Identification of Missense ADGRV1 Mutation as a Candidate Genetic Cause of Familial Febrile Seizure 4


1
Department of Pediatrics, College of Medicine, The Catholic University of Korea, Seoul 06591, Korea
2
Departments of Pediatrics, Yonsei University College of Medicine, Seoul 06273, Korea
3
Department of Laboratory Medicine, College of Medicine, The Catholic University of Korea, Seoul 06591, Korea
4
Department of Laboratory Medicine, Jeonbuk National University Medical School and Hospital, Jeonju 54907, Korea
*
Authors to whom correspondence should be addressed.
†
These authors have contributed equally to this work.
Children 2020, 7(9), 144; https://doi.org/10.3390/children7090144
Submission received: 26 July 2020
/
Revised: 4 September 2020
/
Accepted: 10 September 2020
/
Published: 18 September 2020
(This article belongs to the Section Child Neurology)
Abstract
:Febrile seizure (FS) is related to a febrile illness (temperature > 38 °C) not caused by an infection of central nervous system, without neurologic deficits in children aged 6–60 months. The family study implied a polygenic model in the families of proband(s) with single FS, however in families with repeated FS, inheritance was matched to autosomal dominance with reduced disease penetrance. A 20 month-old girl showed recurrent FS and afebrile seizures without developmental delay or intellectual disability. The seizures disappeared after 60 months without anti-seizure medication. The 35 year-old proband’s mother also experienced five episodes of simple FS and two episodes of unprovoked seizures before 5 years old. Targeted exome sequencing was conducted along with epilepsy/seizure-associated gene-filtering to identify the candidate causative mutation. As a result, a heterozygous c.2039A>G of the ADGRV1 gene leading to a codon change of aspartic acid to glycine at the position 680 (rs547076322) was identified. This protein’s glycine residue is highly conserved, and its allele frequency is 0.00002827 in the gnomAD population database. ADGRV1 mutation may have an influential role in the occurrence of genetic epilepsies, especially those with febrile and afebrile seizures. Further investigation of ADGRV1 mutations is needed to prove that it is a significant susceptible gene for febrile and/or afebrile seizures in early childhood.
1. Introduction
The international League Against Epilepsy defined febrile seizures (FSs) as a seizure occurring in children aged 6 to 60 months related to a febrile illness (temperature > 38 °C) not caused by an infection of the central nervous system (CNS), without previous afebrile seizures or neurologic deficits [1,2]. The mean prevalence of FS in children aged 6–60 months according to hospital visit rates in South Korea was 6.92% (6.12% for girls; 7.67% for boys) during the period 2009–2013 [3]. About a third of patients with experience of FS will have a second attack, and 50% of those will have a third episode. Past history of FS during childhood has been reported in 5 to 15% of epileptic patients, with a higher risk of evolving epilepsy found in children < 14 years with preceding complex FS [4]. The risk of unprovoked seizures is 5 to 7 times higher in children who experience FS compared to the general population and is estimated to be 2 to 5% [5,6]. Prolonged FS and febrile status epilepticus are considered as potential risk factors in subsequently developing epilepsy [7]. Simple FS seem to be genetically complex disorders affected by variations in several susceptibility genes [8]. Children with a familial history of FS show a three-fold or higher risk of recurrence than the general population experiencing FSs. A positive familial history within first-degree relatives is regarded as a compatible risk factor for recurrent FSs than recent infections, fever, and perinatal exposure [9,10]. The family study implied a polygenic model in families of proband(s) with a single FS, however, in the families with repeated FSs, the inheritance was matched to autosomal dominance with reduced disease penetrance [11].
To date, the genetic heterogeneity of familial FSs (termed FEB1–FEB11) has been described in different studies [12,13,14,15,16,17,18,19,20,21,22,23,24,25]. Particularly, ADGRV1 (OMIM *602851) encoding adhesion G protein–coupled receptor (aGPCR) V1, a large calcium-binding protein widely expressed in the CNS, was previously reported as a genetic cause of afebrile and febrile seizures [26]. In this study, targeted exome sequencing was conducted along with epilepsy/seizure-associated gene-filtering to identify the candidate causative mutation in a Korean family with febrile and afebrile seizures.
