Gb) raw reads were generated. Removal of adapter sequences, low-quality reads, and ambiguous sequences resulted in 297,049,842 clean reads (Q20 > 97.73%) with an average length of 100 nucleotides. WW generated more raw reads when compared to spring wood samples (Table 1). The de novo assembling of clean reads produced 129,428 and 104,388 contigs for WW and SW cambial tissues, respectively. Clustering and assembly of these contigs resulted in 64,142 and 45,671 unigenes for WW and SW with an average length of 960 and 842 nucleotides, respectively. Among the unigenes, all unigenes were sub-classified according to nucleotide length (Figure S1). A total of 40,814 unigenes were greater than 1 Kbs, and 58,654 unigenes were greater than 500 nucleotides in length. The mean length of the contigs (~340 bp) was shorter than that of the unigenes (>1000 bp) in most previously reported studies. The paired-end reads resulted in longer unigenes (average of ~900 bp) than those reported in previous transcriptome studies on trees [45,46]. The mean length of unigenes (900 nucleotides) was less than those reported in previous studies on tetraploid Paulownia australis and drought exposed P. tomentosa [23,47]. Most of our assembled unigenes showed homology to nucleotide sequences deposited in six public nucleotide databases. The unmatched unigenes are most likely to represent Paulownia-specific genes especially related to the winter and spring seasons.
3.2. Functional Annotation of Paulownia Cambial Transcriptome
A total of 61,639 transcripts were annotated by performing a BLAST search of the sequences across six databases as described above. The BLAST search using 61,639 transcripts showed 72.47% similarity to non-redundant (Nr) protein database (Tables S1 and S2).
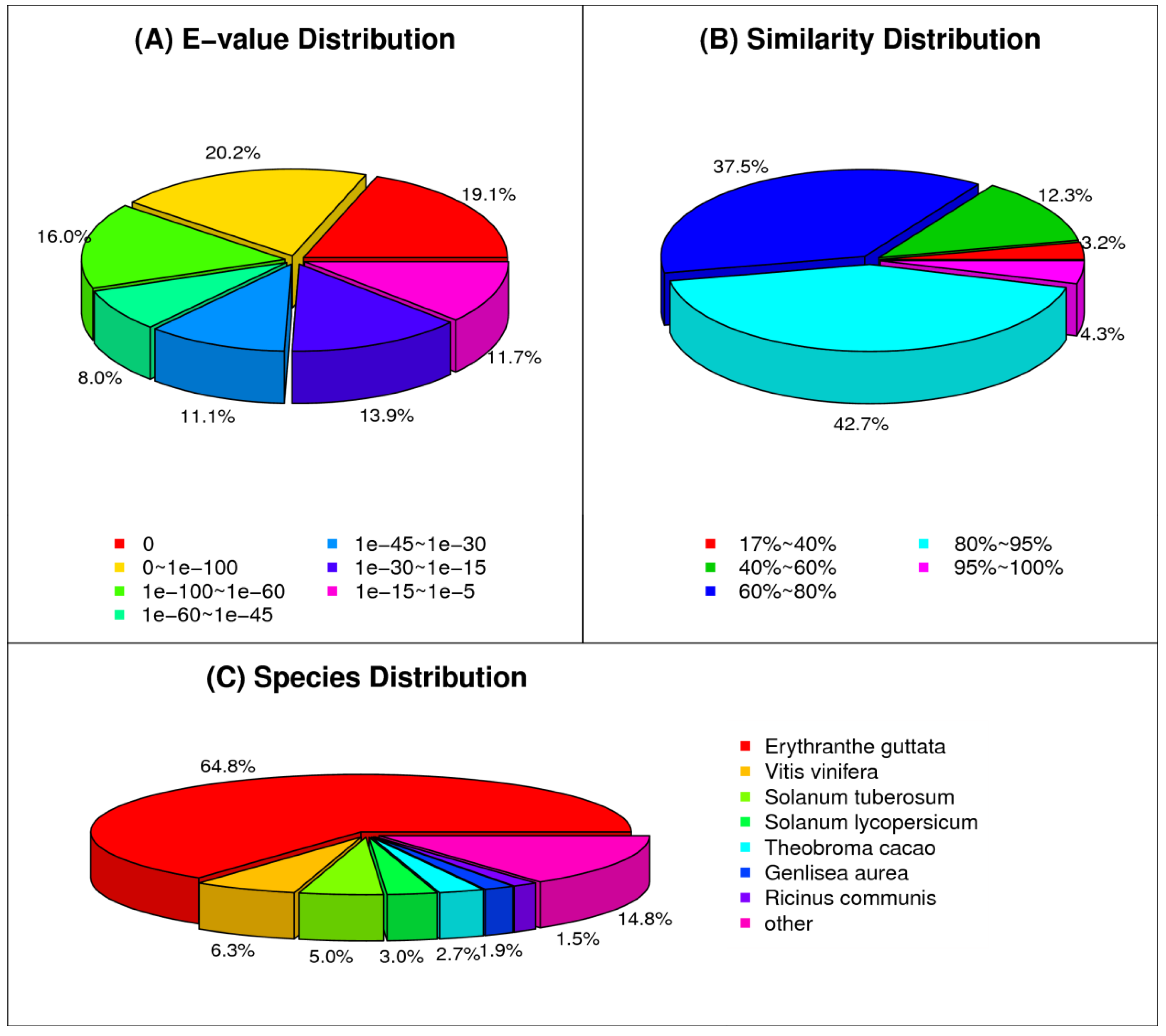
Of the annotated sequences in the Nr database, 39.3% of the mapped unigenes had very significant homology to known sequences (e-value 10–100), 35.1% showed significant homology (10–100; e-value 10–30), and 25.6% showed weak homology (e-value 10–30 to 10–5) (Figure 2A). We also performed the sequence conservation analysis of Paulownia transcripts with proteomes of all sequenced plant species. As depicted in Figure 2B, the E-value distribution analysis of transcripts showed that 47.0% of unigenes had a similarity of more than 80% with plant species. We used a BLAST search to study the relationship of Paulownia with other plant species to identify proteins and pathways that would be unique to Paulownia. The sequence conservation analysis of transcripts showed homology to nucleotide sequences from Erythranthe guttata (64.8%), followed by Vitis vinifera (6.3%), Solanum tuberosum (5.0%), Solanum lycopersicum (3.0%), Theobroma cacao (2.7%), and others (Figure 2C). Erythranthe guttata, a yellow bee-pollinated annual or perennial plant, is a model organism for biological studies. Paulownia transcripts shared strong homology with Erythranthe species, and this could be due to close phylogenetic relationships between these two species [48]. However, transcripts from a drought-related transcriptomic study of Paulownia [28,47] showed homology to Vitis vinifera (45–48%), which could be due to the selection of seasonal abiotic tissue for cambial transcriptome study.
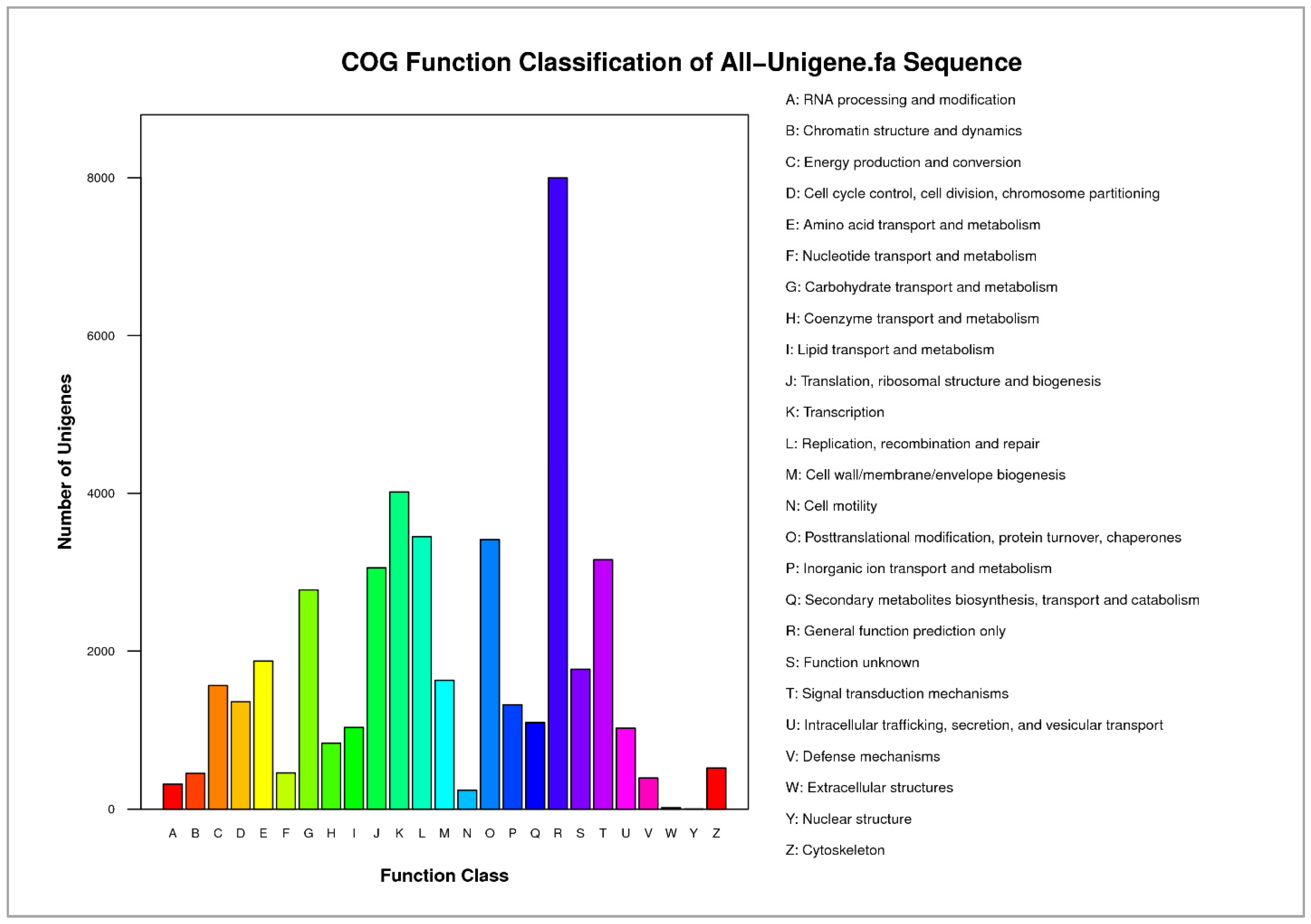
Further analysis of 43,780 transcripts using the COG database classified these transcripts into 25 different protein families with potential functions in transcription, replication and recombination, posttranslational modification, signal transduction, and others.
