Clinical Importance of the Human Umbilical Artery Potassium Channels




1
CICS-UBI, Health Sciences Research Centre, University of Beira Interior, 6200-506 Covilhã, Portugal
2
FCS-UBI, Faculty of Health Sciences, Department of Medical Sciences, University of Beira Interior, 6200-506 Covilhã, Portugal
3
UDI-IPG, Research Unit for Inland Development, Department of Social Sciences and Communication, Polytechnic Institute of Guarda, 6300-654 Guarda, Portugal
*
Author to whom correspondence should be addressed.
Cells 2020, 9(9), 1956; https://doi.org/10.3390/cells9091956
Submission received: 15 July 2020
/
Revised: 21 August 2020
/
Accepted: 24 August 2020
/
Published: 25 August 2020
(This article belongs to the Special Issue MiNICAD Special Issue: Ion Channels and Human Diseases)
Abstract
:Potassium (K+) channels are usually predominant in the membranes of vascular smooth muscle cells (SMCs). These channels play an important role in regulating the membrane potential and vessel contractility—a role that depends on the vascular bed. Thus, the activity of K+ channels represents one of the main mechanisms regulating the vascular tone in physiological and pathophysiological conditions. Briefly, the activation of K+ channels in SMC leads to hyperpolarization and vasorelaxation, while its inhibition induces depolarization and consequent vascular contraction. Currently, there are four different types of K+ channels described in SMCs: voltage-dependent K+ (KV) channels, calcium-activated K+ (KCa) channels, inward rectifier K+ (Kir) channels, and 2-pore domain K+ (K2P) channels. Due to the fundamental role of K+ channels in excitable cells, these channels are promising therapeutic targets in clinical practice. Therefore, this review discusses the basic properties of the various types of K+ channels, including structure, cellular mechanisms that regulate their activity, and new advances in the development of activators and blockers of these channels. The vascular functions of these channels will be discussed with a focus on vascular SMCs of the human umbilical artery. Then, the clinical importance of K+ channels in the treatment and prevention of cardiovascular diseases during pregnancy, such as gestational hypertension and preeclampsia, will be explored.
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Potassium (K+) channels are usually predominant in the membranes of vascular smooth muscle cells (SMCs). These channels play an important role in regulating the resting membrane potential (MP) and vessel contractility—a role that depends on the vascular bed. Therefore, the physiological MP range in SMCs is essential to understand the K+ channel’s role in smooth muscle (SM) [1]. Overall, vascular SMCs have an MP ranging from −40 and −60 mV, depending on the type of blood vessel [2,3]. Thus, since MP is considerably more positive than the K+ equilibrium potential (EK; about −85 mV), it appears that the permeability of K+ does not completely dominate MP conductance. When EK is substantially more negative than MP, the opening of K+-channels induces the K+ efflux into the extracellular medium, which can cause hyperpolarization or repolarization of the cell membrane, closure of the voltage-gated Ca2+-channels (VGCCs), decrease in calcium (Ca2+)-entry into the cell, and vasodilatation. On the contrary, the closing of K+-channels can cause depolarization, opening of VGCCs, increase of intracellular Ca2+, and vasoconstriction [1,2,4,5,6,7,8].
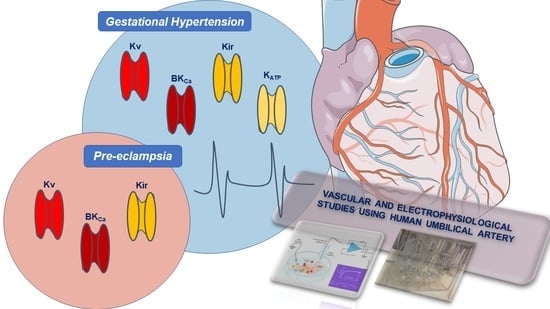
In the vascular SMCs, the presence of the various K+-channels varies and there is no homogeneous distribution. For example, there are 100–500 ATP-sensitive K+ (KATP) and inward rectifier K+ (Kir) channels per cell, while voltage-dependent K+ (KV) and Ca2+-activated K+ (KCa) channels are around 1000 to 10,000 per cell. Moreover, the electrophysiological measurements using the patch-clamp technique showed that the amplitudes of the basal potassium currents in each channel are very different [1,5]. Currently, there are four different types of K+ channels described in SMCs from the human umbilical artery: KV, KCa, Kir, and 2-pore domain K+ (K2P) channels (Figure 1) [9,10]. However, the presence of intermediate-conductance Ca2+-activated K+ channels (IKCa), one subtype of KCa, has never been demonstrated [9,10].
