Nasopharyngeal Carcinoma Progression: Accumulating Genomic Instability and Persistent Epstein–Barr Virus Infection

 , ,
, , 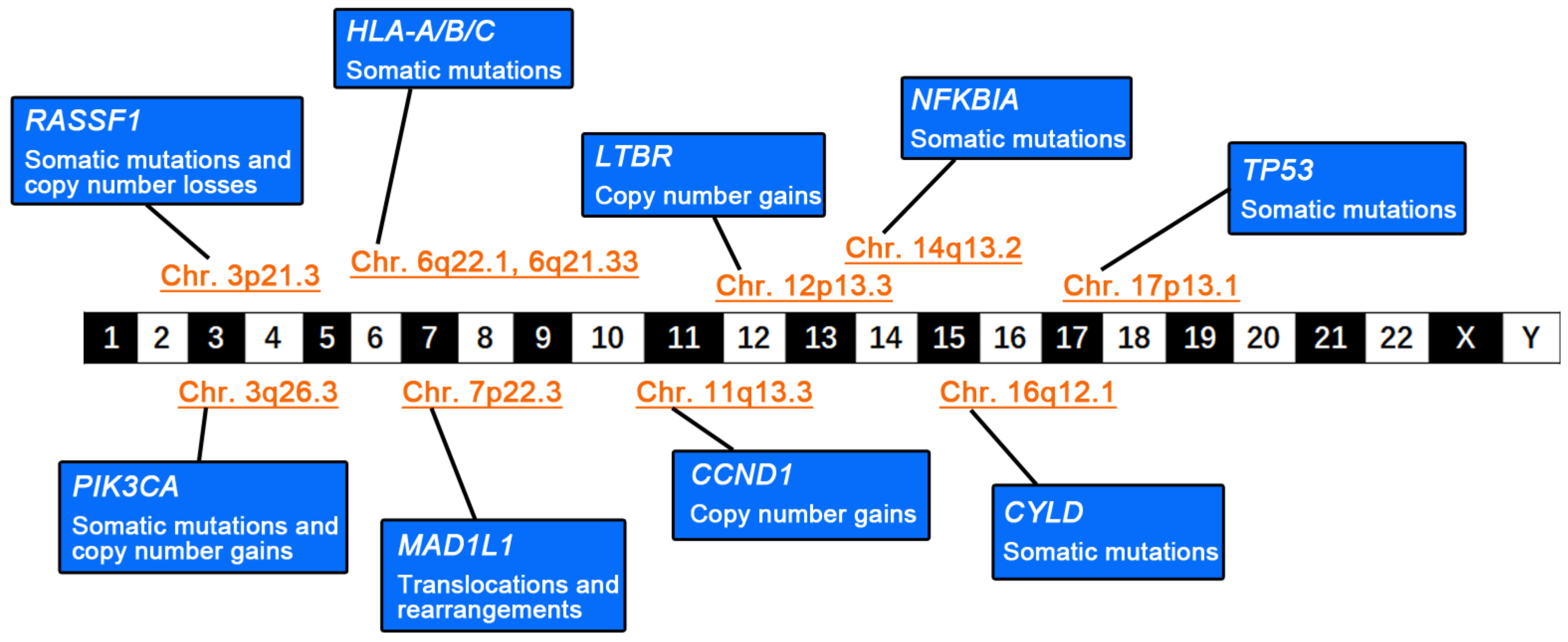{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Characteristics of Genomic Instability in NPC
2.1. Diverse Somatic Mutations
2.2. High-Frequency CNVs
2.3. Gene Translocations and Rearrangements
2.4. Unique Genomic Alterations in Relapse Lesions
2.5. Deleterious Germline Variants
3. Synergy of Host Genomic Instability and EBV Infection
3.1. Host Genomic Alterations Enable Stable EBV Infection
3.2. EBV Genes and Their Products Promote Host Genomic Instability
4. The Clinical Translational Value of Genomic Instability in NPC
4.1. Targeted Therapeutic Strategies for Mutated Genes
4.2. Genomic Instability and Cancer Immunotherapy
5. Conclusions and Perspective
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chen, Y.-P.; Chan, A.T.C.; Le, Q.-T.; Blanchard, P.; Sun, Y.; Ma, J. Nasopharyngeal carcinoma. Lancet 2019, 394, 64–80. [Google Scholar] [CrossRef]
- Badoual, C. Update from the 5th Edition of the World Health Organization Classification of Head and Neck Tumors: Oropharynx and Nasopharynx. Head Neck Pathol. 2022, 16, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Shanmugaratnam, K.; Sobin, L.H. The World Health Organization histological classification of tumours of the upper respiratory tract and ear. A commentary on the second edition. Cancer 1993, 71, 2689–2697. [Google Scholar] [CrossRef]
- Hau, P.M.; Lung, H.L.; Wu, M.; Tsang, C.M.; Wong, K.L.; Mak, N.K.; Lo, K.W. Targeting Epstein-Barr Virus in Nasopharyngeal Carcinoma. Front. Oncol. 2020, 10, 600. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.C.W.; Hui, E.P.; Lo, K.W.; Lam, W.K.J.; Johnson, D.; Li, L.; Tao, Q.; Chan, K.C.A.; To, K.F.; King, A.D.; et al. Nasopharyngeal carcinoma: An evolving paradigm. Nat. Rev. Clin. Oncol. 2021, 18, 679–695. [Google Scholar] [CrossRef]
- Dai, W.; Zheng, H.; Cheung, A.K.; Lung, M.L. Genetic and epigenetic landscape of nasopharyngeal carcinoma. Chin. Clin. Oncol. 2016, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, P.; Zhang, X.; Xu, J.; Xu, J.; Yu, S.; Wang, D.; Dong, W.; Cao, X.; Yan, H.; et al. Mutational landscape of nasopharyngeal carcinoma based on targeted next-generation sequencing: Implications for predicting clinical outcomes. Mol. Med. 2022, 28, 55. [Google Scholar] [CrossRef]
- Zhu, G.; Pan, C.; Bei, J.X.; Li, B.; Liang, C.; Xu, Y.; Fu, X. Mutant p53 in Cancer Progression and Targeted Therapies. Front. Oncol. 2020, 10, 595187. [Google Scholar] [CrossRef] [PubMed]
- Bruce, J.P.; To, K.F.; Lui, V.W.Y.; Chung, G.T.Y.; Chan, Y.Y.; Tsang, C.M.; Yip, K.Y.; Ma, B.B.Y.; Woo, J.K.S.; Hui, E.P.; et al. Whole-genome profiling of nasopharyngeal carcinoma reveals viral-host co-operation in inflammatory NF-kappaB activation and immune escape. Nat. Commun. 2021, 12, 4193. [Google Scholar] [CrossRef]
- Tsang, C.M.; Lui, V.W.Y.; Bruce, J.P.; Pugh, T.J.; Lo, K.W. Translational genomics of nasopharyngeal cancer. Semin. Cancer Biol. 2020, 61, 84–100. [Google Scholar] [CrossRef]
- Li, Y.Y.; Chung, G.T.; Lui, V.W.; To, K.F.; Ma, B.B.; Chow, C.; Woo, J.K.; Yip, K.Y.; Seo, J.; Hui, E.P.; et al. Exome and genome sequencing of nasopharynx cancer identifies NF-kappaB pathway activating mutations. Nat. Commun. 2017, 8, 14121. [Google Scholar] [CrossRef]
- Massoumi, R. CYLD: A deubiquitination enzyme with multiple roles in cancer. Future Oncol. 2011, 7, 285–297. [Google Scholar] [CrossRef]
- Park, H.H. Structure of TRAF Family: Current Understanding of Receptor Recognition. Front. Immunol. 2018, 9, 1999. [Google Scholar] [CrossRef]
- Lin, D.C.; Meng, X.; Hazawa, M.; Nagata, Y.; Varela, A.M.; Xu, L.; Sato, Y.; Liu, L.Z.; Ding, L.W.; Sharma, A.; et al. The genomic landscape of nasopharyngeal carcinoma. Nat. Genet. 2014, 46, 866–871. [Google Scholar] [CrossRef]
- Lo, K.W.; Chung, G.T.; To, K.F. Deciphering the molecular genetic basis of NPC through molecular, cytogenetic, and epigenetic approaches. Semin. Cancer Biol. 2012, 22, 79–86. [Google Scholar] [CrossRef]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef]
- Gustin, J.P.; Cosgrove, D.P.; Park, B.H. The PIK3CA gene as a mutated target for cancer therapy. Curr. Cancer Drug. Targets 2008, 8, 733–740. [Google Scholar] [CrossRef]
- Miled, N.; Yan, Y.; Hon, W.C.; Perisic, O.; Zvelebil, M.; Inbar, Y.; Schneidman-Duhovny, D.; Wolfson, H.J.; Backer, J.M.; Williams, R.L. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science 2007, 317, 239–242. [Google Scholar] [CrossRef]
- Kang, S.; Bader, A.G.; Zhao, L.; Vogt, P.K. Mutated PI 3-kinases—Cancer targets on a silver platter. Cell Cycle 2005, 4, 578–581. [Google Scholar] [CrossRef]
- Lo, A.K.; Huang, D.P.; Lo, K.W.; Chui, Y.L.; Li, H.M.; Pang, J.C.; Tsao, S.W. Phenotypic alterations induced by the Hong Kong-prevalent Epstein-Barr virus-encoded LMP1 variant (2117-LMP1) in nasopharyngeal epithelial cells. Int. J. Cancer 2004, 109, 919–925. [Google Scholar] [CrossRef]
- Mainou, B.A.; Raab-Traub, N. LMP1 strain variants: Biological and molecular properties. J. Virol. 2006, 80, 6458–6468. [Google Scholar] [CrossRef] [PubMed]
