Frequent Germline and Somatic Single Nucleotide Variants in the Promoter Region of the Ribosomal RNA Gene in Japanese Lung Adenocarcinoma Patients

 , , , , ,
, , , , ,  ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Cell Culture
2.3. Microdissection and DNA Extraction
2.4. Sanger Sequencing
2.5. Deep Sequencing Using Illumina MiSeq
2.6. Quantitative Real-Time Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.7. Statistical Analysis
3. Results
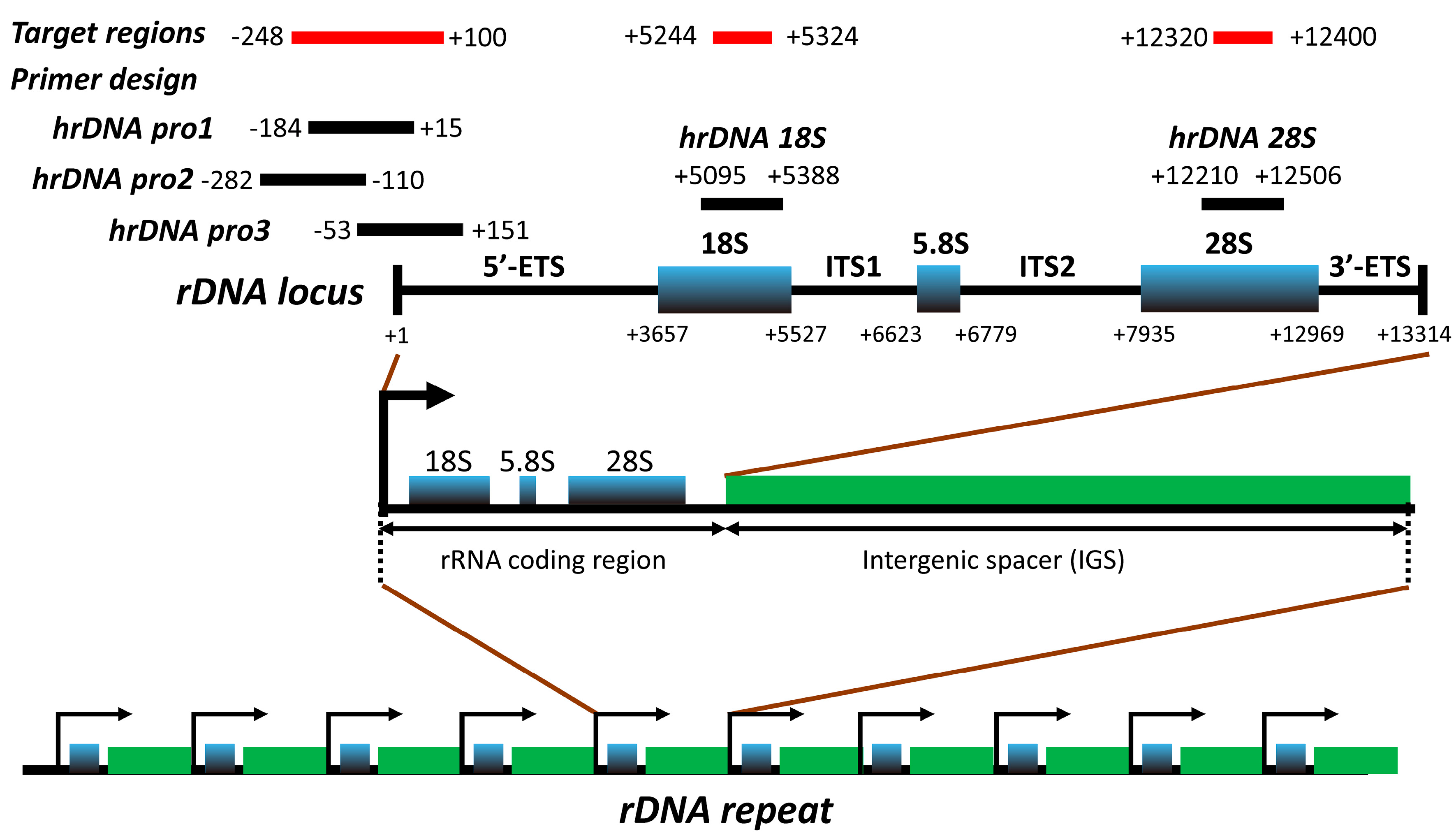
3.1. Assessment and Validation of the Assay Design for Identification of rDNA Target Region
3.2. Patient Characteristics
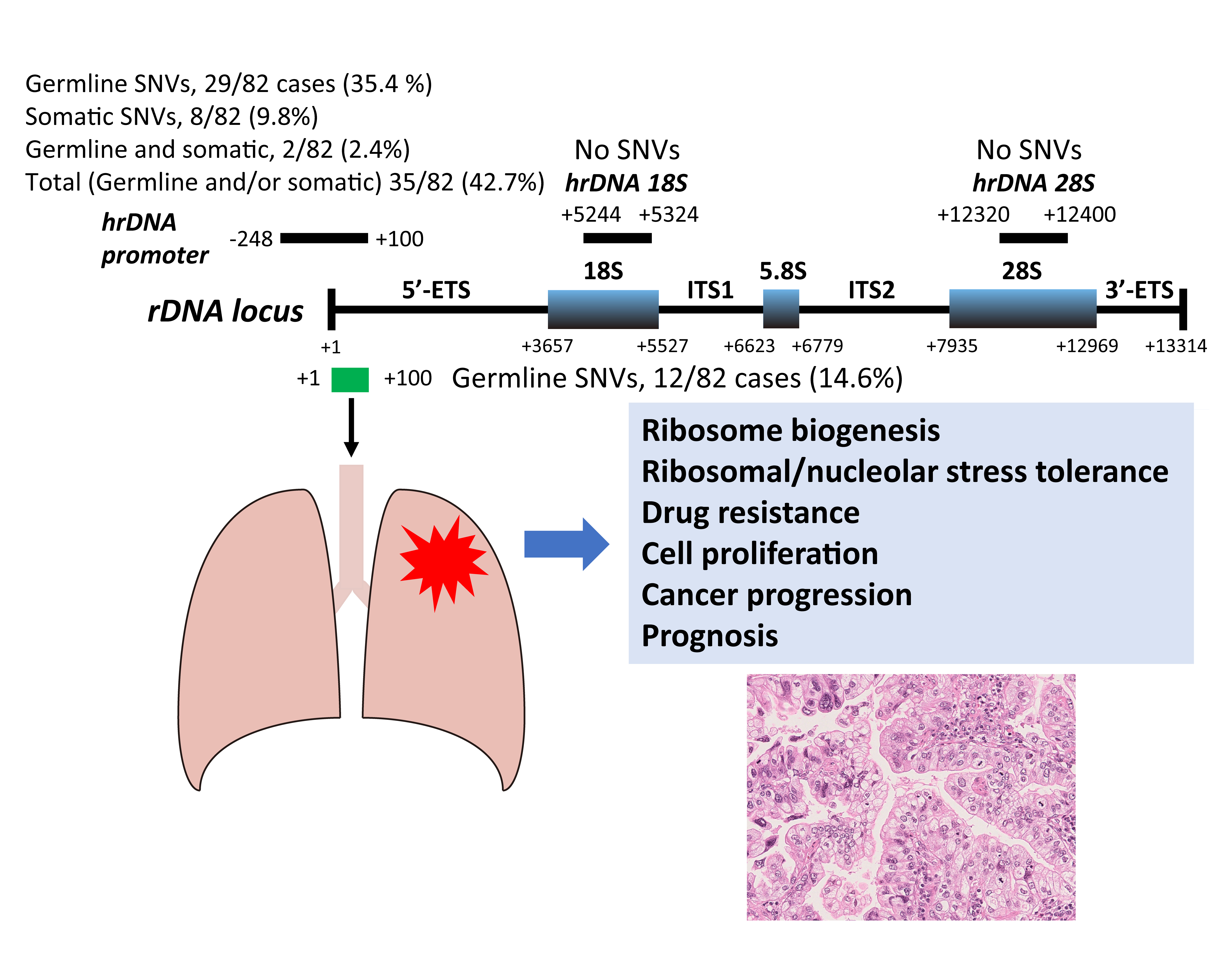
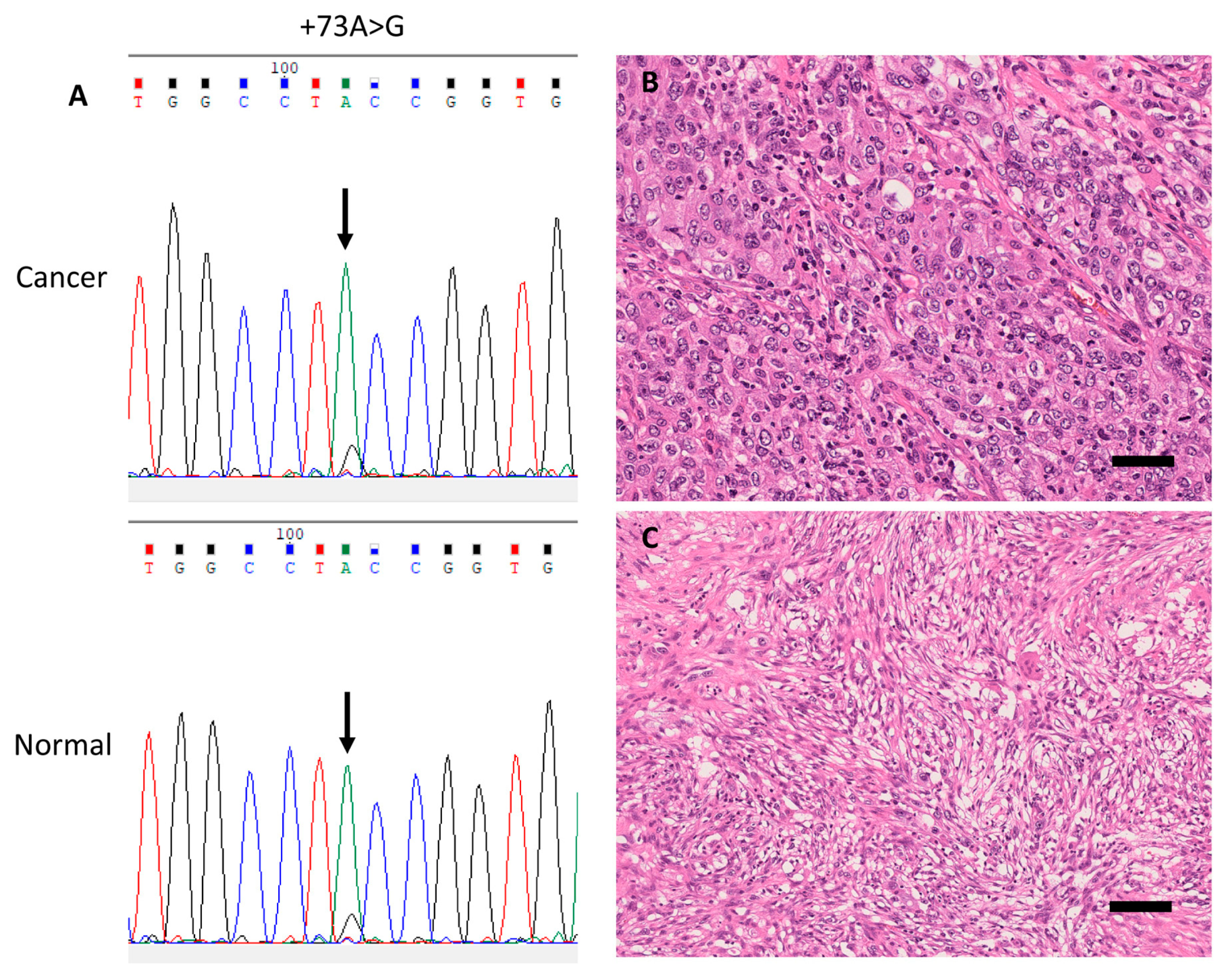
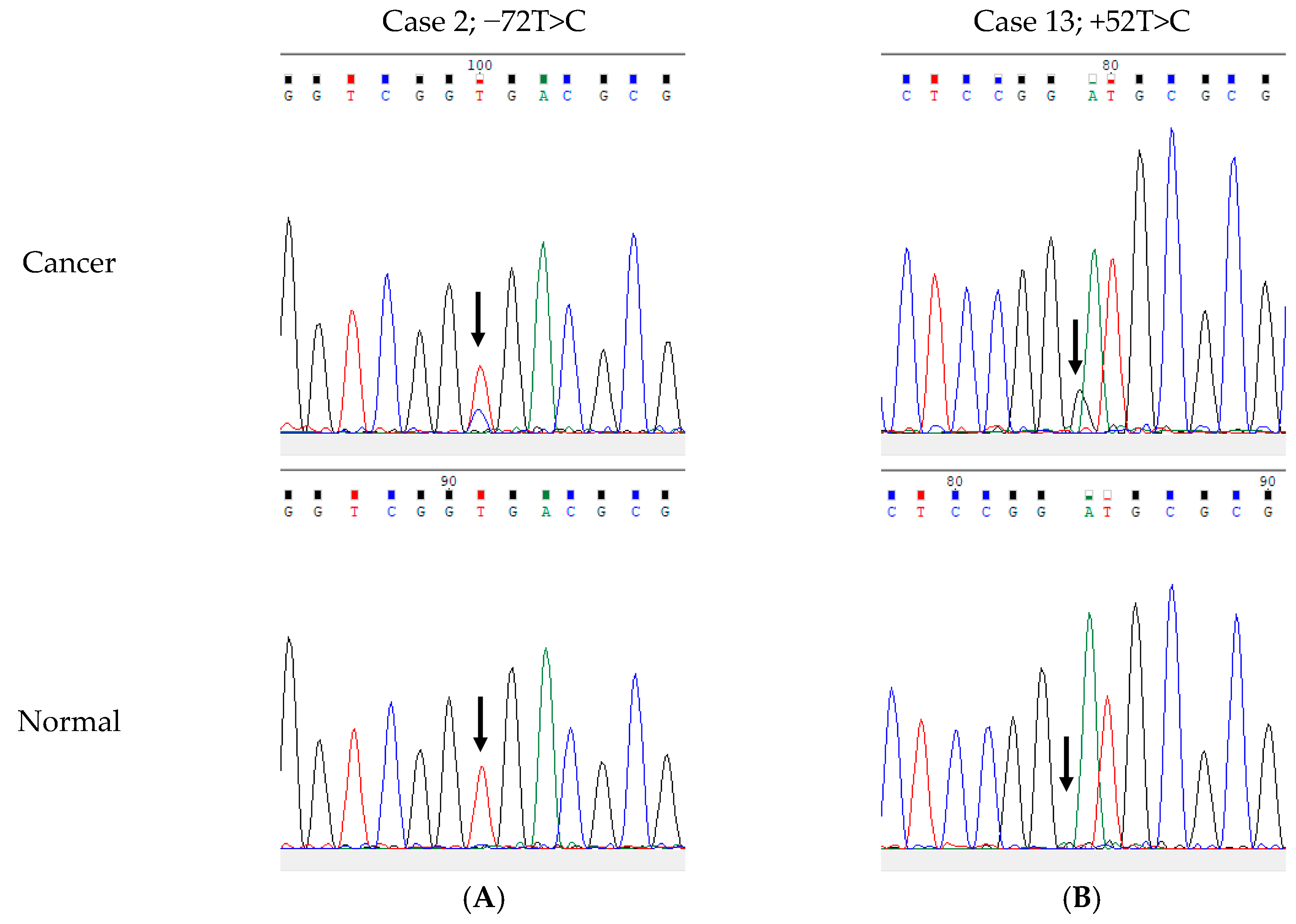
3.3. Germline and Somatic SNVs in the rDNA Promoter Region Among 82 LUAC Patients
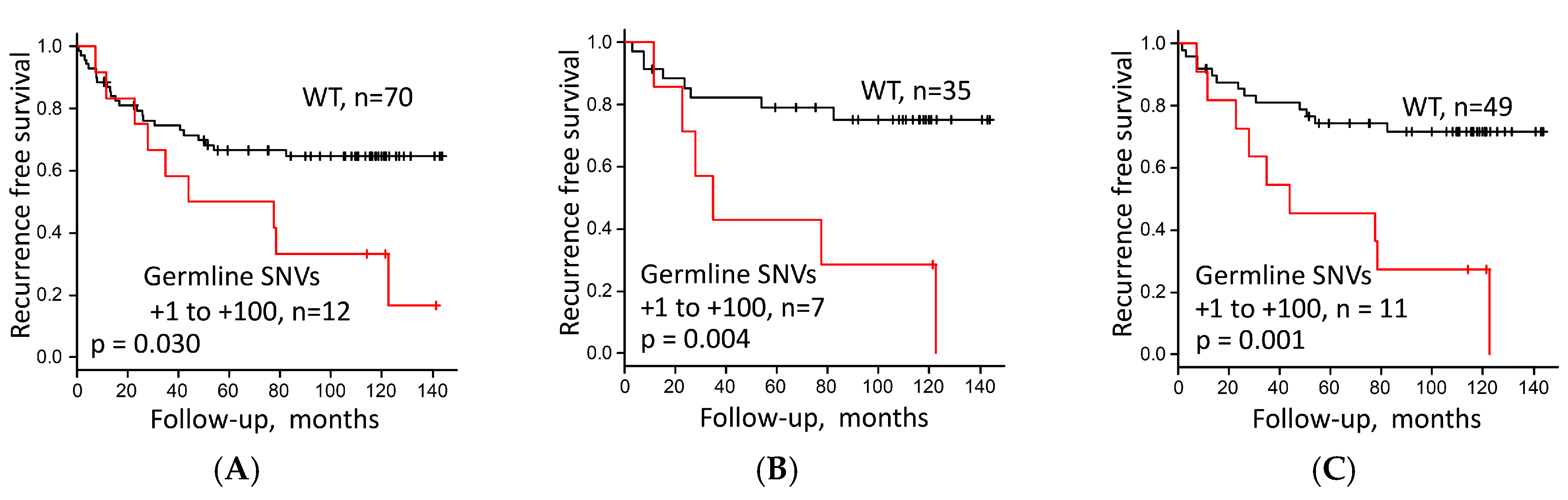
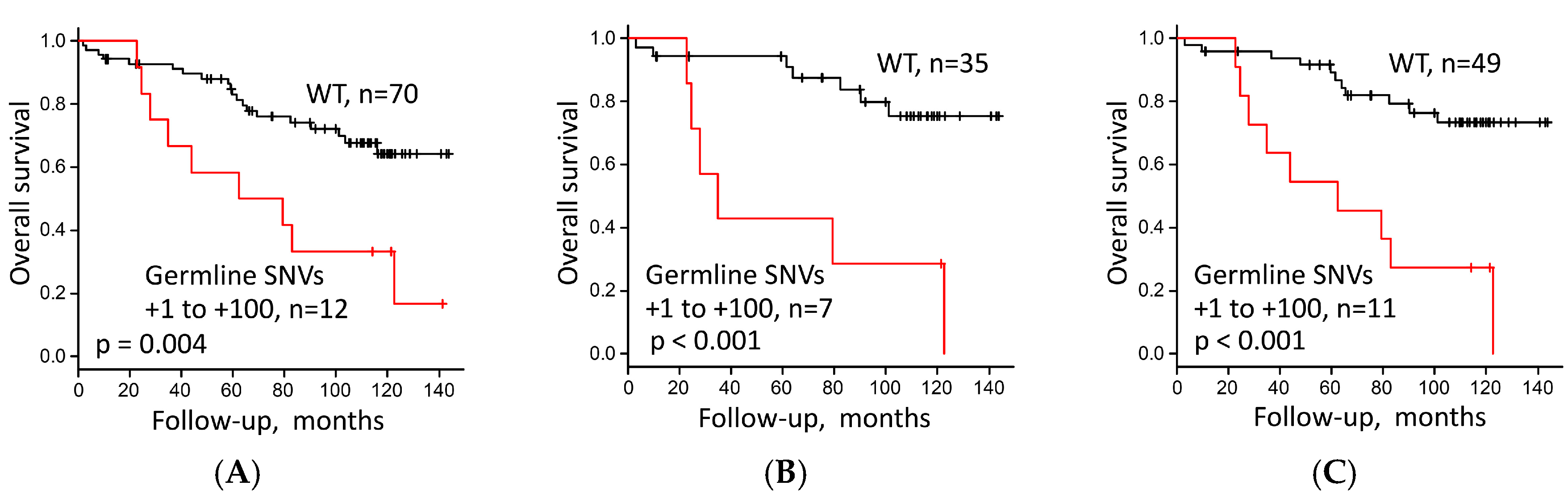
3.4. Survival Analysis of LUAC Patients
3.5. rDNA Promoter SNVs in Publicly Available Databases
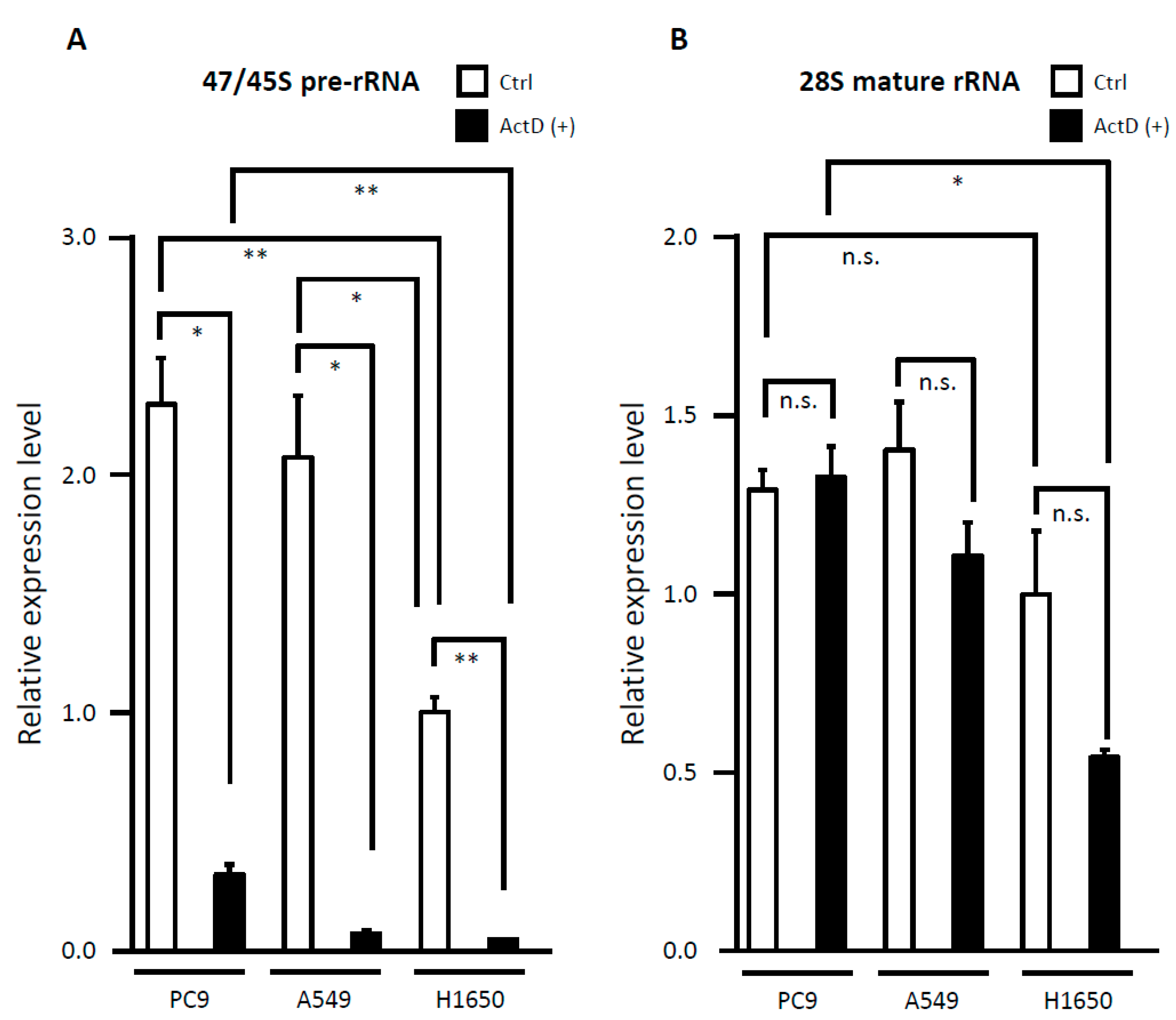
3.6. Evaluation of rRNA Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Inamura, K. Lung cancer: Understanding its molecular pathology and the 2015 WHO classification. Front. Oncol. 2017, 7, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collisson, E.A.; Campbell, J.D.; Brooks, A.N.; Berger, A.H.; Lee, W.; Chmielecki, J.; Beer, D.G.; Cope, L.; Creighton, C.J.; Danilova, L.; et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar]
