
Citius, Altius, Fortius—Advanced Mass Spectrometry in Service of Forensic Analysis

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Mass Spectrometry in Forensic Analysis
2.1. Mass Spectrometry and Analysis of Falsified Medicines
2.1.1. Falsified Lifestyle Drugs as a Global Problem
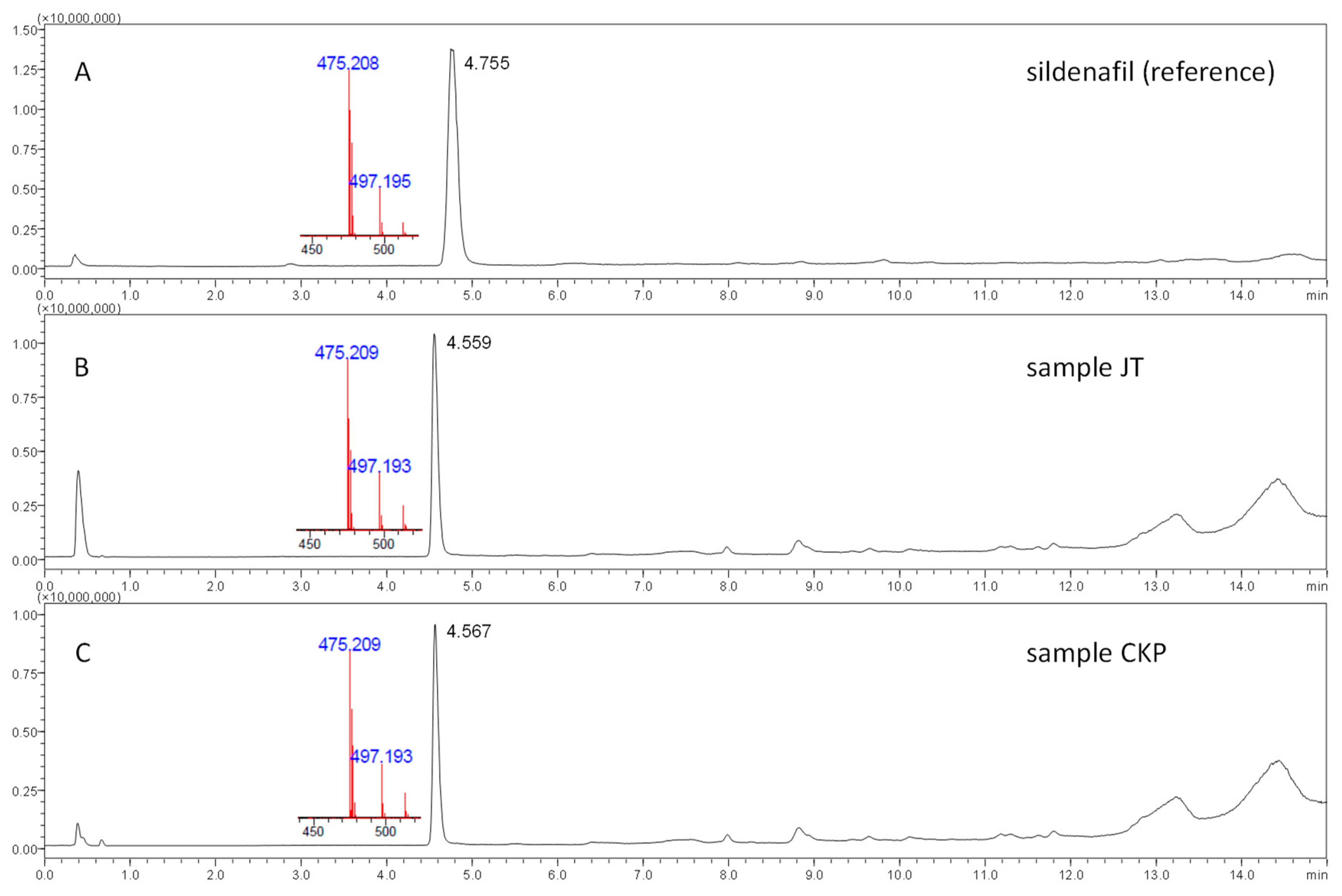
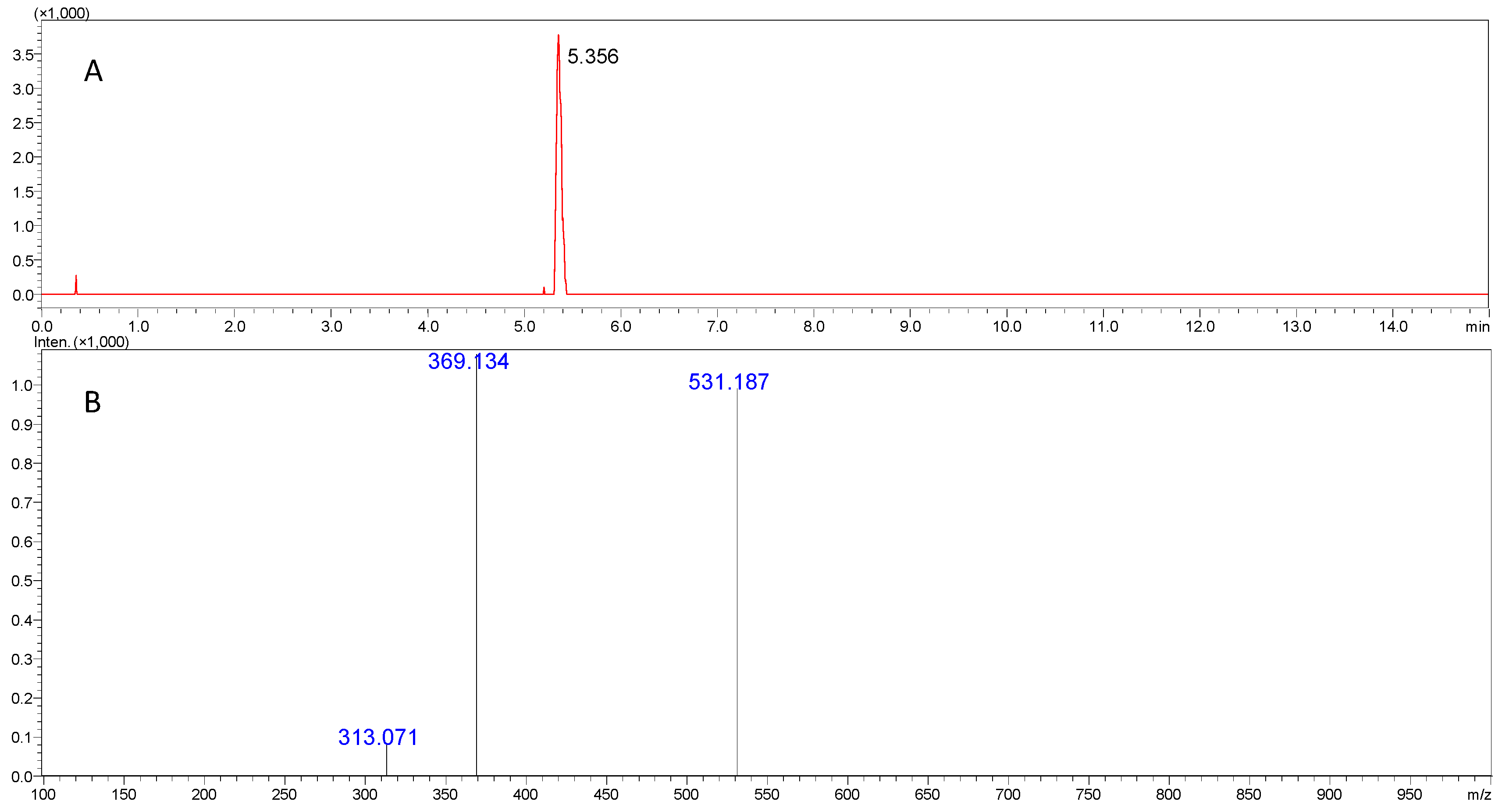
2.1.2. Technical Advances in Falsified Drug Analysis
2.1.3. Falsification of Supplements in Doping Cases
2.2. Forensic Applications of IRMS
2.2.1. The Origin of Isotopic Differences
2.2.2. Food Authentication by IRMS
2.2.3. Endogenous and Exogenous Compounds Distinguished by IRMS
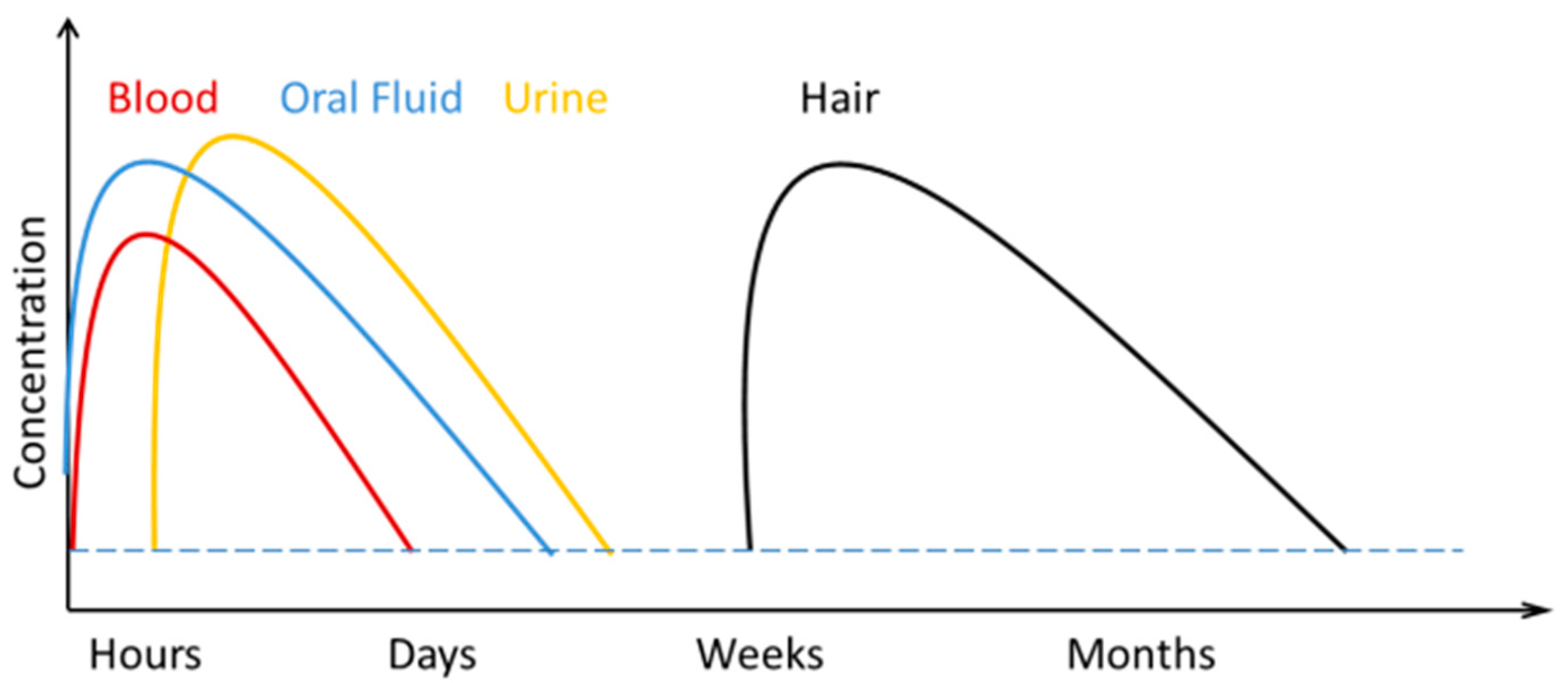
2.3. Alternative Matrices in Toxicological Analysis
2.3.1. Forensic Hair Analysis by Mass Spectrometry
2.3.2. Special MS Techniques in Hair Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
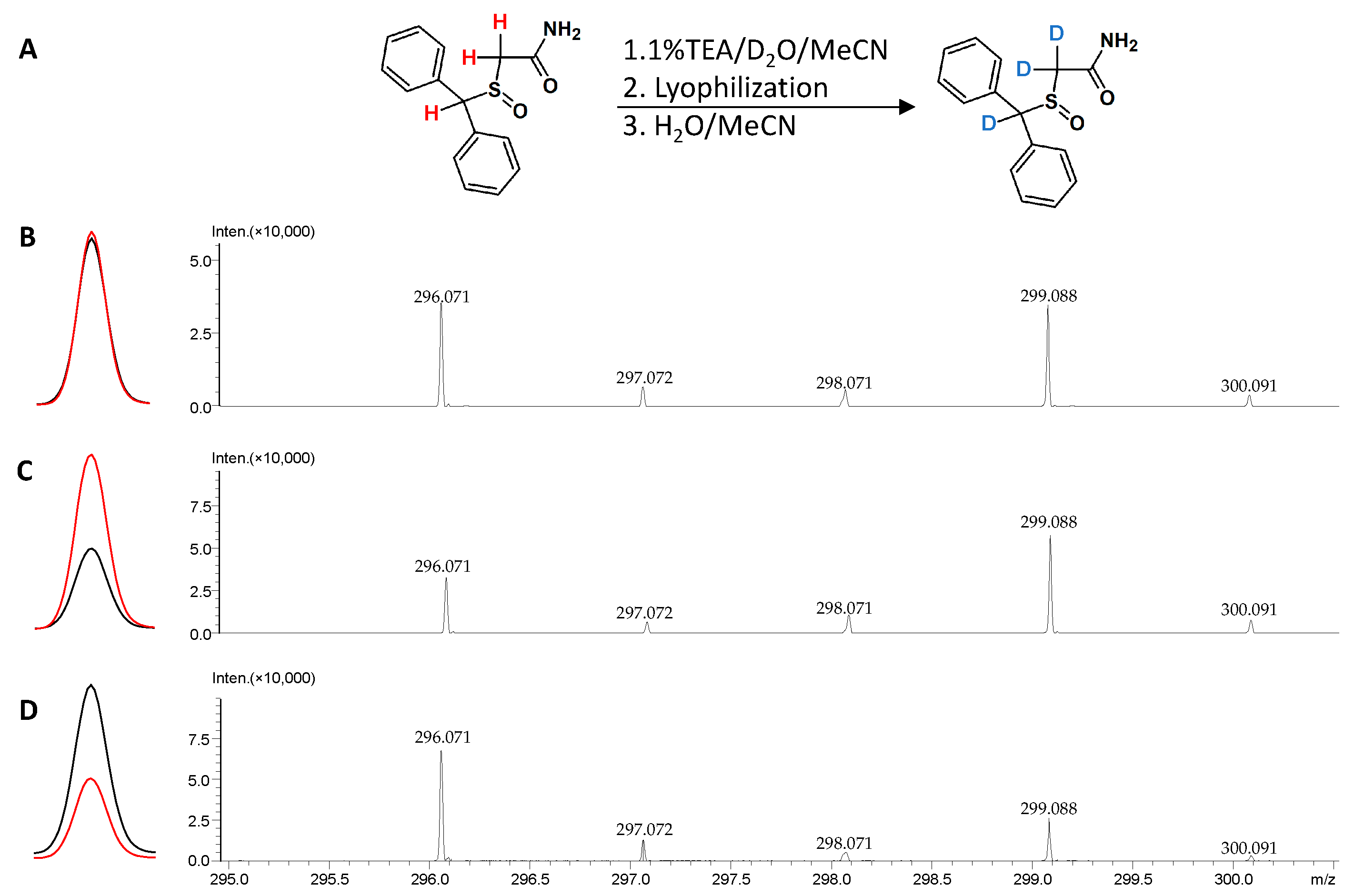{kind=link}
| Drug Name | Abbreviation | Cut off Level ng/mL | Approximate Urine Detection Times | Approximate Saliva Detection Times | Cut off Level in Hair ng/mg |
|---|---|---|---|---|---|
| Amphetamine | AMP | 1000 | 2–4 Days | 1–3 Days | 0.2 |
| Barbiturates | BAR | 300 | 3–10 Days | 1–2 Days | |
| Benzodiazepine | BZO | 300 | 1–14 Days | 1–2 Days | 0.05 |
| Buprenorphine | BUP | 10 | 3–5+ Days | 1–2 Days | 0.01 |
| Cocaine | COC | 150 | 1–7 Days | 1–2 Days | 0.5 |
| Ecstasy | MDMA | 500 | 1–3 Days | 1–2 Days | 0.2 |
| Ethyl Glucuronide | EtG | 500 | 1–3 Days | 6–24 h | 0.3 |
| Fentanyl | FEN | 50 or 100 | 1–4 Days | 1–3 Days | |
| Heroin metabolite | 6-MAM 6-AM | 10 | 1–2 Days | 8 h | 0.2 |
| Marijuana Tetrahydrocannabinol | THC | 50 | 3–30 Days | 6–12 h | 0.0002 |
| Methadone Metabolite | EDDP | 300 | 1–7 Days | 1–2 Days | 0.05 |
| Methadone | MTD | 300 | 1–7 Days | 1–2 Days | 0.2 |
| Methamphetamine | mAMP, MET | 500 | 1–4 Days | 1–3 Days | 0.2 |
| Morphine | MOP, MOR, MOP-300 | 300 | 1–3 Days | 1–2 Days | 0.2 |
| Nicotine Metabolite | COT | 200 | 2–4 Days | 2–4 Days | |
| Opiates | OPI MOP NOR | 2000 | 1–3 Days | 1–2 Days | 0.2 |
| Oxycodone | OXY | 100 | 1–3 Days | 1–2 Days | |
| Phencyclidine | PCP | 25 | 2–30 Days | 1–2 Days | |
| Propoxyphene | PPX | 300 | 1–7 Days | 1–2 Days | |
| Tramadol | TRA | 100 | 13 Days | 1–2 Days | |
| Tricyclic Antidepressants | TCA | 1000 ng/mL | 1–14 Days * | 1–2 Days |
2.3.3. Unusual Matrices for Special Toxicological Cases
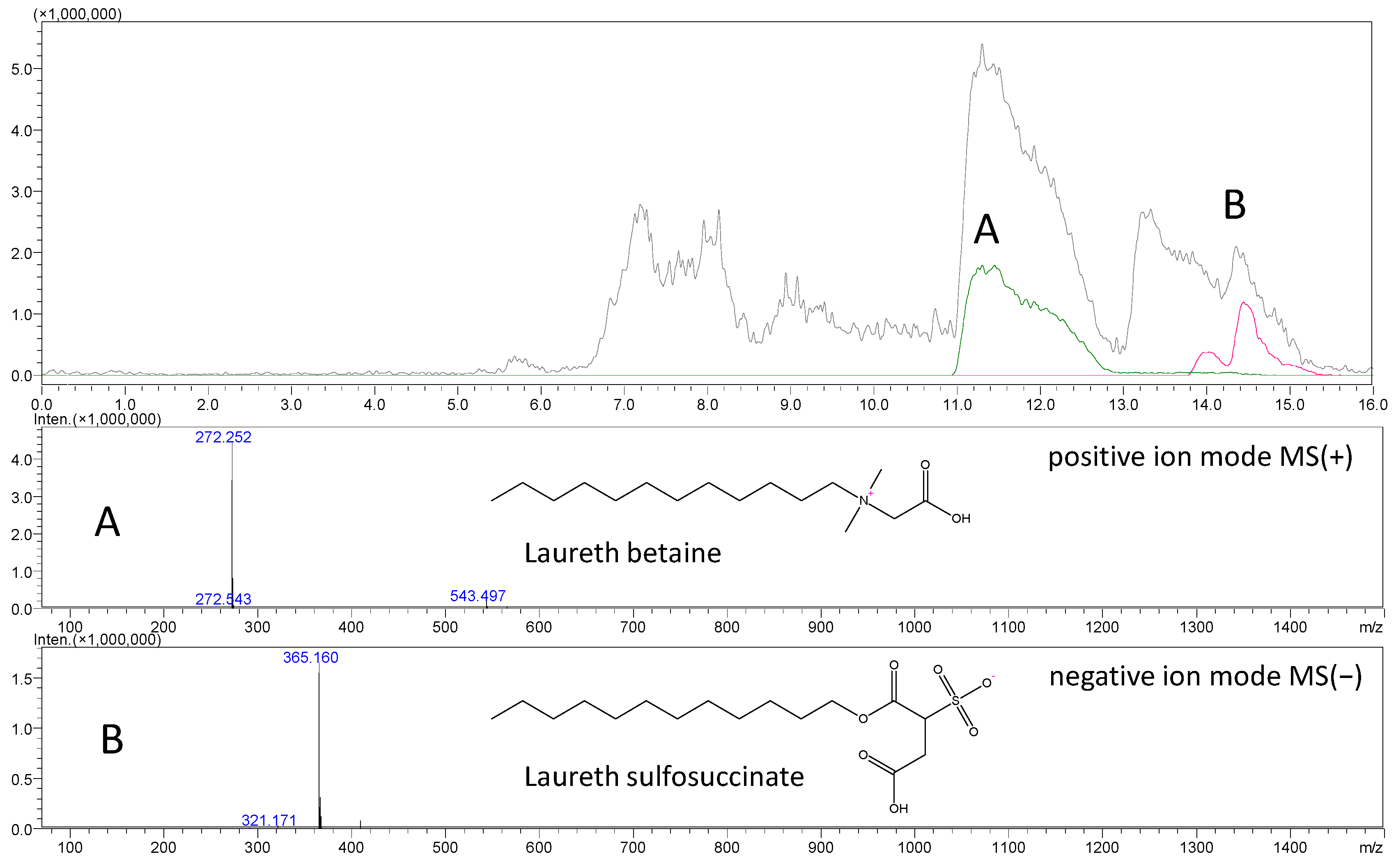
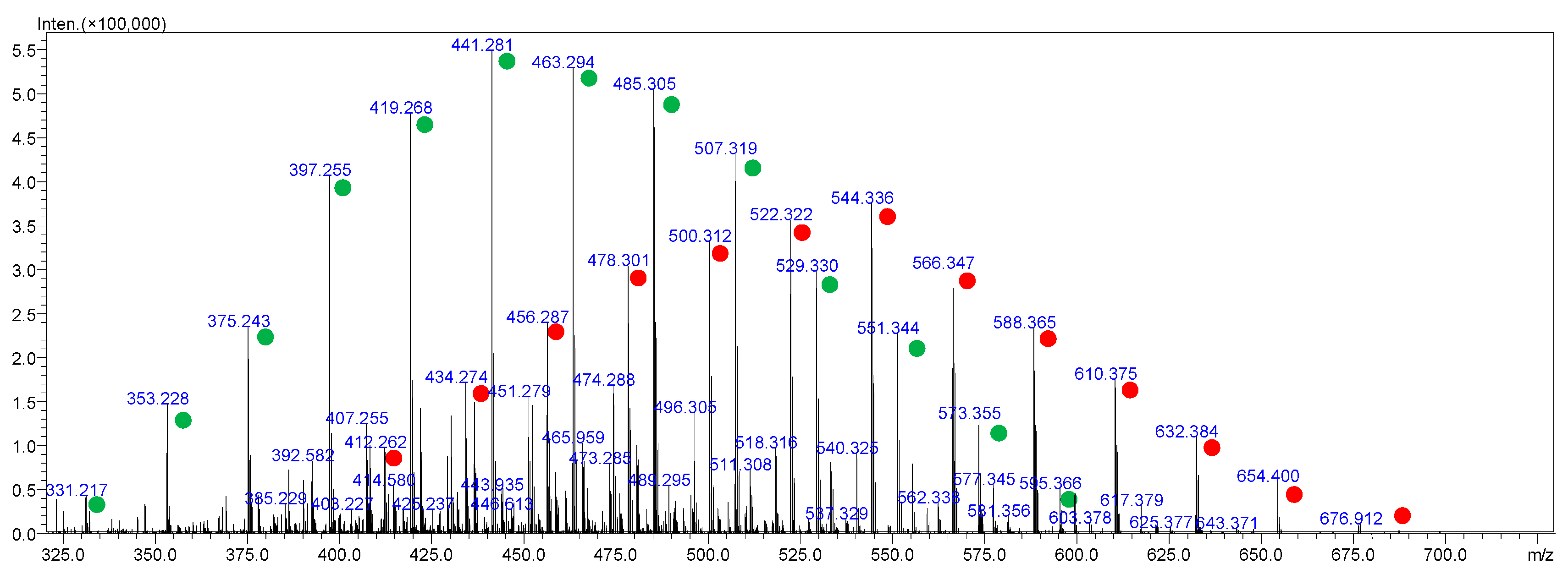
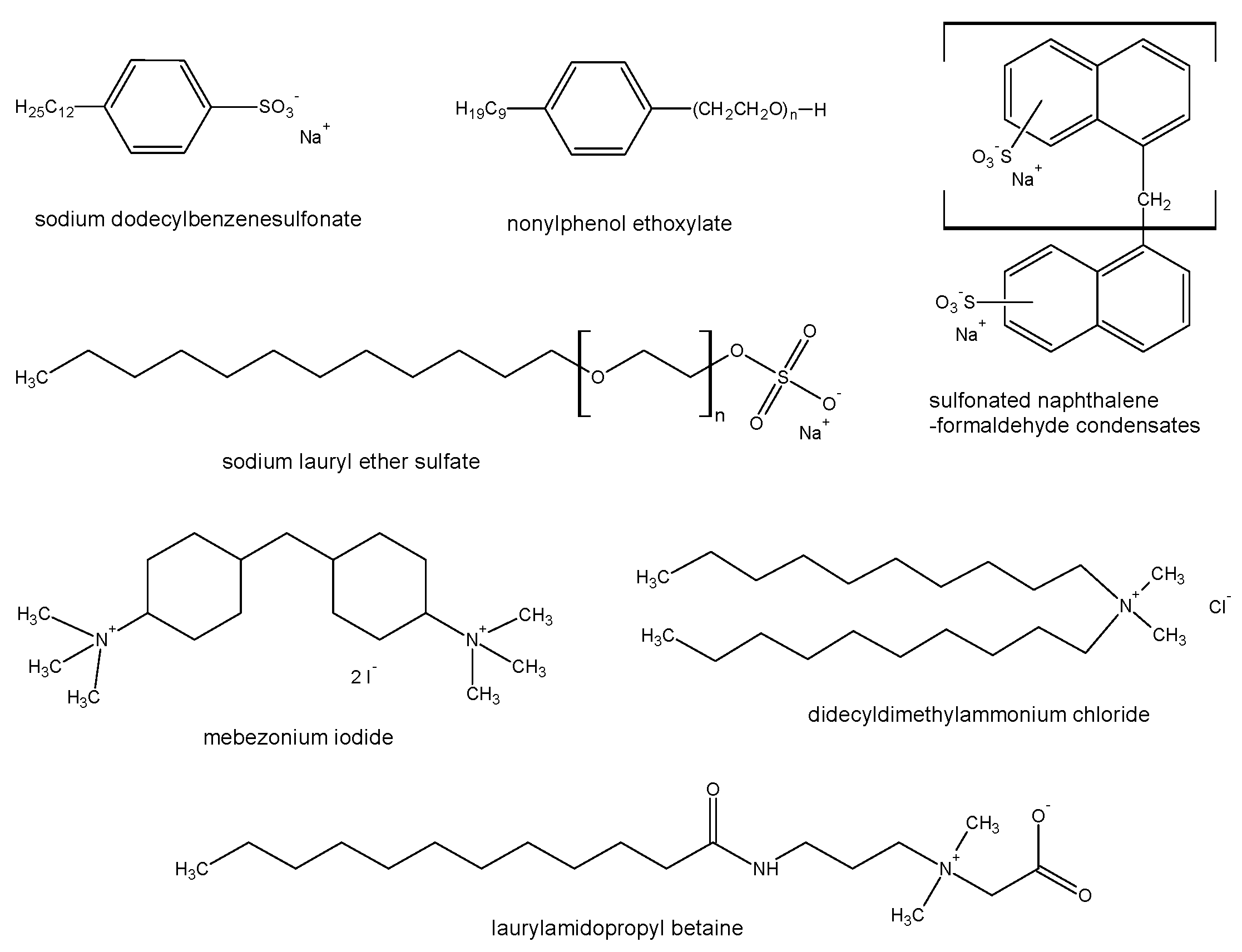