- Hutajulu, S.H.; Hoebe, E.K.; Verkuijlen, S.A.; Fachiroh, J.; Hariwijanto, B.; Haryana, S.M.; Stevens, S.J.; Greijer, A.E.; Middeldorp, J.M. Conserved mutation of Epstein-Barr virus-encoded BamHI-A Rightward Frame-1 (BARF1) gene in Indonesian nasopharyngeal carcinoma. Infect. Agent. Cancer 2010, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Ko, J.Y.; Chen, P.J.; Shu, C.H.; Hsu, M.T.; Tsai, S.F.; Lin, C.H. Chromosomal aberrations in nasopharyngeal carcinoma analyzed by comparative genomic hybridization. Genes. Chromosomes Cancer 1999, 25, 169–175. [Google Scholar] [CrossRef]
- Hui, A.B.; Lo, K.W.; Leung, S.F.; Teo, P.; Fung, M.K.; To, K.F.; Wong, N.; Choi, P.H.; Lee, J.C.; Huang, D.P. Detection of recurrent chromosomal gains and losses in primary nasopharyngeal carcinoma by comparative genomic hybridisation. Int. J. Cancer 1999, 82, 498–503. [Google Scholar] [CrossRef]
- Hui, A.B.; Or, Y.Y.; Takano, H.; Tsang, R.K.; To, K.F.; Guan, X.Y.; Sham, J.S.; Hung, K.W.; Lam, C.N.; van Hasselt, C.A.; et al. Array-based comparative genomic hybridization analysis identified cyclin D1 as a target oncogene at 11q13.3 in nasopharyngeal carcinoma. Cancer Res. 2005, 65, 8125–8133. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Zeng, Z.; Qi, P.; Li, X.; Guo, C.; Xiong, F.; Xiang, B.; Zhou, M.; Liao, Q.; Yu, J.; et al. Identification of genomic alterations in nasopharyngeal carcinoma and nasopharyngeal carcinoma-derived Epstein-Barr virus by whole-genome sequencing. Carcinogenesis 2018, 39, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Fu, L.; Zhang, L.Y.; Kwong, D.L.; Yan, L.; Guan, X.Y. Tumor suppressor genes on frequently deleted chromosome 3p in nasopharyngeal carcinoma. Chin. J. Cancer 2012, 31, 215–222. [Google Scholar] [CrossRef]
- Chan, A.S.; To, K.F.; Lo, K.W.; Ding, M.; Li, X.; Johnson, P.; Huang, D.P. Frequent chromosome 9p losses in histologically normal nasopharyngeal epithelia from southern Chinese. Int. J. Cancer 2002, 102, 300–303. [Google Scholar] [CrossRef]
- Song, M.S.; Song, S.J.; Ayad, N.G.; Chang, J.S.; Lee, J.H.; Hong, H.K.; Lee, H.; Choi, N.; Kim, J.; Kim, H.; et al. The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC-Cdc20 complex. Nat. Cell Biol. 2004, 6, 129–137. [Google Scholar] [CrossRef]
- Dallol, A.; Cooper, W.N.; Al-Mulla, F.; Agathanggelou, A.; Maher, E.R.; Latif, F. Depletion of the ras association domain family 1, isoform A-associated novel microtubule-associated protein, C190RF5/MAP1S, causes mitotic abnormalities. Cancer Res. 2007, 67, 492–500. [Google Scholar] [CrossRef]
- van der Weyden, L.; Tachibana, K.K.; Gonzalez, M.A.; Adams, D.J.; Ng, B.L.; Petty, R.; Venkitaraman, A.R.; Arends, M.J.; Bradley, A. The RASSF1A isoform of RASSF1 promotes microtubule stability and suppresses tumorigenesis. Mol. Cell. Biol. 2005, 25, 8356–8367. [Google Scholar] [CrossRef]
- Liang, Y.Y.; Deng, X.B.; Lin, X.T.; Jiang, L.L.; Huang, X.T.; Mo, Z.W.; Yuan, Y.W.; Teh, M.T. RASSF1A inhibits PDGFB-driven malignant phenotypes of nasopharyngeal carcinoma cells in a YAP1-dependent manner. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein-Barr virus infection and nasopharyngeal carcinoma. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160270. [Google Scholar] [CrossRef]
- Tsang, C.M.; Yip, Y.L.; Lo, K.W.; Deng, W.; To, K.F.; Hau, P.M.; Lau, V.M.; Takada, K.; Lui, V.W.; Lung, M.L.; et al. Cyclin D1 overexpression supports stable EBV infection in nasopharyngeal epithelial cells. Proc. Natl. Acad. Sci. USA 2012, 109, E3473–E3482. [Google Scholar] [CrossRef]
- Liggett, W.H., Jr.; Sidransky, D. Role of the p16 tumor suppressor gene in cancer. J. Clin. Oncol. 1998, 16, 1197–1206. [Google Scholar] [CrossRef]
- Lu, B.; Jiang, R.; Xie, B.; Wu, W.; Zhao, Y. Fusion genes in gynecologic tumors: The occurrence, molecular mechanism and prospect for therapy. Cell Death Dis. 2021, 12, 783. [Google Scholar] [CrossRef]
- Chung, G.T.Y.; Lung, R.W.M.; Hui, A.B.Y.; Yip, K.Y.L.; Woo, J.K.S.; Chow, C.; Tong, C.Y.K.; Lee, S.D.; Yuen, J.W.F.; Lun, S.W.M.; et al. Identification of a recurrent transforming UBR5-ZNF423 fusion gene in EBV-associated nasopharyngeal carcinoma. J. Pathol. 2013, 231, 158–167. [Google Scholar] [CrossRef]
- Zhong, Q.; Liu, Z.H.; Lin, Z.R.; Hu, Z.D.; Yuan, L.; Liu, Y.M.; Zhou, A.J.; Xu, L.H.; Hu, L.J.; Wang, Z.F.; et al. The RARS-MAD1L1 Fusion Gene Induces Cancer Stem Cell-like Properties and Therapeutic Resistance in Nasopharyngeal Carcinoma. Clin. Cancer Res. 2018, 24, 659–673. [Google Scholar] [CrossRef]
- Yuan, L.; Liu, Z.H.; Lin, Z.R.; Xu, L.H.; Zhong, Q.; Zeng, M.S. Recurrent FGFR3-TACC3 fusion gene in nasopharyngeal carcinoma. Cancer Biol. Ther. 2014, 15, 1613–1621. [Google Scholar] [CrossRef]
- You, R.; Liu, Y.P.; Lin, D.C.; Li, Q.; Yu, T.; Zou, X.; Lin, M.; Zhang, X.L.; He, G.P.; Yang, Q.; et al. Clonal Mutations Activate the NF-kappaB Pathway to Promote Recurrence of Nasopharyngeal Carcinoma. Cancer Res. 2019, 79, 5930–5943. [Google Scholar] [CrossRef]
- Campion, N.J.; Ally, M.; Jank, B.J.; Ahmed, J.; Alusi, G. The molecular march of primary and recurrent nasopharyngeal carcinoma. Oncogene 2021, 40, 1757–1774. [Google Scholar] [CrossRef]
- Cho, W.C.S.; Tse, K.P.; Ngan, R.K.C.; Cheuk, W.; Ma, V.W.S.; Yang, Y.T.; Yip, T.T.C.; Tan, K.T.; Chen, S.J. Genomic characterization reveals potential biomarkers in nasopharyngeal carcinoma patients with relapse. Expert. Rev. Mol. Diagn. 2020, 20, 1149–1159. [Google Scholar] [CrossRef]
- Lung, H.L.; Cheung, A.K.; Xie, D.; Cheng, Y.; Kwong, F.M.; Murakami, Y.; Guan, X.Y.; Sham, J.S.; Chua, D.; Protopopov, A.I.; et al. TSLC1 is a tumor suppressor gene associated with metastasis in nasopharyngeal carcinoma. Cancer Res. 2006, 66, 9385–9392. [Google Scholar] [CrossRef]