- La Fleur, L.; Falk-Sörqvist, E.; Smeds, P.; Berglund, A.; Sundström, M.; Mattsson, J.S.; Brandén, E.; Koyi, H.; Isaksson, J.; Brunnström, H.; et al. Mutation patterns in a population-based non-small cell lung cancer cohort and prognostic impact of concomitant mutations in KRAS and TP53 or STK11. Lung Cancer 2019, 130, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Lindeman, N.I.; Cagle, P.T.; Aisner, D.L.; Arcila, M.E.; Beasley, M.B.; Bernicker, E.H.; Colasacco, C.; Dacic, S.; Hirsch, F.R.; Kerr, K.; et al. Updated molecular testing guideline for the selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors: Guideline from the College of American Pathologists, the International Association for the study of lung cancer and the Association for Molecular Pathology. Arch. Pathol. Lab. Med. 2018, 142, 321–346. [Google Scholar] [PubMed] [Green Version]
- Piraino, S.W.; Furney, S.J. Beyond the exome: The role of non-coding somatic mutations in cancer. Ann. Oncol. 2016, 27, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazzo, A.F.; Lee, E.S. Non-coding RNA: What is functional and what is junk? Front. Genet. 2015, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-coding RNAs and their integrated networks. J. Integr. Bioinform. 2019, 16, 20190027. [Google Scholar] [CrossRef] [PubMed]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef]