Interestingly, only a few transcripts exhibited their potential role in the extracellular structure and nuclear structure (17 and 4 transcripts, respectively). Importantly, many unigenes have been assigned to a wide range of COG classifications (Figure 3), indicating that a wide diversity of transcripts were involved in wood formation as in Chinese fir [31].
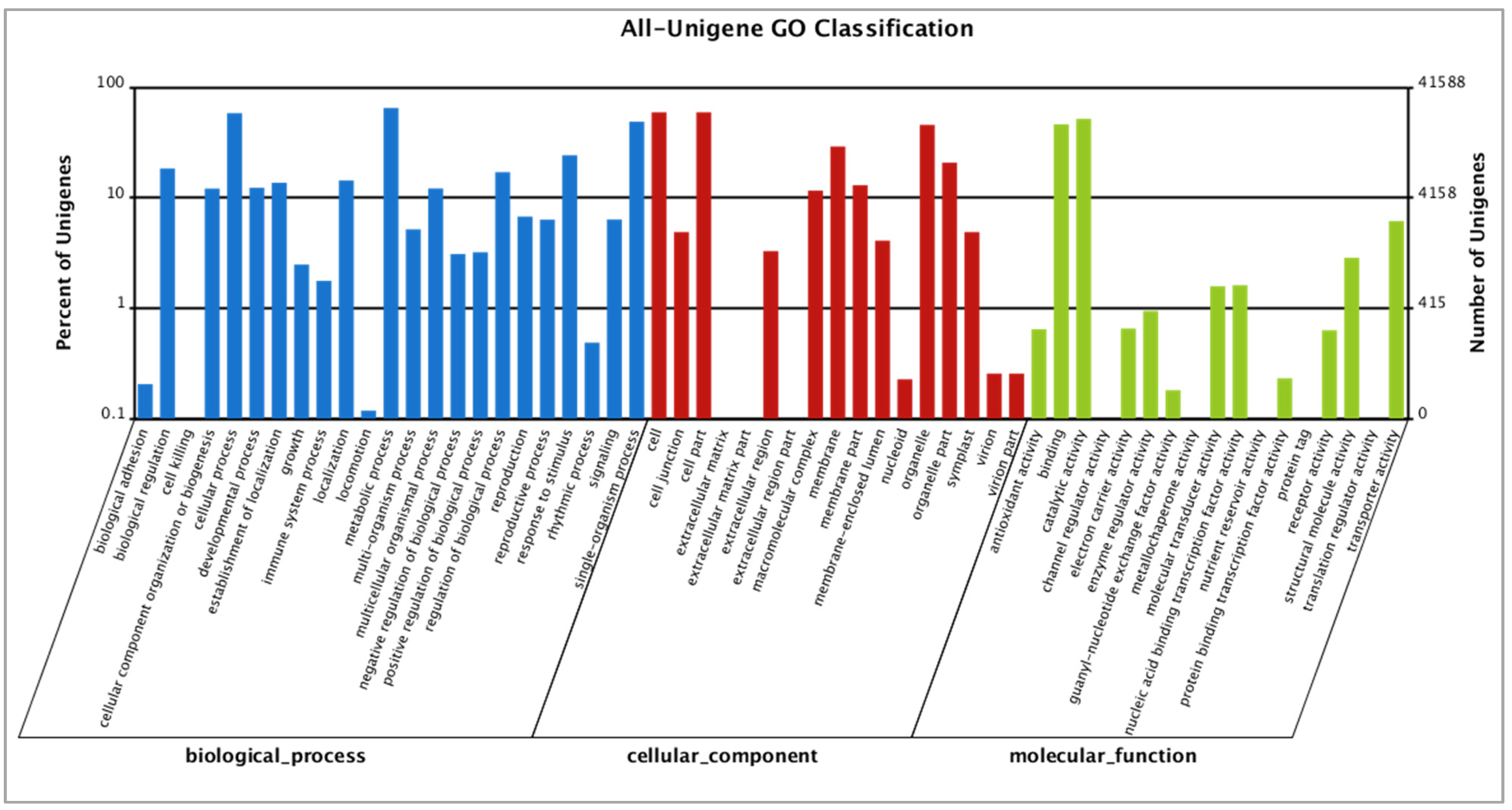
The Gene Ontology (GO) classification classified 42,588 out of 62,639 transcripts into ontologies related to molecular functions, cellular components, and biological processes (Figure 4). We identified a significantly higher number of transcripts involved in metabolic processes (19,418) and related to cellular processes (18,047) when compared to others, such as rhythmic processes. The most represented category for cellular components was cells (GO: 0005623; 18,186 genes), followed by organelles (GO:0043226; 14,053 genes). Genes and pathways putatively responsible for dormant winter and active spring growth in Paulownia were identified in this study. In Populus, PtrHB7, a class III HD-Zip gene, is known to play a critical role in the regulation of vascular cambium differentiation [49] and homeobox gene ARBORKNOX1 regulates the shoot apical meristem and the vascular cambium [50]. In our study, Unigene2201, Unigene3374, and Unigene4121 which were downregulated in their expressions, belong to GO:0005488 (molecular function: binding) and are homologs of the KNOX gene. KNOX family gene KNAT7 negatively regulates secondary wall formation in Arabidopsis and Populus [51]. Since KNAT7 is a negative regulator of secondary wall biosynthesis, these Paulownia homologs might positively regulate cambium growth during the active spring season.
KEGG annotation yielded a total of 35,471 (57.5%) transcripts that were mapped to 128 KEGG pathways. The top three KEGG enriched pathways were metabolic pathways (7943 transcripts; 22.39%; ko01100), biosynthesis of secondary metabolites (3768 transcripts; 10.62%; ko01110), and plant-pathogen interaction (1852 transcripts; 5.22%; ko04626 (Tables S3 and S4). With the help of the KEGG database, we could further analyze the metabolic pathways and functions of gene products, which can help in studying the complex biological behaviors of genes. The majority of representative unigenes were annotated to metabolic pathways, biosynthesis of secondary metabolites, plant-pathogen interaction, plant hormone signaling, spliceosome, and phenylpropanoid biosynthesis using the KEGG database, which led us to conclude that most of the genes identified in this study are involved in cambial differentiation and wood formation.
3.3. Transcriptional Profiling of Cambial Tissues in Winter and Spring
A total of 10,099 (12.33%) transcripts were found to be significantly differentially expressed between two tissue samples. Of these differentially expressed genes (DEGs), 2688 (26.61%) transcripts were found to be upregulated (>1.6 fold) in the spring season, whereas 7411 (73.39%) were downregulated (<−1.6 fold) when compared to the winter season (Figure S2). Hierarchical clustering of the DEGs identified in winter and spring conditions led to the detailed overall structure of clustering. This indirectly indicated that more genes were upregulated, active, and required during the senescent winter season to keep tissues dormant. Out of 2688 genes, the top 20 genes with log2Fold change >8.00 are summarized in Table 2. This included APC/C cyclosome complex, phosphoenolpyruvate carboxykinase, different classes of heat shock proteins, actin depolymerization factor, anaphase-promoting complex subunit (>12-fold expression), etc. Similarly, many key genes, including synthases such as galactinol synthase (<−12-fold expression), rosmarinate synthase, and valencene synthase, kinases such as receptor-like protein kinase, serine/threonine-protein kinase, CBL-interacting protein kinase, and hormone-regulated genes such as auxin efflux carrier family protein and ethylene-responsive transcription factor were downregulated (Table 3). The cell cycle is one of the most important biological processes in the cambial zone and plays a central role in regulating the growth and development of organisms, including plants. The anaphase-promoting complex/cyclosome (APC/C; homolog in our study Unigene8688), a well-known ubiquitin ligase, acts to accomplish basic cell-cycle control. The APC/C must be turned off at the end of the G1 phase to allow the S phase cyclins to accumulate and cells to begin DNA replication [52]. This is very key during the spring season for cell multiplication and growth. The Cyclin U2 (Unigene22553), one of the major cyclins involved in cell cycle control, like cyclins A and B on maximum gene expression in the poplar cambium zone [53], was upregulated in Paulownia. The high abundance of cyclin transcripts in active cambium during the spring season also reflected a positive correlation between cambium cell division and key cell cycle gene expression.