- Lung, H.L.; Bangarusamy, D.K.; Xie, D.; Cheung, A.K.; Cheng, Y.; Kumaran, M.K.; Miller, L.; Liu, E.T.; Guan, X.Y.; Sham, J.S.; et al. THY1 is a candidate tumour suppressor gene with decreased expression in metastatic nasopharyngeal carcinoma. Oncogene 2005, 24, 6525–6532. [Google Scholar] [CrossRef]
- Rege, T.A.; Hagood, J.S. Thy-1, a versatile modulator of signaling affecting cellular adhesion, proliferation, survival, and cytokine/growth factor responses. Biochim. Biophys. Acta 2006, 1763, 991–999. [Google Scholar] [CrossRef]
- Tang, X.R.; Li, Y.Q.; Liang, S.B.; Jiang, W.; Liu, F.; Ge, W.X.; Tang, L.L.; Mao, Y.P.; He, Q.M.; Yang, X.J.; et al. Development and validation of a gene expression-based signature to predict distant metastasis in locoregionally advanced nasopharyngeal carcinoma: A retrospective, multicentre, cohort study. Lancet Oncol. 2018, 19, 382–393. [Google Scholar] [CrossRef]
- Mi, J.L.; Xu, M.; Liu, C.; Wang, R.S. Identification of novel biomarkers and small-molecule compounds for nasopharyngeal carcinoma with metastasis. Medicine 2020, 99, e21505. [Google Scholar] [CrossRef]
- Si, J.; Huang, B.; Lan, G.; Zhang, B.; Wei, J.; Deng, Z.; Li, Y.; Qin, Y.; Li, B.; Lu, Y.; et al. Comparison of whole exome sequencing in circulating tumor cells of primitive and metastatic nasopharyngeal carcinoma. Transl. Cancer Res. 2020, 9, 4080–4092. [Google Scholar] [CrossRef] [PubMed]
- Ramroop, J.R.; Gerber, M.M.; Toland, A.E. Germline Variants Impact Somatic Events during Tumorigenesis. Trends Genet. 2019, 35, 515–526. [Google Scholar] [CrossRef]
- Dai, W.; Zheng, H.; Cheung, A.K.; Tang, C.S.; Ko, J.M.; Wong, B.W.; Leong, M.M.; Sham, P.C.; Cheung, F.; Kwong, D.L.; et al. Whole-exome sequencing identifies MST1R as a genetic susceptibility gene in nasopharyngeal carcinoma. Proc. Natl. Acad. Sci. USA 2016, 113, 3317–3322. [Google Scholar] [CrossRef]
- Sasaki, M.M.; Skol, A.D.; Bao, R.; Rhodes, L.V.; Chambers, R.; Vokes, E.E.; Cohen, E.E.; Onel, K. Integrated genomic analysis suggests MLL3 is a novel candidate susceptibility gene for familial nasopharyngeal carcinoma. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1222–1228. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Hsu, W.L.; Coghill, A.E.; Yu, K.J.; Wang, C.P.; Lou, P.J.; Liu, Z.; Jones, K.; Vogt, A.; Wang, M.; et al. Whole-Exome Sequencing of Nasopharyngeal Carcinoma Families Reveals Novel Variants Potentially Involved in Nasopharyngeal Carcinoma. Sci. Rep. 2019, 9, 9916. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Chung, D.L.; Chow, L.K.; Yu, V.Z.; Lei, L.C.; Leong, M.M.; Chan, C.K.; Ko, J.M.; Lung, M.L. Clinical Outcome-Related Mutational Signatures Identified by Integrative Genomic Analysis in Nasopharyngeal Carcinoma. Clin. Cancer Res. 2020, 26, 6494–6504. [Google Scholar] [CrossRef] [PubMed]
- Hildesheim, A.; Wang, C.P. Genetic predisposition factors and nasopharyngeal carcinoma risk: A review of epidemiological association studies, 2000-2011: Rosetta Stone for NPC: Genetics, viral infection, and other environmental factors. Semin. Cancer Biol. 2012, 22, 107–116. [Google Scholar] [CrossRef]
- Müller, H.; Lukas, J.; Schneider, A.; Warthoe, P.; Bartek, J.; Eilers, M.; Strauss, M. Cyclin D1 expression is regulated by the retinoblastoma protein. Proc. Natl. Acad. Sci. USA 1994, 91, 2945–2949. [Google Scholar] [CrossRef] [PubMed]
- Yip, Y.L.; Pang, P.S.; Deng, W.; Tsang, C.M.; Zeng, M.; Hau, P.M.; Man, C.; Jin, Y.; Yuen, A.P.; Tsao, S.W. Efficient immortalization of primary nasopharyngeal epithelial cells for EBV infection study. PLoS ONE 2013, 8, e78395. [Google Scholar] [CrossRef]
- Tsang, C.M.; Deng, W.; Yip, Y.L.; Zeng, M.S.; Lo, K.W.; Tsao, S.W. Epstein-Barr virus infection and persistence in nasopharyngeal epithelial cells. Chin. J. Cancer 2014, 33, 549–555. [Google Scholar] [CrossRef]
- Fang, C.Y.; Lee, C.H.; Wu, C.C.; Chang, Y.T.; Yu, S.L.; Chou, S.P.; Huang, P.T.; Chen, C.L.; Hou, J.W.; Chang, Y.; et al. Recurrent chemical reactivations of EBV promotes genome instability and enhances tumor progression of nasopharyngeal carcinoma cells. Int. J. Cancer 2009, 124, 2016–2025. [Google Scholar] [CrossRef]
- Wu, C.C.; Liu, M.T.; Chang, Y.T.; Fang, C.Y.; Chou, S.P.; Liao, H.W.; Kuo, K.L.; Hsu, S.L.; Chen, Y.R.; Wang, P.W.; et al. Epstein-Barr Virus DNase (BGLF5) induces genomic instability in human epithelial cells. Nucleic Acids Res. 2010, 38, 1932–1949. [Google Scholar] [CrossRef]
- Chiu, S.H.; Wu, C.C.; Fang, C.Y.; Yu, S.L.; Hsu, H.Y.; Chow, Y.H.; Chen, J.Y. Epstein-Barr virus BALF3 mediates genomic instability and progressive malignancy in nasopharyngeal carcinoma. Oncotarget 2014, 5, 8583–8601. [Google Scholar] [CrossRef]
- Huang, S.Y.; Wu, C.C.; Cheng, Y.J.; Chou, S.P.; Jiang, Y.J.; Chu, K.C.; Tsai, C.H.; Lin, S.F.; Chen, J.Y. Epstein-Barr virus BRLF1 induces genomic instability and progressive malignancy in nasopharyngeal carcinoma cells. Oncotarget 2017, 8, 78948–78964. [Google Scholar] [CrossRef]
- Chang, Y.H.; Lee, C.P.; Su, M.T.; Wang, J.T.; Chen, J.Y.; Lin, S.F.; Tsai, C.H.; Hsieh, M.J.; Takada, K.; Chen, M.R. Epstein-Barr virus BGLF4 kinase retards cellular S-phase progression and induces chromosomal abnormality. PLoS ONE 2012, 7, e39217. [Google Scholar] [CrossRef]