- Lambert, M.; Benmoussa, A.; Provost, P. Small non-coding RNAs derived from eukaryotic ribosomal RNA. Noncoding RNA 2019, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Schwarzacher, H.G.; Wachtler, F. The nucleolus. Anat. Embryol. 1993, 188, 515–536. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.L.; Herrmann, H. Functional architecture of the cell nucleus. BBA Mol. Cell Res. 2008, 1783, 2041–2043. [Google Scholar] [CrossRef] [Green Version]
- Sirri, V.; Urcuqui-Inchima, S.; Roussel, P.; Hernandez-Verdun, D. Nucleolus: The fascinating nuclear body. Histochem. Cell Biol. 2008, 129, 13–31. [Google Scholar] [CrossRef] [Green Version]
- Montanaro, L.; Treré, D.; Derenzini, M. Nucleolus, ribosomes and cancer. Am. J. Pathol. 2008, 173, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Sulima, S.O.; Kampen, K.R.; De Keersmaecker, K. Cancer biogenesis in ribosomopathies. Cells 2019, 8, 229. [Google Scholar] [CrossRef] [Green Version]
- Bohnsack, K.E.; Bohnsack, M.T. Uncovering the assembly pathway of human ribosomes and its emerging links to disease. EMBO J. 2019, 38, e100278. [Google Scholar] [CrossRef]
- Shiao, Y.H.; Lupascu, S.T.; Gu, Y.D.; Kasprzak, W.; Hwang, C.J.; Fields, J.R.; Leighty, R.M.; Quiñones, O.; Shapiro, B.A.; Alvord, W.G.; et al. An intergenic non-coding rRNA correlated with expression of the rRNA and frequency of an rRNA single nucleotide polymorphism in lung cancer cells. PLoS ONE 2009, 4, e7505. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Lemos, B. Ribosomal DNA copy number amplification and loss in human cancers is linked to tumor genetic context, nucleolus activity, and proliferation. PLoS Genet. 2017, 13, e1006994. [Google Scholar] [CrossRef]