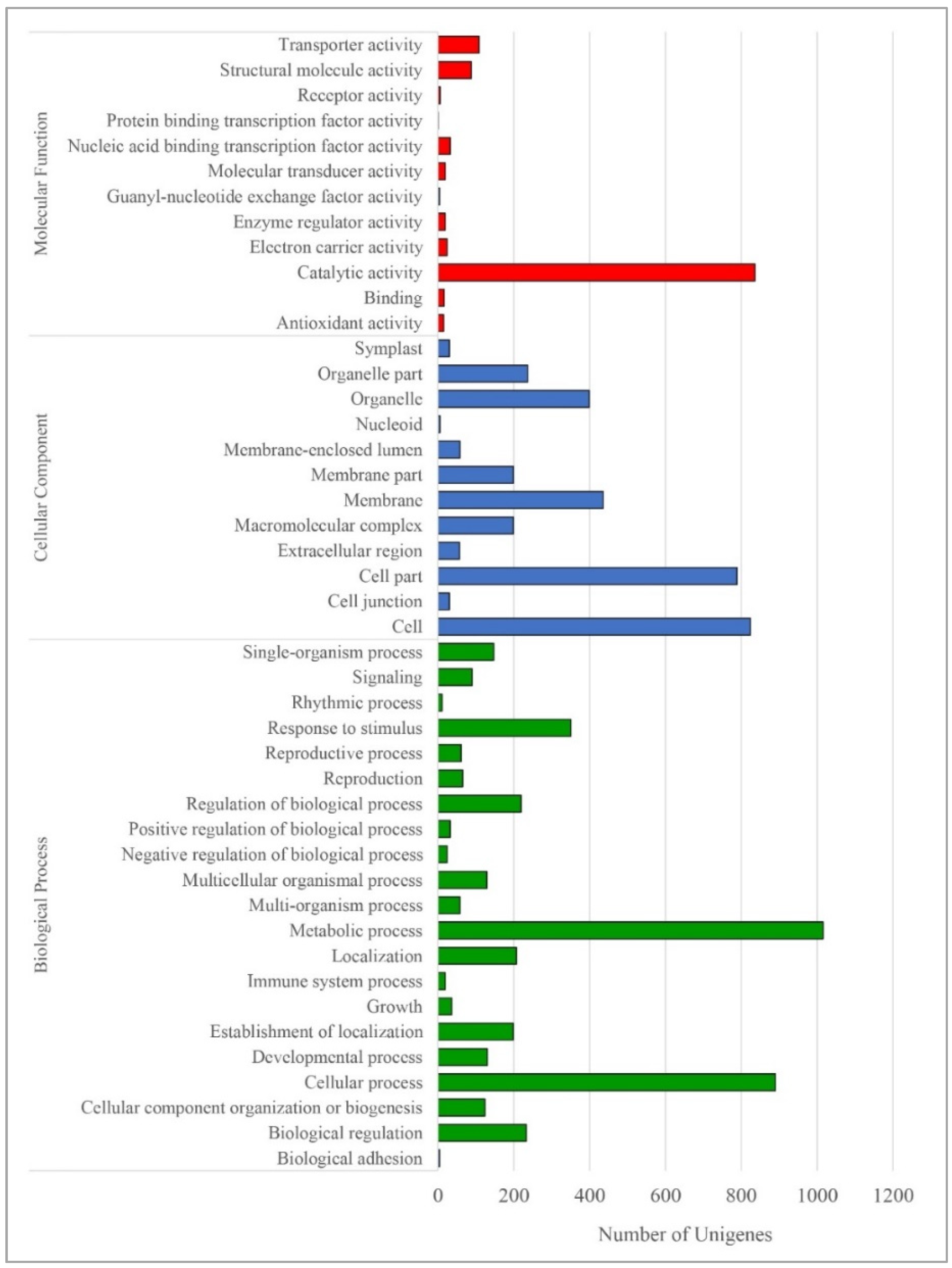
As shown in Figure 5, GO analysis indicated that most of the differentially expressed transcripts for biological processes were involved in the metabolic process (1016), cellular process (890), and response to stimulus (350). Similarly, GO cellular component analysis revealed that a large number of transcripts encoded functions related to cell (824), cell part (789), membrane (436), and organelle (399) synthesis. Meanwhile, GO molecular function analysis showed that the DEGs predominantly contributed to catalytic activity (836), followed by transporter activity and structural molecule activity. KEGG enrichment analysis of DEG showed that these genes were involved in various pathways in the Paulownia plant during seasonal changes (Table 4). Most of the DEGs were enriched in metabolic pathways (ko01100; 1387), biosynthesis of secondary metabolites (ko01110; 827), and plant hormone signal transduction (ko04075; 320). It was also found that starch and sucrose metabolism (ko00500; 172; Figure S3) and phenylpropanoid biosynthesis (ko00940; 106; Figure S4), which correspond to the production of several key wood-forming genes, were within the top 25 most DEG enriched KEGG pathways (Table 4). Nineteen (Unigene11539, Unigene12164, Unigene12788, Unigene16018, Unigene17615, Unigene18048, Unigene18594, Unigene18926, Unigene22808, Unigene24462, Unigene24837, Unigene3634, Unigene4753, Unigene6221, Unigene891, and Unigene9670 (K00430), Unigene16856 (K11188), Unigene22599 and Unigene25305 (K03782)) out of 497 unigenes (total unigenes) involved in lignin synthesis in the phenylpropanoid biosynthesis pathway (Ko00940) were identified and differentially regulated (Figure S4). Lignin plays a vital role in keeping the structural integrity of the cell wall and protecting plants from pathogens [54], as well as a main component of wood. Of these 19, different types of peroxidases (Unigene11539, Unigene18926, Unigene4753 and Unigene6221) were upregulated during winter. Recently, a notable remodeling of the transcriptome was reported in Norway Spruce, where monolignol biosynthesis genes showed high expression during the period of secondary cell wall formation as well as the second peak in midwinter. Interestingly, this midwinter peak expression did not trigger lignin deposition [55]. These genes could be preparing for the biosynthesis and distribution of guaiacyl (G), p-hydroxyl phenol (H), and syringyl (S) lignin in developing biomass as soon as the onset of spring.
3.4. Expression of Lignocellulosic Pathway Genes and Their Validation
Wood, the secondary xylem, is produced from the activity of vascular cambium that is composed of two meristematic initials, fusiform and ray initials [56], with the sequential developmental process including differentiation of vascular cambium cells into secondary xylem mother cells, cell expansion, and massive deposition of secondary walls (where a number of genes involved in vascular tissue differentiation and secondary wall biosynthesis are) [57]. When the wood compression starts, the expression of a number of genes involved in the synthesis of lignocellulosic components (cellulose, hemicellulose, and lignin) and lignans was upregulated in maritime pine [58]. In addition, the onset of wood formation undergoes three periods: winter shrinkage, spring rehydration (32–47 days), and summer transpiration in the stem [59].
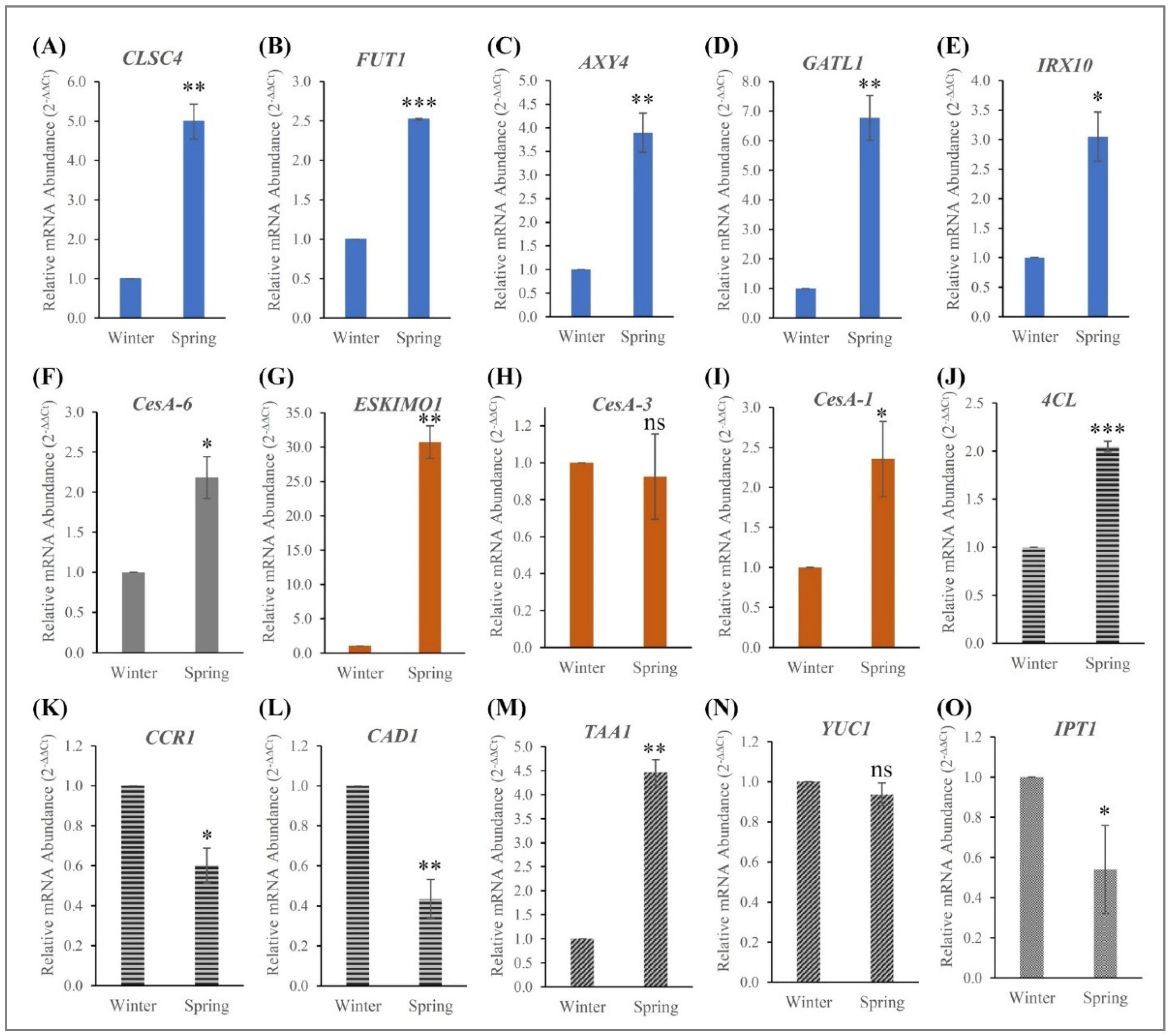
In order to explore the roles of cell wall- and hormone-related genes for the seasonal cues, fifteen candidate genes were identified from previous studies (Table 5). They are involved in cellulose (CesA1, CesA3, CesA6 [60]), hemicellulose (CSLC4 [61], FUT1 [62,63], AXY4 [64], GATL1 [65], IRX10 [66], ESKIMO1 [67]), lignin (4CL [68], CCR1 [69], CAD1 [70]), auxin (TAA1 [71], YUC1 [72,73]), and cytokinin (IPT1 [29] synthesis/pathways). RT-qPCR was employed to study the expression of these wood formation genes in Paulownia during the winter and spring seasons (Figure 6). Cellulose is synthesized in plant cell walls by large membrane-bound protein complexes proposed to contain several copies of the catalytic subunit of the cellulose synthase, CesA. Here, we found CesA1 and CesA6 were upregulated during spring while CesA3 was moderately downregulated during the winter season. In hybrid aspen, expression analyses of the CesA family showed a specific location in normal wood undergoing xylogenesis, while PttCesA2 seems to be activated on the opposite side of a tension wood [60]. However, in Arabidopsis, the expression levels of the three primary cell wall genes (AtCesA2, AtCesA5, AtCesA6) was increased, but not AtCesA3, AtCesA9 or secondary cell wall AtCesA7 [74]. Our results, along with these studies, indicated that the expression of major primary wall CesA genes accelerate the primary wall CesA complex.
Several proteins encoded by the cellulose synthase-like (CSL) gene family, including CSLA proteins, which synthesize β-(1→4)-linked mannans, and CSLC proteins, which are thought to synthesize the β-(1→4)-linked glucan backbone of xyloglucan are known to be involved in the synthesis of cell-wall polysaccharides [61]. Higher expression of CLSC4 in Paulownia during the spring season indicated that it might involve cellulose synthesis. The fucosyltransferase (FUT1) is an enzyme that transfers an L-fucose sugar from a GDP-fucose (guanosine diphosphate-fucose) donor substrate to an acceptor substrate. The Arabidopsis mur1 (AtFUT1) mutant study [63] exhibited a dwarf growth habit and decreased wall strength indicating the indispensable role of FUT1 function in wood formation. Another key gene family of O-acetyl substituents seems to be very important for various plant tissues and during plant development [75], suggesting an important functional role in the plant. Mutants lacking AXY4 transcript resulted in a complete lack of O-acetyl substituents on xyloglucan in several tissues, except seeds [64]. Biosynthesis of xylan in woody plants is a major pathway for plant biomass. Populus genes PdGATL1.1 and PdGATL1.2, the closest orthologs to the Arabidopsis PARVUS/GATL1 gene, have been shown to be important for xylan synthesis but may also have a role(s) in the synthesis of other wall polymers [65]. The expression of GATL1 homolog was six-fold increased (Figure 6) in the spring season tissue of Paulownia, implying more xylan biosynthesis. Collapsed xylem phenotypes of Arabidopsis [76] and Physcomitrella patens [66] mutants (irx10) identified mutants deficient in cellulose deposition in the secondary cell wall due to lack of synthesis of the glucuronoxylan. Acetyl transferases are involved in cellulose biosynthesis in plants. In Arabidopsis, the ESKIMO1 (ESK1) gene participated in several functions, and esk1 mutants indicated that ESK1 is necessary for the synthesis of functional xylem vessels in forming secondary cell wall components [67]. Our RNA seq data indicated that all the genes associated with cellulose and hemicellulose synthesis were expressed during the spring season to complete wood formation.