The human umbilical artery (HUA) is a unique vessel that transports deoxygenated blood. This artery does not have nerve endings, so the regulation of its tonus is completely dependent on the locally released vasoactive mediators, substances carried by the bloodstream, and some ions, such as K+ and Ca2+ [11,12]. Therefore, HUA is much more susceptible to hemodynamic changes caused by vascular disorders [13]. Moreover, its SMCs appear to play an important role in the control of fetoplacental blood flow. Moreover, vascular SMCs are responsible for the vascular tone by responding to various hormonal and hemodynamic stimuli [9,14]. In this sense, being a fetal structure, the umbilical cord (UC) can be used as a window to learn about maternal dysfunctions and their impacts on fetal well-being [13], namely, regarding HUA K+-channels. The elucidation of the exact mechanisms regulating HUA contractile status is extremely important to detect potential targets for the treatment of pregnancy-related pathologies, mainly gestational hypertension and preeclampsia.
Therefore, this review discusses the basic properties of the various types of K+-channels, including the structure and cellular mechanisms that regulate their activity and new advances in the development of activators and blockers of these channels. The vascular functions of these channels will be discussed with a focus on vascular human umbilical artery smooth muscle cells (HUASMCs). Then, the clinical importance of K+-channels in the treatment and prevention of cardiovascular diseases (CVDs) during pregnancy, such as gestational hypertension and preeclampsia, will be explored.
2. The Importance of the K+ Channels in Physiological Regulation of HUA
Ion flux is one of the pathways responsible for regulating the arterial SM tonus. In this process, the activation of K+-channels plays a crucial role because their activation is mainly responsible for SM relaxation [7,15]. The K+ channels are the most abundant in SMCs and play a crucial role in the regulation of MP because they directly control K+ concentrations and indirectly control Ca2+ concentrations [1]. More specifically, the activation of these channels in vascular SMCs hyperpolarizes the cell membrane, inducing the closure of VGCC and thus decreasing the intracellular Ca2+ concentration ([Ca2+]i), giving rise to a vasorelaxation [2,4,16]. In addition, this channel activation can be originated through some vasoactive substances (e.g., nitric oxide (NO), natriuretic peptides (NP), and prostacyclins), which can activate kinase proteins [17].
Focusing on the HUA K+ channels, the presence of KV, large-conductance Ca2+-activated K+ channels (BKCa), small-conductance Ca2+-activated K+ (SKCa), and Kir, KATP, and K2P channels was demonstrated. On the other hand, the presence of IKCa channels has never been reported [15]. In addition, in HUA, the activation of K+-channels is one of the main mechanisms involved in its vasorelaxation. The Kv channels regulate MP in response to membrane depolarization, while BKCa channels respond to changes in intracellular Ca2+ levels [15,18,19,20,21]. The activity of the several types of K+-channels can be altered by several physiological factors, including intracellular Ca2+, cyclic nucleotides, and various signal transduction mechanisms. In HUA, the cyclic guanosine monophosphate (cGMP)-induced vasodilatation is mediated by K+-channels activation [9], and this effect is lower when HUA is contracted by potassium chloride (60 mmol/L) or serotonin [22]. Additionally, Cairrao et al. (2010) demonstrated that K+ currents (IK) are stimulated by NP, and this effect corresponds to the activation of BKCa and Kv channels through an increase of cGMP and protein kinase G (PKG) activation [23]. The same authors also observed that K+ currents (IK) are not stimulated by NO donors. In this sense, they concluded that the activation of BKCa and Kv channels is dependent on differences in the spatiotemporal distribution of intracellular cGMP. These results suggest that when a cyclase is activated, besides its hydrosolubility, intracellular cGMP is not uniformly distributed within the cell, and it is probably clustered in specific sites. Moreover, the authors found that phosphodiesterases (PDE; e.g., PDE3 and PDE5) are involved in this compartmentalization, regulating the pools of particulate and cytosolic cGMP. These results are suggestive that the particulate pool but not the soluble one, seems to be controlled by PDE5, while PDE3 appears to control the soluble cGMP pool exclusively [24,25]. Thus, these results are extremely important for the development of new drugs, since we can control the effect on the ion channels not only with drugs that act in K+ channels, but also with drugs that act in PKG and PDE.
In summary, further studies on the functional role and the expression of K+-channels in HUA are needed to clarify the mechanisms involved in relaxation. The expression and functionality of these channels may change in vascular- or pregnancy-related pathologies. Therefore, further studies are needed to understand how K+-channels can be an important pharmacological target in the development of new treatments, mainly in pregnancy hypertensive disorders.