- Brierley, J.D.; Gospodarowicz, M.K.; Wittekind, C. TNM Classification of Malignant Tumours; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Sato, M.; Watanabe, S.; Tanaka, H.; Nozaki, K.; Arita, M.; Takahashi, M.; Shoji, S.; Ichikawa, K.; Kondo, R.; Aoki, N.; et al. Retrospective analysis of antitumor effects and biomarkers for nivolumab in NSCLC patients with EGFR mutations. PLoS ONE 2019, 14, e0215292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travis, W.D.; Brambilla, E.; Burke, A.P.; Marx, A.; Nicholson, A.G. WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart, 4th ed.; IARC: Lyon, France, 2015. [Google Scholar]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization classification of lung tumors: Impact of genetic, clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motoyama, T.; Hojo, H.; Watanabe, H. Comparison of seven cell lines derived from human gastric carcinomas. Acta Pathol. Jpn. 1986, 36, 65–83. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant review with the integrative genomics viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, Y. Investigation of the freely-available easy-to-use software “EZR” (Easy R) for medical statistics. Bone Marrow Transpl. 2013, 48, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Firth, D. Bias reduction of maximum likelihood estimates. Biometrika 1993, 80, 27–38. [Google Scholar] [CrossRef]
- Cox Regression with Firth’s Penalized Likelihood. Available online: https://CRAN.R-project.org/package=coxphf or https://cran.r-project.org/web/packages/coxphf/coxphf.pdf (accessed on 10 October 2018).