- Shumilov, A.; Tsai, M.H.; Schlosser, Y.T.; Kratz, A.S.; Bernhardt, K.; Fink, S.; Mizani, T.; Lin, X.; Jauch, A.; Mautner, J.; et al. Epstein-Barr virus particles induce centrosome amplification and chromosomal instability. Nat. Commun. 2017, 8, 14257. [Google Scholar] [CrossRef]
- Saridakis, V.; Sheng, Y.; Sarkari, F.; Holowaty, M.N.; Shire, K.; Nguyen, T.; Zhang, R.G.; Liao, J.; Lee, W.; Edwards, A.M.; et al. Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. Mol. Cell 2005, 18, 25–36. [Google Scholar] [CrossRef]
- Sivachandran, N.; Sarkari, F.; Frappier, L. Epstein-Barr nuclear antigen 1 contributes to nasopharyngeal carcinoma through disruption of PML nuclear bodies. PLoS Pathog. 2008, 4, e1000170. [Google Scholar] [CrossRef]
- Chang, H.R.; Munkhjargal, A.; Kim, M.J.; Park, S.Y.; Jung, E.; Ryu, J.H.; Yang, Y.; Lim, J.S.; Kim, Y. The functional roles of PML nuclear bodies in genome maintenance. Mutat. Res. 2018, 809, 99–107. [Google Scholar] [CrossRef]
- Frappier, L. EBNA1. Curr. Top. Microbiol. Immunol. 2015, 391, 3–34. [Google Scholar] [CrossRef]
- Sivachandran, N.; Cao, J.Y.; Frappier, L. Epstein-Barr virus nuclear antigen 1 Hijacks the host kinase CK2 to disrupt PML nuclear bodies. J. Virol. 2010, 84, 11113–11123. [Google Scholar] [CrossRef]
- Chen, Y.R.; Liu, M.T.; Chang, Y.T.; Wu, C.C.; Hu, C.Y.; Chen, J.Y. Epstein-Barr virus latent membrane protein 1 represses DNA repair through the PI3K/Akt/FOXO3a pathway in human epithelial cells. J. Virol. 2008, 82, 8124–8137. [Google Scholar] [CrossRef]
- Liu, M.T.; Chen, Y.R.; Chen, S.C.; Hu, C.Y.; Lin, C.S.; Chang, Y.T.; Wang, W.B.; Chen, J.Y. Epstein-Barr virus latent membrane protein 1 induces micronucleus formation, represses DNA repair and enhances sensitivity to DNA-damaging agents in human epithelial cells. Oncogene 2004, 23, 2531–2539. [Google Scholar] [CrossRef]
- Liu, M.T.; Chang, Y.T.; Chen, S.C.; Chuang, Y.C.; Chen, Y.R.; Lin, C.S.; Chen, J.Y. Epstein-Barr virus latent membrane protein 1 represses p53-mediated DNA repair and transcriptional activity. Oncogene 2005, 24, 2635–2646. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Pang, P.S.; Tsang, C.M.; Hau, P.M.; Yip, Y.L.; Cheung, A.L.; Tsao, S.W. Epstein-Barr virus-encoded latent membrane protein 1 impairs G2 checkpoint in human nasopharyngeal epithelial cells through defective Chk1 activation. PLoS ONE 2012, 7, e39095. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.N.; Tsai, C.L.; Tse, K.P.; Chang, H.Y.; Chang, Y.S. The Epstein-Barr virus oncogene product, latent membrane protein 1, induces the downregulation of E-cadherin gene expression via activation of DNA methyltransferases. Proc. Natl. Acad. Sci. USA 2002, 99, 10084–10089. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.L.; Li, H.P.; Lu, Y.J.; Hsueh, C.; Liang, Y.; Chen, C.L.; Tsao, S.W.; Tse, K.P.; Yu, J.S.; Chang, Y.S. Activation of DNA methyltransferase 1 by EBV LMP1 Involves c-Jun NH(2)-terminal kinase signaling. Cancer Res. 2006, 66, 11668–11676. [Google Scholar] [CrossRef] [PubMed]
- Man, C.; Rosa, J.; Lee, L.T.; Lee, V.H.; Chow, B.K.; Lo, K.W.; Doxsey, S.; Wu, Z.G.; Kwong, Y.L.; Jin, D.Y.; et al. Latent membrane protein 1 suppresses RASSF1A expression, disrupts microtubule structures and induces chromosomal aberrations in human epithelial cells. Oncogene 2007, 26, 3069–3080. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, N.; Brennan, P.; Gaubatz, S.; Sanij, E.; Hertzog, P.; Wolvetang, E.; Ghysdael, J.; Rowe, M.; Hara, E. Epstein-Barr virus LMP1 blocks p16INK4a-RB pathway by promoting nuclear export of E2F4/5. J. Cell Biol. 2003, 162, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Hino, R.; Uozaki, H.; Murakami, N.; Ushiku, T.; Shinozaki, A.; Ishikawa, S.; Morikawa, T.; Nakaya, T.; Sakatani, T.; Takada, K.; et al. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res. 2009, 69, 2766–2774. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.Z.; Li, W.F.; Chen, L.; Luo, W.; Chen, Y.Y.; Liu, L.Z.; Sun, Y.; Lin, A.H.; Liu, M.Z.; Ma, J. How does intensity-modulated radiotherapy versus conventional two-dimensional radiotherapy influence the treatment results in nasopharyngeal carcinoma patients? Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 661–668. [Google Scholar] [CrossRef]
- Peng, Z. Current status of gendicine in China: Recombinant human Ad-p53 agent for treatment of cancers. Hum. Gene Ther. 2005, 16, 1016–1027. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Yee-Lin, V.; Pooi-Fong, W.; Soo-Beng, A.K. Nutlin-3, A p53-Mdm2 Antagonist for Nasopharyngeal Carcinoma Treatment. Mini Rev. Med. Chem. 2018, 18, 173–183. [Google Scholar] [CrossRef]
- Fan, X.; Wang, Y.; Song, J.; Wu, H.; Yang, M.; Lu, L.; Weng, X.; Liu, L.; Nie, G. MDM2 inhibitor RG7388 potently inhibits tumors by activating p53 pathway in nasopharyngeal carcinoma. Cancer Biol. Ther. 2019, 20, 1328–1336. [Google Scholar] [CrossRef]
- Shinohara, T.; Uesugi, M. [In-vivo activation of the p53 pathway by small-molecule antagonists of MDM2]. Tanpakushitsu Kakusan Koso 2007, 52, 1816–1817. [Google Scholar]
- Voon, Y.L.; Ahmad, M.; Wong, P.F.; Husaini, R.; Ng, W.T.; Leong, C.O.; Lane, D.P.; Khoo, A.S. Nutlin-3 sensitizes nasopharyngeal carcinoma cells to cisplatin-induced cytotoxicity. Oncol. Rep. 2015, 34, 1692–1700. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule phosphatidylinositol 3-kinase inhibitors prescribed for the treatment of malignancies. Pharmacol. Res. 2021, 168, 105579. [Google Scholar] [CrossRef]