3. Diversity of K+ Channels in HUA
3.1. Voltage-Dependent K+ (Kv) Channels
The voltage-dependent K+ channels, or Kv channels, are channels present in SMCs that are activated by depolarization. When a depolarization of SMCs occurs, even if small, there is an activation of L-type VGCCs, which leads to an increase in the Ca2+-influx and stimulation of the contractile apparatus. However, the influx of Ca2+ through L-type VGCCs associated with membrane depolarization can also inhibit Kv channels. There is the reason for why the activity of the Kv channels is important in regulating the excitability of SMCs and maintaining basal tone [2,7,20,26].
At the structural level, the Kv channels consist of four α-subunits, each containing six transmembrane α-helical segments, S1–S6, and a membrane-reentering P-loop (P) [27]. This ion-conduction pore is lined by four S5-P-S6 sequences, while the four S1–S4 segments, each containing positively charged amino acid residues (arginine and lysine) in membrane-spanning domain S4, detect changes in membrane potential and work as the voltage sensor [27,28,29]. Accessory subunits interact with the pore-forming α-subunits and modulate channel function and interactions with scaffolding and other proteins in macromolecular signaling complexes [2]. Kv channels are a family of 40 proteins classified into 12 subtypes (KV1-12) that can assemble as homo- or heterotetramers. This diversity generates a vast array of Kv current phenotypes that can differ in their kinetics, amplitude, and responses to different modulators. Tissue- and species-specific expression of Kv channel isoforms have also been described, which further contributes to Kv current heterogeneity [30]. The presence of several families of Kv channels has been demonstrated in vascular SMCs, namely, KV1, KV2, KV3, KV4, KV6, KV7, KV9, and KV11, with KV1 (KV1.1, KV1.2, KV1.3, KV1.5, KV1.6), -2 (KV2.1), and -7 (KV 7.1–7.5) being particularly important (see review in [2]). Moreover, the surface abundance of Kv channels in arterial SMCs is regulated by several mechanisms (see review in [30]).
At the pharmacological level, Jackson et al. (2018) [2] and Dogan et al. (2019) [1]) have recently extensively reviewed the pharmacology of Kv channels in vascular SMCs. Briefly, the main Kv blockers are 4-aminopyridine (4-AP), tetraethylammonium (TEA), barium ion (Ba2+), glibenclamide, margatoxin, charybdotoxin, diphenylphosphine oxide-1 linopirdine, XE991, and HMR1556, of which the most widely used is 4-AP. Regarding the activators/agonists, there are only a few, and they are specific to Kv7 and Kv11 (please see the review in [2]).
Regarding the Kv functional presence in HUA, it seems that these channels are not involved in determining basal contractile since the Kv blocker 4-AP had no effect on basal tonus of the HUA rings [18,21]. However, the relaxations induced by NO [31], testosterone [21], or levosimendan [32] are partially inhibited by 4-AP in HUA rings contracted with thromboxane mimetic U46619, serotonin, histamine, and KCl 60 mmol/L. Furthermore, Milesi et al. (2003), by patch-clamp recordings, demonstrated that IK was partially inhibited by 4-AP [18]. Using a depolarizing voltage step protocol, the authors determined the presence of members of the Kv family sensitive to 4-AP [18]. Accordingly, our research group reported similar results in primary cultured SMCs obtained from HUA. IK is stimulated by atrial natriuretic peptide (ANP), and this effect corresponds to the activation of BKCa and Kv channels through an increase of cGMP and PKG activation [23] (Figure 2). Recently, Martin et al. (2014) demonstrated the existence of members of Kv subfamilies in HUASMCs, namely, KV1 (except KV1.4, which presents inactivation), KV2, or KV3 [15]. However, the authors highlighted the importance of more studies with specific blockers to identify the different types of Kv channels.
In summary, the Kv channels are the main determinants of vascular tone, and their presence in SMCs and HUASMCs is demonstrated. However, some differences at the family level were found. Members of the Kv subfamilies (Kv1, Kv2, Kv3, Kv4, Kv6, Kv7, Kv9, and Kv11) were found in SMCs, while in HUASMCs, it was only possible to identify the subfamilies (Kv1, Kv2, and Kv3). Thus, the presence and importance of Kv1 in HUASMCs make this subfamily of channels the main candidate for pharmacological studies in the fight against vascular diseases, mainly the hypertensive disorders of pregnancy. In this sense, the blocker 4-aminopyridine (4-AP) is the most used, but new activators of these channels have also been introduced to the market. Due to the importance of these channels, and the vast knowledge that exists about them, it is clear that modulation of Kv channels is a promising alternative for the treatment of vascular diseases, but it also plays a crucial role in the clinical practice of obstetrics and gynecology as a new pharmacological approach.