- Haltiner, M.M.; Smale, S.T.; Tjian, R. Two distinct promoter elements in the human rRNA gene identified by linker scanning mutagenesis. Mol. Cell. Biol. 1986, 6, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Eichler, D.C.; Craig, N. Processing of eukaryotic ribosomal RNA. Prog. Nucleic Acid Res. Mol. Biol. 1994, 49, 197–239. [Google Scholar]
- Zhang, S.; Wang, J.; Tseng, H. Basonuclin regulates a subset of ribosomal RNA genes in HaCaT cells. PLoS ONE 2007, 2, e902. [Google Scholar] [CrossRef] [Green Version]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for validation of next-generation sequencing-based oncology panels: A joint consensus recommendation of the Association for Molecular Pathology and College of American Pathologists. J. Mol. Diagn. 2017, 19, 341–365. [Google Scholar] [CrossRef] [Green Version]
- ICGC Data Portal. Available online: https://dcc.icgc.org/ (accessed on 1 October 2020).
- NBDC Human Database. Available online: https://humandbs.biosciencedbc.jp/en/ (accessed on 10 October 2020).
- European Genome-phenome Archive. Available online: https://www.ebi.ac.uk/ega/ (accessed on 10 October 2020).
- GDC Data Portal-National Cancer Institute. Available online: https://portal.gdc.cancer.gov/ (accessed on 1 October 2020).
- Genome Browser. Available online: https://gnomad.broadinstitute.org/ (accessed on 19 October 2020).
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- UCSC Genome Browser Lift over Tool. Available online: https://genome.ucsc.edu/cgi-bin/hgLiftOver (accessed on 21 October 2020).
- Perry, R.P.; Kelley, D.E. Inhibition of RNA synthesis by actinomycin D: Characteristic dose–response of different RNA species. J. Cell. Physiol. 1970, 76, 127–139. [Google Scholar] [CrossRef]
- Iapalucci-Espinoza, S.; Franze-Fernández, M.T. Effect of protein synthesis inhibitors and low concentrations of actinomycin D on ribosomal RNA synthesis. FEBS Lett. 1979, 107, 281–284. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Hu, Z.; Feng, Y.; Hu, X.; Yuan, J.; Zhao, S.D.; Zhang, Y.; Yang, L.; Shan, W.; He, Q.; et al. Comprehensive genomic characterization of long non-coding RNAs across human cancers. Cancer Cell 2015, 28, 529–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Li, X.; Zhi, H.; Zhang, Y.; Wang, P.; Wang, Y.; Shang, S.; Fang, Y.; Shen, W.; Ning, S.; et al. Comprehensive characterization of somatic mutations impacting lncRNA expression for pan-cancer. Mol. Ther. Nucleic Acids 2019, 18, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhan, A.; Soleimani, M.; Mandal, S.S. Long noncoding RNA and cancer: A new paradigm. Cancer Res. 2017, 77, 3965–3981. [Google Scholar] [CrossRef] [Green Version]
- Pirogov, S.A.; Gvozdev, V.A.; Klenov, M.S. Long noncoding RNAs and stress response in the nucleolus. Cells 2019, 8, 668. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Arora, A.; Begg, C.B.; Shen, R. Using somatic variant richness to mine signals from rare variants in the cancer genome. Nat. Commun. 2019, 10, 5506. [Google Scholar] [CrossRef]
- Batalini, F.; Peacock, E.G.; Stobie, L.; Robertson, A.; Garber, J.; Weitzel, J.N.; Tung, N.M. Li-Fraumeni syndrome: Not a straightforward diagnosis anymore-the interpretation of pathogenic variants of low allele frequency and the differences between germline PVs, mosaicism, and clonal hematopoiesis. Breast Cancer Res. 2019, 21, 107. [Google Scholar] [CrossRef]
- Bomba, L.; Walter, K.; Soranzo, N. The impact of rare and low-frequency genetic variants in common disease. Genome Biol. 2017, 18, 77. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Kweon, S.S.; Tanikawa, C.; Jia, W.H.; Xiang, Y.B.; Cai, Q.; Zeng, C.; Schmit, S.L.; Shin, A.; Matsuo, K.; et al. Large-scale genome-wide association study of East Asians identifies loci associated with risk for colorectal cancer. Gastroenterology 2019, 156, 1455–1466. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Mukherjee, S.; Landi, M.T.; Bosse, Y.; Joubert, P.; Zhu, D.; Gorlov, I.; Xiao, X.; Han, Y.; Gorlova, O.; et al. Protein-altering germline mutations implicate novel genes related to lung cancer development. Nat. Commun. 2020, 11, 2220. [Google Scholar] [CrossRef]
- Craig, D.J.; Morrison, T.; Khuder, S.A.; Crawford, E.L.; Wu, L.; Xu, J.; Blomquist, T.M.; Willey, J.C. Technical advance in targeted NGS analysis enables identification of lung cancer risk-associated low frequency TP53, PIK3CA, and BRAF mutations in airway epithelial cells. BMC Cancer 2019, 19, 1081. [Google Scholar] [CrossRef]