2. Case Presentations
A 20 month-old girl (II-2 in Figure 1a) was referred to the department of pediatric neurology after four episodes of FS. Her mother witnessed generalized tonic–clonic seizures that lasted under 5 min with a fever of 38 °C–40 °C. By the time the patient was transported to the emergency department, she had fully recovered. At 21 months of age, she came to our emergency department after generalized tonic–clonic seizure that lasted for 20 min without fever. The seizure had ceased and her mental state had recovered when she arrived at the emergency room. Two hours later, her seizure recurred and the seizure was terminated after an injection of lorazepam. In brain magnetic resonance images (MRI), no obvious abnormalities for her age including an appropriate myelination pattern were observed (Figure 1b). The interictal electroencephalogram (EEG) was normal (Figure 1c). The results of metabolic laboratory testing including plasma amino acid, thyroid function tests, lactate/pyruvate, urine organic acid, and blood gases were normal. We observed her without anti-seizure medication at the outpatient clinic. After that, she showed two more simple FSs at the age of 24 months and 35 months. At 36 months old, she was admitted because of a prolonged FS. The seizure apparently lasted for 25 min and ceased after the administration of intravenous lorazepam in the emergency department. Later, a fever of 39.5 °C was noted, and adenoviral infection was diagnosed. Her growth and developmental milestones were proper for her age. Her height (92 cm), weight (13 kg), and head circumstance (47 cm) were 50 percentiles, respectively. Febrile and afebrile seizures had not occurred after the age of 3 and her development was appropriate for her age. Except for a history of seizures in her mother (I-2 in Figure 1a), there was no family history of developmental delay, intellectual disability, and epilepsy. The 35 year-old proband’s mother experienced five episodes of simple FS and two episodes of unprovoked seizures before 5 years old. The mother’s computed tomography scan and EEG were normal and she has remained seizure free since 5 years old without anti-seizure medication. The mother’s development was completely normal and she completed her college education.
3. Molecular Analysis
3.1. Targeted Exome Sequencing
The study protocol was approved by the Institutional Review Board of the Catholic University of Korea. Written informed consent was collected from the parents on behalf of their children for the publication of recognizable data or images included in this report before the blood sampling, and clinical data were obtained from the proband and her parents. To resolve the potential genetic cause, the genomic DNA of the proband was studied by targeted exome sequencing using TruSight One Sequencing Panel (Illumina, Inc., San Diego, CA, USA), which covers rare inherited disease-associated regions of the exome with a comprehensive coverage of > 4800 disease-associated genes. Massively parallel sequencing was conducted using the Illumina HiSeq2500 (Illumina, Inc.) to generate paired-end reads of 150 nucleotides. Exome sequences were estimated for all modes of inheritance and the variants filtered initially for rare variants (allele frequency < 0.01) from the public sequence databases (gnomAD, https://gnomad.broadinstitute.org). The remaining base change and small indels located on epilepsy/seizure-associated genes were selected according to web-based genetic databases (OMIM, https://www.omim.org/; ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/). The candidate missense mutations were estimated to be pathogenic or have a damaging effect by in silico analysis for conservation level as well as for functional effect.
3.2. mRNA Expression Analysis for ADGRV1 Mutation
To determine the pathogenicity of the candidate ADGRV1 mutation, quantitative reverse transcription polymerase chain reaction (RT-PCR) was performed with complementary DNA synthesized from RNA isolated from whole blood on a CFX96™ Real Time PCR Detection System (BioRad, Hercules, CA, USA) using the 5′ reporter FAM™ dye-labeled and 3′ quencher Minor groove binder (MGB)-labeled probes (Applied Biosystems, Foster City, CA, USA). Gene expression level was normalized to GAPDH endogenous control and analyzed according to the relative quantification method (2−ΔΔCt). Three independent experiments were performed. Mean difference in gene expression between p.Asp680Gly and the wild-type of the ADGRV1 were estimated by Student’s t-test. The p value < 0.05 was considered to indicate a statistically significant difference.