2.4. Surfactants in Forensic Toxicology—A New Analytical Challenge
2.4.1. Mass Spectrometry in Surfactant Analysis
2.4.2. Surfactants in Forensic Cases
2.5. Derivatization as a Tool in Mass Spectrometry Analysis in Forensic Chemistry
- compounds absorbing light in the UV-Vis range, introducing chromophores into the molecule;
- fluorogenic reagents, compounds which do not exhibit fluorescence on their own, but they acquire this ability under the influence of reactions with appropriate compounds;
- fluorescent reagents introducing fluorophores;
- reagents capable of redox reactions;
- reagents that introduce a permanent charge to the molecule
2.5.1. Derivatization in Cyanide Analysis
2.5.2. Derivatization of Propofol
2.5.3. Derivatization of Carbonyl Compounds
2.5.4. Derivatization in Analysis of Neurotoxins
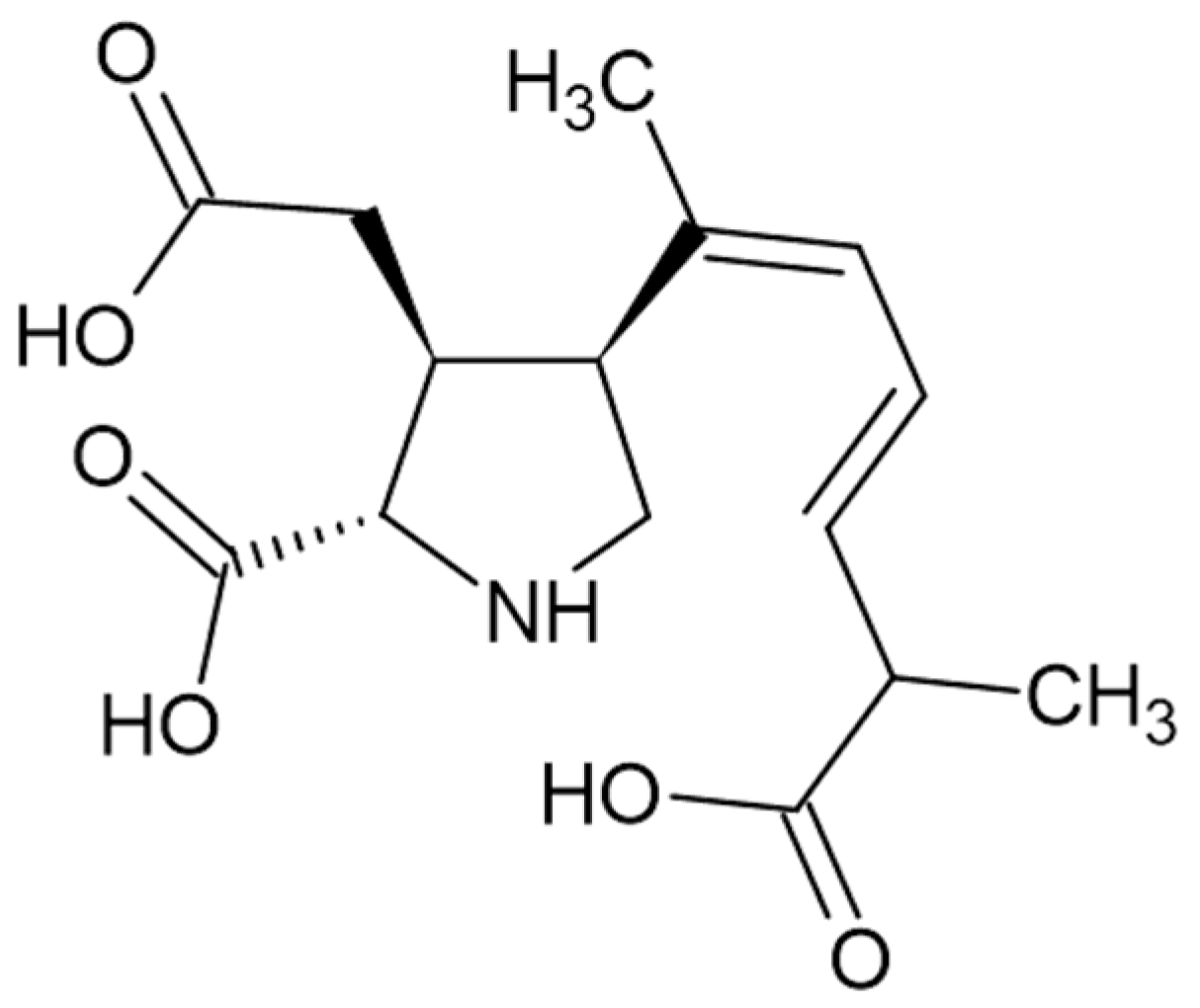
2.5.5. Derivatization of Alkaloids: Atropine
2.6. Internal Standards in Quantitative LCMS Analysis in Forensic Toxicology
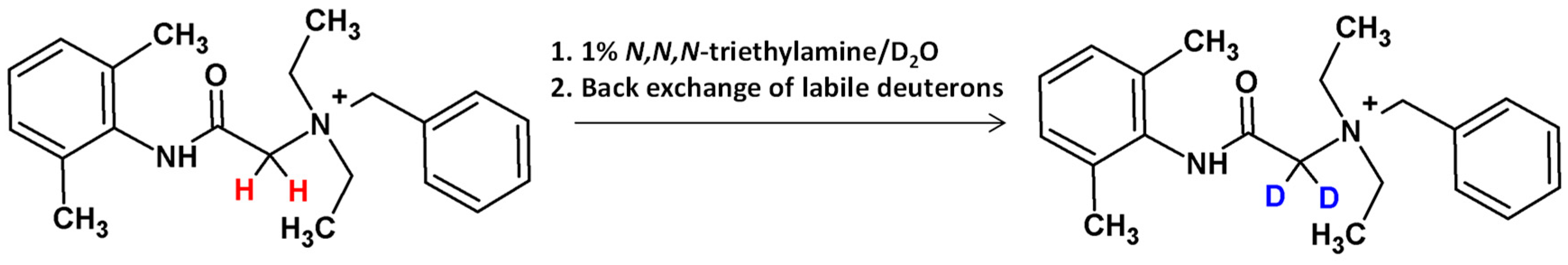
2.6.1. Isotope Labeling: Hydrogen-Deuterium Exchange
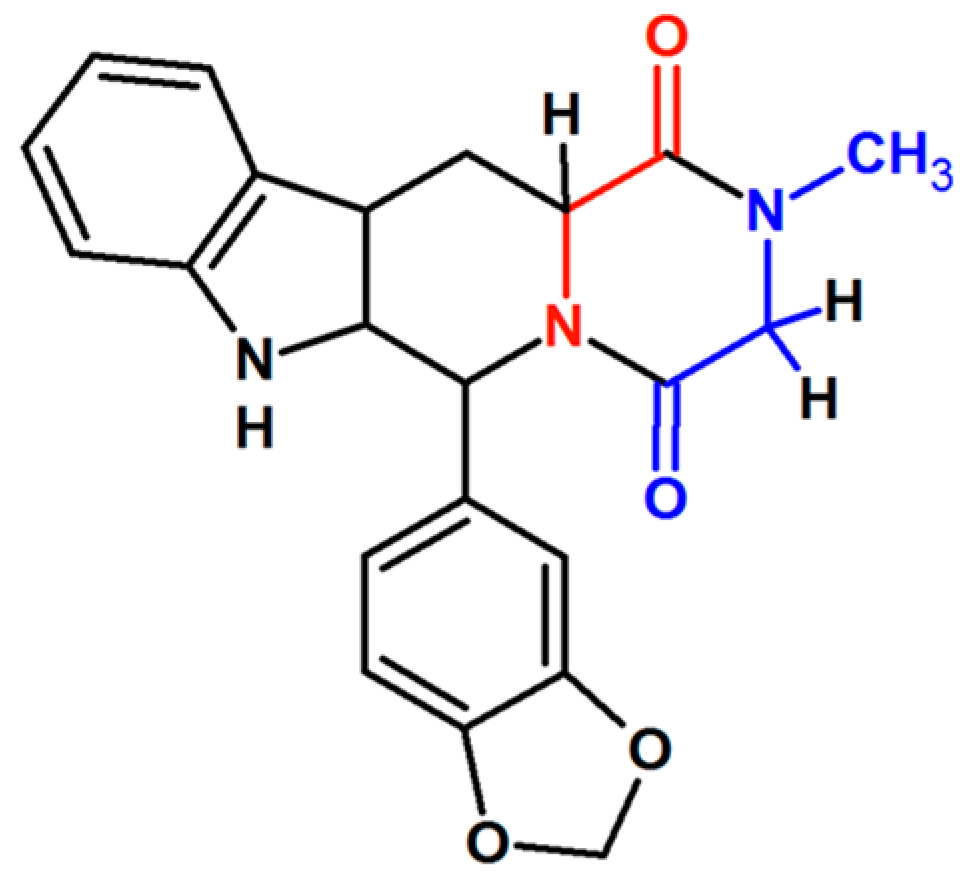
2.6.2. Isotopically Labeled Standards: Glycine Deerivatives
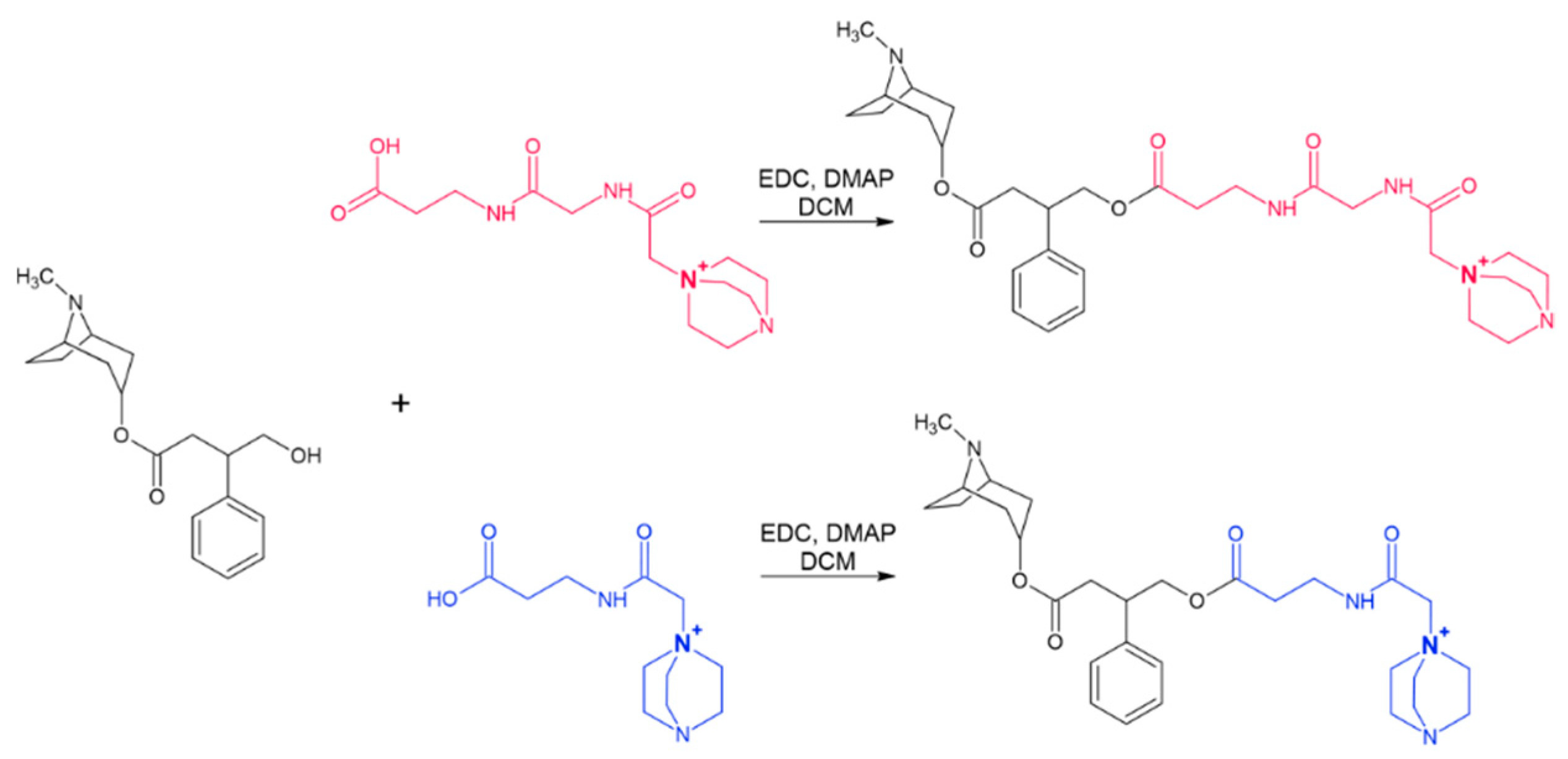
2.6.3. Denatonium Benzoate
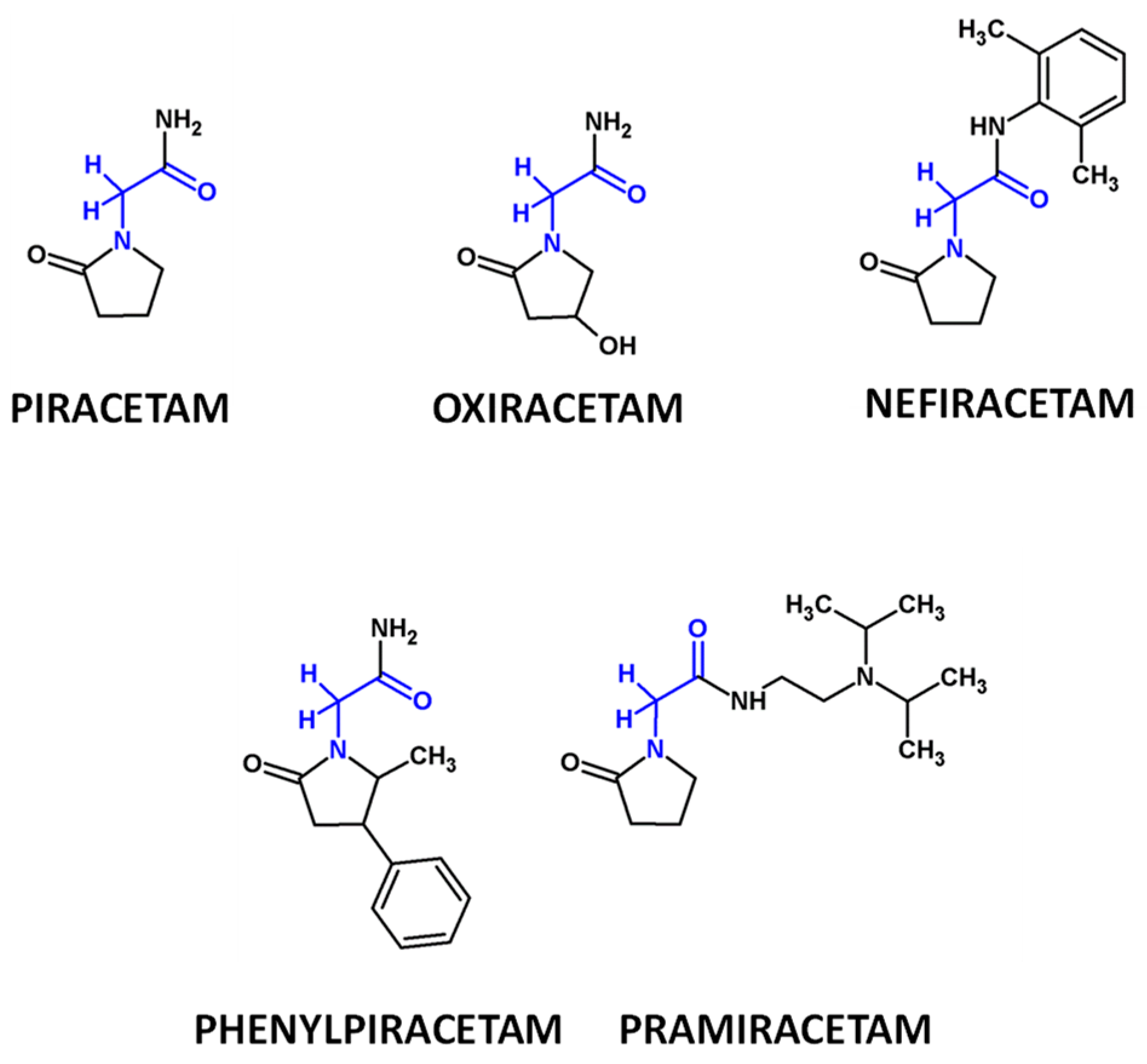
2.6.4. Tadalafil and Racetams
2.6.5. Armodafinil
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, H.M.; McDaniel, T.J.; Fedick, P.W.; Mulligan, C.C. The current role of mass spectrometry in forensics and future prospects. Anal. Methods 2020, 12, 3974–3997. [Google Scholar] [CrossRef]
- Hamidi, S. Assessment of Undeclared Synthetic Drugs in Dietary Supplements in an Analytical View: A Comprehensive Review. Crit. Rev. Anal. Chem. 2021, 1–11. [Google Scholar] [CrossRef]
- Giebułtowicz, J.; Rużycka, M.; Fudalej, M.; Krajewski, P.; Wroczyński, P. LC-MS/MS method development and validation for quantitative analyses of 2-aminothiazoline-4-carboxylic acid—A new cyanide exposure marker in post mortem blood. Talanta 2016, 150, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Shokry, E.; Marques, J.G.; Ragazzo, P.C.; Pereira, N.Z.; Filho, N.R.A. Earwax as an alternative specimen for forensic analysis. Forensic Toxicol. 2017, 35, 348–358. [Google Scholar] [CrossRef] [Green Version]
- Musshoff, F.; Schwarz, G.; Sachs, H.; Skopp, G.; Franz, T. Concentration distribution of more than 100 drugs and metabolites in forensic hair samples. Int. J. Leg. Med. 2020, 134, 989–995. [Google Scholar] [CrossRef]