- Xu, C.; Gu, X.; Padmanabhan, R.; Wu, Z.; Peng, Q.; DiCarlo, J.; Wang, Y. smCounter2: An accurate low-frequency variant caller for targeted sequencing data with unique molecular identifiers. Bioinformatics 2019, 35, 1299–1309. [Google Scholar] [CrossRef] [Green Version]
- Sater, V.; Viailly, P.J.; Lecroq, T.; Prieur-Gaston, É.; Bohers, É.; Viennot, M.; Ruminy, P.; Dauchel, H.; Vera, P.; Jardin, F. UMI-VarCal: A new UMI-based variant caller that efficiently improves low-frequency variant detection in paired-end sequencing NGS libraries. Bioinformatics 2020, 36, 2718–2724. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetic gene silencing in cancer: The DNA hypermethylome. Hum. Mol. Genet. 2007, 16, 50–59. [Google Scholar] [CrossRef]
- Raval, A.; Sridhar, K.J.; Patel, S.; Turnbull, B.B.; Greenberg, P.L.; Mitchell, B.S. Reduced rRNA expression and increased rDNA promoter methylation in CD34+ cells of patients with myelodysplastic syndromes. Blood 2012, 120, 4812–4818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uemura, M.; Zheng, Q.; Koh, C.M.; Nelson, W.G.; Yegnasubramanian, S.; De Marzo, A.M. Overexpression of ribosomal RNA in prostate cancer is common but not linked to rDNA promoter hypomethylation. Oncogene 2012, 31, 1254–1263. [Google Scholar] [CrossRef] [Green Version]
- Bacalini, M.G.; Pacilli, A.; Giuliani, C.; Penzo, M.; Treré, D.; Pirazzini, C.; Salvioli, S.; Franceschi, C.; Montanaro, L.; Garagnani, P. The nucleolar size is associated to the methylation status of ribosomal DNA in breast carcinomas. BMC Cancer 2014, 14, 361. [Google Scholar] [CrossRef] [Green Version]
- Karahan, G.; Sayar, N.; Gozum, G.; Bozkurt, B.; Konu, O.; Yulug, I.G. Relative expression of rRNA transcripts and 45S rDNA promoter methylation status are dysregulated in tumors in comparison with matched-normal tissues in breast cancer. Oncol. Rep. 2015, 33, 3131–3145. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Wang, Y.; Lv, Q.; Zhang, J.; Wang, Q.; Gao, F.; Hou, H.; Zhang, H.; Zhang, W.; Li, L.; et al. Overexpression of Ribosomal RNA in the Development of Human Cervical Cancer Is Associated with rDNA Promoter Hypomethylation. PLoS ONE 2016, 11, e0163340. [Google Scholar] [CrossRef] [Green Version]
- Ha, S.; Zhou, H.; Gautam, M.; Song, Y.; Wang, C. Reduced ribosomal RNA expression and unchanged ribosomal DNA promoter methylation in oral squamous cell carcinoma. Mol. Genet. Genom. Med. 2019, 7, e00783. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, K.; Majumder, S.; Datta, J.; Motiwala, T.; Bai, S.; Sharma, S.M.; Frankel, W.; Jacob, S.T. Role of human ribosomal RNA (rRNA) promoter methylation and of methyl-CpG-binding protein MBD2 in the suppression of rRNA gene expression. J. Biol. Chem. 2004, 279, 6783–6793. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.H. Hypermethylation of ribosomal DNA in human breast carcinoma. Br. J. Cancer 2000, 82, 514–517. [Google Scholar]
- Powell, M.A.; Mutch, D.G.; Rader, J.S.; Herzog, T.J.; Huang, T.H.; Goodfellow, P.J. Ribosomal DNA methylation in patients with endometrial carcinoma: An independent prognostic marker. Cancer 2002, 94, 2941–2952. [Google Scholar] [CrossRef]
- Chan, M.W.; Wei, S.H.; Wen, P.; Wang, Z.; Matei, D.E.; Liu, J.C.; Liyanarachchi, S.; Brown, R.; Nephew, K.P.; Yan, P.S.; et al. Hypermethylation of 18S and 28S ribosomal DNAs predicts progression-free survival in patients with ovarian cancer. Clin. Cancer Res. 2005, 11, 7376–7383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, D.; Lu, Y.J.; Fang, C.; Pritchard-Jones, K.; Shipley, J. Nascent pre-rRNA overexpression correlates with an adverse prognosis in alveolar rhabdomyosarcoma. Genes Chromosomes Cancer 2006, 45, 839–845. [Google Scholar] [CrossRef]
- Tsoi, H.; Lam, K.C.; Dong, Y.; Zhang, X.; Lee, C.K.; Zhang, J.; Ng, S.C.; Ng, S.S.M.; Zheng, S.; Chen, Y.; et al. Pre-45s rRNA promotes colon cancer and is associated with poor survival of CRC patients. Oncogene 2017, 36, 6109–6118. [Google Scholar] [CrossRef] [Green Version]
- Hein, N.; Hannan, K.M.; George, A.J.; Sanij, E.; Hannan, R.D. The nucleolus: An emerging target for cancer therapy. Trends Mol. Med. 2013, 19, 643–654. [Google Scholar] [CrossRef]
- Perry, R.P.; Kelley, D.E. Persistent synthesis of 5S RNA when production of 28S and 18S ribosomal RNA is inhibited by low doses of actinomycin D. J. Cell. Physiol. 1968, 72, 235–246. [Google Scholar] [CrossRef]
- Kapoor, N.R.; Ahuja, R.; Shukla, S.K.; Kumar, V. The HBx protein of hepatitis B virus confers resistance against nucleolar stress and anti-cancer drug-induced p53 expression. FEBS Lett. 2013, 587, 1287–1292. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Pederson, T. The nucleolus stress response is coupled to an ATR-Chk1-mediated G2 arrest. Mol. Biol. Cell 2013, 24, 1334–1342. [Google Scholar] [CrossRef]