3.5. Analysis of Hormone-Specific Genes and Their Validation
Plant growth regulator auxins play a key role in regulating wood formation through their action on cambial activity and xylem development [77]. Auxin has been shown to be required for maintaining the cambium in a meristematic state as depleting the auxin in cambium leads to differentiation of cambial cells into axial parenchyma cells [78]. Cytokinins, on the other hand, have a well-established function in cell division during growth and development, and they are called central regulators of cambial activity [79]. The interaction between auxin and cytokinin seems to be essential for the induction of phenylalanine ammonialyase activity in support of lignification [80]. TAA1, which performs the first two reactions in the auxin pathway, is a Trp aminotransferase that converts Trp to IPA in the IPA auxin biosynthesis branch in Arabidopsis [73]. Higher-order mutants in TAA1 showed auxin-related multiple phenotypes. Later, it was identified that the TAA1 gene was essential for hormone crosstalk with ethylene for plant development [71]. Later, new putative functions of IAA production via IPyA and transport were identified [81].
Another group of auxin biosynthesis gene family, YUCCA flavin monooxygenases, control the formation of floral organs and vascular tissues in Arabidopsis [72]. When the TAA family of aminotransferases converts tryptophan to indole-3-pyruvate (IPA), YUCCA (YUC) family participates in converting IPA to IAA [73]. In addition, the authors found that YUC and TAA work genetically in the same pathway and that YUC is downstream of TAA. From our transcriptome and gene expression studies, we observed TAA1 was strongly expressed, but YUC1 expression was negligible during the spring season. Our study identified several transcripts involved in auxin biosynthesis through the tryptophan pathway for cell enlargement and plant growth (Figure S5).
In Arabidopsis, cambial activity responded to small changes in cytokinin levels indicating that cytokinins are central regulators of cambium activity [79]. Isopentenyltransferase, the rate-limiting step of cytokinin biosynthesis, is an important enzyme playing key roles in meristem maintenance and organ development. Arabidopsis quadruple mutants lacking AtIPT1, AtIPT3, AtIPT5, and AtIPT7 were unable to form cambium and showed reduced thickening of the root and stem, though single mutant atipt3 showed moderately decreased levels of cytokinins without any other recognizable morphological changes. Similarly, increased cytokinin biosynthesis stimulates the cambial cell division rate and increases the production of trunk biomass in transgenic Populus trees [29]. Surprisingly, IPT1 expression was high in winter and moderately reduced in spring. There could be many other members in the IPT family that complements the function of cambial development. Auxin and cytokinin display distinct distribution profiles across the cambium, and elevated cytokinin content leads to an increased cambial auxin concentration [29]. Together, it is very interesting to see the interaction of lignocellulosic pathways genes along with major hormone-regulated genes and their cross-talks to maintain the balance of cambial activities for quality wood formation with alternative seasonal changes (Figure S6).
3.6. Analysis of Simple Sequence Repeats (SSRs)
SSR markers are very useful for multiple applications in plant genetics because of their co-dominance, high level of polymorphism, multi-allelic variance, and abundance, and cross-species transferability [82,83]. In the present study, SSR was identified utilizing the transcriptome of Paulownia cambial tissues because EST-SSR markers have a relatively higher transferability than genomic SSRs [84]. Recent studies showed that abundant EST-SSRs from RNA-seq have agronomic potential and constitute a scientific basis for future studies on the identification, classification, molecular verification, and innovation of germplasms in hawthorn and Lei bamboo [85,86].
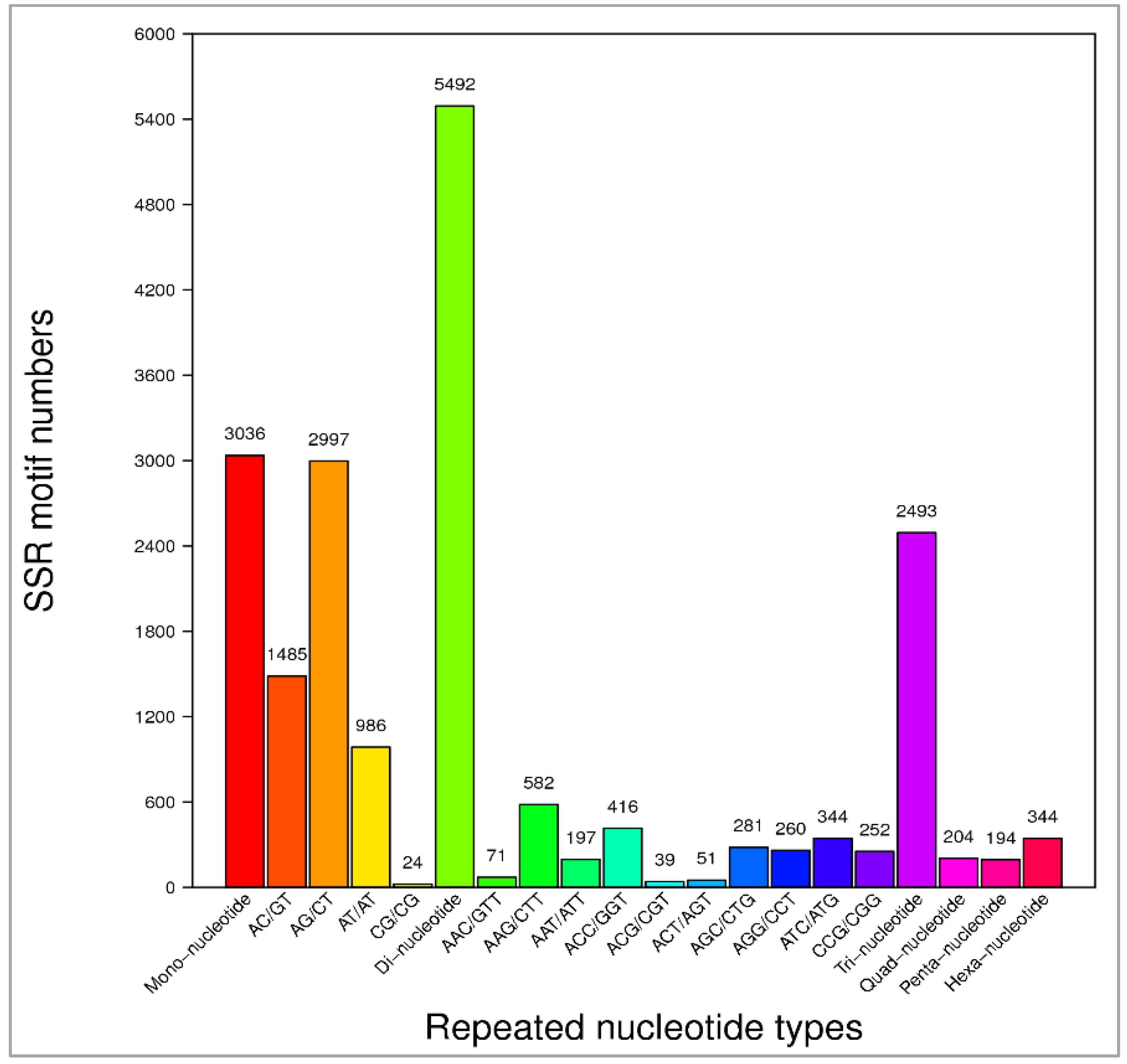
We identified 11,338 SSRs from the annotated 61,639 unigenes. We detected 3036 mononucleotides, 5492 dinucleotides, 2493 trinucleotides, 204 tetranucleotides, 194 pentanucleotides, and 344 hexanucleotides motifs (Figure 7). Among the dinucleotide and trinucleotide SSRs, AG/CT repeats represented 2997 SSRs, and AAG/CTT repeats represented 582 SSRs. In mononucleotide, dinucleotide, trinucleotide, quadnucleotide, pentanucleotide, and hexanucleotide repeat categories, the occurrences of repeats were twelve, six, five, five, four, and four, respectively (Table S4). Finally, 6773 oligonucleotide pairs were generated for these identified SSR markers. SSRs and SNPs are the most useful and robust molecular markers for genetics and plant breeding applications [87]. This study provided a set of SSR markers that could be used, for example, in diversity analysis of Paulownia species. In addition, Paulownia tree breeding programs will benefit from the availability of these SSR markers identified from our RNAseq data. Mononucleotide SSRs would be excluded because of the frequent homopolymer errors found in sequencing data and fewer polymorphisms; dinucleotides (46.6%) and trinucleotides (21.2%) contributed most in Paulownia. This is consistent with the EST-SSRs distributions reported in other plant species [88,89]. In plants, SNPs are predominantly beneficial in the construction of high-resolution genetic maps, positional cloning, marker-assisted selection (MAS) of important genes, genome-wide linkage disequilibrium associate analysis, and species origin, relationship, and evolutionary studies [90].
4. Conclusions
Paulownia is a fast-growing, multipurpose timber tree suitable for use as a dedicated lignocellulosic bioenergy crop. To understand the genes involved in the formation of woody biomass related to seasonal cues, a de novo transcriptome study was conducted on vascular cambium tissue from senescent winter vascular cambium tissue and actively growing spring vascular cambium tissue. To the best of our knowledge, this is the first transcriptome-based study on P. elongata, as well as the first transcriptome study performed on Paulownia vascular cambium tissue focusing on seasonal difference. A set of transcripts was specifically expressed in the same set of tissues at two different time points. The transcript abundance data confirms the differential pattern of expression of cellulosic, hemicellulosic, lignin biosynthesis specific, and hormone pathway-specific genes. By analyzing the transcriptome from two different temporal treatments (winter and spring), representing two distinct physiological states of the plant, DEGs were identified from both treatments. Cell division is one of the key processes taking place in the cambial zone, and the majority of the cell cycle genes were upregulated during the active stage. The onset of cambial activity began between the end of March and the beginning of April as the increased vacuolization of meristematic cells, and the mitotic activity occurs. However, our current study showed that more genes were downregulated in the spring season. Overall, data from this study will serve as a benchmark for further analysis of molecular mechanisms of wood formation in Paulownia and other trees.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/d13090423/s1, Figure S1. Length distribution of all Paulownia unigenes, Figure S2. Statistics of differentially expressed genes, Figure S3. Unigenes involved in starch and sucrose metabolism, Figure S4. Unigenes involved in H-, G-, S-lignin biosynthesis pathway, Figure S5. Unigenes involved in auxin biosynthesis pathway, Figure S6. Unigenes involved in cytokinin biosynthesis pathway, Table S1 Annotation BLASTX hits of unigenes in Nr database, Table S2 GO categories of unigenes, Table S3 COG function classsification of unigenes, Table S4 KEGG annotation of unigenes.