- Woźniak, M.K.; Jaszczak, E.; Wiergowski, M.; Polkowska, Ż.; Namieśnik, J.; Biziuk, M. Meconium analysis as a promising diagnostic tool for monitoring fetal exposure to toxic substances: Recent trends and perspectives. TrAC Trends Anal. Chem. 2018, 109, 124–141. [Google Scholar] [CrossRef]
- Pascale, R.; Acquavia, M.A.; Onzo, A.; Cataldi, T.R.I.; Calvano, C.D.; Bianco, G. Analysis of surfactants by mass spectrometry: Coming to grips with their diversity. Mass Spectrom. Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.P.; Jackson, G.P. Isotope ratio mass spectrometry in forensic science applications. Forensic Chem. 2019, 13, 100154. [Google Scholar] [CrossRef]
- Damont, A.; Legrand, A.; Cao, C.; Fenaille, F.; Tabet, J. Hydrogen/deuterium exchange mass spectrometry in the world of small molecules. Mass Spectrom. Rev. 2021, e21765. [Google Scholar] [CrossRef]
- Legrand, P.; Dembele, O.; Alamil, H.; Lamoureux, C.; Mignet, N.; Houzé, P.; Gahoual, R. Structural identification and absolute quantification of monoclonal antibodies in suspected counterfeits using capillary electrophoresis and liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2022, 414, 2699–2712. [Google Scholar] [CrossRef]
- Janvier, S.; De Spiegeleer, B.; Vanhee, C.; Deconinck, E. Falsification of biotechnology drugs: Current dangers and/or future disasters? J. Pharm. Biomed. Anal. 2018, 161, 175–191. [Google Scholar] [CrossRef]
- Deconinck, E. Trends in the analysis of falsified and illegal medicines. Talanta 2019, 203, 328–329. [Google Scholar] [CrossRef]
- Hart, I.M.B.; Ohana, D.; Venhuis, B.J. Current challenges in the detection and analysis of falsified medicines. J. Pharm. Biomed. Anal. 2021, 197, 113948. [Google Scholar] [CrossRef]
- Zhang, H.; Hua, D.; Huang, C.; Samal, S.K.; Xiong, R.; Sauvage, F.; Braeckmans, K.; Remaut, K.; De Smedt, S.C. Materials and Technologies to Combat Counterfeiting of Pharmaceuticals: Current and Future Problem Tackling. Adv. Mater. 2020, 32, e1905486. [Google Scholar] [CrossRef]
- Muschietti, L.; Redko, F.; Ulloa, J. Adulterants in selected dietary supplements and their detection methods. Drug Test. Anal. 2020, 12, 861–886. [Google Scholar] [CrossRef]
- Zou, W.-B.; Yin, L.-H.; Jin, S.-H. Advances in rapid drug detection technology. J. Pharm. Biomed. Anal. 2018, 147, 81–88. [Google Scholar] [CrossRef]
- Rebiere, H.; Guinot, P.; Chauvey, D.; Brenier, C. Fighting falsified medicines: The analytical approach. J. Pharm. Biomed. Anal. 2017, 142, 286–306. [Google Scholar] [CrossRef]