- Kaboli, P.J.; Salimian, F.; Aghapour, S.; Xiang, S.X.; Zhao, Q.J.; Li, M.X.; Wu, X.; Du, F.K.; Zhao, Y.S.; Shen, J.; et al. Akt-targeted therapy as a promising strategy to overcome drug resistance in breast cancer—A comprehensive review from chemotherapy to immunotherapy. Pharmacol. Res. 2020, 156, 104806. [Google Scholar] [CrossRef]
- China Anti-cancer Association Tumor Drug Clinical Research; Breast Cancer Expert Committee; National Tumor Quality Control Center; Tumor Pathology Committee of China Anti-cancer Association; Boao Institute of Oncology Innovation. [Expert consensus on the clinical application of PI3K/AKT/mTOR inhibitors in the treatment of advanced breast cancer]. Zhonghua Zhong Liu Za Zhi 2022, 44, 673–692. [Google Scholar] [CrossRef]
- Ma, B.B.; Goh, B.C.; Lim, W.T.; Hui, E.P.; Tan, E.H.; Lopes Gde, L.; Lo, K.W.; Li, L.; Loong, H.; Foster, N.R.; et al. Multicenter phase II study of the AKT inhibitor MK-2206 in recurrent or metastatic nasopharyngeal carcinoma from patients in the mayo phase II consortium and the cancer therapeutics research group (MC1079). Investig. New Drugs 2015, 33, 985–991. [Google Scholar] [CrossRef]
- Yang, F.; Qian, X.J.; Qin, W.; Deng, R.; Wu, X.Q.; Qin, J.; Feng, G.K.; Zhu, X.F. Dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 has a therapeutic potential and sensitizes cisplatin in nasopharyngeal carcinoma. PLoS ONE 2013, 8, e59879. [Google Scholar] [CrossRef]
- Wong, C.H.; Loong, H.H.; Hui, C.W.; Lau, C.P.; Hui, E.P.; Ma, B.B.; Chan, A.T. Preclinical evaluation of the PI3K-mTOR dual inhibitor PF-04691502 as a novel therapeutic drug in nasopharyngeal carcinoma. Investig. New Drugs 2013, 31, 1399–1408. [Google Scholar] [CrossRef]
- Bar-Sela, G.; Kuten, A.; Ben-Eliezer, S.; Gov-Ari, E.; Ben-Izhak, O. Expression of HER2 and C-KIT in nasopharyngeal carcinoma: Implications for a new therapeutic approach. Mod. Pathol. 2003, 16, 1035–1040. [Google Scholar] [CrossRef]
- Zhang, J.W.; Qin, T.; Hong, S.D.; Zhang, J.; Fang, W.F.; Zhao, Y.Y.; Yang, Y.P.; Xue, C.; Huang, Y.; Zhao, H.Y.; et al. Multiple oncogenic mutations related to targeted therapy in nasopharyngeal carcinoma. Chin. J. Cancer 2015, 34, 177–183. [Google Scholar] [CrossRef]
- Huang, P.Y.; Hong, M.H.; Zhang, X.; Mai, H.Q.; Luo, D.H.; Zhang, L. C-KIT overexpression and mutation in nasopharyngeal carcinoma cell lines and reactivity of Imatinib on these cell lines. Chin. J. Cancer 2010, 29, 131–135. [Google Scholar] [CrossRef]
- Roberts, P.J.; Stinchcombe, T.E. KRAS mutation: Should we test for it, and does it matter? J. Clin. Oncol. 2013, 31, 1112–1121. [Google Scholar] [CrossRef]
- Chen, X.; Liang, R.; Zhu, X. Anti-EGFR therapies in nasopharyngeal carcinoma. Biomed. Pharmacother. 2020, 131, 110649. [Google Scholar] [CrossRef]
- Pinheiro, G.; Pereira, T.; Dias, C.; Freitas, C.; Hespanhol, V.; Costa, J.L.; Cunha, A.; Oliveira, H.P. Identifying relationships between imaging phenotypes and lung cancer-related mutation status: EGFR and KRAS. Sci. Rep. 2020, 10, 3625. [Google Scholar] [CrossRef]
- Jin, X.; Yan, J.; Chen, C.; Chen, Y.; Huang, W.K. Integrated Analysis of Copy Number Variation, Microsatellite Instability, and Tumor Mutation Burden Identifies an 11-Gene Signature Predicting Survival in Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 721505. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Buttner, R.; Longshore, J.W.; Lopez-Rios, F.; Merkelbach-Bruse, S.; Normanno, N.; Rouleau, E.; Penault-Llorca, F. Implementing TMB measurement in clinical practice: Considerations on assay requirements. ESMO Open. 2019, 4, e000442. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Pedrero, M.; Lefebvre, C.; Marabelle, A.; Soria, J.C.; Postel-Vinay, S. Mutational Landscape and Sensitivity to Immune Checkpoint Blockers. Clin. Cancer Res. 2016, 22, 4309–4321. [Google Scholar] [CrossRef]
- Ali, S.M.; Yao, M.; Yao, J.; Wang, J.; Cheng, Y.; Schrock, A.B.; Chirn, G.W.; Chen, H.; Mu, S.; Gay, L.; et al. Comprehensive genomic profiling of different subtypes of nasopharyngeal carcinoma reveals similarities and differences to guide targeted therapy. Cancer 2017, 123, 3628–3637. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.L.; Wu, M.L. Spatiotemporal homogeneity and distinctness of the T-cell receptor beta-chain repertoires in Epstein-Barr virus-associated primary and metastatic nasopharyngeal carcinomas. Int. J. Cancer 2018, 143, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Yang, Y.; Ma, Y.; Hong, S.; Lin, L.; He, X.; Xiong, J.; Li, P.; Zhao, H.; Huang, Y.; et al. Camrelizumab (SHR-1210) alone or in combination with gemcitabine plus cisplatin for nasopharyngeal carcinoma: Results from two single-arm, phase 1 trials. Lancet Oncol. 2018, 19, 1338–1350. [Google Scholar] [CrossRef]
- Wang, F.H.; Wei, X.L.; Feng, J.; Li, Q.; Xu, N.; Hu, X.C.; Liao, W.; Jiang, Y.; Lin, X.Y.; Zhang, Q.Y.; et al. Efficacy, Safety, and Correlative Biomarkers of Toripalimab in Previously Treated Recurrent or Metastatic Nasopharyngeal Carcinoma: A Phase II Clinical Trial (POLARIS-02). J. Clin. Oncol. 2021, 39, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Chen, X.; Wang, A.; Zhao, H.; Lin, Q.; Bao, H.; Zhang, Y.; Hong, S.; Tang, W.; Huang, Y.; et al. Copy number loss in granzyme genes confers resistance to immune checkpoint inhibitor in nasopharyngeal carcinoma. J. Immunother. Cancer 2021, 9, e002014. [Google Scholar] [CrossRef] [PubMed]