Author Contributions
Conceptualization, N.J.; data curation, T.S.; formal analysis, A.A.; funding acquisition, N.J.; investigation, Z.D.P., C.B. and U.K.R.; methodology, Z.D.P., T.S., B.N.V. and U.K.R.; project administration, B.N.V.; validation, U.K.R.; writing—original draft, Z.D.P. and T.S.; writing—review & editing, A.A. and N.J. All authors have read and agreed to the published version of the manuscript.
Funding
Capacity building USDA NIFA grant ‘Developing a sustainable bioenergy system: Paulownia production for fuel, chemicals, and materials’ CSREES Award #2014-01293 (P.I: N. Joshee).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article or supplementary material.
Acknowledgments
ZP received financial assistance in the form of a graduate research assistantship (GRA) through the capacity building USDA NIFA grant ‘Developing a sustainable bioenergy system: Paulownia production for fuel, chemicals, and materials’ CSREES Award #2014-01293 (P.I: N. Joshee).
Conflicts of Interest
Authors declare that no competing interests exist.
References
- Zhu, Z.-H.; Chao, C.-J.; Lu, X.-Y.; Xiong, Y.G. Paulownia in China: Cultivation and Utilization; Asian Network for Biological Sciences and International Development Research Centre and International Development Research Centre: Ottawa, ON, Canada, 1986; pp. 1–65. [Google Scholar]
- Clatterbuck, K.W.; Hodges, D.G. Tree Crops for Marginal Farmland-Paulownia; The University of Tennessee Extension Service: Knoxville, TN, USA, 2004; pp. 1–32. Available online: https://trace.tennessee.edu/cgi/viewcontent.cgi?article=1002&context=utk_agexfores (accessed on 26 May 2021).
- El-Showk, S.; El-Showk, N. The Paulownia Tree. An Alternative for Sustainable Forestry. 2003. Available online: www.cropdevelopment.org (accessed on 30 August 2021).
- Akyildiz, M.H.; Kol Sahin, H. Some technological properties and uses of paulownia (Paulownia tomentosa Steud.) wood. J. Environ. Biol. 2010, 31, 351–355. [Google Scholar]
- Li, P.; Oda, J. Flame retardancy of paulownia wood and its mechanism. J. Mater. Sci. 2007, 42, 8544–8550. [Google Scholar] [CrossRef] [Green Version]
- Yadav, N.K.; Vaidya, B.N.; Henderson, K.; Lee, J.F.; Stewart, W.M.; Dhekney, S.A.; Joshee, N. A Review of Paulownia biotechnology: A short rotation, fast growing multipurpose bioenergy tree. Am. J. Plant. Sci. 2013, 4, 2070–2082. [Google Scholar] [CrossRef] [Green Version]
- Tisserat, B.; Joshee, N.; Mahapatra, A.K.; Selling, G.W.; Finkenstadt, V. Physical and mechanical properties of extruded poly(lactic acid)-based Paulownia elongata biocomposites. Ind. Crop. Prod. 2013, 44, 88–96. [Google Scholar] [CrossRef]
- Tisserat, B.; Reifschneider, L.; Joshee, N.; Finkenstadt, V. Properties of high density polyethylene—Paulownia wood flour composites via injection molding. Bioresources 2013, 8, 4440–4458. [Google Scholar] [CrossRef] [Green Version]
- Tisserat, B.; Reifschneider, L.; Joshee, N.; Finkenstadt, V. Evaluation of Paulownia elongata wood polyethylene composites. J. Thermoplast. Compos. Mater. 2015, 28, 1301–1320. [Google Scholar] [CrossRef]
- Vaughn, S.F.; Dinelli, F.D.; Tisserat, B.; Joshee, N.; Vaughan, M.M.; Peterson, S.C. Creeping bentgrass growth in sand-based root zones with or without biochar. Sci. Hortic. 2015, 197, 592–596. [Google Scholar] [CrossRef]
- Stewart, W.M.; Vaidya, B.N.; Mahapatra, A.K.; Terrill, T.H.; Joshee, N. Potential use of multipurpose paulownia elongata tree as an animal feed resource. Am. J. Plant. Sci. 2018, 9, 1212–1227. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.-H.; Zhong, R. Molecular control of wood formation in trees. J. Exp. Bot. 2015, 66, 4119–4131. [Google Scholar] [CrossRef] [Green Version]
- Samuels, A.L.; Kaneda, M.; Rensing, K. The cell biology of wood formation: From cambial divisions to mature secondary xylem. Can. J. Bot. 2006, 84, 631–639. [Google Scholar] [CrossRef]
- Song, J.; Lu, S.; Chen, Z.-Z.; Lourenco, R.; Chiang, V.L. Genetic transformation of populus trichocarpa genotype nisqually-1: A functional genomic tool for woody plants. Plant. Cell Physiol. 2006, 47, 1582–1589. [Google Scholar] [CrossRef] [Green Version]
- Shim, D.; Ko, J.-H.; Kim, W.-C.; Wang, Q.; Keathley, D.E.; Han, K.-H. A molecular framework for seasonal growth-dormancy regulation in perennial plants. Hortic. Res. 2014, 1, 14059. [Google Scholar] [CrossRef] [Green Version]
- Joshee, N. Paulownia: A multipurpose tree for rapid lignocellulosic biomass production. In Handbook of Bioenergy Crop Plants; Kole, C., Joshi, C.P., Shonnard, D., Eds.; Taylor & Francis: Boca Raton, FL, USA, 2012; pp. 671–686. [Google Scholar]
- Aspeborg, H.; Schrader, J.; Coutinho, P.M.; Stam, M.; Kallas, A.; Djerbi, S.; Nilsson, P.; Denman, S.; Amini, B.; Sterky, F.; et al. Carbohydrate-active enzymes involved in the secondary cell wall biogenesis in hybrid aspen. Plant. Physiol. 2005, 137, 983–997. [Google Scholar] [CrossRef] [Green Version]
- Dharmawardhana, P.; Brunner, A.M.; Strauss, S.H. Genome-wide transcriptome analysis of the transition from primary to secondary stem development in Populus trichocarpa. BMC Genom. 2010, 11, 150. [Google Scholar] [CrossRef] [Green Version]
- Pavy, N.; Boyle, B.; Nelson, C.; Paule, C.; Giguère, I.; Caron, S.; Parsons, L.; Dallaire, N.; Bedon, F.; Bérubé, H.; et al. Identification of conserved core xylem gene sets: Conifer cDNA microarray development, transcript profiling and computational analyses. New Phytol. 2008, 180, 766–786. [Google Scholar] [CrossRef]
- Wang, M.; Qi, X.; Zhao, S.; Zhang, S.; Lu, M.-Z. Dynamic changes in transcripts during regeneration of the secondary vascular system in Populus tomentosa Carr. revealed by cDNA microarrays. BMC Genom. 2009, 10, 215. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, O.; Nahal, H.; Foong, J.; Provart, N.J.; Campbell, M.M. Expansion and diversification of the populus R2R3-MYB family of transcription factors. Plant Physiol. 2009, 149, 981–993. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Fan, G.; Zhao, Z.; Deng, M. Compatible solute, transporter protein, transcription factor, and hormone-related gene expression provides an indicator of drought stress in Paulownia fortunei. Funct. Integr. Genom. 2014, 14, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Fan, G.; Zhao, Z.; Deng, M. Transcriptome expression profiling in response to drought stress in paulownia australis. Int. J. Mol. Sci. 2014, 15, 4583–4607. [Google Scholar] [CrossRef] [Green Version]
- Mou, H.-Q.; Lu, J.; Zhu, S.-F.; Lin, C.-L.; Tian, G.-Z.; Xu, X.; Zhao, W.-J. Transcriptomic analysis of paulownia infected by paulownia witches’-broom phytoplasma. PLoS ONE 2013, 8, e77217. [Google Scholar] [CrossRef]
- Li, B.; Zhai, X.; Cao, Y.; Zhao, H.; Wang, Z.; Liu, H.; Fan, G. Transcriptome and small RNA sequencing analysis revealed roles of PaWB-related miRNAs and genes in Paulownia fortunei. Forests 2018, 9, 397. [Google Scholar] [CrossRef] [Green Version]
- Nitsch, J. Photoperiodism in woody plants. J. Am. Soc. Hort. Sci. 1957, 70, 526–544. [Google Scholar]
- Espinosa-Ruiz, A.; Saxena, S.; Schmidt, J.; Mellerowicz, E.; Miskolczi, P.; Bakó, L.; Bhalerao, R.P. Differential stage-specific regulation of cyclin-dependent kinases during cambial dormancy in hybrid aspen. Plant. J. 2004, 38, 603–615. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Deng, T.; Liu, Z.; Wang, X. Time-coursed transcriptome analysis identifies key expressional regulation in growth cessation and dormancy induced by short days in Paulownia. Sci. Rep. 2019, 9, 16602. [Google Scholar] [CrossRef]
- Immanen, J.; Nieminen, K.; Smolander, O.-P.; Kojima, M.; Serra, J.A.; Koskinen, P.; Zhang, J.; Elo, A.; Mähönen, A.P.; Street, N. Cytokinin and auxin display distinct but interconnected distribution and signaling profiles to stimulate cambial activity. Curr. Biol. 2016, 26, 1990–1997. [Google Scholar] [CrossRef] [Green Version]
- Nieminen, K.; Blomster, T.; Helariutta, Y.; Mähönen, A.P. Vascular cambium development. Arab. Book 2015, 13, e0177. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.; He, Y.; Zhang, Y.; Guo, J.; Zhang, L. Genome-wide identification and profiling of microRNAs in Paulownia to-mentosa cambial tissues in response to seasonal changes. Gene 2018, 677, 32–40. [Google Scholar] [CrossRef]
- Saminathan, T.; Nimmakayala, P.; Manohar, S.; Malkaram, S.; Almeida, A.; Cantrell, R.; Tomason, Y.; Abburi, L.; Rahman, M.A.; Vajja, V.G.; et al. Differential gene expression and alternative splicing between diploid and tetraploid watermelon. J. Exp. Bot. 2015, 66, 1369–1385. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, D480–D484. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Boil. 1999, 99, 138–148. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Tarazona, S.; García-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef] [Green Version]
- Doering, A.; Lathe, R.; Persson, S. An Update on xylan synthesis. Mol. Plant. 2012, 5, 769–771. [Google Scholar] [CrossRef] [Green Version]
- Jia, X.-L.; Wang, G.-L.; Xiong, F.; Yu, X.-R.; Xu, Z.-S.; Wang, F.; Xiong, A.-S. De novo assembly, transcriptome characterization, lignin accumulation and anatomic characteristics: Novel insights into lignin biosynthesis during celery leaf development. Sci. Rep. 2015, 5, srep08259. [Google Scholar] [CrossRef] [Green Version]