- Bottoni, P.; Caroli, S. Fake pharmaceuticals: A review of current analytical approaches. Microchem. J. 2019, 149, 104053. [Google Scholar] [CrossRef]
- Deconinck, E.; Sacré, P.-Y.; Courselle, P.; De Beer, J.O. Chromatography in the Detection and Characterization of Illegal Pharmaceutical Preparations. J. Chromatogr. Sci. 2013, 51, 791–806. [Google Scholar] [CrossRef] [Green Version]
- Bolla, A.S.; Patel, A.R.; Priefer, R. The silent development of counterfeit medications in developing countries—A systematic review of detection technologies. Int. J. Pharm. 2020, 587, 119702. [Google Scholar] [CrossRef]
- Evans-Nguyen, K.; Stelmack, A.R.; Clowser, P.C.; Holtz, J.M.; Mulligan, C.C. Fieldable mass spectrometry for forensic science, homeland security, and defense applications. Mass Spectrom. Rev. 2020, 40, 628–646. [Google Scholar] [CrossRef] [PubMed]
- Forbes, T.P.; Sisco, E. Recent advances in ambient mass spectrometry of trace explosives. Analyst 2018, 143, 1948–1969. [Google Scholar] [CrossRef]
- Minzière, V.R.; Werner, D.; Schneider, D.; Manganelli, M.; Jung, B.; Weyermann, C.; Gassner, A. Combined Collection and Analysis of Inorganic and Organic Gunshot Residues. J. Forensic Sci. 2020, 65, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Luo, Y.; Wang, Y.; Zhang, Q.; Yang, X. Comparative analysis of aged documents by desorption electrospray ionization–Mass spectrometry (DESI-MS) imaging. J. Forensic Sci. 2022. [Google Scholar] [CrossRef]
- Gupta, S.; Samal, N. Application of direct analysis in real-time mass spectrometry (DART-MS) in forensic science: A comprehensive review. Egypt. J. Forensic Sci. 2022, 12, 17. [Google Scholar] [CrossRef]
- Mogollón, N.G.S.; Quiroz-Moreno, C.D.; Prata, P.S.; De Almeida, J.R.; Cevallos, A.S.; Torres-Guiérrez, R.; Augusto, F. New Advances in Toxicological Forensic Analysis Using Mass Spectrometry Techniques. J. Anal. Methods Chem. 2018, 2018, 4142527. [Google Scholar] [CrossRef] [Green Version]
- Maurer, H.H. Mass Spectrometry for Research and Application in Therapeutic Drug Monitoring or Clinical and Forensic Toxicology. Ther. Drug Monit. 2018, 40, 389–393. [Google Scholar] [CrossRef]
- Wu, A.H.; Gerona, R.; Armenian, P.; French, D.; Petrie, M.; Lynch, K.L. Role of liquid chromatography–high-resolution mass spectrometry (LC-HR/MS) in clinical toxicology. Clin. Toxicol. 2012, 50, 733–742. [Google Scholar] [CrossRef]
- Substandard and Falsified Medical Products. Available online: https://www.who.int/news-room/fact-sheets/detail/substandard-and-falsified-medical-products (accessed on 25 April 2022).