4. Results
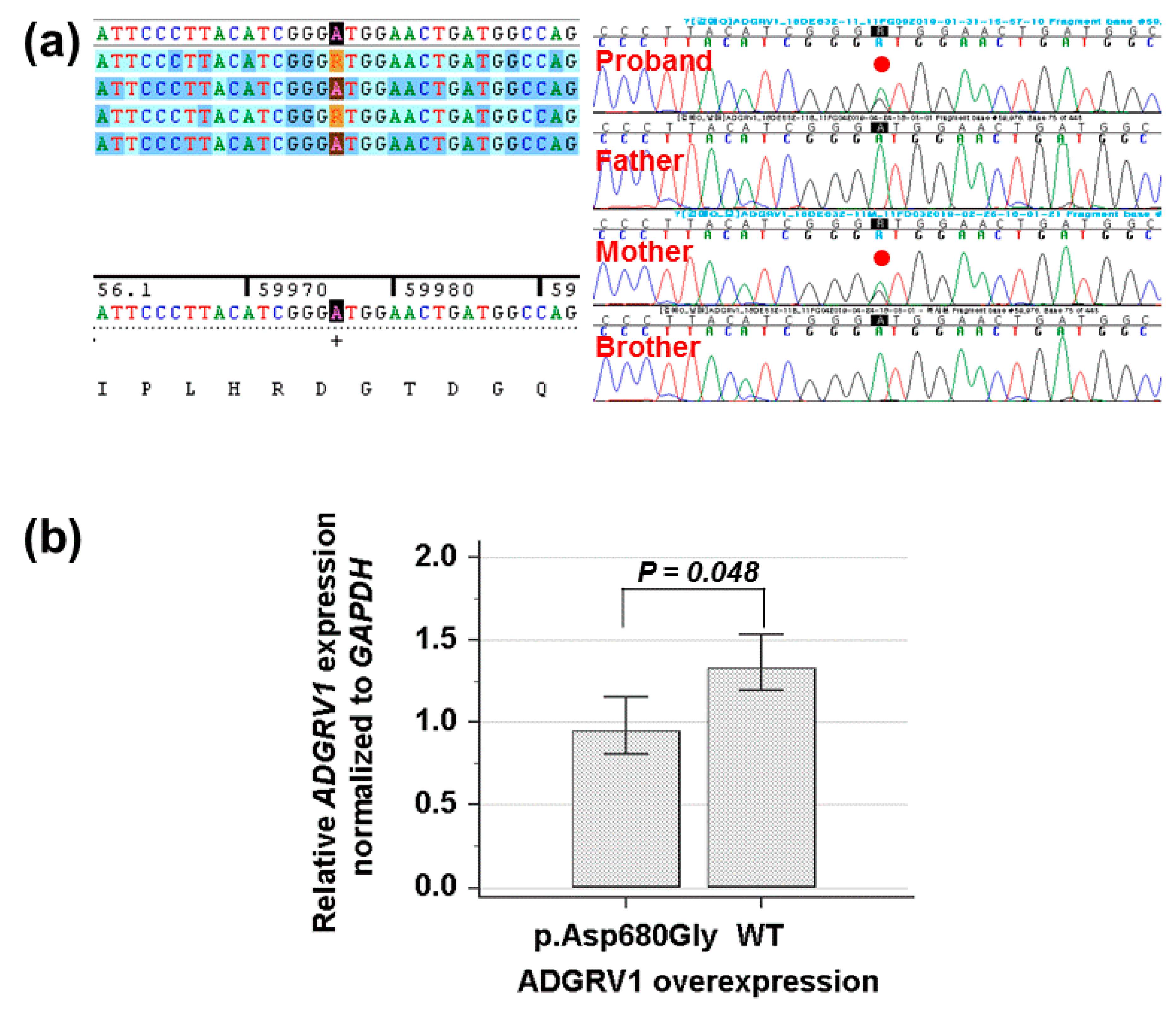
By estimating the sequence quality along all sequences, 18.9 million reads 150 bp in read length were produced from the patient sample. The % bases above average 30× were achieved for 99.2% of the target region and the mean read depth (×) was 170 bp. Targeted exome sequencing identified a heterozygous c.2039A>G of the ADGRV1 gene leading to a codon change of aspartic acid to glycine at position 680 (NM_032119.3: c.2039A>G, p.Asp680Gly; rs547076322) had not been reported previously to be related to afebrile and/or febrile seizures in the proband. To confirm the mutation segregated with affected members, Sanger sequencing was conducted and revealed that this mutation as a heterozygous state was present in the proband and her mother, respectively (Figure 2a). Cross-species sequence comparisons (phastCons, SiPhy, and GERP) of amino acid sequences of ADGRV1 protein revealed that this mutated site was highly conserved in vertebrates (phastCons 0.994 > cut-off of 0.8, SiPhy 16.285 > 12.17, and GERP 5.87 > 4.4) [27]. The exome database of gnomAD showed a rare allele frequency of 0.00002827. In addition, quantitative RT-PCR revealed that the expression of ADGRV1 mutant was weaker than that of the wild type (p = 0.048) (Figure 2b). Particularly, (likely) pathogenic SCN1A mutations were not identified in this family.