3.2. Calcium-Activated K+ (Kca) Channels
The KCa channels are important effectors in the control of vascular tone and blood pressure [33]. They mediate membrane hyperpolarization in response to elevations of intracellular Ca2+ and thus counteract SM contractility [34]. To date, three broad categories of KCa channels have been identified and classified according to their conductance [35]: large-conductance KCa channels, 100–300 pS (BKCa) [36], intermediate-conductance KCa channels, 25–100 pS (IKCa) [34,37], and small-conductance KCa channels, 2–25 pS (SKCa) [34,38,39]. Each of them has important physiological and pathophysiological functions important to the CV system. BKCa channels are predominantly expressed in vascular SMCs and the inner mitochondrial membrane of cardiomyocytes. The activation of these channels leads to vasodilation and cardioprotection in situations such as cardiac ischemia. Concerning SKCa, its channels can be largely found in the nervous and CV systems. Their activation is essential for the hyperpolarization of the cell membrane. The IKCa channels are expressed in SMCs and cardiac fibroblasts but are predominantly expressed in ECs and proliferating vascular SMCs. Their functions include proliferation, migration, vasodilation of SMCs, and cardiac fibrosis [35].
In the vascular SMCs, the dominant KCa channels are the BKCa channels (also designated BK, Slo, or MaxiK channels due to their primordial contribution to vascular tone control [40,41]. Its activation begins with the depolarization of the membrane and intracellular binding of Ca2+ and Mg2+ ions. On the other hand, these channels can also contribute to MP modulation in small myogenic vessels [5,16,20,42,43]. After the activation of BKCa channels, the cell membrane hyperpolarizes and triggers VGCC closure—a role that is opposite to that of these Ca2+-channels in vasoconstriction [41,44]. Some investigations have shown that the BKCa channels are activated by highly localized Ca2+ sparks produced by transient Ca2+-release events from the sarcoplasmic reticulum and/or Ca2+-influx through VGCCs. The resulting spontaneous transient outward currents (STOCs) produce hyperpolarization/repolarization and thereby counteract depolarization-activated VGCC activity. As a consequence of the VGCC inactivation, there is a decrease in [Ca2+]i, with consequence inactivation of myosin light-chain kinase, thus leading to relaxation [44,45,46,47,48].
Therefore, the BKCa channels have the function of regulating membrane voltage and [Ca2+]i, a function that is regulated by a diverse set of vasodilators and vasoconstrictors. In partnership with other ion channels, kinases, phosphatases, and regulatory proteins, the BKCa channels appear to be organized into macromolecular complexes that facilitate rapid and specific activation through different signal transduction pathways [41]. Some SM vasodilators, such as β2-adrenergic agonists, NO, prostaglandin I2, arachidonic acid, calcitonin gene-related peptide, and carbon monoxide, can activate these channels [5,48,49]. On the other hand, potent vasoconstrictors, such as angiotensin II, 5-HT, phenylephrine, and thromboxane A2, can inhibit them [48,49,50,51]. Overall, most agonist actions involve intracellular signaling pathways that include (1) Ca2+ sparks, which appear to be involved in vasodilator action through a negative feedback mechanism, (2) kinases that phosphorylate the channel protein, and (3) G-proteins that regulate channel activity independently of downstream kinases [48,49]. It has also been shown that BKCa channels can play a contributory role in altering the vascular tone in pathophysiological states (e.g., hypertension, stroke, atherosclerosis, diabetes, and CV surgery complications), and that is why they are also possible therapeutic targets in the treatment of these CVDs [35,41,52,53].
At the structural level, the examination of BKCa channel protein has been carried out by both X-ray crystallography and cryoelectronic microscopy [54,55,56]. These channels have a pore-forming α-subunit and a regulatory β-subunit [20,39,57]. The α-subunits contain six membrane-spanning domains (S1–S6), including voltage sensing (S1–S4; designated as VSD) and pore-gate (S5–S6, designated as PGD) domains [58]. However, the α-subunits, which are produced from a single gene (slo) by alternative splicing [20,59,60], contain an additional seventh transmembrane region (S0) that confers the N-terminus to the extracellular side and a large cytoplasmic domain containing ~800 amino acids [48,58]. Moreover, there are four β-subunit isoforms (β1–4), each with two transmembrane domains that may be associated with the α-subunits in a 1:1 ratio [43,61,62]. Among the four β isoforms, the β1 subunit is predominant in vascular SM [20,43,63,64]. The major function of the β-subunits is to enhance the Ca2+ sensitivity of the channel, and thus increase STOC activity and repolarizations and thereby facilitate relaxations [35,44]. The properties of the channels can be further modified by association with γ-subunits [52,65]. Concerning SKCa and IKCa, the main difference with the BKCa channel is that the Ca2+ sensor of SKCa and IKCa channels is calmodulin (CaM), which is constitutively bound to the calmodulin-binding domain (CaM-BD) in the C terminus and thus functions as a β-subunit that endows these channels with Ca2+ sensitivity [34].