- Janvier, S.; Cheyns, K.; Canfyn, M.; Goscinny, S.; De Spiegeleer, B.; Vanhee, C.; Deconinck, E. Impurity profiling of the most frequently encountered falsified polypeptide drugs on the Belgian market. Talanta 2018, 188, 795–807. [Google Scholar] [CrossRef]
- Goryainov, S.V.; Ivlev, V.A.; Orlova, S.V.; Nikitina, E.A.; Sheremeta, A.V.; Vasil’Ev, V.G.; Kalabin, G.A. Modern Methods for Identification and Quantitative Analysis of Undeclared Phosphodiesterase-5 Inhibitors as Pharmaceutically Active Substances in Dietary Supplements (Review). Pharm. Chem. J. 2022, 55, 1395–1400. [Google Scholar] [CrossRef]
- Lee, J.H.; Han, J.H.; Kim, S.; Kim, N.S.; Yoon, C.-Y.; Kim, J.; Baek, S.Y. Screening sexual performance enhancing compounds and their analogues in counterfeit and illicit erectile dysfunction drugs by high-performance liquid chromatography and liquid chromatography-tandem mass spectrometry. J. Forensic Leg. Med. 2021, 82, 102224. [Google Scholar] [CrossRef]
- Yusop, A.Y.M.; Xiao, L.; Fu, S. Liquid chromatography-high-resolution mass spectrometry analysis of erectile dysfunction drugs and their analogues in food products. Forensic Sci. Int. 2021, 322, 110748. [Google Scholar] [CrossRef] [PubMed]
- Qian, Q.; Li, S.-L.; Sun, E.; Zhang, K.-R.; Tan, X.-B.; Wei, Y.-J.; Fan, H.-W.; Cui, L.; Jia, X.-B. Metabolite profiles of icariin in rat plasma by ultra-fast liquid chromatography coupled to triple-quadrupole/time-of-flight mass spectrometry. J. Pharm. Biomed. Anal. 2012, 66, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, M.; Borioni, A.; Prestinaci, F.; Sestili, I.; Gaudiano, M.C. A NIR, 1H-NMR, LC-MS and chemometrics pilot study on the origin of carvedilol drug substances: A tool for discovering falsified active pharmaceutical ingredients. Anal. Methods 2022, 14, 1396–1405. [Google Scholar] [CrossRef] [PubMed]
- Falco, L.F.G.; Melo, C.F.O.R.; de Oliveira, D.N.; Guerreiro, T.M.; Catharino, R.R. An LDI-MSI approach for targeted and untargeted differentiation and assessment of pharmaceutical formulations. Talanta 2018, 197, 92–97. [Google Scholar] [CrossRef]
- Sisco, E.; Forbes, T.P. Forensic applications of DART-MS: A review of recent literature. Forensic Chem. 2020, 22, 100294. [Google Scholar] [CrossRef]
- Lehmann, E.L.; Arruda, M.A.Z. Minimalist strategies applied to analysis of forensic samples using elemental and molecular analytical techniques—A review. Anal. Chim. Acta 2019, 1063, 9–17. [Google Scholar] [CrossRef]
- dos Santos, M.K.; Gleco, E.; Davidson, J.T.; Jackson, G.P.; Limberger, R.P.; Arroyo, L.E. DART-MS/MS screening for the determination of 1,3-dimethylamylamine and undeclared stimulants in seized dietary supplements from Brazil. Forensic Chem. 2018, 8, 134–145. [Google Scholar] [CrossRef]
- Fabresse, N.; Gheddar, L.; Kintz, P.; Knapp, A.; Larabi, I.A.; Alvarez, J.-C. Analysis of pharmaceutical products and dietary supplements seized from the black market among bodybuilders. Forensic Sci. Int. 2021, 322, 110771. [Google Scholar] [CrossRef]
- Smit, D.L.; Hon, O.; Venhuis, B.J.; Heijer, M.D.; Ronde, W. Baseline characteristics of the HAARLEM study: 100 male amateur athletes using anabolic androgenic steroids. Scand. J. Med. Sci. Sports 2019, 30, 531–539. [Google Scholar] [CrossRef]
- Kaklamanos, G.; Aprea, E.; Theodoridis, G. Mass spectrometry: Principles and instrumentation. In Chemical Analysis of Food. Techniques and Applications, 2nd ed.; Pico, Y., Ed.; Academic Press: London, UK, 2020; pp. 525–552. ISBN 9780128132661. [Google Scholar] [CrossRef]
- Aceto, M. Food Forensics. In Comprehensive Analytical Chemistry; Elsevier: Amsterdam, The Netherlands, 2015; Volume 68, pp. 441–514. ISBN 9780444633408. [Google Scholar] [CrossRef]
- Rodrigues, C.; Maia, R.; Lauteri, M.; Brugnoli, E.; Máguas, C. Stable Isotope Analysis. Compr. Anal. Chem. 2013, 60, 77–99. [Google Scholar] [CrossRef]
- Muccio, Z.; Jackson, G.P. Isotope ratio mass spectrometry. Analyst 2008, 134, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Tian, L.; Chen, B.; Jin, B.; Tian, B.; Xie, L.; Rogers, K.; Lin, G. Verification of imported red wine origin into China using multi isotope and elemental analyses. Food Chem. 2019, 301, 125137. [Google Scholar] [CrossRef] [PubMed]
- Voica, C.; Iordache, A.M.; Miricioiu, M.G.; Magdas, D.A.; Cristea, G. Preliminary Study on Characterization of Edible Oils Using Inductively Coupled Plasma Mass Spectrometry (ICP-MS) and Isotope Ratio Mass Spectrometry (IRMS). Rev. Chim. 2020, 71, 283–289. [Google Scholar] [CrossRef]
- Wadood, S.A.; Boli, G.; Yimin, W. Geographical traceability of wheat and its products using multielement light stable isotopes coupled with chemometrics. Biol. Mass Spectrom. 2018, 54, 178–188. [Google Scholar] [CrossRef]
- Jin, X.; Zhang, L.; Wu, S.; Huang, M.; Yu, W.; Zhang, S. Developing an authentication approach using SPME-GC-IRMS based on compound-specific δ 13C analysis of six typical volatiles in wine. Food Qual. Saf. 2020, 5, fyaa031. [Google Scholar] [CrossRef]
- Ciepielowski, G.; Pacholczyk-Sienicka, B.; Frączek, T.; Klajman, K.; Paneth, P.; Albrecht, Ł. Comparison of quantitative NMR and IRMS for the authentication of ‘Polish Vodka’. J. Sci. Food Agric. 2018, 99, 263–268. [Google Scholar] [CrossRef] [Green Version]
- Brailsford, A.D.; Majidin, W.N.M.; Wojek, N.; Cowan, D.A.; Walker, C. IRMS delta values (13C) of nandrolone and testosterone products available in the UK: Implications for anti-doping. Drug Test. Anal. 2018, 10, 1722–1727. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Lee, D.-H.; Go, A.; Park, M.; Choe, S.; In, S.; Kim, E.; Lee, H.; Shin, K.-H.; Han, E. Differentiation of endogenous and exogenous γ-Hydroxybutyrate in rat and human urine by GC/C/IRMS. Int. J. Leg. Med. 2019, 133, 1785–1794. [Google Scholar] [CrossRef]
- Benson, S.J.; Lennard, C.J.; Maynard, P.; Hill, D.M.; Andrew, A.S.; Roux, C. Forensic analysis of explosives using isotope ratio mass spectrometry (IRMS)—Preliminary study on TATP and PETN. Sci. Justice 2009, 49, 81–86. [Google Scholar] [CrossRef]
- Bontempo, L.; Paolini, M.; Franceschi, P.; Ziller, L.; García-González, D.L.; Camin, F. Characterisation and attempted differentiation of European and extra-European olive oils using stable isotope ratio analysis. Food Chem. 2018, 276, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Circi, S.; Ingallina, C.; Vista, S.; Capitani, D.; Di Vecchia, A.; Leonardi, G.; D’Achille, G.; Centauri, L.; Camin, F.; Mannina, L. A Multi-Methodological Protocol to Characterize PDO Olive Oils. Metabolites 2018, 8, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinkovič, L.; Ogrinc, N.; Potočnik, D.; Meglič, V. Isotope Fingerprints of Common and Tartary Buckwheat Grains and Milling Fractions: A Preliminary Study. Foods 2022, 11, 1414. [Google Scholar] [CrossRef] [PubMed]
- Ishida-Fujii, K.; Goto, S.; Uemura, R.; Yamada, K.; Sato, M.; Yoshida, N. Botanical and Geographical Origin Identification of Industrial Ethanol by Stable Isotope Analyses of C, H, and O. Biosci. Biotechnol. Biochem. 2005, 69, 2193–2199. [Google Scholar] [CrossRef] [Green Version]
- Iannella, L.; Botrè, F.; Colamonici, C.; Curcio, D.; De La Torre, X. Development and validation of a method to confirm the exogenous origin of prednisone and prednisolone by GC-C-IRMS. Drug Test. Anal. 2019, 11, 1615–1628. [Google Scholar] [CrossRef]
- Tobias, H.J.; Brenna, J.T. Cryofocus fast gas chromatography combustion isotope ratio mass spectrometry for rapid detection of synthetic steroid use in sport doping. Analyst 2018, 143, 1124–1132. [Google Scholar] [CrossRef]
- Howa, J.D.; Barnette, J.E.; Chesson, L.A.; Lott, M.J.; Ehleringer, J.R. TATP isotope ratios as influenced by worldwide acetone variation. Talanta 2018, 181, 125–131. [Google Scholar] [CrossRef]
- Chesson, L.A.; Berg, G.E. The use of stable isotopes in postconflict forensic identification. WIREs Forensic Sci. 2021, 4, 1–28. [Google Scholar] [CrossRef]
- Frederick, D.L. Toxicology Testing in Alternative Specimen Matrices. Clin. Lab. Med. 2012, 32, 467–492. [Google Scholar] [CrossRef]
- Barbosa, J.; Faria, J.; Carvalho, F.; Pedro, M.; Queirós, O.; Moreira, R.; Dinis-Oliveira, R.J. Hair as an alternative matrix in bioanalysis. Bioanalysis 2013, 5, 895–914. [Google Scholar] [CrossRef]
- Gallardo, E.; Queiroz, J.A. The role of alternative specimens in toxicological analysis. Biomed. Chromatogr. 2008, 22, 795–821. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, A.G.; Christodoulou, M.C.; Stavrou, I.J.; Kapnissi-Christodoulou, C.P. Analysis of cannabinoids in conventional and alternative biological matrices by liquid chromatography: Applications and challenges. J. Chromatogr. A 2021, 1651, 462277. [Google Scholar] [CrossRef] [PubMed]
- Pirone, J. Drug Testing: An Overview of SAMHSA’s New Oral Fluid Testing Guidelines. Occupational Health & Safety Magazine. Available online: https://ohsonline.com/Articles/2020/03/01/An-Overview-of-SAMHSAs-New-Oral-Fluid-Testing-Guidelines.aspx?Page=1 (accessed on 15 June 2022).
- Ferreira, C.; Paulino, C.; Quintas, A. Extraction Procedures for Hair Forensic Toxicological Analysis: A Mini-Review. Chem. Res. Toxicol. 2019, 32, 2367–2381. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wang, J.F.; Wang, Y. Influence of cosmetic hair treatments on hair of methamphetamine abuser: Bleaching, perming and coloring. Ecotoxicol. Environ. Saf. 2021, 222, 112542. [Google Scholar] [CrossRef]
- Agius, R. Utility of coloured hair for the detection of drugs and alcohol. Drug Test. Anal. 2014, 6 (Suppl. 1), 110–119. [Google Scholar] [CrossRef] [Green Version]
- Sporkert, F.; Kharbouche, H.; Augsburger, M.P.; Klemm, C.; Baumgartner, M.R. Positive EtG findings in hair as a result of a cosmetic treatment. Forensic Sci. Int. 2012, 218, 97–100. [Google Scholar] [CrossRef]
- Kintz, P.; Gheddar, L.; Raul, J. Simultaneous testing for anabolic steroids in human hair specimens collected from various anatomic locations has several advantages when compared with the standard head hair analysis. Drug Test. Anal. 2021, 13, 1445–1451. [Google Scholar] [CrossRef]
- Fernández, M.D.M.R.; Wille, S.M.; Di Fazio, V.; Samyn, N. Time course detection of dihydrocodeine in body hair after a single dose. Forensic Sci. Int. 2019, 302, 109864. [Google Scholar] [CrossRef]
- Cooper, G.A.; Kronstrand, R.; Kintz, P. Society of Hair Testing guidelines for drug testing in hair. Forensic Sci. Int. 2012, 218, 20–24. [Google Scholar] [CrossRef]
- Available online: http://www.ewdts.org/ewdts-guidelines.html (accessed on 25 May 2022).
- Wang, T.; Shen, B.; Wu, H.; Gu, J.; Shen, M.; Xiang, P. Segmental Analysis of R/S-Methamphetamine and R/S-Amphetamine in Abusers’ Head Hair. J. Anal. Toxicol. 2020, 44, 596–600. [Google Scholar] [CrossRef]
- Busardò, F.P.; Pichini, S.; Zaami, S.; Pacifici, R.; Kintz, P. Hair testing of GHB: An everlasting issue in forensic toxicology. Clin. Chem. Lab. Med. 2017, 56, 198–208. [Google Scholar] [CrossRef]
- Kong, R.; Li, L.; Liu, W.; Xiang, P.; Zhao, J. Rapid Characterization of Drugs in a Single Hair using Thermal Desorption Ionization Mass Spectrometry. Anal. Methods 2022, 14, 806–812. [Google Scholar] [CrossRef] [PubMed]
- D’Ovidio, C.; Bonelli, M.; Rosato, E.; Tartaglia, A.; Ulusoy, H.I.; Samanidou, V.; Furton, K.G.; Kabir, A.; Ali, I.; Savini, F.; et al. Novel Applications of Microextraction Techniques Focused on Biological and Forensic Analyses. Separations 2022, 9, 18. [Google Scholar] [CrossRef]
- Kernalléguen, A.; Enjalbal, C.; Alvarez, J.-C.; Belgacem, O.; Léonetti, G.; Lafitte, D.; Pélissier-Alicot, A.-L. Synthetic cannabinoid isomers characterization by MALDI-MS3 imaging: Application to single scalp hair. Anal. Chim. Acta 2018, 1041, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Duvivier, W.F.; van Beek, T.A.; Nielen, M.W.F. Critical comparison of mass analyzers for forensic hair analysis by ambient ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2016, 30, 2331–2340. [Google Scholar] [CrossRef]
- Society of Toxicological and Forensic Chemistry (GTFCh). Available online: https://www.gtfch.org/cms/index.php/en/ (accessed on 14 May 2022).