- Pauly, M.; Gille, S.; Liu, L.; Mansoori, N.; de Souza, A.J.; Schultink, A.; Xiong, G. Hemicellulose biosynthesis. Planta 2013, 238, 627–642. [Google Scholar] [CrossRef]
- Quang, T.H.; Hallingback, H.R.; Gyllenstrand, N.; Von Arnold, S.; Clapham, D. Expression of genes of cellulose and lignin synthesis in Eucalyptus urophylla and its relation to some economic traits. Trees 2011, 26, 893–901. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Novaes, E.; Drost, D.R.; Farmerie, W.G.; Pappas, G.J.; Grattapaglia, D.; Sederoff, R.R.; Kirst, M. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genom. 2008, 9, 312. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Wang, Y.; Diao, G.; Jiang, J.; Yang, C. Isolation and characterization of expressed sequence tags (ESTs) from cambium tissue of birch (betula platyphylla suk). Plant. Mol. Biol. Rep. 2010, 28, 438–449. [Google Scholar] [CrossRef]
- Xu, E.; Fan, G.; Niu, S.; Zhao, Z.; Deng, M.; Dong, Y. Transcriptome-wide profiling and expression analysis of diploid and autotetraploid paulownia tomentosa × paulownia fortunei under drought stress. PLoS ONE 2014, 9, e113313. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Li, R.; Shang, F. The complete chloroplast genome of Paulownia elongata and phylogenetic implications in Lamiales. Mitochondrial DNA Part B 2019, 4, 2067–2068. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Song, D.; Sun, J.; Wang, X.; Li, L. PtrHB7, a class III HD-zip gene, plays a critical role in regulation of vascular cambium differentiation in populus. Mol. Plant. 2013, 6, 1331–1343. [Google Scholar] [CrossRef] [Green Version]
- Groover, A.T.; Mansfield, S.D.; DiFazio, S.; Dupper, G.; Fontana, J.R.; Millar, R.; Wang, Y. The populus homeobox gene ARBORKNOX1 reveals overlapping mechanisms regulating the shoot apical meristem and the vascular cambium. Plant. Mol. Biol. 2006, 61, 917–932. [Google Scholar] [CrossRef]
- Li, E.; Bhargava, A.; Qiang, W.; Friedmann, M.C.; Forneris, N.; Savidge, R.A.; Johnson, L.A.; Mansfield, S.; Ellis, B.E.; Douglas, C.J. The class II KNOX gene KNAT7 negatively regulates secondary wall formation in Arabidopsis and is functionally conserved in Populus. New Phytol. 2012, 194, 102–115. [Google Scholar] [CrossRef]
- Pines, J. Cubism and the cell cycle: The many faces of the APC/C. Nat. Rev. Mol. Cell Biol. 2011, 12, 427–438. [Google Scholar] [CrossRef]
- Hertzberg, M.; Aspeborg, H.; Schrader, J.; Andersson, A.; Erlandsson, R.; Blomqvist, K.; Bhalerao, R.; Uhlen, M.; Teeri, T.T.; Lundeberg, J.; et al. A transcriptional roadmap to wood formation. Proc. Natl. Acad. Sci. USA 2001, 98, 14732–14737. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Liu, Q.; Tan, M.; Yi, H.; Deng, X. Lignin deposition converts juice sacs to “Brown Thorns” in a citrus triploid hybrid. J. Am. Soc. Hortic. Sci. 2008, 133, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Jokipii-Lukkari, S.; Delhomme, N.; Schiffthaler, B.; Mannapperuma, C.; Prestele, J.; Nilsson, O.; Street, N.R.; Tuominen, H. Transcriptional roadmap to seasonal variation in wood formation of norway spruce. Plant. Physiol. 2018, 176, 2851–2870. [Google Scholar] [CrossRef] [Green Version]
- Mauseth, J.D. Plant Anatomy; The Benjamin/Cummings: San Francisco, CA, USA, 1988; p. 560. [Google Scholar]
- Zhong, R.; Ye, Z.-H. Secondary cell walls: Biosynthesis, patterned deposition and transcriptional regulation. Plant. Cell Physiol. 2015, 56, 195–214. [Google Scholar] [CrossRef] [Green Version]
- Villalobos, D.P.; Moreno, R.B.; Said, E.-S.S.; Cañas, R.A.; Osuna, D.; Van Kerckhoven, S.H.E.; Bautista, R.; Claros, M.G.; Canovas, F.M.; Cantón, F.R. Reprogramming of gene expression during compression wood formation in pine: Coordinated modulation of S-adenosylmethionine, lignin and lignan related genes. BMC Plant. Biol. 2012, 12, 100. [Google Scholar] [CrossRef] [Green Version]
- Turcotte, A.; Morin, H.; Krause, C.; Deslauriers, A.; Thibeault-Martel, M. The timing of spring rehydration and its relation with the onset of wood formation in black spruce. Agric. For. Meteorol. 2009, 149, 1403–1409. [Google Scholar] [CrossRef]
- Djerbi, S.; Aspeborg, H.; Nilsson, P.; Sundberg, B.; Mellerowicz, E.; Blomqvist, K.; Teeri, T.T. Identification and expression analysis of genes encoding putative cellulose synthases (CesA) in the hybrid aspen, Populus tremula (L.) P. tremuloides (Michx.). Cellulose 2004, 11, 301–312. [Google Scholar] [CrossRef]
- Davis, J.; Brandizzi, F.; Liepman, A.H.; Keegstra, K. Arabidopsis mannan synthase CSLA9 and glucan synthase CSLC4 have opposite orientations in the Golgi membrane. Plant. J. 2010, 64, 1028–1037. [Google Scholar] [CrossRef]
- Perrin, R.M.; DeRocher, A.E.; Bar-Peled, M.; Zeng, W.; Norambuena, L.; Orellana, A.; Raikhel, N.V.; Keegstra, K. Xyloglucan fucosyltransferase, an enzyme involved in plant cell wall biosynthesis. Science 1999, 284, 1976–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanzin, G.; Madson, M.; Carpita, N.C.; Raikhel, N.V.; Keegstra, K.; Reiter, W.-D. The mur2 mutant of Arabidopsis thaliana lacks fucosylated xyloglucan because of a lesion in fucosyltransferase AtFUT. Proc. Natl. Acad. Sci. USA 2002, 99, 3340–3345. [Google Scholar] [CrossRef] [Green Version]
- Gille, S.; de Souza, A.J.; Xiong, G.; Benz, M.; Cheng, K.; Schultink, A.; Reca, I.-B.; Pauly, M. O-Acetylation of arabidopsis hemicellulose xyloglucan requires AXY4 or AXY4L, proteins with a TBL and DUF231 domain. Plant. Cell 2011, 23, 4041–4053. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Zhou, G.; Avci, U.; Gu, X.; Jones, C.; Yin, Y.; Xu, Y.; Hahn, M. Two poplar glycosyltransferase genes, PdGATL1.1 and PdGATL1.2, are functional orthologs to PARVUS/AtGATL1 in arabidopsis. Mol. Plant. 2009, 2, 1040–1050. [Google Scholar] [CrossRef] [Green Version]
- Hörnblad, E.; Ulfstedt, M.; Ronne, H.; Marchant, A. Partial functional conservation of IRX10 homologs in physcomitrella patens and Arabidopsis thaliana indicates an evolutionary step contributing to vascular formation in land plants. BMC Plant. Biol. 2013, 13, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefebvre, V.; Fortabat, M.-N.; Ducamp, A.; North, H.M.; Maia-Grondard, A.; Trouverie, J.; Boursiac, Y.; Mouille, G.; Durand-Tardif, M. ESKIMO1 disruption in arabidopsis alters vascular tissue and impairs water transport. PLoS ONE 2011, 6, e16645. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.-J.; Harding, S.A.; Lung, J.; Popko, J.L.; Ralph, J.; Stokke, D.D.; Tsai, C.-J.; Chiang, V.L. Repression of lignin biosynthesis promotes cellulose accumulation and growth in transgenic trees. Nat. Biotechnol. 1999, 17, 808–812. [Google Scholar] [CrossRef] [Green Version]
- Goujon, T.; Ferret, V.; Mila, I.; Pollet, B.; Ruel, K.; Burlat, V.; Joseleau, J.-P.; Barrière, Y.; Lapierre, C.; Jouanin, L. Down-regulation of the AtCCR1 gene in Arabidopsis thaliana: Effects on phenotype, lignins and cell wall degradability. Planta 2003, 217, 218–228. [Google Scholar] [CrossRef]
- Bouvier d’Yvoire, M.; Bouchabke-Coussa, O.; Voorend, W.; Antelme, S.; Cézard, L.; Legée, F.; Lebris, P.; Legay, S.; Whitehead, C.; McQueen-Mason, S.J. Disrupting the cinnamyl alcohol dehydrogenase 1 gene (Bd CAD 1) leads to altered lignification and improved saccharification in Brachypodium distachyon. Plant J. 2013, 73, 496–508. [Google Scholar] [CrossRef]
- Stepanova, A.; Robertson-Hoyt, J.; Yun, J.; Benavente, L.M.; Xie, D.-Y.; Dolezal, K.; Schlereth, A.; Jürgens, G.; Alonso, J.M. TAA1-mediated auxin biosynthesis is essential for hormone crosstalk and plant development. Cell 2008, 133, 177–191. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Dai, X.; Zhao, Y. Auxin biosynthesis by the YUCCA flavin monooxygenases controls the formation of floral organs and vascular tissues in Arabidopsis. Genes Dev. 2006, 20, 1790–1799. [Google Scholar] [CrossRef] [Green Version]
- Won, C.; Shen, X.; Mashiguchi, K.; Zheng, Z.; Dai, X.; Cheng, Y.; Kasahara, H.; Kamiya, Y.; Chory, J.; Zhao, Y. Conversion of tryptophan to indole-3-acetic acid by tryptophan aminotransferases of arabidopsis and yuccas in arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 18518–18523. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Zhang, R.; Feng, S.; Wang, Y.; Wang, Y.; Fan, C.; Li, Y.; Liu, Z.; Schneider, R.; Xia, T. Three AtCesA6-like members enhance biomass production by distinctively promoting cell growth in Arabidopsis. Plant Biotechnol. J. 2018, 16, 976–988. [Google Scholar] [CrossRef] [Green Version]
- Liners, F.; Gaspar, T.; Van Cutsem, P. Acetyl- and methyl-esterification of pectins of friable and compact sugar-beet calli: Consequences for intercellular adhesion. Planta 1994, 192, 545–556. [Google Scholar] [CrossRef]
- Turner, S.R.; Somerville, C.R. Collapsed xylem phenotype of Arabidopsis identifies mutants deficient in cellulose deposition in the secondary cell wall. Plant. Cell 1997, 9, 689–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundberg, B.; Uggla, C.; Tuominen, H.; Savidge, R.; Barnett, J.; Napier, R. Cell and Molecular Biology of Wood Formation; BIOS Scientific Publishers: Oxford, UK; pp. 169–188.