At the pharmacological level, BKCa channels depend on their association with auxiliary subunits. The BKCa blockers are TEA (<1 mM), charybdotoxin, iberiotoxin, penitrem A, and paxillin, but only the last three compounds are selective for the BKCa channels [1,39]. On the other hand, these channels can be activated by agents that are called BKCa openers or BKCa activators. The BKCa openers comprise a large series of synthetic benzimidalone derivatives (e.g., NS1619 and NS004), biaryl amines (e.g., mefenamic and flufenamic acids), biarylureas (e.g., NS1609), pyridyl amines, natural modulators (e.g., resveratrol and dedydrosoyasaponin-1 (DHS-1)), and flavonoids [39,52]. Concerning SKCa and IKCa, the neurotoxin apamin, a fully selective inhibitor of SKCa and IKCa, can be blocked by charybdotoxin and the small molecule TRAM-34 [52].
Regarding the presence of KCa in HUA, Radenkovic et al. (2007) demonstrated that the SKCa channels appear to be present and functional at HUA [66]. However, Martin et al. (2014) suggested that SKCa appears to be in endothelial cells (ECs) and not SMCs as they did not find any small or intermediate conductance activated by an increase in (Ca2+)i [15]. However, if SKCa channels are present in HUASMCs, their role does not seem to be very relevant since NO-induced relaxation did not activate these channels [31], and they also did not contribute to IK in HUASMCs [18,23]. Regarding the role of IKCa channels in HUA, their presence has not been demonstrated. On the other hand, BKCa are fundamental in regulating the contractile state of HUA. Milesi et al. (2003) were among the first authors to demonstrate the presence of these channels in HUA. The authors demonstrated that TEA (5 mmol/L) induced contraction of HUA, and that chloretin (50µmol/L), an activator of BKCa channels, induced relaxation of them [18]. According to this investigation, other authors have also suggested the involvement of BKCa in the relaxation of HUA induced by NO [31], testosterone [21,23,67], levosimendan [32], and (-)-carveol [68] since this relaxation was inhibited by TEA. Additionally, Lovren et al. (2000) [31] demonstrated that BKCa channels are activated by NO at the single-channel level through a mechanism that probably involves PKG activation [69]. This hypothesis was confirmed years later by Cairrao et al. (2010), who demonstrated that the relaxation induced by testosterone in HUA is due to the opening of the BKCa channels through PKG-dependent action that is activated by increasing the intracellular levels of cGMP [23]. The electrophysiological experiments performed by the authors demonstrated reductions in IK in the presence of TEA 100 µmol/L (35.9%) and iberiotoxin 0.1 µmol/L (36.4%) [23]. These findings are also supported by electrophysiological studies in fresh HUASMCs, in which reductions in IK in the presence of 1 mmol/L TEA (±71%), 200 nmol/L iberiotoxin (±65%) [18], and paxilin 500 nmol/L [70] (±85%) were observed. The characterization of BKCa properties at the single-channel level was also in line with the results of previous investigations [18,70]. Thus, these results show that IK in HUASMCs are mainly constituted by K+ exit through BKCa channels, together with Kv channels [15,23] (Figure 2). Overall, the authors found that the BKCa channels have all the biophysical properties typical of these channels in other tissues (for example, high selectivity to K+ or an increase in the probability of opening due to membrane depolarization and increased (Ca2+)i [15].
In summary, there are some differences between the presence of the KCa channels in SMCs and HUASMCs. Concerning the SKCa channels, they are present in SMCs [35] and HUA. However, it is not yet known whether the SKCa channels are in ECs or SMCs of the artery, but the evidence seems to point to a location at the endothelial level. Regarding IKCa channels, the investigations demonstrate the presence of these channels in proliferative SMCs [35], but there is no scientific evidence in the literature to report their presence in HUASMCs. In contrast, concerning BKCa channels, the studies clearly demonstrate the presence of these channels in SMCs and HUASMCs, and their role in regulating vascular tone is well established. While the mechanisms by which BKCa channels contribute to vascular dysfunction are still unclear, on the other hand, their role in clinical practice is well known. In this sense, the BKCa channels are the more promising type of KCa channels that can be used as pharmacological targets for the prevention or reversal of depolarization, contraction, and maintenance of resting tone. Like Kv channels, there are several blockers identified and the modulation of these channels can be a promising alternative for the treatment of vascular diseases. At HUA, there is no certainty about the presence of SKCa and IKCa. However, if the presence of these channels is proven in HUA, it is possible that their physiological relevance will not be significant and, therefore, their pharmacological importance will be small. Thus, in the development of a new pharmacological approach, BKCa channels should be the main therapeutic target to be used, for example, for the treatment of hypertensive diseases of pregnancy.