- Wabuyele, S.L.; Colby, J.M.; McMillin, G.A. Detection of Drug-Exposed Newborns. Ther. Drug Monit. 2018, 40, 166–185. [Google Scholar] [CrossRef]
- Szeto, H.H. Kinetics of Drug Transfer to the Fetus. Clin. Obstet. Gynecol. 1993, 36, 246–254. [Google Scholar] [CrossRef]
- Joya, X.; Marchei, E.; Salat-Batlle, J.; García-Algar, O.; Calvaresi, V.; Pacifici, R.; Pichini, S. Fetal exposure to ethanol: Relationship between ethyl glucuronide in maternal hair during pregnancy and ethyl glucuronide in neonatal meconium. Clin. Chem. Lab. Med. 2016, 54, 427–435. [Google Scholar] [CrossRef]
- McMillin, G.A.; Wood, K.E.; Strathmann, F.G.; Krasowski, M.D. Patterns of Drugs and Drug Metabolites Observed in Meconium. Ther. Drug Monit. 2015, 37, 568–580. [Google Scholar] [CrossRef]
- Bévalot, F.; Gustin, M.P.; Cartiser, N.; Gaillard, Y.; Le Meur, C.; Fanton, L.; Guitton, J.; Malicier, D. Using bone marrow matrix to analyze meprobamate for forensic toxicological purposes. Int. J. Leg. Med. 2013, 127, 915–921. [Google Scholar] [CrossRef]
- Petrochilou, E.; Athanaselis, S.; Nikolaou, P.; Papadodima, S.; Panderi, I.; Spiliopoulou, C.; Papoutsis, I. Synovial fluid as an alternative specimen for quantification of drugs of abuse by GC–MS. Forensic Toxicol. 2019, 37, 496–503. [Google Scholar] [CrossRef]
- Badmus, S.O.; Amusa, H.K.; Oyehan, T.A.; A Saleh, T. Environmental risks and toxicity of surfactants: Overview of analysis, assessment, and remediation techniques. Environ. Sci. Pollut. Res. 2021, 28, 62085–62104. [Google Scholar] [CrossRef]
- Sasi, S.; Rayaroth, M.P.; Aravindakumar, C.T.; Aravind, U.K. Alcohol ethoxysulfates (AES) in environmental matrices. Environ. Sci. Pollut. Res. Int. 2021, 28, 34167–34186. [Google Scholar] [CrossRef] [PubMed]
- Lasek, F.; Leitner, N.K.V.; Rauwel, G.; Blanchier, L.; Castel, O.; Ayraud-Thevenot, S.; Deborde, M. Discharge of biocidal products from healthcare activities into a sewage system—A case study at a French university hospital. Environ. Sci. Pollut. Res. Int. 2018, 26, 4938–4951. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Lu, H.; Zhu, L. Molecular mechanism of antibiotic resistance induced by mono-and twin-chained quaternary ammonium compounds. Sci. Total Environ. 2022, 832, 155090. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, L.Y.; Pashirova, T.N.; Doktorovova, S.; Fernandes, A.R.; Sanchez-Lopez, E.; Silva, A.M.; Souto, S.B.; Souto, E.B. Cationic Surfactants: Self-Assembly, Structure-Activity Correlation and Their Biological Applications. Int. J. Mol. Sci. 2019, 20, 5534. [Google Scholar] [CrossRef] [Green Version]
- Kawamoto, O.; Ishikawa, T.; Oritani, S.; Kuramoto, Y.; Michiue, T.; Maeda, H. Death following the ingestion of detergent: An autopsy case with special regard to the histochemical findings. Forensic Sci. Med. Pathol. 2012, 9, 208–213. [Google Scholar] [CrossRef]
- Kumar, M.; Bhatia, R.; Rawal, R.K. Applications of various analytical techniques in quality control of pharmaceutical excipients. J. Pharm. Biomed. Anal. 2018, 157, 122–136. [Google Scholar] [CrossRef]
- Bazel, Y.R.; Antal, I.P.; Lavra, V.M.; Kormosh, Z.A.; Bazel, Y. Methods for the determination of anionic surfactants. J. Anal. Chem. 2014, 69, 211–236. [Google Scholar] [CrossRef]
- Heinig, K.; Vogt, C.; Werner, G. Separation of ionic and neutral surfactants by capillary electrophoresis and high-performance liquid chromatography. J. Chromatogr. A 1996, 745, 281–292. [Google Scholar] [CrossRef]
- Villar, M.; Callejón, M.; Jiménez, J.; Alonso, E.; Guiraúm, A. New rapid methods for determination of total LAS in sewage sludge by high performance liquid chromatography (HPLC) and capillary electrophoresis (CE). Anal. Chim. Acta 2009, 634, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Maurer, H.H. Current role of liquid chromatography–mass spectrometry in clinical and forensic toxicology. Anal. Bioanal. Chem. 2007, 388, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Katagi, M.; Miki, A.; Tsuchihashi, H. Forensic Toxicological Determination of Surfactant by Liquid Chromatography/Electrospray lonization Mass Spectrometry and Liquid Chromatography/Electrospray lonization Tandem Mass Spectrometry. J. Health Sci. 2003, 49, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Kudo, K.; Tsuchihashi, H.; Ikeda, N. Meeting challenges in forensic toxicology in Japan by liquid chromatography/mass spectrometry. Anal. Chim. Acta 2003, 492, 83–104. [Google Scholar] [CrossRef]
- Bongiorno, D.; Ceraulo, L.; Ruggirello, A.; Liveri, V.T.; Basso, E.; Seraglia, R.; Traldi, P. Surfactant self-assembling in gas phase: Electrospray ionization- and matrix-assisted laser desorption/ionization-mass spectrometry of singly charged AOT clusters. Biol. Mass Spectrom. 2005, 40, 1618–1625. [Google Scholar] [CrossRef]
- Ceraulo, L.; Giorgi, G.; Liveri, V.T.; Bongiorno, D.; Indelicato, S.; Di Gaudio, F.; Indelicato, S. Mass Spectrometry of Surfactant Aggregates. Eur. J. Mass Spectrom. 2011, 17, 525–541. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Zhang, Y.; Zhai, J.; Chen, X.; Du, Z.; Li, W.; Bai, H. Characterization and analysis of non-ionic surfactants by supercritical fluid chromatography combined with ion mobility spectrometry-mass spectrometry. Anal. Bioanal. Chem. 2019, 411, 2759–2765. [Google Scholar] [CrossRef]
- Pan, J.; Ji, Y.; Du, Z.; Zhang, J. Rapid characterization of commercial polysorbate 80 by ultra-high performance supercritical fluid chromatography combined with quadrupole time-of-flight mass spectrometry. J. Chromatogr. A 2016, 1465, 190–196. [Google Scholar] [CrossRef]
- Hitosugi, M.; Tsukada, C.; Yamauchi, S.; Matsushima, K.; Furukawa, S.; Morita, S.; Nagai, T. An autopsy case of fatal repellent air freshener poisoning. Leg. Med. 2015, 17, 360–363. [Google Scholar] [CrossRef]
- Usui, K.; Hayashizaki, Y.; Okubo, Y.; Hashiyada, M.; Funayama, M. A case of death after ingestion of an agrochemical spreading agent. Forensic Sci. Int. 2011, 210, 117–121. [Google Scholar] [CrossRef]
- Tay, M.; Fang, G.; Chia, P.L.; Li, S.F.Y. Rapid screening for detection and differentiation of detergent powder adulteration in infant milk formula by LC–MS. Forensic Sci. Int. 2013, 232, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Kuepper, U.; Musshoff, F.; Madea, B. A fully validated isotope dilution HPLC-MS/MS method for the simultaneous determination of succinylcholine and succinylmonocholine in serum and urine samples. Biol. Mass Spectrom. 2008, 43, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Kirschbaum, K.M.; Grellner, W.; Rochholz, G.; Musshoff, F.; Madea, B. Liquid chromatography-tandem mass spectrometry detection of the quaternary ammonium compound mebezonium as an active ingredient in t61. J. Anal. Toxicol. 2011, 35, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massey, K.A.; Snelling, A.M.; Nicolaou, A. Quantitative analysis of surfactant deposits on human skin by liquid chromatography/electrospray ionisation tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2010, 24, 1371–1376. [Google Scholar] [CrossRef]
- Onzo, A.; Acquavia, M.A.; Cataldi, T.R.I.; Ligonzo, M.; Coviello, D.; Pascale, R.; Martelli, G.; Bondoni, M.; Scrano, L.; Bianco, G. Coceth sulfate characterization by electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2020, 34, e8884. [Google Scholar] [CrossRef]
- Zecchi, R.; Franceschi, P.; Tigli, L.; Ricci, F.; Boscaro, F.; Pioselli, B.; Mileo, V.; Murgia, X.; Bianco, F.; Salomone, F.; et al. Mass spectrometry imaging as a tool for evaluating the pulmonary distribution of exogenous surfactant in premature lambs. Respir. Res. 2019, 20, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hird, S.J.; Lau, B.P.-Y.; Schuhmacher, R.; Krska, R. Liquid chromatography-mass spectrometry for the determination of chemical contaminants in food. TrAC Trends Anal. Chem. 2014, 59, 59–72. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.-Y.; Li, S.; Zhu, Q.-F.; Wang, Q.; Hussain, D.; Feng, Y.-Q. Derivatization for liquid chromatography-electrospray ionization-mass spectrometry analysis of small-molecular weight compounds. TrAC Trends Anal. Chem. 2019, 119, 115608. [Google Scholar] [CrossRef]
- Kamińska, A.; Krawczyk, M.; Chwatko, G. Chemical derivatization in high-performance liquid chromatography. Wiad. Chem. 2016, 70, 833–834. [Google Scholar]
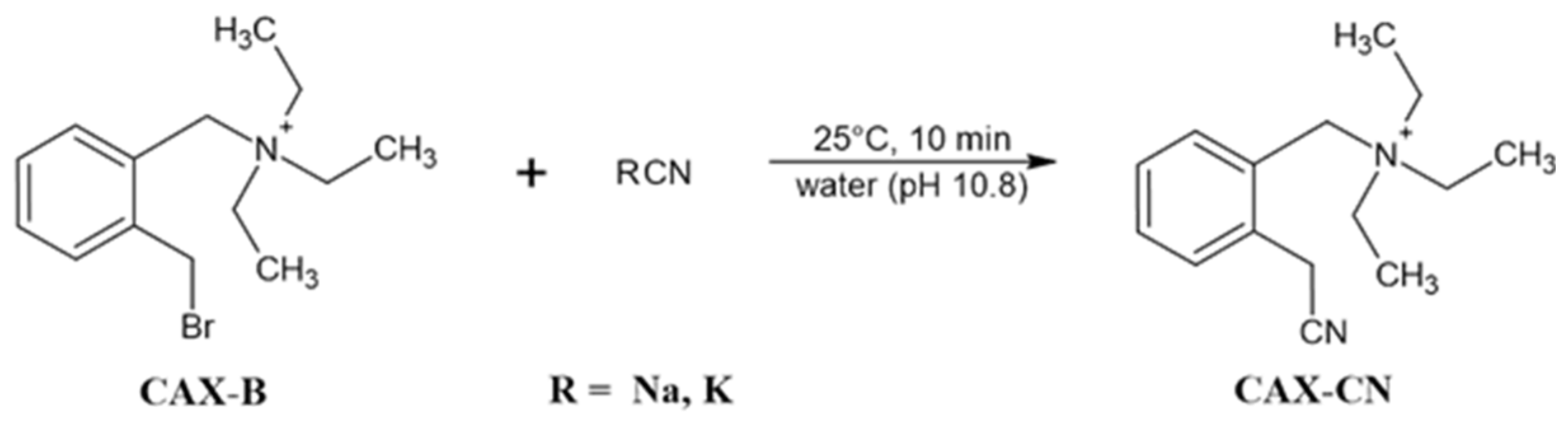
- Madmon, M.; Shifrovich, A.; Tamar, S.Y.; Weissberg, A. Simple and fast determination of free cyanide in drinking water by liquid chromatography electrospray ionization tandem mass spectrometry following “in vial” derivatization. Int. J. Mass Spectrom. 2021, 463, 116553. [Google Scholar] [CrossRef]
- Vaiano, F.; Mari, F.; Busardò, F.P.; Bertol, E. Enhancing the sensitivity of the LC-MS/MS detection of propofol in urine and blood by azo-coupling derivatization. Anal. Bioanal. Chem. 2014, 406, 3579–3587. [Google Scholar] [CrossRef] [PubMed]
- Vaiano, F.; Busardò, F.P.; Pascali, J.; Fioravanti, A.; Mortali, C.; Mari, F.; Bertol, E. Hair testing of propofol by liquid chromatography-tandem mass spectrometry and azo-coupling derivatization. Drug Test. Anal. 2017, 9, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Tang, H.; Wang, Y. Progress and Challenges in Quantifying Carbonyl-Metabolomic Phenomes with LC-MS/MS. Molecules 2021, 26, 6147. [Google Scholar] [CrossRef] [PubMed]
- Athanasiadou, I.; Angelis, Y.S.; Lyris, E.; Georgakopoulos, C. Chemical derivatization to enhance ionization of anabolic steroids in LC-MS for doping-control analysis. TrAC Trends Anal. Chem. 2013, 42, 137–156. [Google Scholar] [CrossRef]
- Kowalska, M.; Popiel, D.; Walter, M.; Bąchor, R.; Biernat, M.; Cebrat, M.; Kijewska, M.; Kuczer, M.; Modzel, M.; Kluczyk, A. Veni, Vidi, Vici: Immobilized Peptide-Based Conjugates as Tools for Capture, Analysis, and Transformation. Chemosensors 2022, 10, 31. [Google Scholar] [CrossRef]
- Aviram, L.Y.; Loewenthal, D.; Weissberg, A.; Marder, D.; Gura, S.; Chapman, S.; Gez, R.; Lazar, S.; Dagan, S. Determination of free G-type nerve agents in blood: In situ derivatization on a dried blood spot (DBS) paper followed by LC–MS/MS analysis. Forensic Toxicol. 2020, 38, 327–339. [Google Scholar] [CrossRef]
- Weissberg, A.; Madmon, M.; Elgarisi, M.; Dagan, S. Aqueous extraction followed by derivatization and liquid chromatography–mass spectrometry analysis: A unique strategy for trace detection and identification of G-nerve agents in environmental matrices. J. Chromatogr. A 2018, 1577, 24–30. [Google Scholar] [CrossRef]
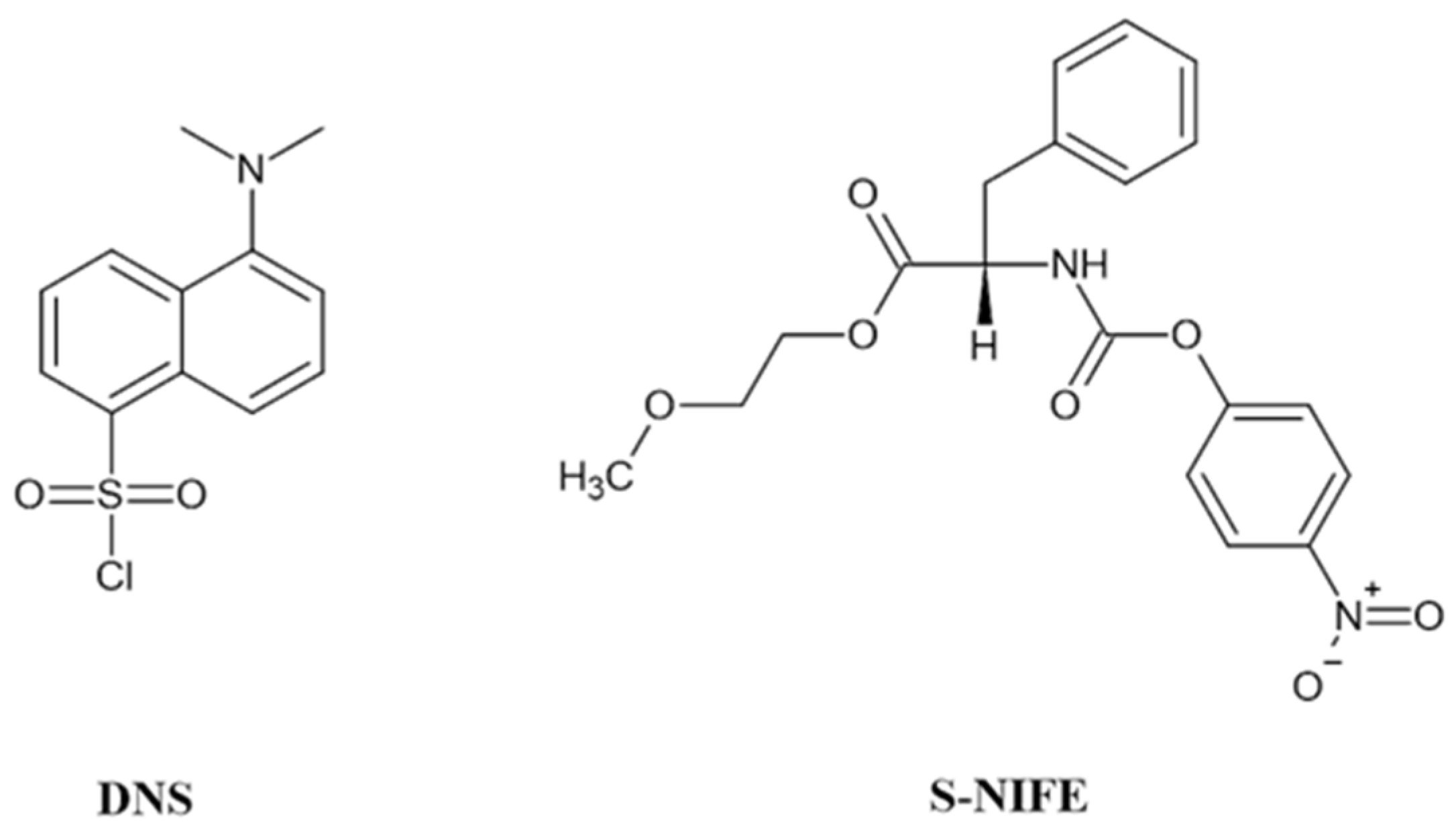
- Beach, D.G.; Liu, H.; Quilliam, M.A. Sensitive determination of domoic acid in mussel tissue using dansyl chloride derivatization and liquid chromatography-mass spectrometry. Anal. Methods 2014, 7, 1000–1007. [Google Scholar] [CrossRef] [Green Version]
- Patočka, J.; Jelínková, R. Atropine and atropine-like substances usable in warfare. Mil. Med Sci. Lett. 2017, 86, 58–69. [Google Scholar] [CrossRef]
- World Health Organization Model List of Essential Medicines—22nd List; Licence: CC BY-NC-SA 3.0 IGO; World Health Organization: Geneva, Switzerland, 2021.