- Pecina-Slaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Chen, F.M.; Zhang, Y.X.; Li, X.F.; Gao, J.F.; Ma, H.; Wang, X.L.; Li, Y.; Li, C.; Zhang, Y.N.; Zhang, Y.T.; et al. The Prognostic Value of Deficient Mismatch Repair in Stage II-IVa Nasopharyngeal Carcinoma in the Era of IMRT. Sci. Rep. 2020, 10, 9690. [Google Scholar] [CrossRef]
- Wu, X.; Gu, Z.; Chen, Y.; Chen, B.; Chen, W.; Weng, L.; Liu, X. Application of PD-1 Blockade in Cancer Immunotherapy. Comput. Struct. Biotechnol. J. 2019, 17, 661–674. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Hellmann, M.D.; Hamid, O.; Tsai, K.K.; Loo, K.L.; Gubens, M.A.; Rosenblum, M.; Harview, C.L.; Taube, J.M.; Handley, N.; et al. Liver Metastasis and Treatment Outcome with Anti-PD-1 Monoclonal Antibody in Patients with Melanoma and NSCLC. Cancer Immunol. Res. 2017, 5, 417–424. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Mehra, R.; Seiwert, T.Y.; Gupta, S.; Weiss, J.; Gluck, I.; Eder, J.P.; Burtness, B.; Tahara, M.; Keam, B.; Kang, H.; et al. Efficacy and safety of pembrolizumab in recurrent/metastatic head and neck squamous cell carcinoma: Pooled analyses after long-term follow-up in KEYNOTE-012. Br. J. Cancer 2018, 119, 153–159. [Google Scholar] [CrossRef]
- Feng, B.; Hess, J. Immune-Related Mutational Landscape and Gene Signatures: Prognostic Value and Therapeutic Impact for Head and Neck Cancer. Cancers 2021, 13, 1162. [Google Scholar] [CrossRef]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef]
- Xu, J.Y.; Wei, X.L.; Wang, Y.Q.; Wang, F.H. Current status and advances of immunotherapy in nasopharyngeal carcinoma. Ther. Adv. Med. Oncol. 2022, 14, 17588359221096214. [Google Scholar] [CrossRef]
- Mandal, R.; Senbabaoglu, Y.; Desrichard, A.; Havel, J.J.; Dalin, M.G.; Riaz, N.; Lee, K.W.; Ganly, I.; Hakimi, A.A.; Chan, T.A.; et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight 2016, 1, e89829. [Google Scholar] [CrossRef]
- Everly, D.N., Jr.; Mainou, B.A.; Raab-Traub, N. The ID proteins contribute to the growth of rodent fibroblasts during LMP1-mediated transformation. Virology 2008, 376, 258–269. [Google Scholar] [CrossRef]
- Shi, Y.; Tao, Y.; Jiang, Y.; Xu, Y.; Yan, B.; Chen, X.; Xiao, L.; Cao, Y. Nuclear epidermal growth factor receptor interacts with transcriptional intermediary factor 2 to activate cyclin D1 gene expression triggered by the oncoprotein latent membrane protein 1. Carcinogenesis 2012, 33, 1468–1478. [Google Scholar] [CrossRef]
- Xu, Y.; Shi, Y.; Yuan, Q.; Liu, X.; Yan, B.; Chen, L.; Tao, Y.; Cao, Y. Epstein-Barr Virus encoded LMP1 regulates cyclin D1 promoter activity by nuclear EGFR and STAT3 in CNE1 cells. J. Exp. Clin. Cancer Res. 2013, 32, 90. [Google Scholar] [CrossRef]
- Yang, X.; He, Z.; Xin, B.; Cao, L. LMP1 of Epstein-Barr virus suppresses cellular senescence associated with the inhibition of p16INK4a expression. Oncogene 2000, 19, 2002–2013. [Google Scholar] [CrossRef]
- Lo, A.K.; Lo, K.W.; Ko, C.W.; Young, L.S.; Dawson, C.W. Inhibition of the LKB1-AMPK pathway by the Epstein-Barr virus-encoded LMP1 promotes proliferation and transformation of human nasopharyngeal epithelial cells. J. Pathol. 2013, 230, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.K.; Dawson, C.W.; Lo, K.W.; Yu, Y.; Young, L.S. Upregulation of Id1 by Epstein-Barr virus-encoded LMP1 confers resistance to TGFbeta-mediated growth inhibition. Mol. Cancer 2010, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Guo, L.; Tao, Y.; Zhou, S.; Wang, Z.; Luo, W.; Hu, D.; Li, Z.; Xiao, L.; Tang, M.; et al. Latent membrane protein 1 of Epstein-Barr virus regulates p53 phosphorylation through MAP kinases. Cancer Lett. 2007, 255, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Fries, K.L.; Miller, W.E.; Raab-Traub, N. Epstein-Barr virus latent membrane protein 1 blocks p53-mediated apoptosis through the induction of the A20 gene. J. Virol. 1996, 70, 8653–8659. [Google Scholar] [CrossRef]
- Yang, J.; Deng, X.; Deng, L.; Gu, H.; Fan, W.; Cao, Y. Telomerase activation by Epstein-Barr virus latent membrane protein 1 is associated with c-Myc expression in human nasopharyngeal epithelial cells. J. Exp. Clin. Cancer Res. 2004, 23, 495–506. [Google Scholar]
- Kondo, S.; Seo, S.Y.; Yoshizaki, T.; Wakisaka, N.; Furukawa, M.; Joab, I.; Jang, K.L.; Pagano, J.S. EBV latent membrane protein 1 up-regulates hypoxia-inducible factor 1alpha through Siah1-mediated down-regulation of prolyl hydroxylases 1 and 3 in nasopharyngeal epithelial cells. Cancer Res. 2006, 66, 9870–9877. [Google Scholar] [CrossRef]
- Yang, L.; Liu, L.; Xu, Z.; Liao, W.; Feng, D.; Dong, X.; Xu, S.; Xiao, L.; Lu, J.; Luo, X.; et al. EBV-LMP1 targeted DNAzyme enhances radiosensitivity by inhibiting tumor angiogenesis via the JNKs/HIF-1 pathway in nasopharyngeal carcinoma. Oncotarget 2015, 6, 5804–5817. [Google Scholar] [CrossRef]
- Wakisaka, N.; Kondo, S.; Yoshizaki, T.; Murono, S.; Furukawa, M.; Pagano, J.S. Epstein-Barr virus latent membrane protein 1 induces synthesis of hypoxia-inducible factor 1 alpha. Mol. Cell Biol. 2004, 24, 5223–5234. [Google Scholar] [CrossRef]
- Wakisaka, N.; Murono, S.; Yoshizaki, T.; Furukawa, M.; Pagano, J.S. Epstein-barr virus latent membrane protein 1 induces and causes release of fibroblast growth factor-2. Cancer Res. 2002, 62, 6337–6344. [Google Scholar]