3.3. Inward Rectifier K+ (Kir) Channels
The inward rectifier K+ (Kir) channels are a “special” class of K+ channels that can regulate an immensity of physiological processes since they control MP of excitable and nonexcitable cells [71]. At the functional level, these channels act as biological diodes as they are the only ones that can mediate the inward K+ flow by hyperpolarizing membrane voltages more quickly than the outward K+ flow at depolarizing voltages [72]. For a driving force of a given magnitude, the inward flow of K+ ions is greater than the outward flow. Hence, in these channels, the K+ flow can enter the cell (K+ circulates in the opposite direction). In this sense, the functional role of the Kir channels is critically dependent on their degree of rectification [7,73,74]. However, other K+ channels can also carry inward currents when Em < EK, just like Kir channels. These channels are not activated by MP and are not controlled by voltage-gated channels. Instead, its activation is dependent on the difference between MP and EK [7,75]. Some studies have strongly pointed out that the physiological role of these channels in vascular SM is related to MP maintenance and resting tone in small diameter coronary and cerebral arteries [76]. Kir channels were first described about 30 years ago in vascular SMCs as being present in only certain small-diameter cerebral and submucosal arterial SM and in coronary arterial SM [76].
Structurally, Kir channels have four subunits forming two transmembrane (TM) spanning helices, called TM1 and TM2, that are connected to the pore region containing the selectivity filter for K+ ions, with intracellular C- and N-terminal domains. [77,78]. TM1 (the outer helix) makes contact with TM2 (the inner helix), which lines the pore of the channel and forms an “inverted tepee” in a closed channel conformation [1,75,79]. These channels are encoded by members of the Kir gene family. The most relevant Kir expressed in the vasculature belong to the Kir2.x (strong inward rectifier), and K6.x (weakly inward rectifier) subfamilies [1]. Four isoforms of Kir2.x family have been identified, Kir2.1 to Kir2.4, however in vascular SMCs, only Kir2.1, 2.2, and 2.4 have been identified [20]. Kir2.2 has only been detected in cerebral arteries [80], and Kir2.4 expression has only been detected in SMCs of the human pulmonary artery [81]. In vascular SMCs, Kir2.1 is the dominant isoform expressed, particularly in mice cerebral, coronary, and mesenteric arteries [1,8,82]. Additional studies are required to characterize the expression and function of each Kir subtype.
At the pharmacological level, Kir channels can be blocked by extracellular Ba2+ and cesium (Cs+) ions, in addition to small molecule inhibitors being developed for specific channels. Modulation by other factors, including magnesium, extracellular K+ concentration, intracellular H+, ATP, phosphorylation by protein kinases A and C (PKA and PKC), and protein–protein interactions, is distinct to each Kir subfamily [1,8,78,83].
Regarding the Kir in HUA, V. Milesi has unpublished results that show that the blocker of Kir channels, Ba2+ (500 µmol/L), induces contraction of unstimulated HUA rings, which suggests that Kir channels may be involved in the maintenance of the basal contractile state of HUA. However, another study by Radenkovic et al. (2007) showed that the same blocker, Ba2+ (3 µmol/L), did not affect the top of bradykinin contractions of HUA rings coming from normal pregnancies, suggesting, on the contrary, that Kir channels are not involved in this mechanism [66]. However, the same authors found that this agent increased the bradykinin-induced strength in those HUA rings from pregnant women with gestational diabetes mellitus, which emphasized the clinical importance of the different K+ channels in pathological situations in pregnancy. More recently, Zhu et al. (2016) observed in SMCs from rat umbilical vein by patch-clamp that barium chloride did not affect IK [84].
In summary, Kir channels are important in MP control, and their presence has been demonstrated in SMCs, with the Kir2.1 isoform being the most dominantly expressed. However, concerning the Kir channels in HUA, the studies seem to suggest their presence, but the studies carried out on HUA are very few, and, therefore, there is insufficient scientific evidence to classify these channels as potential pharmacological targets. Thus, further studies are needed to clarify its role in clinical practice. Since the initial approaches involved the use of the Ba2+ ion, it is suggested that other methodologies, such as protein kinase modulation, for example, be approached in future studies.