- Carlier, J.; Escard, E.; Peoc’H, M.; Boyer, B.; Romeuf, L.; Faict, T.; Guitton, J.; Gaillard, Y. Atropine Eye Drops: An Unusual Homicidal Poisoning. J. Forensic Sci. 2014, 59, 859–864. [Google Scholar] [CrossRef]
- Le Garff, E.; Delannoy, Y.; Mesli, V.; Hédouin, V.; Tournel, G. Forensic features of a fatal Datura poisoning case during a robbery. Forensic Sci. Int. 2016, 261, e17–e21. [Google Scholar] [CrossRef] [PubMed]
- Mutebi, R.R.; Ario, A.R.; Nabatanzi, M.; Kyamwine, I.B.; Wibabara, Y.; Muwereza, P.; Eurien, D.; Kwesiga, B.; Bulage, L.; Kabwama, S.N.; et al. Large outbreak of Jimsonweed (Datura stramonium) poisoning due to consumption of contaminated humanitarian relief food: Uganda, March–April 2019. BMC Public Health 2022, 22, 623. [Google Scholar] [CrossRef] [PubMed]
- Skulska, A.; Kała, M. Consequences of medical and non-medical use of atropine and fentanyl analogues. Probl. Forensic Sci. 2007, LXXI, 303–331. [Google Scholar]
- National Report to the EMCDDA: France. New Development, Trends and In-Depth Information on Selected Issues. Available online: https://www.emcdda.europa.eu/system/files/publications/958/NR2005FranceEN.pdf (accessed on 8 May 2022).
- Koželj, G.; Perharič, L.; Stanovnik, L.; Prosen, H. Simple validated LC–MS/MS method for the determination of atropine and scopolamine in plasma for clinical and forensic toxicological purposes. J. Pharm. Biomed. Anal. 2014, 96, 197–206. [Google Scholar] [CrossRef]
- Moriyama, K.; Takami, Y.; Uozumi, N.; Okuda, A.; Yamashita, M.; Yokomizo, R.; Shimada, K.; Egawa, T.; Kamei, T.; Takayanagi, K. Assessment of drug content uniformity of atropine sulfate triturate by liquid chromatography-tandem mass spectrometry, X-ray powder diffraction, and Raman chemical imaging. J. Pharm. Health Care Sci. 2016, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Crapnell, R.D.; Banks, C.E. Electroanalytical overview: The detection of the molecule of murder atropine. Talanta Open 2021, 4, 100073. [Google Scholar] [CrossRef]
- Rbeida, O.; Christiaens, B.; Hubert, P.; Lubda, D.; Boos, K.-S.; Crommen, J.; Chiap, P. Integrated on-line sample clean-up using cation exchange restricted access sorbent for the LC determination of atropine in human plasma coupled to UV detection. J. Pharm. Biomed. Anal. 2005, 36, 947–954. [Google Scholar] [CrossRef]
- Namera, A.; Yashiki, M.; Hirose, Y.; Yamaji, S.; Tani, T.; Kojima, T. Quantitative analysis of tropane alkaloids in biological materials by gas chromatography–mass spectrometry. Forensic Sci. Int. 2002, 130, 34–43. [Google Scholar] [CrossRef]
- Aehle, E.; Dräger, B. Tropane alkaloid analysis by chromatographic and electrophoretic techniques: An update. J. Chromatogr. B 2010, 878, 1391–1406. [Google Scholar] [CrossRef] [PubMed]
- Koželj, G.; Prosen, H. Thermal (In) stability of Atropine and Scopolamine in the GC-MS Inlet. Toxics 2021, 9, 156. [Google Scholar] [CrossRef]
- Kuczer, M.; Kluczyk, A.; Bąchor, R.; Szewczuk, Z. Derywatyzacja jako metoda zwiększania czułości pomiarów w chromatografii cieczowej sprzężonej ze spektrometrią mas. In Proceedings of the IV Ogólnopolska Konferencja Fizykochemiczne Badania Śladów Kryminalistycznych, Bronisławów, Poland, 20–23 November 2018. [Google Scholar]
- Setner, B.; Rudowska, M.; Klem, E.; Cebrat, M.; Szewczuk, Z. Peptides derivatized with bicyclic quaternary ammonium ionization tags. Sequencing via tandem mass spectrometry. Biol. Mass Spectrom. 2014, 49, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Bąchor, R.; Mielczarek, P.; Rudowska, M.; Silberring, J.; Szewczuk, Z. Sensitive detection of charge derivatized peptides at the attomole level using nano-LC-ESI–MRM analysis. Int. J. Mass Spectrom. 2014, 362, 32–38. [Google Scholar] [CrossRef]
- Kim, S.; Lee, M.S.; Kim, M.; Ko, B.J.; Lee, H.S.; Lee, S. Derivatization-assisted LC-MS/MS method for simultaneous quantification of endogenous gamma-hydroxybutyric acid and its metabolic precursors and products in human urine. Anal. Chim. Acta 2021, 1194, 339401. [Google Scholar] [CrossRef] [PubMed]
- Hess, C.; Murach, J.; Krueger, L.; Scharrenbroch, L.; Unger, M.; Madea, B.; Sydow, K. Simultaneous detection of 93 synthetic cannabinoids by liquid chromatography-tandem mass spectrometry and retrospective application to real forensic samples. Drug Test. Anal. 2016, 9, 721–733. [Google Scholar] [CrossRef]
- Al-Zahrani, M.A.; Al-Asmari, A.I.; Al-Zahrani, F.F.; Torrance, H.J.; Watson, D.G. Quantification of cannabinoids in human hair using a modified derivatization procedure and liquid chromatography–tandem mass spectrometry. Drug Test. Anal. 2021, 13, 1095–1107. [Google Scholar] [CrossRef]
- Tsumura, Y.; Kiguchi, A.; Komatsuzaki, S.; Ieuji, K. A novel method to distinguish β-methylphenylethylamines from isomeric α-methylphenylethylamines by liquid chromatography coupled to electrospray ionization mass spectrometry. Forensic Toxicol. 2019, 38, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Anselmo, C.D.S.; Silveira, C.M.D.V.; Sardela, V.F.; Pereira, H.M.G. Chiral Analysis of Amphetamine and Methamphetamine in Urine by Liquid Chromatography-Tandem Mass Spectrometry Applying Mosher Derivatization. Chromatographia 2020, 84, 47–52. [Google Scholar] [CrossRef]
- Borden, S.A.; Palaty, J.; Termopoli, V.; Famiglini, G.; Cappiello, A.; Gill, C.G.; Palma, P. Mass Spectrometry Analysis of Drugs of Abuse: Challenges and Emerging Strategies. Mass Spectrom. Rev. 2020, 37, 258–280. [Google Scholar] [CrossRef]
- Englander, S.W.; Sosnick, T.R.; Englander, J.J.; Mayne, L. Mechanisms and uses of hydrogen exchange. Curr. Opin. Struct. Biol. 1996, 6, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Milne, J.S.; Mayne, L.; Englander, S.W. Primary structure effects on peptide group hydrogen exchange. Proteins Struct. Funct. Bioinform. 1993, 17, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Junk, T.; Catallo, W.J. Hydrogen isotope exchange reactions involving C–H (D, T) bonds. Chem. Soc. Rev. 1997, 26, 401–406. [Google Scholar] [CrossRef]
- Ho, J.; Coote, M.L.; Easton, C.J. Validation of the Distal Effect of Electron-Withdrawing Groups on the Stability of Peptide Enolates and Its Exploitation in the Controlled Stereochemical Inversion of Amino Acid Derivatives. J. Org. Chem. 2011, 76, 5907–5914. [Google Scholar] [CrossRef] [PubMed]
- Lygo, B.; Humphreys, L.D. Enantioselective synthesis of α-carbon deuterium-labelled l-α-amino acids. Tetrahedron Lett. 2002, 43, 6677–6679. [Google Scholar] [CrossRef]
- Lankiewicz, L.; Nyassé, B.; Fransson, B.; Grehn, L.; Ragnarsson, U. Synthesis of amino acid derivatives substituted in the backbone with stable isotopes for application in peptide synthesis. J. Chem. Soc. Perkin Trans. 1 1994, 17, 2503–2510. [Google Scholar] [CrossRef]
- Rose, J.E.; Leeson, P.D.; Gani, D. Stereospecific synthesis of α-deuteriated α-amino acids: Regiospecific deuteriation of chiral 3-isopropyl-2,5-dimethoxy-3,6-dihydropyrazines. J. Chem. Soc. Perkin Trans. 1 1995, 2, 1563–1565. [Google Scholar] [CrossRef]
- Jung, P.G.; Kim, B.; Park, S.-R.; So, H.-Y.; Shi, L.H.; Kim, Y. Determination of serum cortisol using isotope dilution?liquid chromatography?mass spectrometry as a candidate reference method. Anal. Bioanal. Chem. 2004, 380, 782–788. [Google Scholar] [CrossRef]
- Gries, W.; Küpper, K.; Leng, G. Rapid and sensitive LC–MS–MS determination of 2-mercaptobenzothiazole, a rubber additive, in human urine. Anal. Bioanal. Chem. 2015, 407, 3417–3423. [Google Scholar] [CrossRef]
- Brun, V.; Masselon, C.; Garin, J.; Dupuis, A. Isotope dilution strategies for absolute quantitative proteomics. J. Proteom. 2009, 72, 740–749. [Google Scholar] [CrossRef]
- Guo, K.; Ji, C.; Li, L. Stable-Isotope Dimethylation Labeling Combined with LC−ESI MS for Quantification of Amine-Containing Metabolites in Biological Samples. Anal. Chem. 2007, 79, 8631–8638. [Google Scholar] [CrossRef]
- Di Palma, S.; Raijmakers, R.; Heck, A.J.R.; Mohammed, S. Evaluation of the Deuterium Isotope Effect in Zwitterionic Hydrophilic Interaction Liquid Chromatography Separations for Implementation in a Quantitative Proteomic Approach. Anal. Chem. 2011, 83, 8352–8356. [Google Scholar] [CrossRef]
- Turowski, M.; Yamakawa, N.; Meller, J.; Kimata, K.; Ikegami, T.; Hosoya, K.; Tanaka, N.; Thornton, E.R. Deuterium Isotope Effects on Hydrophobic Interactions: The Importance of Dispersion Interactions in the Hydrophobic Phase. J. Am. Chem. Soc. 2003, 125, 13836–13849. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; Zhang, Z.-P.; Kellogg, G.; Karnes, H.T. Evaluation of Deuterium Isotope Effects in Normal-Phase LC-MS-MS Separations Using a Molecular Modeling Approach. J. Chromatogr. Sci. 2004, 42, 383–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogusz, M.J.; Maier, R.-D.; Driessen, S. Morphine, Morphine-3-Glucuronide, Morphine-6-Glucuronide, and 6-Monoacetylmorphine Determined by Means of Atmospheric Pressure Chemical Ionization-Mass Spectrometry-Liquid Chromatography in Body Fluids of Heroin Victims. J. Anal. Toxicol. 1997, 21, 346–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogusz, M.J.; Maier, R.-D.; Erkens, M.; Driessen, S. Determination of morphine and its 3-and 6-glucuronides, codeine, codeine-glucuronide and 6-monoacetylmorphine in body fluids by liquid chromatography atmospheric pressure chemical ionization mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 1997, 703, 115–127. [Google Scholar] [CrossRef]
- Tai, S.S.-C.; Christensen, R.; Paule, R.; Sander, L.; Welch, M. The Certification of Morphine and Codeine in a Human Urine Standard Reference Material. J. Anal. Toxicol. 1994, 18, 7–12. [Google Scholar] [CrossRef]
- Slawson, M.H.; Crouch, D.J.; Andrenyak, D.M.; Rollins, U.E.; Lu, J.K.; Bailey, P.L. Determination of morphine, morphine-3-glucuronide, and morphine-6-glucuronide in plasma after intravenous and intrathecal morphine administration using HPLC with electrospray ionization and tandem mass spectrometry. J. Anal. Toxicol. 1999, 23, 468–473. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Bartlett, M.G. Collision-induced dissociation mass spectra of cocaine, and its metabolites and pyrolysis products. Biol. Mass Spectrom. 1998, 33, 961–967. [Google Scholar] [CrossRef]
- Sosnoff, C.S.; Ann, Q.; Bernert, J.T.; Powell, M.K.; Miller, B.B.; Henderson, L.O.; Hannon, W.H.; Fernhoff, P.; Sampson, E.J. Analysis of Benzoylecgonine in Dried Blood Spots by Liquid Chromatography-Atmospheric Pressure Chemical Ionization Tandem Mass Spectrometry. J. Anal. Toxicol. 1996, 20, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Singh, G.; Arora, V.; Fenn, P.T.; Mets, B.; Blair, I.A. A Validated Stable Isotope Dilution Liquid Chromatography Tandem Mass Spectrometry Assay for the Trace Analysis of Cocaine and Its Major Metabolites in Plasma. Anal. Chem. 1999, 71, 2021–2027. [Google Scholar] [CrossRef]
- Breindahl, T.; Andreasen, K. Determination of 11-nor-Δ9-tetrahydrocannabinol-9-carboxylic acid in urine using high-performance liquid chromatography and electrospray ionization mass spectrometry. J. Chromatogr. B 1999, 732, 155–164. [Google Scholar] [CrossRef]
- Bogusz, M.J.; Kruger, K.-D.; Maier, R.-D. Analysis of Underivatized Amphetamines and Related Phenethylamines with High-Performance Liquid Chromatography-Atmospheric Pressure Chemical Ionization Mass Spectrometry. J. Anal. Toxicol. 2000, 24, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goff, M.L.; Miller, M.L.; Paulson, J.D.; Lord, W.D.; Richards, E.; Omori, A.I. Effects of 3,4-Methylenedioxymethamphetamine in Decomposing Tissues on the Development of Parasarcophaga ruficornis (Diptera: Sarcophagidae) and Detection of the Drug in Postmortem Blood, Liver Tissue, Larvae, and Puparia. J. Forensic Sci. 1997, 42, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Bogusz, M.J.; Maier, R.-D.; Krüger, K.-D.; Kohls, U. Determination of Common Drugs of Abuse in Body Fluids Using One Isolation Procedure and Liquid Chromatography-Atmospheric-Pressure Chemical-Ionization Mass Spectrometry*. J. Anal. Toxicol. 1998, 22, 549–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bąchor, R.; Setner, B.; Kluczyk, A.; Stefanowicz, P.; Szewczuk, Z. The unusual hydrogen-deuterium exchange of α-carbon protons in N-substituted glycine-containing peptides. Biol. Mass Spectrom. 2014, 49, 43–49. [Google Scholar] [CrossRef]
- Bąchor, R.; Kluczyk, A.; Stefanowicz, P.; Szewczuk, Z. Preparation of novel deuterated cyclosporin A standards for quantitative LC-MS analysis. Biol. Mass Spectrom. 2017, 52, 817–822. [Google Scholar] [CrossRef]
- Bąchor, R.; Konieczny, A.; Szewczuk, Z. Preparation of Isotopically Labelled Standards of Creatinine Via H/D Exchange and Their Application in Quantitative Analysis by LC-MS. Molecules 2020, 25, 1514. [Google Scholar] [CrossRef] [Green Version]
- Bąchor, R.; Dębowski, D.; Łęgowska, A.; Stefanowicz, P.; Rolka, K.; Szewczuk, Z. Convenient preparation of deuterium-labeled analogs of peptides containing N -substituted glycines for a stable isotope dilution LC-MS quantitative analysis. J. Pept. Sci. 2015, 21, 819–825. [Google Scholar] [CrossRef]
- Bąchor, R.; Kluczyk, A.; Stefanowicz, P.; Szewczuk, Z. Facile synthesis of deuterium-labeled denatonium cation and its application in the quantitative analysis of Bitrex by liquid chromatography-mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 6557–6561. [Google Scholar] [CrossRef] [Green Version]
- Ng, L.-K.; Hupé, M.; Harnois, J.; Lawrence, A.H. Direct Injection Gas Chromatographic/Mass Spectrometric Analysis for Denatonium Benzoate in Specific Denatured Alcohol Formulations. Anal. Chem. 1998, 70, 4389–4393. [Google Scholar] [CrossRef]
- Commission Implementing Regulation (EU) No 162/2013 of 21 February 2013 ammending the Annex to Regulation (EC) No 3199/93 on the mutual recognition of procedures for the complete denaturing of alcohol for the purposes of exemption from excise duty. Off. J. Eur. Union 2013, 56, 55–61. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2013:124:FULL:EN:PDF (accessed on 1 August 2022).