- Savidge, R.A. The role of plant hormones in higher plant cellular differentiation. II. Experiments with the vascular cambium, and sclereid and tracheid differentiation in the pine, Pinus contorta. J. Mol. Histol. 1983, 15, 447–466. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto-Kitano, M.; Kusumoto, T.; Tarkowski, P.; Kinoshita-Tsujimura, K.; Václavíková, K.; Miyawaki, K.; Kakimoto, T. Cytokinins are central regulators of cambial activity. Proc. Natl. Acad. Sci. USA 2008, 105, 20027–20031. [Google Scholar] [CrossRef] [Green Version]
- Bevan, M.; Northcote, D.H. The interaction of auxin and cytokinin in the induction of phenylalanine ammonia-lyase in suspension cultures of Phaseolus vulgaris. Planta 1979, 147, 77–81. [Google Scholar] [CrossRef]
- Stepanova, A.; Yun, J.; Robles, L.M.; Novak, O.; He, W.; Guo, H.; Ljung, K.; Alonso, J.M. The arabidopsis YUCCA1 flavin monooxygenase functions in the indole-3-pyruvic acid branch of auxin biosynthesis. Plant. Cell 2011, 23, 3961–3973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbará, T.; Palma-Silva, C.; Paggi, G.; Bered, F.; Fay, M.F.; Lexer, C. Cross-species transfer of nuclear microsatellite markers: Potential and limitations. Mol. Ecol. 2007, 16, 3759–3767. [Google Scholar] [CrossRef]
- Powell, W.; Machray, G.C.; Provan, J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1996, 1, 215–222. [Google Scholar] [CrossRef]
- Varshney, R.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Cai, K.; Zhu, L.; Zhang, K.; Li, L.; Zhao, Z.; Zeng, W.; Lin, X. Development and characterization of EST-SSR markers from RNA-seq data in phyllostachys violascens. Front. Plant. Sci. 2019, 10, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Dong, W.; Lyu, T.; Lyu, Y. An RNA sequencing transcriptome analysis and development of EST-SSR markers in chinese hawthorn through illumina sequencing. Forest 2019, 10, 82. [Google Scholar] [CrossRef] [Green Version]
- Hiremath, P.J.; Kumar, A.; Penmetsa, R.V.; Farmer, A.; Schlueter, J.; Chamarthi, S.K.; Whaley, A.M.; Carrasquilla-Garcia, N.; Gaur, P.; Upadhyaya, H.D.; et al. Large-scale development of cost-effective SNP marker assays for diversity assessment and genetic mapping in chickpea and comparative mapping in legumes. Plant. Biotechnol. J. 2012, 10, 716–732. [Google Scholar] [CrossRef] [Green Version]
- Ahn, Y.-K.; Tripathi, S.; Cho, Y.-I.; Kim, J.-H.; Lee, H.-E.; Kim, D.-S.; Woo, J.-G.; Cho, M.-C. De novo transcriptome assembly and novel microsatellite marker information in Capsicum annuum varieties Saengryeg 211 and Saengryeg 213. Bot. Stud. 2013, 54, 58. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yu, G.; Shi, B.; Wang, X.; Qiang, H.; Gao, H. Development and characterization of simple sequence repeat (SSR) markers based on RNA-sequencing of medicago sativa and in silico mapping onto the m. truncatula genome. PLoS ONE 2014, 9, e92029. [Google Scholar] [CrossRef] [PubMed]
- Shahinnia, F.; Sayed-Tabatabaei, B.E. Conversion of barley SNPs into PCR-based markers using dCAPS method. Genet. Mol. Biol. 2009, 32, 564–567. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Six-year-old Paulownia trees growing at the ‘Paulownia Demonstration Plot’ at FVSU. Paulownia trees in dormant state during winter (A) and active state during spring (B). Cambial tissue sampling from a paulownia twig (C); normal branch (left), branch after bark removal (middle), and scraping wood forming cambial tissue for RNA extraction (right).
Figure 1.
Six-year-old Paulownia trees growing at the ‘Paulownia Demonstration Plot’ at FVSU. Paulownia trees in dormant state during winter (A) and active state during spring (B). Cambial tissue sampling from a paulownia twig (C); normal branch (left), branch after bark removal (middle), and scraping wood forming cambial tissue for RNA extraction (right).

Figure 2.
Statistics of homology search of unigenes against Non-redundant (NR) protein database. Distribution of top BLASTX hits with cut-off e-value of <1.0 × 10−5 (A), similarity (B), and species distributions (C) of all unigenes.
Figure 2.
Statistics of homology search of unigenes against Non-redundant (NR) protein database. Distribution of top BLASTX hits with cut-off e-value of <1.0 × 10−5 (A), similarity (B), and species distributions (C) of all unigenes.

Figure 3.
Histogram representation of clusters of orthologous groups (COG). The horizontal coordinates are function classes of COG, and the vertical coordinates are numbers of unigenes in one class. The notation on the right is the full name of the functions in the x-axis. Histogram representation of classification of the clusters of orthologous groups (COG) for the total aligned 43,780 unigenes (53.43%) into 25 functional groups.
Figure 3.
Histogram representation of clusters of orthologous groups (COG). The horizontal coordinates are function classes of COG, and the vertical coordinates are numbers of unigenes in one class. The notation on the right is the full name of the functions in the x-axis. Histogram representation of classification of the clusters of orthologous groups (COG) for the total aligned 43,780 unigenes (53.43%) into 25 functional groups.

Figure 4.
GO classification analysis of unigenes. GO functions are showed on the x-axis. The right y-axis shows the number of genes that have the GO function, and the left y-axis shows the percentage. Unigenes in the winter and spring season are classified into biological processes, cellular components, and molecular functions. In total, 41,588 (50.76% of all unigenes) were assigned to 48 GO categories.
Figure 4.
GO classification analysis of unigenes. GO functions are showed on the x-axis. The right y-axis shows the number of genes that have the GO function, and the left y-axis shows the percentage. Unigenes in the winter and spring season are classified into biological processes, cellular components, and molecular functions. In total, 41,588 (50.76% of all unigenes) were assigned to 48 GO categories.

Figure 5.
GO function analysis of the differentially expressed genes. GO function analysis results for the differentially expressed genes in cambial tissues due to winter and spring seasons into biological processes, cellular components, and molecular functions.
Figure 5.
GO function analysis of the differentially expressed genes. GO function analysis results for the differentially expressed genes in cambial tissues due to winter and spring seasons into biological processes, cellular components, and molecular functions.

Figure 6.
Relative mRNA expression of key genes involved in winter and spring seasons (A–O). Expression of genes involved in cell wall synthesis (CSLC4, FUT1, AXY4, GATL1, IRX10, CesA-6, ESKIMO1, CesA-3, CesA-1, 4CL, CCR1, and CAD1) (A–L) and hormone synthesis (TAA1, YUC1, IPT1) (M–O). Expression quantity of the calibrator sample (winter tissue) was set to 1. Data are the mean ± SD. Student’s t-test was used to compare significant changes in spring tissues compared to winter tissues. *, p < 0.1; **, p < 0.01; ***, p < 0.001; ns, no significance.
Figure 6.
Relative mRNA expression of key genes involved in winter and spring seasons (A–O). Expression of genes involved in cell wall synthesis (CSLC4, FUT1, AXY4, GATL1, IRX10, CesA-6, ESKIMO1, CesA-3, CesA-1, 4CL, CCR1, and CAD1) (A–L) and hormone synthesis (TAA1, YUC1, IPT1) (M–O). Expression quantity of the calibrator sample (winter tissue) was set to 1. Data are the mean ± SD. Student’s t-test was used to compare significant changes in spring tissues compared to winter tissues. *, p < 0.1; **, p < 0.01; ***, p < 0.001; ns, no significance.

Figure 7.
Simple sequence repeat (SSR) marker variation statistics. Number of motifs are given against each repeated nucleotide category from mono-nucleotides to hexa-nucleotides.
Figure 7.
Simple sequence repeat (SSR) marker variation statistics. Number of motifs are given against each repeated nucleotide category from mono-nucleotides to hexa-nucleotides.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of the sequence assembly after Illumina sequencing and statistics of contigs and unigenes (n = 3). The values are given as Mean ± SD from three replications.
Table 1.
Summary of the sequence assembly after Illumina sequencing and statistics of contigs and unigenes (n = 3). The values are given as Mean ± SD from three replications.
| Winter Wood | Spring Wood | |
|---|---|---|
| Total raw reads | 51,005,253 ± 2,639,904 | 52,359,187 ± 737,515 |
| Total clean reads | 49,766,422 ± 2,349,991 | 50,689,696 ± 1,090,856 |
| Percentage of reads | 97.60 ± 0.52 | 96.64 ± 0.63 |
| Q20 Percentage | 97.96 ± 0.36 | 97.51 ± 0.35 |
| Contigs | ||
| Total Number | 129,428 ± 610 | 104,388 ± 1779 |
| Total Length (nt) | 43,730,308 ± 513,387 | 35,501,692 ± 721,304 |
| Mean Length (nt) | 338 ± 3 | 340 ± 1 |
| N50 | 605 ± 7 | 642 ± 4 |
| Unigenes | ||
| Total Number | 64,142 ± 1229 | 45,671 ± 1225 |
| Total Length (nt) | 61,610,800 ± 2,101,797 | 38,465,680 ± 1,457,846 |
| Mean Length (nt) | 960 ± 14 | 842 ± 9 |
| N50 | 1551 ± 25 | 1354 ± 22 |
Table 2.