ATP-Sensitive K+ (KATP) Channels
The ATP-sensitive K+ (KATP) channels were first identified in the cardiac muscle in 1983 [85,86]. After that time, the presence of these channels was also identified in other cells, including vascular SMCs [1,5,7,20,87,88,89], and its role has been studied at different levels, including CVDs [90]. The KATP channels belong to the Kir superfamily and conduct weak inward rectifier IK [90]. Overall, KATP channels act as metabolic sensors and maintain homeostasis in acute metabolic changes, including hyperglycemia, hypoglycemia, ischemia, and hypoxia [91]. Therefore, the KATP channels are designated as having an ability to decrease its activity when the intracellular concentration of ATP increases. This activity, interestingly, is modulated by several signaling pathways that are independent of ATP sensitivity [7].
At the structural level, the KATP channels are a macromolecular complex (~900 kDa) consisting of 4 pore-forming subunits (Kir6.1 encoded by KCNJ8 or Kir6.2 encoded by KCNJ11) and 4 regulatory sulfonylurea receptor (SUR) ATP-binding cassette subunits (subfamily C: SUR1, SUR2A, or SUR2B). Different combinations of Kir6.1 or Kir6.2 and SUR1 or SUR2 variants (SUR2A or SUR2B) in cell lines constitute KATP channels with distinct electrophysiological and pharmacological properties that correspond to the various KATP channels in native tissues [91]. In vascular SM, the combination Kir6.2/SUR2B is likely the most widespread, although Kir6.1/SUR2B may also be present in this tissue [92,93]. KATP channels are inhibited by ATP and ADP (channels close) and activated by Mg-ADP and Mg-ATP (channels increase activity), allowing the cell to engage the cellular metabolic state (ATP/ADP ratio) to electrical activity of the cell membrane [90,94].
At the pharmacological level, KATP channels are blocked by antidiabetic sulfonylurea drugs such as tolbutamide, glibenclamide, and glimepiride [95]. Glibenclamide is the most frequently used inhibitor in studies performed in arterial SMCs and tissues. Some artery contractility studies have elucidated that the half-inhibitory concentration of glibenclamide oscillates between 20 and 200 nM [5,20], and, for tolbutamide, the half-inhibitory concentration is 350 µmol/L [20]. External Ba2+ can also block the KATP channels, with a half-inhibitory concentration of 100 µmol/L at –80 mV [5,20]. Furthermore, 4-AP also induced a small inhibition (10%) at 1 mmol/L [1]. Several antihypertensive drugs seem to act through K+-channel activation and have been designated as K+-channel openers. These openers exhibit extreme chemical diversity and comprise a few different structural classes. The most used are nicorandil, cromakalim, pinacidil, minoxidil sulfate, aprikalim, diazoxide, and lemakalim [95].
Regarding the KATP in HUA, currently, there is still no consensus of their participation in the contraction/relaxation of intact HUA rings [15,21,66]. Several studies have shown that the KATP blocker glibenclamide (1 and 10 µmol/L) did not affect HUA rings when applied on top of bradykinin [66], histamine, KCl, or serotonin-induced contractions [21]. Moreover, glibenclamide (10 µmol/L) did not modify NO- or levosimendan-induced relaxations, ruling out the participation of KATP channels in such contractile and relaxing mechanisms [31,32]. In contrast, glibenclamide partially impaired relaxation in the human umbilical vein (HUV) rings caused by sodium sulfide (Na2S), suggesting that part of the relaxation is due to the increased open time of the KATP channels [96]. Concerning IK in HUASMCs, it has been reported by whole-cell patch-clamp techniques that KATP channels do not contribute to the total current because glibenclamide does not affect IK [23]. Furthermore, Zhu et al. (2016) also observed on SMCs from rat umbilical vein by patch-clamp that glibenclamide did not affect IK [84]. In contrast, Bai et al. (2013) noted that when using a high K+ concentration (140 mmol/L) and perfusing a K+-channel-specific opener, pinacidil, the IK increased [97]. More recently, Li et al. (2018), using the whole-cell patch-clamp technique, also demonstrated in HUASMCs that the application of pinacidil (10 µmol/L) increased IK, but glibenclamide (10 µmol/L) completely inhibited the K+ resulting from the application of pinacidil [88]. The authors also identified mRNA expression levels of Kir6.1, Kir6.2, and SUR2B subunits in these cells [88].
In summary, KATP channels play an important role in regulating vascular tone, and their presence has been demonstrated in SMCs and HUASMCs. Moreover, differences in subunits were not found since, in both cell types, levels of mRNA expression of Kir6.1, Kir6.2, and SUR2B subunits were found. At the pharmacological level, the blocker glibenclamide is the most used, including in studies on HUA. However, the role of KATP channels in the physiological regulation of HUA is not clear, and, therefore, the role of these channels in clinical practice is still unknown. More pharmacological studies are needed, but it is assumed that due to little physiological relevance, the role of KATP at the pharmacological level will not be relevant.