5. Discussion
Seizures in FEB 4 are related to febrile episodes in childhood with no evidence of defined pathologiccause or CNS infection. Simple FS is common, affecting 2–5% of children aged from 6 to 60 months. The tendency of developing epilepsy following simple FSs is not common. Complex FS is characterized as focal onset, with a duration of >15 min, and/or >one seizure in 24 h and related to an increased incidence of epilepsy. Channelopathies caused by mutation in ligand-gated or voltage-gated channels lead to idiopathic epilepsies such as FS, benign neonatal or infantile epilepsy, and autosomal dominant nocturnal frontal-lobe epilepsy. FEB 4 locus comprise the ADGRV1 gene, the causative gene for audiogenic reflex seizures in the Frings mouse (Adgrv1) [28]. In this study, Avlgr1 (Algr1b, Algr1d, and AVlgr1e) mRNA appeared dorminantly in the neuroepithelium of the developing mouse brain. Knockout mice without exons 2–4 of Avlgr1b were prone to experience audiogenic reflex seizure, although obvious cerebral histological abnormalities were not proven [29]. Differing from previous patients with ultra-rare ADGRV1 variants [26,30], our patient showed febrile seizures with afebrile seizures.
To the best of our knowledge, we reported a first Korean family who showed FS and afebrile seizures with ADGRV1 mutation, but without developmental delay/and or intellectual disability and the seizure disappeared after 60 months without anti-seizure medication. The identified nucleotide change in ADGRV1 replaces aspartic acid with glycine at position 680 of the ADGRV1 protein (p.Asp680Gly). This protein’s glycine residue is highly conserved, and has not been reported for individuals with ADGRV1-related FEB 4 (Table 1). In a previous study, mutation screening for the ADGRV1 detected several missense mutations with allele frequency < 0.0005 in 48 families with FSs or afebrile seizures [26]. Although different missense mutations (p.Tyr674Cys and p.Val754Ala) at the near codons have been predicted to be benign or pathogenic, the segregation of the mutation in our family supports its pathogenicity as in autosomal dominant manner. However, this prediction has not been confirmed by functional analysis, and the clinical manifestation of the observed mutation is certain only in two-generation family members showing afebrile and febrile seizures. Thus, this ADGRV1 mutation may be a candidate as a genetic cause of afebrile and febrile seizures in FEB 4.
On the other hand, ADGRV1 variation plays a part in developing epilepsy with myoclonic seizures, although the inheritance manner may be different in various patients [30]. The function of ADGRV1 is unestablished, but multiple calcium exchanger b-repeats in the ectodomain imply a contribution to protein–protein interaction that may be calcium mediated [31]. Particularly, the sequence of the ADGRV1/VLGR1 gene harbors the same EAR (epilepsy-related repeat) domain located in the LGI1 (leucine-rich glioma-inactivated 1, 604619) gene which is a causative gene in autosomal dominant lateral temporal lobe epilepsy with auditory features (ADLTE; 600512) [32]. ADGRV1 and LGI1 encode proteins that share a seven-fold repeated 44-residue motif homology domain, which has been named an EAR domain (Figure 3a) [32]. Scheel et al. suggested that the EAR domain plays a critical part in developing epilepsy by attaching to an unknown antiepileptic ligand or by interfering, with synaptogenesis or axon guidance [32]. The pathogenesis of ADGRV1 haploinsufficiency leading to seizures remains not well established, but animal studies have provided early insight into the nature of the mechanism. Comprehending the signaling pathways downstream of aGPCRs is not only essential for drug discovery, but also for achieving a fundamental understanding of receptor function [33]. ADGRV1, the largest aGPCR, has been found to couple to Gαi and signal to protein kinases A and C via Gαq and Gαs (Figure 3b). Lobe-Philippot et al. reported that ADGRV1 is needed for GABAergic interneuron development in the auditory cortex [34]. Dysfunction of cortical GABAnergic neurons can be a potential epileptogenic mechanism in humans, thus it can be a therapeutic target. In this family, the proband and her mother showed recurrent febrile seizures without other neurological comorbidity, and the seizure disappeared after the age of 5 without anti-seizure medication. ADGRV1 can be a susceptible gene for the mutation that can lead to febrile and afebrile seizures. On the other hand, whole-exome sequencing in combination with a genome-wide association study might be the most straightforward way to get some insight into the genetics of FS, which addresses the issues of phenocopies and genetic heterogeneity by sheer statistical power and sample size [35,36].
6. Conclusions
ADGRV1 may be related to a range of self-limited, mild febrile or infantile seizure without intellectual disability and developmental delay. ADGRV1 mutation may play an influential role in the occurrence of genetic epilepsies, especially those with febrile and afebrile seizures. If children under the age of 5 with no other neurological symptoms have febrile and other types of seizures, including those with no fevers, the ADGRV1 test will be useful like other genetic testings in finding the causes and prognosis of seizures. This may provide reassurance for the family towards an individual patient based on their genetic profile instead of empirical trials of anti-seizure medication. Further research is needed to establish an association between ADGRV1 mutations and febrile/afebrile seizures.