List of top 20 upregulated known genes.
| Unigene/Contig | Length | WW Expression | SW Expression | log2Fold Change (SW/WW) | Probability | Gene |
|---|---|---|---|---|---|---|
| Unigene8688 | 995 | 0.01 | 42.67 | 12.06 | 0.9997 | Anaphase-promoting complex, cyclosome, subunit 4 |
| Unigene11861 | 477 | 0.01 | 27.14 | 11.41 | 0.9994 | Heat shock protein |
| Unigene6740 | 2339 | 0.01 | 17.72 | 10.79 | 0.9989 | Phosphoenolpyruvate carboxykinase |
| Unigene1612 | 1191 | 0.01 | 11.95 | 10.22 | 0.9978 | Cysteine-type peptidase activity |
| Unigene3797 | 247 | 0.01 | 8.11 | 9.66 | 0.9953 | Calcium-binding domain |
| Unigene10820 | 977 | 0.03 | 24.79 | 9.54 | 0.9992 | Class IV heat shock protein |
| Unigene17843 | 560 | 0.01 | 6.50 | 9.34 | 0.9926 | Pericarp peroxidase |
| Unigene1476 | 971 | 0.01 | 6.17 | 9.27 | 0.9918 | Sulfated surface glycoprotein |
| Unigene11860 | 940 | 0.10 | 58.64 | 9.24 | 0.9996 | Class I heat shock protein |
| Unigene34010 | 359 | 0.01 | 5.87 | 9.20 | 0.9910 | Photosystem II oxygen-evolving complex protein 2 precursor |
| Unigene15576 | 532 | 0.01 | 5.42 | 9.08 | 0.9893 | Gibberellin-regulated protein |
| Unigene35083 | 779 | 0.82 | 359.96 | 8.78 | 0.9995 | Heat shock protein |
| Unigene1551 | 276 | 0.01 | 4.14 | 8.69 | 0.9814 | Large subunit ribosomal protein |
| Unigene8720 | 500 | 0.01 | 4.08 | 8.67 | 0.9808 | Actin-depolymerizing factor |
| Unigene19860 | 1028 | 0.01 | 4.01 | 8.65 | 0.9801 | Leucine-rich repeat extensin |
| Unigene4065 | 510 | 0.01 | 4.00 | 8.64 | 0.9801 | Actin depolymerization factor |
| Unigene4063 | 527 | 0.06 | 23.35 | 8.60 | 0.9986 | Aquaporin PIP2 |
| Unigene3966 | 521 | 0.01 | 3.62 | 8.50 | 0.9754 | Tubulin/FtsZ family |
| Unigene8766 | 606 | 0.01 | 3.59 | 8.49 | 0.9750 | Class II heat shock protein |
| Unigene11017 | 503 | 0.01 | 3.54 | 8.47 | 0.9742 | Cyclophilin peptidyl-prolyl cis-trans isomerase |
Table 3.
List of top 20 downregulated genes.
| Unigene/Contig | Length | WW Expression | SW Expression | log2Fold Change (SW/WW) | Probability | Gene |
|---|---|---|---|---|---|---|
| Unigene22837 | 205 | 73.99 | 0.01 | −12.85 | 0.9998 | Galactinol synthase |
| Unigene6926 | 252 | 21.15 | 0.01 | −11.05 | 0.9991 | MATE efflux family protein |
| CL7319.Contig2 | 890 | 16.20 | 0.01 | −10.66 | 0.9987 | Coproporphyrinogen-III oxidase |
| Unigene13375 | 584 | 14.56 | 0.01 | −10.51 | 0.9985 | Rosmarinate synthase |
| Unigene10966 | 1645 | 11.55 | 0.01 | −10.17 | 0.9977 | Receptor-like protein kinase |
| Unigene10726 | 1857 | 8.84 | 0.01 | −9.79 | 0.9961 | Valencene synthase |
| CL698.Contig3 | 3901 | 6.65 | 0.01 | −9.38 | 0.9929 | Pectin methyltransferase |
| CL8528.Contig1 | 1476 | 6.63 | 0.01 | −9.37 | 0.9929 | Root phototropism protein |
| CL7708.Contig2 | 888 | 6.54 | 0.01 | −9.35 | 0.9927 | Splicing factor U2af large subunit |
| CL889.Contig1 | 1018 | 6.46 | 0.01 | −9.33 | 0.9925 | Tropinone reductase homolog |
| CL7009.Contig1 | 1844 | 6.39 | 0.01 | −9.32 | 0.9924 | Auxin efflux carrier family protein |
| Unigene13264 | 1617 | 6.04 | 0.01 | −9.24 | 0.9915 | Ethylene-responsive transcription factor |
| Unigene15599 | 228 | 5.86 | 0.01 | −9.20 | 0.9909 | Nitrate transporter |
| CL8637.Contig1 | 863 | 4.75 | 0.01 | −8.89 | 0.9859 | DNA repair protein RadA |
| CL799.Contig5 | 3446 | 4.72 | 0.01 | −8.88 | 0.9857 | Serine/threonine-protein kinase |
| Unigene1474 | 1021 | 4.55 | 0.01 | −8.83 | 0.9845 | Xanthoxin dehydrogenase |
| Unigene25603 | 518 | 4.52 | 0.01 | −8.82 | 0.9844 | SPX domain-containing membrane protein |
| Unigene6422 | 1304 | 45.72 | 0.10 | −8.79 | 0.9993 | Ethylene-responsive transcription factor |
| CL822.Contig5 | 1916 | 4.24 | 0.01 | −8.73 | 0.9823 | Putative dual-specificity protein phosphatase |
| Unigene13350 | 451 | 4.13 | 0.01 | −8.69 | 0.9813 | CBL-interacting protein kinase |
Table 4.
Top 25 DEG enriched KEGG pathways.
| Pathway | Number of DEGs Genes | p-Value | Pathway ID |
|---|---|---|---|
| Metabolic pathways | 1387 | 1.32E−12 | ko01100 |
| Biosynthesis of secondary metabolites | 827 | 7.55E−10 | ko01110 |
| Plant hormone signal transduction | 320 | 1.58E−05 | ko04075 |
| Plant-pathogen interaction | 266 | 0.2430222 | ko04626 |
| Ribosome | 204 | 0.200625 | ko03010 |
| Spliceosome | 178 | 0.9468082 | ko03040 |
| Starch and sucrose metabolism | 172 | 1.22E−06 | ko00500 |
| Protein processing in E.R. | 162 | 0.3255727 | ko04141 |
| Carbon metabolism | 161 | 0.04321163 | ko01200 |
| RNA transport | 151 | 0.986695 | ko03013 |
| Glycerophospholipid metabolism | 138 | 0.1244536 | ko00564 |
| Endocytosis | 134 | 0.6458048 | ko04144 |
| Biosynthesis of amino acids | 123 | 0.7258124 | ko01230 |
| Glycolysis/Gluconeogenesis | 115 | 5.48E−05 | ko00010 |
| Phenylpropanoid biosynthesis | 106 | 1.50E−06 | ko00940 |
| Circadian rhythm—plant | 105 | 5.10E−09 | ko04712 |
| Ether lipid metabolism | 104 | 0.02664109 | ko00565 |
| Ubiquitin mediated proteolysis | 99 | 0.7266876 | ko04120 |
| Pentose and glucuronate interconversions | 93 | 1.57E−05 | ko00040 |
| Purine metabolism | 92 | 0.9911015 | ko00230 |
| Amino sugar and nucleotide sugar metabolism | 86 | 0.006516035 | ko00520 |
| Pyrimidine metabolism | 78 | 0.992583 | ko00240 |
| mRNA surveillance pathway | 77 | 0.9938776 | ko03015 |
| Flavonoid biosynthesis | 70 | 8.18E−07 | ko00941 |
| RNA degradation | 68 | 0.940714 | ko03018 |
Table 5.
Wood-Forming Genes Selected for RT-qPCR Expression Validation.
| Gene Name | Unigene/Contig | Macromolecule | Enzyme/Protein Name | Activity | Function |
|---|---|---|---|---|---|
| CESA3 | Unigene9908 | Cellulose | Cellulose synthase A catalytic subunit 3 [UDP-forming] | Cellulose synthase | Catalytic subunit of cellulose synthase terminal complexes required for cell wall formation. |
| CESA1 | Unigene21132 | ” | ” | ” | ” |
| CESA6 | Unigene13924 | ” | ” | ” | ” |
| CSLC4 | CL7362.Contig2 | Hemicellulose | Xyloglucan glycosyltransferase 4 | Glucan synthesis | Involved in the synthesis of the xyloglucan backbone. |
| FUT1 | Unigene2841 | Hemicellulose | Galactoside 2-alpha-L-fucosyltransferase | Fucosyl transferase | Addition of the terminal fucosyl residue on xyloglucan side chains. |
| AXY4 | Unigene14391 | Hemicellulose | Protein ALTERED XYLOGLUCAN 4 | Acetyl transferase | Involved in xyloglucan-specific O-acetylation in roots and rosette leaves. |
| GATL1 | Unigene14440 | Hemicellulose | Galacuronosyltransferase-like 1 | Xylan synthase | Family 8 glycosyl transferase that contributes to xylan biosynthesis. |
| IRX10 | Unigene2644 | Hemicellulose | Beta-1,4-xylosyltransferase | Xylan synthase | Synthesis of the hemicellulose glucuronoxylan, a major component of secondary cell walls. |
| ESKIMO1 | CL7514.Contig1 | Hemicellulose | Protein ESKIMO 1 | Acetyl transferase | Xylan acetyltransferase required for 2-O- and 3-O-monoacetylation of xylosyl residues in xylan. |
| 4CL | CL764.Contig3 | Lignin | 4-coumarate--CoA ligase 1 | Monolignol synthesis | Produces CoA thioesters of a variety of hydroxy- and methoxy-substituted cinnamic acids. |
| CCR1 | CL6693.Contig1 | Lignin | Cinnamoyl-CoA reductase 1 | Monolignol synthesis | Involved in monolignol biosynthesis, the conversion of cinnamoyl-CoAs into cinnamaldehydes. |
| CAD1 | Unigene17183 | Lignin | Cinnamyl alcohol dehydrogenase 1 | Monolignol synthesis | Involved in lignin biosynthesis. Catalyzes the final step specific for the production of lignin monomers. |
| TAA1 | CL8952.Contig1 | Auxin | L-tryptophan--pyruvate aminotransferase 1 | Auxin synthesis | Performs first two reactions in auxin pathway. |
| YUC1 | CL1596.Contig1 | Auxin | Indole-3-pyruvate monooxygenase YUCCA1 | Auxin synthesis | Catalyzes the N-oxidation of tryptamine to form N-hydroxyl tryptamine. |
| IPT1 | Unigene8131 | Cytokinin | Adenylate isopentenyltransferase 1, chloroplastic | Cytokinin synthesis | Catalyzes the transfer of an isopentenyl group from dimethylallyl diphosphate (DMAPP) to ATP, ADP, and AMP. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Perry, Z.D.; Saminathan, T.; Arun, A.; Vaidya, B.N.; Basu, C.; Reddy, U.K.; Joshee, N. Transcriptome Analysis of Cambium Tissue of Paulownia Collected during Winter and Spring. Diversity 2021, 13, 423. https://doi.org/10.3390/d13090423
AMA Style
Perry ZD, Saminathan T, Arun A, Vaidya BN, Basu C, Reddy UK, Joshee N. Transcriptome Analysis of Cambium Tissue of Paulownia Collected during Winter and Spring. Diversity. 2021; 13(9):423. https://doi.org/10.3390/d13090423
Chicago/Turabian StylePerry, Zachary D., Thangasamy Saminathan, Alok Arun, Brajesh N. Vaidya, Chhandak Basu, Umesh K. Reddy, and Nirmal Joshee. 2021. "Transcriptome Analysis of Cambium Tissue of Paulownia Collected during Winter and Spring" Diversity 13, no. 9: 423. https://doi.org/10.3390/d13090423
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.