- Léger, K.; Bär, D.; Savić, N.; Santoro, R.; Hottiger, M.O. ARTD2 activity is stimulated by RNA. Nucleic Acids Res. 2014, 42, 5072–5082. [Google Scholar] [CrossRef] [Green Version]
- Yan, Q.; Zhu, C.; Guang, S.; Feng, X. The functions of non-coding RNAs in rRNA regulation. Front. Genet. 2019, 10, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, D.W.; Hassan, M.Q.; Pratap, J.; Galindo, M.; Zaidi, S.K.; Lee, S.H.; Yang, X.; Xie, R.; Javed, A.; Underwood, J.M.; et al. Mitotic occupancy and lineage-specific transcriptional control of rRNA genes by Runx2. Nature 2007, 445, 442–446. [Google Scholar] [CrossRef]
- Pistoni, M.; Verrecchia, A.; Doni, M.; Guccione, E.; Amati, B. Chromatin association and regulation of rDNA transcription by the Ras-family protein RasL11a. EMBO J. 2010, 29, 1215–1224. [Google Scholar] [CrossRef] [Green Version]
- Donati, G.; Brighenti, E.; Vici, M.; Mazzini, G.; Treré, D.; Montanaro, L.; Derenzini, M. Selective inhibition of rRNA transcription downregulates E2F-1: A new p53-independent mechanism linking cell growth to cell proliferation. J. Cell Sci. 2011, 124, 3017–3028. [Google Scholar] [CrossRef] [Green Version]
- Bursac, S.; Brdovcak, M.C.; Donati, G.; Volarevic, S. Activation of the tumor suppressor p53 upon impairment of ribosome biogenesis. Biochim. Biophys. Acta 2014, 1842, 817–830. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Ichinose, Y.; Ohta, M.; Hata, E.; Tsubota, N.; Tada, H.; Watanabe, Y.; Wada, H.; Tsuboi, M.; Hamajima, N.; et al. A randomized trial of adjuvant chemotherapy with uracil-tegafur for adenocarcinoma of the lung. N. Engl. J. Med. 2004, 350, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Yoh, K.; Takamochi, K.; Shukuya, T.; Hishida, T.; Tsuboi, M.; Sakurai, H.; Goto, Y.; Yoshida, K.; Ohde, Y.; Okumura, S.; et al. Pattern of care in adjuvant therapy for resected Stage I non-small cell lung cancer: Real-world data from Japan. Jpn. J. Clin. Oncol. 2019, 49, 63–68. [Google Scholar] [CrossRef]
- Arriagada, R.; Dunant, A.; Pignon, J.P.; Bergman, B.; Chabowski, M.; Grunenwald, D.; Kozlowski, M.; Le Péchoux, C.; Pirker, R.; Pinel, M.I.; et al. Long-term results of the international adjuvant lung cancer trial evaluating adjuvant Cisplatin-based chemotherapy in resected lung cancer. J. Clin. Oncol. 2010, 28, 35–42. [Google Scholar] [CrossRef]
- Bradbury, P.; Sivajohanathan, D.; Chan, A.; Kulkarni, S.; Ung, Y.; Ellis, P.M. Postoperative Adjuvant Systemic Therapy in Completely Resected Non-Small-Cell Lung Cancer: A Systematic Review. Clin. Lung Cancer 2017, 18, 259. [Google Scholar] [CrossRef]
- Derenzini, M.; Trerè, D.; Pession, A.; Montanaro, L.; Sirri, V.; Ochs, R.L. Nucleolar function and size in cancer cells. Am. J. Pathol. 1998, 152, 1291–1297. [Google Scholar]
- Derenzini, M.; Trerè, D.; Pession, A.; Govoni, M.; Sirri, V.; Chieco, P. Nucleolar size indicates the rapidity of cell proliferation in cancer tissues. J. Pathol. 2000, 191, 181–186. [Google Scholar] [CrossRef]
- Pelletier, J.; Thomas, G.; Volarevic, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef]
- Khot, A.; Brajanovski, N.; Cameron, D.P.; Hein, N.; Maclachlan, K.H.; Sanij, E.; Lim, J.; Soong, J.; Link, E.; Blombery, P.; et al. First-in-human RNA polymerase I transcription inhibitor CX-5461 in patients with advanced hematologic cancers: Results of a phase I dose-escalation study. Cancer Discov. 2019, 9, 1036–1049. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, R.; Schneekloth, J.S., Jr.; Panov, K.I.; Hannan, K.M.; Hannan, R.D. Targeting the RNA polymerase I transcription for cancer therapy comes of age. Cells 2020, 9, 266. [Google Scholar] [CrossRef] [Green Version]
- Ismael, M.; Webb, R.; Ajaz, M.; Kirkby, K.J.; Coley, H.M. The Targeting of RNA polymerase I transcription using CX-5461 in combination with radiation enhances tumour cell killing effects in human solid cancers. Cancers 2019, 11, 1429. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
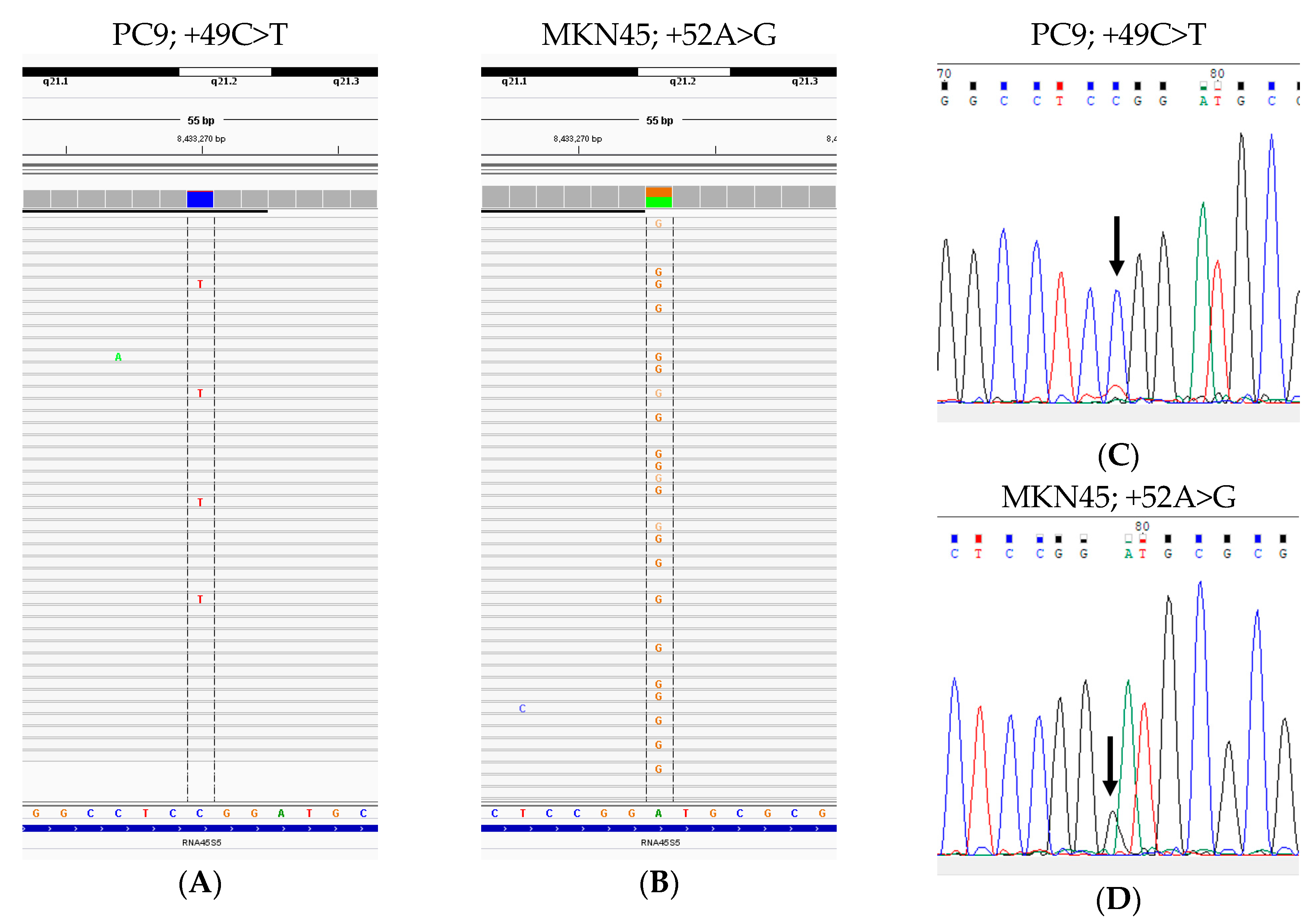
| Cell lines | Tissue of Origin | Histological Type | SNVs 1 |
|---|---|---|---|
| A549 | Lung | Adenocarcinoma | −104T>C, −96C>T |
| H23 | Lung | Adenocarcinoma | −210T>C, −206C>T |
| PC9 | Lung | Adenocarcinoma | +49C>T |
| Caco-2 | Colon | Adenocarcinoma | −186G>C |
| SW480 | Colon | Adenocarcinoma | −96C>T |
| COLO205 | Colon | Adenocarcinoma | −96C>T, −79T>A |
| MKN45 | Stomach | Adenocarcinoma | +52A>G |
| DLD-1 | Colon | Adenocarcinoma | −72T>C |
| Raji | B-lymphocytes | Burkitt’s lymphoma | −96C>T, −85G>C, +49C>T |