Author Contributions
Conceptualization, J.Y.H. and H.J.L.; methodology, Y.-M.L.; formal analysis, J.P.; writing —original draft preparation, J.Y.H.; writing, H.J.L. and J.Y.H.; supervision, J.P. and Y.-M.L. All authors have read and agreed to the published version of manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2020R1F1A1077316).
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- Steering Committee on Quality Improvement and Management, Subcommittee on Febrile Seizures. Febrile seizures: Clinical practice guideline for the long-term management of the child with simple febrile seizures. Pediatrics 2008, 121, 1281–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capovilla, G.; Mastrangelo, M.; Romeo, A.; Vigevano, F. Recommendations for the management of “febrile seizures” Ad hoc Task Force of LICE Guidelines Commission. Epilepsia 2009, 50, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Byeon, J.H.; Kim, G.-H.; Eun, B.-L. Prevalence, incidence, and recurrence of febrile seizures in Korean children based on national registry data. J. Clin. Neurol. 2018, 14, 43–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, J.H. Fever and fever-related epilepsies. Epilepsia 2012, 53 (Suppl. 4), 3–8. [Google Scholar] [CrossRef]
- Offringa, M.; Bossuyt, P.M.M.; Lubsen, J.; Ellenberg, J.H.; Nelson, K.B.; Knudsen, F.U.; Annegers, J.F.; El-Radhi, A.S.M.; Habbema, J.D.F.; Derksen-Lubsen, G.; et al. Risk factors for seizure recurrence in children with febrile seizures: A pooled analysis of individual patient data from five studies. J Pediatr. 1994, 124, 574–584. [Google Scholar] [CrossRef]
- Pavlidou, E.; Hagel, C.; Panteliadis, C. Febrile seizures: recent developments and unanswered questions. Childs Nerv Syst. 2013, 29, 2011–2017. [Google Scholar] [CrossRef]
- Vestergaard, M.; Pedersen, C.B.; Sidenius, P.; Olsen, J.; Christensen, J. The long-term risk of epilepsy after febrile seizures in susceptible subgroups. Am. J. Epidemiol. 2007, 165, 911–918. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, J.; Arinami, T. Molecular genetics of febrile seizures. Epilepsy Res. 2006, 70 (Suppl. 1), S190–S198. [Google Scholar] [CrossRef]
- Fetveit, A. Assessment of febrile seizures in children. Eur. J. Pediatrics 2008, 167, 17–27. [Google Scholar] [CrossRef]
- Hesdorffer, D.C.; Shinnar, S.; Lewis, D.V.; Moshe, S.L.; Nordli, D.R., Jr.; Pellock, J.M.; MacFall, J.; Shinnar, R.C.; Masur, D.; Frank, L.M.; et al. Design and phenomenology of the FEBSTAT study. Epilepsia 2012, 53, 1471–1480. [Google Scholar] [CrossRef] [Green Version]
- Johnson, W.G.; Kugler, S.L.; Stenroos, E.S.; Meulener, M.C.; Rangwalla, I.; Johnson, T.W.; Mandelbaum, D.E. Pedigree analysis in families with febrile seizures. Am. J. Med. Genet. 1996, 61, 345–352. [Google Scholar] [CrossRef]
- Wallace, R.H.; Berkovic, S.F.; Howell, R.A.; Sutherland, G.R.; Mulley, J.C. Suggestion of a major gene for familial febrile convulsions mapping to 8q13-21. J. Med. Genet. 1996, 33, 308–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, E.W.; Dubovsky, J.; Rich, S.S.; O’Donovan, C.A.; Orr, H.T.; Anderson, V.E.; Gil-Nagel, A.; Ahmann, P.; Dokken, C.G.; Schneider, D.T.; et al. Evidence for a novel gene for familial febrile convulsions, FEB2, linked to chromosome 19p in an extended family from the Midwest. Hum. Mol. Genet. 1998, 7, 63–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moulard, B.; Guipponi, M.; Chaigne, D.; Mouthon, D.; Buresi, C.; Malafosse, A. Identification of a new locus for generalized epilepsy with febrile seizures plus (GEFS+) on chromosome 2q24-q33. Am. J. Hum. Genet. 1999, 65, 1396–1400. [Google Scholar] [CrossRef] [Green Version]
- Peiffer, A.; Thompson, J.; Charlier, C.; Otterud, B.; Varvil, T.; Pappas, C.; Barnitz, C.; Gruenthal, K.; Kuhn, R.; Leppert, M. A locus for febrile seizures (FEB3) maps to chromosome 2q23-24. Ann. Neurol. 1999, 46, 671–678. [Google Scholar] [CrossRef]