- Murono, S.; Inoue, H.; Tanabe, T.; Joab, I.; Yoshizaki, T.; Furukawa, M.; Pagano, J.S. Induction of cyclooxygenase-2 by Epstein-Barr virus latent membrane protein 1 is involved in vascular endothelial growth factor production in nasopharyngeal carcinoma cells. Proc. Natl. Acad. Sci. USA 2001, 98, 6905–6910. [Google Scholar] [CrossRef]
- Yoshizaki, T. Promotion of metastasis in nasopharyngeal carcinoma by Epstein-Barr virus latent membrane protein-1. Histol. Histopathol. 2002, 17, 845–850. [Google Scholar] [CrossRef]
- Tsuji, A.; Wakisaka, N.; Kondo, S.; Murono, S.; Furukawa, M.; Yoshizaki, T. Induction of receptor for advanced glycation end products by EBV latent membrane protein 1 and its correlation with angiogenesis and cervical lymph node metastasis in nasopharyngeal carcinoma. Clin. Cancer Res. 2008, 14, 5368–5375. [Google Scholar] [CrossRef]
- Wei, J.; Zhang, J.; Si, Y.; Kanada, M.; Zhang, Z.; Terakawa, S.; Watanabe, H. Blockage of LMP1-modulated store-operated Ca(2+) entry reduces metastatic potential in nasopharyngeal carcinoma cell. Cancer Lett. 2015, 360, 234–244. [Google Scholar] [CrossRef]
- Wang, Z.; Luo, F.; Li, L.; Yang, L.; Hu, D.; Ma, X.; Lu, Z.; Sun, L.; Cao, Y. STAT3 activation induced by Epstein-Barr virus latent membrane protein1 causes vascular endothelial growth factor expression and cellular invasiveness via JAK3 And ERK signaling. Eur. J. Cancer 2010, 46, 2996–3006. [Google Scholar] [CrossRef]
- Horikawa, T.; Yang, J.; Kondo, S.; Yoshizaki, T.; Joab, I.; Furukawa, M.; Pagano, J.S. Twist and epithelial-mesenchymal transition are induced by the EBV oncoprotein latent membrane protein 1 and are associated with metastatic nasopharyngeal carcinoma. Cancer Res. 2007, 67, 1970–1978. [Google Scholar] [CrossRef]
- Zuo, L.L.; Zhang, J.; Liu, L.Z.; Zhou, Q.; Du, S.J.; Xin, S.Y.; Ning, Z.P.; Yang, J.; Yu, H.B.; Yue, W.X.; et al. Cadherin 6 is activated by Epstein-Barr virus LMP1 to mediate EMT and metastasis as an interplay node of multiple pathways in nasopharyngeal carcinoma. Oncogenesis 2017, 6, 402. [Google Scholar] [CrossRef]
- Aga, M.; Bentz, G.L.; Raffa, S.; Torrisi, M.R.; Kondo, S.; Wakisaka, N.; Yoshizaki, T.; Pagano, J.S.; Shackelford, J. Exosomal HIF1alpha supports invasive potential of nasopharyngeal carcinoma-associated LMP1-positive exosomes. Oncogene 2014, 33, 4613–4622. [Google Scholar] [CrossRef]
- Ye, D.; Zhu, J.; Zhao, Q.; Ma, W.; Xiao, Y.; Xu, G.; Zhang, Z. LMP1 Up-regulates Calreticulin to Induce Epithelial-mesenchymal Transition via TGF-beta/Smad3/NRP1 Pathway in Nasopharyngeal Carcinoma Cells. J. Cancer 2020, 11, 1257–1269. [Google Scholar] [CrossRef]
- Zhang, J.; Jia, L.; Liu, T.; Yip, Y.L.; Tang, W.C.; Lin, W.; Deng, W.; Lo, K.W.; You, C.; Lung, M.L.; et al. mTORC2-mediated PDHE1alpha nuclear translocation links EBV-LMP1 reprogrammed glucose metabolism to cancer metastasis in nasopharyngeal carcinoma. Oncogene 2019, 38, 4669–4684. [Google Scholar] [CrossRef]
- Horikawa, T.; Yoshizaki, T.; Kondo, S.; Furukawa, M.; Kaizaki, Y.; Pagano, J.S. Epstein-Barr Virus latent membrane protein 1 induces Snail and epithelial-mesenchymal transition in metastatic nasopharyngeal carcinoma. Br. J. Cancer 2011, 104, 1160–1167. [Google Scholar] [CrossRef]
- Kim, K.R.; Yoshizaki, T.; Miyamori, H.; Hasegawa, K.; Horikawa, T.; Furukawa, M.; Harada, S.; Seiki, M.; Sato, H. Transformation of Madin-Darby canine kidney (MDCK) epithelial cells by Epstein-Barr virus latent membrane protein 1 (LMP1) induces expression of Ets1 and invasive growth. Oncogene 2000, 19, 1764–1771. [Google Scholar] [CrossRef]
- Kondo, S.; Wakisaka, N.; Schell, M.J.; Horikawa, T.; Sheen, T.S.; Sato, H.; Furukawa, M.; Pagano, J.S.; Yoshizaki, T. Epstein-Barr virus latent membrane protein 1 induces the matrix metalloproteinase-1 promoter via an Ets binding site formed by a single nucleotide polymorphism: Enhanced susceptibility to nasopharyngeal carcinoma. Int. J. Cancer 2005, 115, 368–376. [Google Scholar] [CrossRef]
- Zeng, L.; Liu, Y.P.; Tao, Y.G.; Ai, M.D.; Zhao, X.R.; Cao, Y. [Cross-talk between c-Jun/Ets1 involved in EB virus-encoded latent membrane protein 1 regulates expression of matrix metalloproteinase-9 in nasopharyngeal carcinoma]. Zhonghua Zhong Liu Za Zhi 2005, 27, 204–208. [Google Scholar]
- Horikawa, T.; Yoshizaki, T.; Sheen, T.S.; Lee, S.Y.; Furukawa, M. Association of latent membrane protein 1 and matrix metalloproteinase 9 with metastasis in nasopharyngeal carcinoma. Cancer 2000, 89, 715–723. [Google Scholar] [CrossRef]
- Himelstein, B.P.; Lee, E.J.; Sato, H.; Seiki, M.; Muschel, R.J. Transcriptional activation of the matrix metalloproteinase-9 gene in an H-ras and v-myc transformed rat embryo cell line. Oncogene 1997, 14, 1995–1998. [Google Scholar] [CrossRef]
- Luo, X.; Hong, L.; Cheng, C.; Li, N.; Zhao, X.; Shi, F.; Liu, J.; Fan, J.; Zhou, J.; Bode, A.M.; et al. DNMT1 mediates metabolic reprogramming induced by Epstein-Barr virus latent membrane protein 1 and reversed by grifolin in nasopharyngeal carcinoma. Cell Death Dis. 2018, 9, 619. [Google Scholar] [CrossRef]
- Zhang, J.; Jia, L.; Lin, W.; Yip, Y.L.; Lo, K.W.; Lau, V.M.Y.; Zhu, D.; Tsang, C.M.; Zhou, Y.; Deng, W.; et al. Epstein-Barr Virus-Encoded Latent Membrane Protein 1 Upregulates Glucose Transporter 1 Transcription via the mTORC1/NF-kappaB Signaling Pathways. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Xiao, L.; Hu, Z.Y.; Dong, X.; Tan, Z.; Li, W.; Tang, M.; Chen, L.; Yang, L.; Tao, Y.; Jiang, Y.; et al. Targeting Epstein-Barr virus oncoprotein LMP1-mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene 2014, 33, 4568–4578. [Google Scholar] [CrossRef]