| Jurkat | T-lymphocytes | T cell acute lymphoblastic leukemia | −96C>T |
| HeLa | Uterine cervix | Adenocarcinoma, HPV18-positive | −206C>T, −96C>T, −72T>C |
| Characteristics | Group | n (%) |
|---|---|---|
| No. of patients | 82 | |
| Gender | Female | 26 (31.7) |
| Male | 56 (68.3) | |
| Age: Median (range), y | 66.5 [33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87] | |
| ≤55 | 11 (13.4) | |
| >55 | 71 (86.6) | |
| Smoking history | Never | 31 (37.8) |
| Former/current | 51 (62.2) | |
| Stage | I | 50 (61.0) |
| II | 18 (22.0) | |
| III | 11 (13.4) | |
| IV | 3 (3.7) | |
| Nodal and/or distant metastasis | Present | 19 (23.2) |
| Absent | 63 (76.8) | |
| EGFR mutation | Present | 25 (30.5) |
| Exon 19 deletion | 14 (17.1) | |
| Exon 21 L858R | 11 (13.4) | |
| Exon 20 insertion | 0 (0) | |
| Exon 18 G719S | 0 (0) | |
| Absent | 49 (59.8) | |
| NA | 8 (9.8) | |
| Histology | MIA | 8 (9.8) |
| Lepidic | 3 (3.7) | |
| Acinar | 8 (9.8) | |
| Papillary | 34 (41.5) | |
| Micropapillary | 2 (2.4) | |
| Solid | 15 (18.3) | |
| IMA | 8 (9.8) | |
| Fetal | 2 (2.4) | |
| Pleomorphic | 2 (2.4) | |
| SNV in the rDNA promoter region | Germline | 29 (35.4) |
| Somatic | 8 (9.8) | |
| Germline and somatic (at different locus) | 2 (2.4) | |
| Total (Germline and/or somatic) | 35 (42.7) | |
| No SNV | 47 (57.3) |
| Patient | Histology | Sex | Age | Stage | Germline | Somatic | Months Post-Surgery | Alive/Deceased |
|---|---|---|---|---|---|---|---|---|
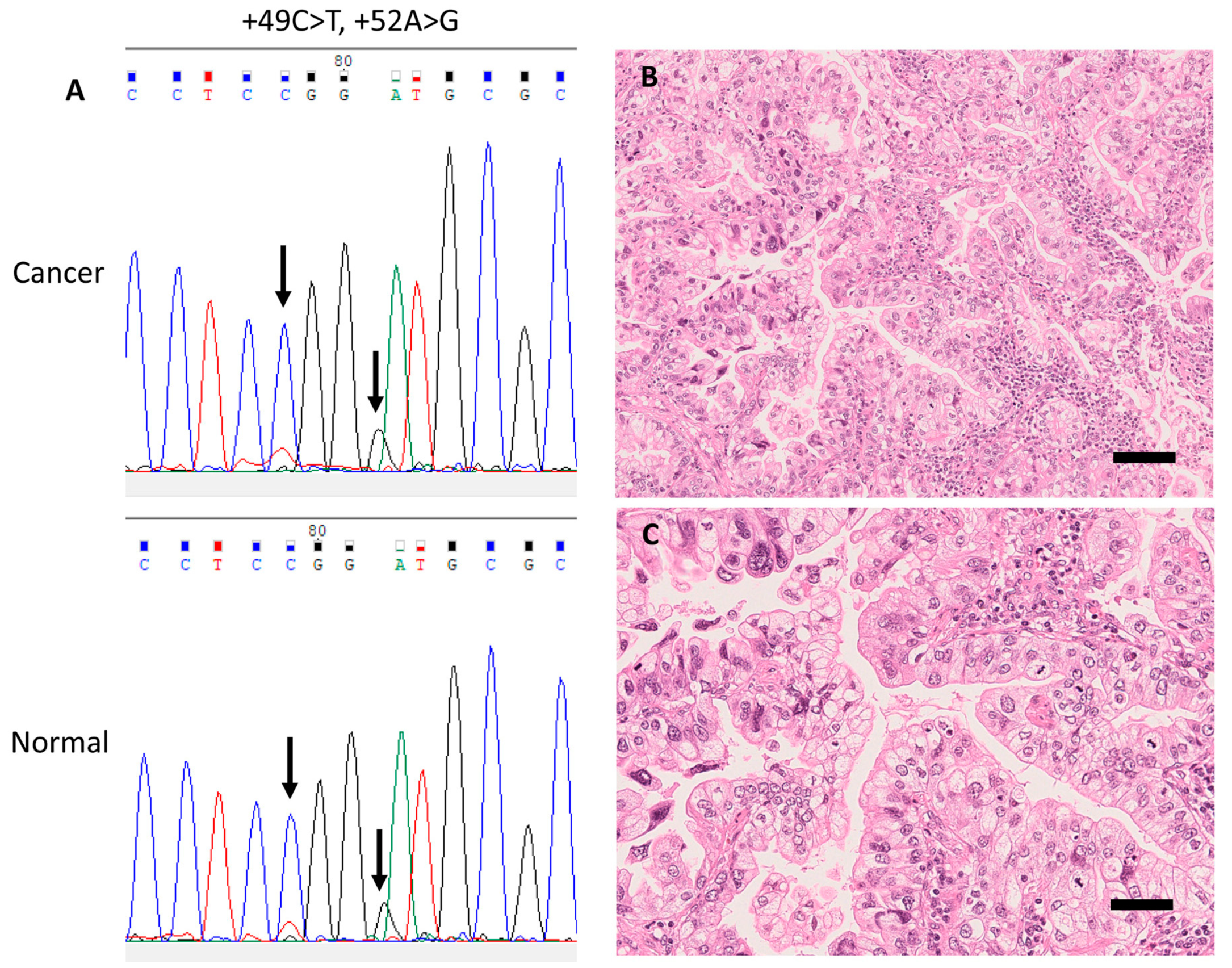
| 1 | IMA | F | 67 | IA2 | −206 1, +49, +52 | 35.3 | Deceased | |
| 2 | Acinar | M | 78 | IIB | +49 | −72 1 | 44.5 | Deceased |
| 3 | Pleomorphic + Solid | M | 68 | IIA | +73 | 63.3 | Deceased | |
| 4 | Acinar | M | 63 | IB | +52 | 124.5 | Deceased | |
| 5 | Papillary | F | 74 | IIB | +52 | 84.1 | Deceased | |
| 6 | Acinar | M | 71 | IA2 | +52 | 80.5 | Deceased | |
| 7 | Acinar (ASQ) | M | 67 | IB | +52 | 24.8 | Deceased | |
| 8 | Papillary | M | 79 | IA | +52 | 22.9 | Deceased | |
| 9 | Papillary | F | 71 | IA3 | +52 | 28.1 | Deceased | |
| 10 | MIA | F | 67 | IA1 | +52 | 143.4 | Alive | |
| 11 | Papillary | M | 62 | IB | +52 | 123.1 | Alive | |
| 12 | Solid | M | 66 | IIB | +52 | 115.6 | Alive | |
| 13 | Papillary | F | 57 | IB | +52 | 145.9 | Alive | |
| 14 | Papillary | M | 79 | IVB | +52 | 50.7 | Alive |
| Variable | Univariate | Multivariate 2 | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p | HR | 95% CI | p | |
| Proportional hazard model for recurrence-free survival | ||||||
| Age (>55/≤55) | n.s. | |||||
| Sex (male/female) | n.s. | |||||
| Smoking status (Former/never) | n.s. | |||||
| pN/N and/or cM (>1/0) 1 | 3.748 | 1.813–7.747 | <0.001 * | - | - | - |
| Stage (III–IV/I–II) | 4.802 | 2.208–10.45 | <0.001 * | 6.106 | 2.587–14.08 | <0.001 * |
| Histology (Invasive/MIA) 2 | 9.212 | 1.303–1166.69 | 0.019 * | 7.267 | 1.006–923.71 | 0.049 * |
| rDNA promoter SNV (Present/Absent) | 2.294 | 1.060–4.964 | 0.035 * | 3.764 | 1.584–8.589 | 0.004 * |
| Proportional hazard model for overall survival | ||||||
| Age (>55/≤55) | n.s. | |||||
| Sex (male/female) | n.s. | |||||
| Smoking status (Former/never) | n.s. | |||||
| pN/N and/or cM (>1/0) 1 | 3.607 | 1.690–7.701 | <0.001 * | - | - | - |
| Stage (III–IV/I–II) | 3.455 | 1.566–7.621 | 0.002 * | 4.745 | 1.951–11.27 | <0.001 * |
| Histology (Invasive/MIA) 2 | 8.097 | 1.140–1206.18 | 0.031 * | 7.192 | 0.985–915.93 | n.s. |
| rDNA promoter SNV (Present/Absent) | 2.995 | 1.360–6.596 | 0.006 * | 5.006 | 2.060–11.88 | <0.001 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohashi, R.; Umezu, H.; Sato, A.; Abé, T.; Kondo, S.; Daigo, K.; Sato, S.; Hara, N.; Miyashita, A.; Ikeuchi, T.; et al. Frequent Germline and Somatic Single Nucleotide Variants in the Promoter Region of the Ribosomal RNA Gene in Japanese Lung Adenocarcinoma Patients. Cells 2020, 9, 2409. https://doi.org/10.3390/cells9112409
Ohashi R, Umezu H, Sato A, Abé T, Kondo S, Daigo K, Sato S, Hara N, Miyashita A, Ikeuchi T, et al. Frequent Germline and Somatic Single Nucleotide Variants in the Promoter Region of the Ribosomal RNA Gene in Japanese Lung Adenocarcinoma Patients. Cells. 2020; 9(11):2409. https://doi.org/10.3390/cells9112409
Chicago/Turabian StyleOhashi, Riuko, Hajime Umezu, Ayako Sato, Tatsuya Abé, Shuhei Kondo, Kenji Daigo, Seijiro Sato, Norikazu Hara, Akinori Miyashita, Takeshi Ikeuchi, and et al. 2020. "Frequent Germline and Somatic Single Nucleotide Variants in the Promoter Region of the Ribosomal RNA Gene in Japanese Lung Adenocarcinoma Patients" Cells 9, no. 11: 2409. https://doi.org/10.3390/cells9112409