- Nakayama, J.; Hamano, K.; Iwasaki, N.; Nakahara, S.; Horigome, Y.; Saitoh, H.; Aoki, T.; Maki, T.; Kikuchi, M.; Migita, T.; et al. Significant evidence for linkage of febrile seizures to chromosome 5q14-q15. Hum. Mol. Genet. 2000, 9, 87–91. [Google Scholar] [CrossRef] [Green Version]
- Baulac, S.; Huberfeld, G.; Gourfinkel-An, I.; Mitropoulou, G.; Beranger, A.; Prud’homme, J.F.; Baulac, M.; Brice, A.; Bruzzone, R.; LeGuern, E. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: A mutation in the gamma2-subunit gene. Nat. Genet. 2001, 28, 46–48. [Google Scholar] [CrossRef]
- Nabbout, R.; Prud’homme, J.F.; Herman, A.; Feingold, J.; Brice, A.; Dulac, O.; LeGuern, E. A locus for simple pure febrile seizures maps to chromosome 6q22–q24. Brain 2002, 125, 2668–2680. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, J.; Yamamoto, N.; Hamano, K.; Iwasaki, N.; Ohta, M.; Nakahara, S.; Matsui, A.; Noguchi, E.; Arinami, T. Linkage and association of febrile seizures to the IMPA2 gene on human chromosome 18. Neurology 2004, 63, 1803–1807. [Google Scholar] [CrossRef]
- Mantegazza, M.; Gambardella, A.; Rusconi, R.; Schiavon, E.; Annesi, F.; Cassulini, R.R.; Labate, A.; Carrideo, S.; Chifari, R.; Canevini, M.P.; et al. Identification of an Nav1.1 sodium channel (SCN1A) loss-of-function mutation associated with familial simple febrile seizures. Proc. Natl. Acad. Sci. USA 2005, 102, 18177–18182. [Google Scholar] [CrossRef] [Green Version]
- Audenaert, D.; Schwartz, E.; Claeys, K.G.; Claes, L.; Deprez, L.; Suls, A.; Van Dyck, T.; Lagae, L.; Van Broeckhoven, C.; Macdonald, R.L.; et al. A novel GABRG2 mutation associated with febrile seizures. Neurology 2006, 67, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Hedera, P.; Ma, S.; Blair, M.A.; Taylor, K.A.; Hamati, A.; Bradford, Y.; Abou-Khalil, B.; Haines, J.L. Identification of a novel locus for febrile seizures and epilepsy on chromosome 21q22. Epilepsia 2006, 47, 1622–1628. [Google Scholar] [CrossRef] [PubMed]
- Nabbout, R.; Baulac, S.; Desguerre, I.; Bahi-Buisson, N.; Chiron, C.; Ruberg, M.; Dulac, O.; LeGuern, E. New locus for febrile seizures with absence epilepsy on 3p and a possible modifier gene on 18p. Neurology 2007, 68, 1374–1381. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.H.; Chen, W.W.; Wang, X.; Zhu, Q.H.; Li, C.; Li, L.; Liu, M.G.; Wang, Q.K.; Liu, J.Y. A novel genetic locus for familial febrile seizures and epilepsy on chromosome 3q26.2-q26.33. Hum. Genet. 2008, 124, 423–429. [Google Scholar] [CrossRef]
- Salzmann, A.; Guipponi, M.; Lyons, P.J.; Fricker, L.D.; Sapio, M.; Lambercy, C.; Buresi, C.; Ouled Amar Bencheikh, B.; Lahjouji, F.; Ouazzani, R.; et al. Carboxypeptidase A6 gene (CPA6) mutations in a recessive familial form of febrile seizures and temporal lobe epilepsy and in sporadic temporal lobe epilepsy. Hum. Mutat. 2012, 33, 124–135. [Google Scholar] [CrossRef]
- Nakayama, J.; Fu, Y.H.; Clark, A.M.; Nakahara, S.; Hamano, K.; Iwasaki, N.; Matsui, A.; Arinami, T.; Ptacek, L.J. A nonsense mutation of the MASS1 gene in a family with febrile and afebrile seizures. Ann. Neurol. 2002, 52, 654–657. [Google Scholar] [CrossRef]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [Green Version]
- Skradski, S.L.; Clark, A.M.; Jiang, H.; White, H.S.; Fu, Y.H.; Ptacek, L.J. A novel gene causing a mendelian audiogenic mouse epilepsy. Neuron 2001, 31, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Yagi, H.; Takamura, Y.; Yoneda, T.; Konno, D.; Akagi, Y.; Yoshida, K.; Sato, M. Vlgr1 knockout mice show audiogenic seizure susceptibility. J. Neurochem. 2005, 92, 191–202. [Google Scholar] [CrossRef]