- Klibi, J.; Niki, T.; Riedel, A.; Pioche-Durieu, C.; Souquere, S.; Rubinstein, E.; Le Moulec, S.; Guigay, J.; Hirashima, M.; Guemira, F.; et al. Blood diffusion and Th1-suppressive effects of galectin-9-containing exosomes released by Epstein-Barr virus-infected nasopharyngeal carcinoma cells. Blood 2009, 113, 1957–1966. [Google Scholar] [CrossRef]
- Xiang, T.; Lin, Y.X.; Ma, W.; Zhang, H.J.; Chen, K.M.; He, G.P.; Zhang, X.; Xu, M.; Feng, Q.S.; Chen, M.Y.; et al. Vasculogenic mimicry formation in EBV-associated epithelial malignancies. Nat. Commun. 2018, 9, 5009. [Google Scholar] [CrossRef]
- Pegtel, D.M.; Subramanian, A.; Sheen, T.S.; Tsai, C.H.; Golub, T.R.; Thorley-Lawson, D.A. Epstein-Barr-virus-encoded LMP2A induces primary epithelial cell migration and invasion: Possible role in nasopharyngeal carcinoma metastasis. J. Virol. 2005, 79, 15430–15442. [Google Scholar] [CrossRef] [PubMed]
- Fotheringham, J.A.; Coalson, N.E.; Raab-Traub, N. Epstein-Barr virus latent membrane protein-2A induces ITAM/Syk- and Akt-dependent epithelial migration through alphav-integrin membrane translocation. J. Virol. 2012, 86, 10308–10320. [Google Scholar] [CrossRef] [PubMed]
- Fotheringham, J.A.; Mazzucca, S.; Raab-Traub, N. Epstein-Barr virus latent membrane protein-2A-induced DeltaNp63alpha expression is associated with impaired epithelial-cell differentiation. Oncogene 2010, 29, 4287–4296. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Lo, K.W.; Wei, W.; Tsao, S.W.; Chung, G.T.Y.; Ibrahim, M.H.; Dawson, C.W.; Murray, P.G.; Paterson, I.C.; Yap, L.F. Oncogenic S1P signalling in EBV-associated nasopharyngeal carcinoma activates AKT and promotes cell migration through S1P receptor 3. J. Pathol. 2017, 242, 62–72. [Google Scholar] [CrossRef]
- Fukuda, M.; Longnecker, R. Latent membrane protein 2A inhibits transforming growth factor-beta 1-induced apoptosis through the phosphatidylinositol 3-kinase/Akt pathway. J. Virol. 2004, 78, 1697–1705. [Google Scholar] [CrossRef]
- Wang, L.; Tian, W.D.; Xu, X.; Nie, B.; Lu, J.; Liu, X.; Zhang, B.; Dong, Q.; Sunwoo, J.B.; Li, G.; et al. Epstein-Barr virus nuclear antigen 1 (EBNA1) protein induction of epithelial-mesenchymal transition in nasopharyngeal carcinoma cells. Cancer 2014, 120, 363–372. [Google Scholar] [CrossRef]
- Lu, J.; Murakami, M.; Verma, S.C.; Cai, Q.; Haldar, S.; Kaul, R.; Wasik, M.A.; Middeldorp, J.; Robertson, E.S. Epstein-Barr Virus nuclear antigen 1 (EBNA1) confers resistance to apoptosis in EBV-positive B-lymphoma cells through up-regulation of survivin. Virology 2011, 410, 64–75. [Google Scholar] [CrossRef]
- Fan, C.; Tang, Y.; Wang, J.; Xiong, F.; Guo, C.; Wang, Y.; Xiang, B.; Zhou, M.; Li, X.; Wu, X.; et al. The emerging role of Epstein-Barr virus encoded microRNAs in nasopharyngeal carcinoma. J. Cancer 2018, 9, 2852–2864. [Google Scholar] [CrossRef]
- Cai, L.; Ye, Y.; Jiang, Q.; Chen, Y.; Lyu, X.; Li, J.; Wang, S.; Liu, T.; Cai, H.; Yao, K.; et al. Epstein-Barr virus-encoded microRNA BART1 induces tumour metastasis by regulating PTEN-dependent pathways in nasopharyngeal carcinoma. Nat. Commun. 2015, 6, 7353, Correction in Nat. Commun. 2020, 11, 3437. [Google Scholar]
- Choy, E.Y.; Siu, K.L.; Kok, K.H.; Lung, R.W.; Tsang, C.M.; To, K.F.; Kwong, D.L.; Tsao, S.W.; Jin, D.Y. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J. Exp. Med. 2008, 205, 2551–2560. [Google Scholar] [CrossRef]
- Wong, T.S.; Chen, S.; Zhang, M.J.; Chan, J.Y.; Gao, W. Epstein-Barr virus-encoded microRNA BART7 downregulates major histocompatibility complex class I chain-related peptide A and reduces the cytotoxicity of natural killer cells to nasopharyngeal carcinoma. Oncol. Lett. 2018, 16, 2887–2892. [Google Scholar] [CrossRef]
- Song, Y.; Li, X.; Zeng, Z.; Li, Q.; Gong, Z.; Liao, Q.; Li, X.; Chen, P.; Xiang, B.; Zhang, W.; et al. Epstein-Barr virus encoded miR-BART11 promotes inflammation-induced carcinogenesis by targeting FOXP1. Oncotarget 2016, 7, 36783–36799. [Google Scholar] [CrossRef]
- Huang, J.; Qin, Y.; Yang, C.; Wan, C.; Dai, X.; Sun, Y.; Meng, J.; Lu, Y.; Li, Y.; Zhang, Z.; et al. Downregulation of ABI2 expression by EBV-miR-BART13-3p induces epithelial-mesenchymal transition of nasopharyngeal carcinoma cells through upregulation of c-JUN/SLUG signaling. Aging 2020, 12, 340–358. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Deng, Y.; Huang, Y.; Ye, J.; Xie, S.; He, Q.; Chen, Y.; Lin, Y.; Liang, R.; Wei, J.; et al. Nasopharyngeal Carcinoma Progression: Accumulating Genomic Instability and Persistent Epstein–Barr Virus Infection. Curr. Oncol. 2022, 29, 6035-6052. https://doi.org/10.3390/curroncol29090475
Liu X, Deng Y, Huang Y, Ye J, Xie S, He Q, Chen Y, Lin Y, Liang R, Wei J, et al. Nasopharyngeal Carcinoma Progression: Accumulating Genomic Instability and Persistent Epstein–Barr Virus Infection. Current Oncology. 2022; 29(9):6035-6052. https://doi.org/10.3390/curroncol29090475
Chicago/Turabian StyleLiu, Xue, Yayan Deng, Yujuan Huang, Jiaxiang Ye, Sifang Xie, Qian He, Yong Chen, Yan Lin, Rong Liang, Jiazhang Wei, and et al. 2022. "Nasopharyngeal Carcinoma Progression: Accumulating Genomic Instability and Persistent Epstein–Barr Virus Infection" Current Oncology 29, no. 9: 6035-6052. https://doi.org/10.3390/curroncol29090475