- Mirgorodskaya, O.A.; Kozmin, Y.P.; Titov, M.I.; Körner, R.; Sönksen, C.P.; Roepstorff, P. Quantitation of peptides and pro-teins by matrix assisted laser desorption/ionization mass spectrometry using 18O-labeled internal standards. Rapid Commun. Mass Spectrom. 2000, 14, 1226–1232. [Google Scholar] [CrossRef]
- Lee, J.H.; Na Park, H.; Park, S.; Lee, Y.-M.; Kang, H. Development of a specific fragment pattern-based quadrupole-Orbitrap mass spectrometry method to screen adulterated products of phosphodiesterase-5 inhibitors and their analogues. Sci. Justice 2019, 59, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, H.N.; Park, O.R.; Kim, N.S.; Park, S.-K.; Kang, H. Screening of illegal sexual enhancement supplements and counterfeit drugs sold in the online and offline markets between 2014 and 2017. Forensic Sci. Int. 2019, 298, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Wilms, W.; Woźniak-Karczewska, M.; Corvini, P.F.-X.; Chrzanowski, Ł. Nootropic drugs: Methylphenidate, modafinil and piracetam—Population use trends, occurrence in the environment, ecotoxicity and removal methods—A review. Chemosphere 2019, 233, 771–785. [Google Scholar] [CrossRef]
- Löscher, W.; Richter, A. Piracetam and levetiracetam, two pyrrolidone derivatives, exert antidystonic activity in a hamster model of paroxysmal dystonia. Eur. J. Pharmacol. 2000, 391, 251–254. [Google Scholar] [CrossRef]
- Winblad, B. Piracetam: A Review of Pharmacological Properties and Clinical Uses. CNS Drug Rev. 2006, 11, 169–182. [Google Scholar] [CrossRef]
- Kim, S.; Park, J.-H.; Myung, S.-W.; Lho, D.-S. Determination of carphedon in human urine by solid-phase microextraction using capillary gas chromatography with nitrogen–phosphorus detection. Analyst 1999, 124, 1559–1562. [Google Scholar] [CrossRef]
- Shirayama, Y.; Yamamoto, A.; Nishimura, T.; Katayama, S.; Kawahara, R. Subsequent exposure to the choline uptake enhancer MKC-231 antagonizes phencyclidine-induced behavioral deficits and reduction in septal cholinergic neurons in rats. Eur. Neuropsychopharmacol. 2007, 17, 616–626. [Google Scholar] [CrossRef]
- Malykh, A.G.; Sadaie, M.R. Piracetam and Piracetam-Like Drugs: From basic science to novel clinical applications to CNS disorders. Drugs 2010, 70, 287–312. [Google Scholar] [CrossRef]
- Keating, G.M.; Raffin, M.J. Modafinil. a review of its use in excessive sleepiness associated with obstructive sleep apnoea/hypopnoea syndrome and shift work sleep disorder. CNS Drugs 2005, 19, 785–803. [Google Scholar] [CrossRef]
- Garnock-Jones, K.P.; Dhillon, S.; Scott, L.J. Armodafinil. CNS Drugs 2009, 23, 793–803. [Google Scholar] [CrossRef]
- Baselt, R.C. Dispositon of Toxic Drugs and Chemicals in Man; Biomedical Publications: Foster City, CA, USA, 1982; pp. 1152–1153. ISBN 978-0-9626523-7-0. [Google Scholar]
- Available online: https://www.wada-ama.org/sites/default/files/prohibited_list_2018_en.pdf (accessed on 23 May 2022).
- Moreno, J.D. Juicing the Brain. Sci. Am. Mind 2006, 17, 66–73. [Google Scholar] [CrossRef]
- Dance, A. Smart drugs: A dose of intelligence. Nature 2016, 531, S2–S3. [Google Scholar] [CrossRef]
- Battleday, R.; Brem, A.-K. Modafinil for cognitive neuroenhancement in healthy non-sleep-deprived subjects: A systematic review. Eur. Neuropsychopharmacol. 2015, 25, 1865–1881. [Google Scholar] [CrossRef]
- Kredlow, M.A.; Keshishian, A.; Oppenheimer, S.; Otto, M.W. The Efficacy of Modafinil as a Cognitive Enhancer: A Systematic Review and Meta-Analysis. J. Clin. Psychopharmacol. 2019, 39, 455–461. [Google Scholar] [CrossRef]
- Roberts, C.A.; Jones, A.; Sumnall, H.; Gage, S.H.; Montgomery, C. How effective are pharmaceuticals for cognitive enhancement in healthy adults? A series of meta-analyses of cognitive performance during acute administration of modafinil, methylphenidate and D-amphetamine. Eur. Neuropsychopharmacol. 2020, 38, 40–62. [Google Scholar] [CrossRef]
- Grocholska, P.; Wieczorek, R.; Bąchor, R. Preparation of Deuterium-Labeled Armodafinil by Hydrogen–Deuterium Exchange and Its Application in Quantitative Analysis by LC-MS. Metabolites 2022, 12, 578. [Google Scholar] [CrossRef]

















| Analytical Method | Benefits | Drawbacks |
|---|---|---|
| Colorimetry | quick, low-cost | limited sensitivity |
| Raman spectroscopy | rapid, specific, portable, fingerprint analysis | requires chemometry for complex samples |
| Nuclear magnetic resonance (NMR) | direct structure elucidation | sensitivity, solvent interference |
| Separation methods | ||
| High-performance thin-layer chromatography (HPTLC) | fingerprint of herbal medicines | resolution |
| Gas chromatography (GC) | analysis of volatiles | requires thermal stability or derivatization |
| High-performance liquid chromatography (HPLC) | analysis of biologicals | solvent consumption |
| Capillary electrophoresis (CE) | charge diversification | limited robustness |
| Separation methods with MS detection | ||
| Gas chromatography-mass spectrometry (GCMS) | efficient, sensitive, databases available | thermal artefacts |
| Liquid chromatography-mass spectrometry (LCMS) | efficient, sensitive, suitable for labile molecules | ion suppression, size |
| Area of Application | Example | Reference |
|---|---|---|
| Wine | [46] | |
| Food origin | Olive oil | [47] |
| Wheat | [48] | |
| Alcohol tampering | Wine | [49] |
| Vodka | [50] | |
| Drug analysis | Testosterone doping | [51] |
| Drugs of abuse | [52] | |
| Explosives | Triacetone triperoxide | [53] |
| No. | Name | Chemical Structure | Application | Reference |
|---|---|---|---|---|
| 1 | 2,4-Dinitrophenylhydrazine |  | Carbonyl compounds | [119] |
| 2 | Girard P reagent |  | Steroids | [119] |
| 3 | Girard T reagent |  | Ketosteroids | [120] |
| 4 | 2-Hydrazino pyridin |  | Ketosteroids | [120] |
| 5 | 2-Hydrazino-1-methyl pyridin |  | Neurosteroids Ketosteroids | [119,120] |
| 6 | Methoxyamine |  | Ketosteroids | [119,120] |
| 7 | 3-nitrophenylhydrazine |  | Malondi-aldehyde | [119] |
| 8 | Picolinic acid |  | Steroids hormones | [120] |
| 9 | Isonicotinoyl azide |  | Dihydroxy- steroids | [120] |
| 10 | AOA-GRG-CMRR |  | Steroids | [121] |
| No. | Internal Standard | Analyzed Compounds | Sample | Reference |
|---|---|---|---|---|
| 1 | morphine-d3 | morphine, morphine-3-glucuronide, morphine-6-glucuronide, and 6-monoacetylmorphine | urine | [162,163] |
| 2 | morphine-d3, codeine-d3 | morphine, codeine | urine | [164] |
| 3 | morphine-d3, morphine-6-glucuronide-d3 | morphine, morphine-3-glucuronide, morphine-6-glucuronide, and | plasma | [165] |
| 4 | cocaine-d3 benzoylecgonine-d3 ecgonine methyl ester-d3 | cocaine and its metabolites | - | [166] |
| 5 | benzoylecgonine-d3 | cocaine and its metabolites | blood spots | [167] |
| 6 | norcocaine-d5 cocaine-d5 benzoylecgonine-d3, ecgonine methyl ester-d3 | cocaine and its metabolites | plasma | [168] |
| 7 | THC-COOH-d3 | THC-COOH | urine | [169] |
| 8 | methamphetamine-d10, amphetamine-d11, 3,4-methylenedioxyethyl-amphetamine-d5, 3,4-methylenedioxymethyl-amphetamine-d5 | Amphetamine, methamphetamine, illicit designer phenethylamines | serum | [170,171] |
| 9 | morphine-d3, morphine-3-glucuronide-d3, morphine-6-glucuronide-d6, codeine-d6, codeine-6-glucuronide-d3, cocaine-d8, benzoylecgonine-d8 | morphine, morphine-3-glucuronide, morphine-6-glucuronide, 6-monoacetylmorphine, codeine, codeine-6-glucuronide, dihydrocodeine, dihydromorphine, buprenorphine, methadone, tramadol, ibogaine, cocaine, benzoylecgonine ecgonine methyl ester lysergic acid diethylamide | serum, blood, urine | [172] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grocholska, P.; Popiel, D.; Walter, M.; Biernat, M.; Cebrat, M.; Kuczer, M.; Modzel, M.; Bąchor, R.; Kluczyk, A. Citius, Altius, Fortius—Advanced Mass Spectrometry in Service of Forensic Analysis. Chemosensors 2022, 10, 324. https://doi.org/10.3390/chemosensors10080324
Grocholska P, Popiel D, Walter M, Biernat M, Cebrat M, Kuczer M, Modzel M, Bąchor R, Kluczyk A. Citius, Altius, Fortius—Advanced Mass Spectrometry in Service of Forensic Analysis. Chemosensors. 2022; 10(8):324. https://doi.org/10.3390/chemosensors10080324
Chicago/Turabian StyleGrocholska, Paulina, Dominik Popiel, Martyna Walter, Monika Biernat, Marek Cebrat, Mariola Kuczer, Maciej Modzel, Remigiusz Bąchor, and Alicja Kluczyk. 2022. "Citius, Altius, Fortius—Advanced Mass Spectrometry in Service of Forensic Analysis" Chemosensors 10, no. 8: 324. https://doi.org/10.3390/chemosensors10080324