- Myers, K.A.; Nasioulas, S.; Boys, A.; McMahon, J.M.; Slater, H.; Lockhart, P.; Sart, D.D.; Scheffer, I.E. ADGRV1 is implicated in myoclonic epilepsy. Epilepsia 2018, 59, 381–388. [Google Scholar] [CrossRef] [Green Version]
- McMillan, D.R.; Kayes Wandover, K.; Richardson, J.; White, P. Very large G protein-coupled receptor-1, the largest known cell surface protein, is highly expressed in the developing central nervous system. J. Biol. Chem. 2002, 277, 785–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheel, H.; Tomiuk, S.; Hofmann, K. A common protein interaction domain links two recently identified epilepsy genes. Hum. Mol. Genet. 2002, 11, 1757–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, R.H.; Hall, R.A. Adhesion G Protein-Coupled Receptors as Drug Targets. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 429–449. [Google Scholar] [CrossRef] [PubMed]
- Libé-Philippot, B.; Michel, V.; de Monvel, J.B.; Le Gal, S.; Dupont, T.; Avan, P.; Métin, C.; Michalski, N.; Petit, C. Auditory cortex interneuron development requires cadherins operating hair-cell mechanoelectrical transduction. Proc. Natl. Acad. Sci. USA 2017, 114, 7765–7774. [Google Scholar]
- Helbig, I.; Ellis, C.A. Personalized medicine in genetic epilepsies—Possibilities, challenges, and new frontiers. Neuropharmacology 2020, 172, 107970. [Google Scholar] [CrossRef]
- Helbig, I.; Barcia, G.; Pendziwiat, M.; Ganesan, S.; Mueller, S.H.; Helbig, K.L.; Vaidiswaran, P.; Xian, J.; Galer, P.D.; Afawi, Z.; et al. Whole-exome and HLA sequencing in Febrile infection-related epilepsy syndrome. Ann. Clin. Transl. Neurol. 2020. [Google Scholar] [CrossRef]
Figure 1.
(a) Pedigree of a Korean family with afebrile (light grey) and febrile seizures (dark grey) carrying ADGRV1 mutation. (b) Brain magnetic resonance imaging was done at the age of 2 in the proband. No abnormal signal intensity or enhanced lesion in the brain parenchyma was seen. No evidence of abnormal finding in the diploic space and cerebrospinal fluid space. (c) Interictal electroencephalogram was normal tracing at the age of 2 in the proband. Focal abnormalities, persistent asymmetries, and potentially epileptogenic discharges were not seen.
Figure 1.
(a) Pedigree of a Korean family with afebrile (light grey) and febrile seizures (dark grey) carrying ADGRV1 mutation. (b) Brain magnetic resonance imaging was done at the age of 2 in the proband. No abnormal signal intensity or enhanced lesion in the brain parenchyma was seen. No evidence of abnormal finding in the diploic space and cerebrospinal fluid space. (c) Interictal electroencephalogram was normal tracing at the age of 2 in the proband. Focal abnormalities, persistent asymmetries, and potentially epileptogenic discharges were not seen.

Figure 2.
(a) Sanger sequencing shows a heterozygous missense mutation (NM_032119.3: c.2039A>G, p.Asp680Gly) of the ADGRV1 gene in the proband and her family member, respectively. (b) Quantitative reverse transcription polymerase chain reaction indicates that the missense mutation (p.Asp680Gly) decreases the mRNA expression level of the ADGRV1, compared to the wild type (p = 0.048).
Figure 2.
(a) Sanger sequencing shows a heterozygous missense mutation (NM_032119.3: c.2039A>G, p.Asp680Gly) of the ADGRV1 gene in the proband and her family member, respectively. (b) Quantitative reverse transcription polymerase chain reaction indicates that the missense mutation (p.Asp680Gly) decreases the mRNA expression level of the ADGRV1, compared to the wild type (p = 0.048).

Figure 3.
(a) Adhesion G protein-coupled receptor structure (GPCR). Calx, calcium exchanger; Lam, laminin; GAIN, GPCR autoproteolysis inducing domain; PBM, PDZ binding motif (b) Adhesion G protein-coupled receptor signaling pathways. GAIN, GPCR autoproteolysis–inducing domain; GPCR, G protein–coupled receptor.
Figure 3.
(a) Adhesion G protein-coupled receptor structure (GPCR). Calx, calcium exchanger; Lam, laminin; GAIN, GPCR autoproteolysis inducing domain; PBM, PDZ binding motif (b) Adhesion G protein-coupled receptor signaling pathways. GAIN, GPCR autoproteolysis–inducing domain; GPCR, G protein–coupled receptor.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Results of in silico analysis of rare missense ADGRV1 mutations with allele frequency < 0.0005 reported by Myers KA et al. and this study.
Table 1.
Results of in silico analysis of rare missense ADGRV1 mutations with allele frequency < 0.0005 reported by Myers KA et al. and this study.
| Base Change | Codon Change | rs ID | gnomAD | Polyphen-2 | Mutation Taster | Grantham (<100) | GERP (>4.4) | Protein Domain | Inheritance |
|---|---|---|---|---|---|---|---|---|---|
| c.2021A>G | p.Tyr674Cys | rs761220696 | 4/17562 (EA) 1/110184 (EU) 5/242476 (All) | Benign (0.001) | Polymorphism | Radical 194 | −5.21 | Calx-β5 | Father |
| * c.2039A>G | p.Asp680Gly | rs547076322 | 7/17866 (EA) 7/247616 (All) | Damaging (1) | Disease causing | Conservative 94 | 5.87 | Calx-β5 | Mother |
| c.2261T>C | p.Val754Ala | rs374609813 | 0/19516 (EA) 22/30544 (SA) 13/128162 (EU) 39/280148 (All) | Damaging (0.682) | Disease causing | Conservative 64 | 5.09 | Extracellular, links Calx-β5 and Calx-β6 | Unknown |
| c.5722G>A | p.Asp1908Asn | rs757418364 | 0/19490 (EA) 9/128042 (EU) 9/279986 (All) | Damaging (0.976) | Disease causing | Conservative 23 | 5.56 | Calx-β13 | Mother |
| c.8266G>A | p.Gly2756Arg | rs546198768 | 0/18314 (EA) 11/117330 (EU) 15/257862 (All) | Damaging (1) | Disease causing | Radical 125 | 5.15 | Calx-β19 | Mother |
| c.9466A>G | p.Ile3156Val | rs372484022 | 0/19532 (EA) 57/30568 (SA) 7/127064 (EU) 69/279132 All) | Benign (0.274) | Disease causing | Conservative 29 | 1.86 | Calx-β22 | Mother |
| c.13228G>A | p.Glu4410Lys | rs371970388 | 0/19532 (EA) 17/126258 (EU) 18/273998 (All) | Damaging (0.676) | Polymorphism | Conservative 56 | 1.39 | Calx-β30 | Unknown |
| c.13495C>T | p.Arg4499Cys | rs567519802 | 0/17780 (EA) 6/29982 (SA) 7/245774 (All) | Damaging (0.998) | Disease causing | Radical 180 | 5.97 | Extracellular, links Calx-β30 and Calx-β31 | Unknown |
| c.13568G>C | p.Ser4523Thr | rs376673439 | 0/17816 (EA) 11/111494 (EU) 12/246276 (All) | Damaging (0.996) | Disease causing | Conservative 58 | 5.78 | Calx-β31 | Father |
* Identified in the present study. Reference sequence number: NM_032119.3 NP_115495.3. Abbreviations: rsID, reference ID in dbSNP; gnomAD, The Genome Aggregation Database; EA, East Asian; EU, non-Finnish Europe; SA, South Asian; All, total population; Polyphen-2, Polymorphism Phenotyping v2;) AF (Allele Frequency); Grantham, Grantham scores for conservative < 100; GERP, Genomic Evolutionary Rate Profiling for conservative > 4.4.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Han, J.Y.; Lee, H.J.; Lee, Y.-M.; Park, J. Identification of Missense ADGRV1 Mutation as a Candidate Genetic Cause of Familial Febrile Seizure 4. Children 2020, 7, 144. https://doi.org/10.3390/children7090144
AMA Style
Han JY, Lee HJ, Lee Y-M, Park J. Identification of Missense ADGRV1 Mutation as a Candidate Genetic Cause of Familial Febrile Seizure 4. Children. 2020; 7(9):144. https://doi.org/10.3390/children7090144
Chicago/Turabian StyleHan, Ji Yoon, Hyun Joo Lee, Young-Mock Lee, and Joonhong Park. 2020. "Identification of Missense ADGRV1 Mutation as a Candidate Genetic Cause of Familial Febrile Seizure 4" Children 7, no. 9: 144. https://doi.org/10.3390/children7090144
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.