3.4. The 2-Pore Domain K+ (K2P) Channels
The 2-pore domain K+ (K2P) channels were first identified in the late 1990s [98,99]. This channel family produces leak-type K+ currents [100] and are involved in controlling and stabilizing MP and the levels of cellular excitability [1,100,101].
Structurally, K2P channels are characterized by having two pore-forming regions (P1–P2) in each channel subunit, surrounded by four transmembrane-spanning (4TMS) helices and a large “cap” domain. Moreover, these channels are different from the remaining K+ channels because they are the smallest, and they do not form tetrameters but functional dimers [1,100,102]. The K2P channels elicit spontaneously active and outwardly rectifying “background leak”-type K+ conductance. However, these channels have no classical time-dependent or voltage-dependent activity [103,104,105,106,107,108].
Currently, in mammals, 15 different K2P channels have been described/identified, each with important physiological roles at different levels. These 15 channels can be divided into six families, according to their biophysical and pharmacological properties [103,104,106,107,109]. The TWIK2 (tandem of P-domains in a weakly inward rectifying K+ channel, K2P6.1), TASK-1 (TWIK-related acid-sensitive K+ channel, K2P3.1), and TREK-1 (TWIK-related K+ channel) families are the ones that presented the highest levels of expression in SMCs [110,111,112]. Unlike most K2P, TWIK2 are highly expressed in all blood vessels studied so far (including resistance-sized vessels), which makes K2P6.1 the main candidate for physiological regulation in the vascular system [111,113]. On the other hand, the contribution of TASK-1 to the regulation of vascular tone is less understood, and it, therefore, needs further investigation [110,114,115].
At the pharmacological level, the K2P channels are very weakly sensitive or insensitive to classical K+ channel blockers (e.g., TEA, Ba2+, Cs+, and 4-AP). Instead, two external blockers of these K+ channels in arterial SM, anandamide 10 µmol/L [114] and zinc ion (Zn+2) 100 µmol/L [114,116], and two openers, halothane [114] and ONO-RS-082 [117], have been reported. Moreover, these channels are sensitive to important biophysical stimuli (e.g., pH, temperature, and mechanical pressure), and some of them are inhibited by the acidification of extracellular fluid [1,100,116,118]. The K2P channels are also targets for drugs (including inhalational and local anesthetics, antipsychotics, antidepressants, and neuroprotective agents), and modulated by G protein signaling pathways and second messengers [103,104,106,107,119,120]. Alternate splicing [121,122,123,124], alternative translation initiation [125], and heterodimerization of K2P channel subunits [126,127,128] make studying these channels even more complex.
To our knowledge, there is no definitive evidence concerning the presence of K2P channels in HUA, although their presence has already been demonstrated in other vessels. However, the fact that there are no specific blockers for these channels makes it difficult to study them in more detail [15]. Martin et al. (2014) reported detecting mRNA for TRAAK and TREK1 in HUASMCs but stressed the need for functional and more specific characterization to confirm its presence. In the same sense, it has been hypothesized that K2P channels are also present in placental vessels, but the evidence is still preliminary [15]. Wareing et al. (2006) detected TASK1 mRNA in the arteries of chorionic plaque and a possible blockade of the channels with anandamide, but these authors also concluded the need for further investigations to be developed, mainly due to the nonspecificity of the compound [129].
In summary, the K2P channels are the most recently identified channels in SMCs. Its physiological role in SMCs is to control and stabilize MP and levels of cellular excitability. Members of the subfamilies of weak inward rectifiers (TWIK-2), acid-sensitive rectifiers (TASK-1), and lipid sensitive mechano-gated channels (TREK-1) have been identified in SMCs. However, in HUASMCs, only the existence of TREK-1 and TRAAK, both belonging to the subfamily of lipid sensitive mechano-gated channels, has been suggested. The fact that there are no specific blockers for these channels makes it difficult to study them in more detail and prove their existence in HUA. In this sense, the next step will be the development of new studies to find specific blockers/activators for these channels. A focus on TREK-1 channels is suggested to find out if these channels can be a pharmacological target in the treatment of vascular diseases.
4. Clinical Importance and Medical Uses of K+-Channels
Gestational hypertension and preeclampsia are hypertensive disorders of pregnancy [130] that affect 10% of all pregnancies in the world [131,132]. Hypertension is the most common medical problem encountered during pregnancy (6–20% of all pregnancies), and it is highly responsible for maternal and perinatal mortality and morbidity. In addition, it is associated with an increased risk of developing CVDs in pregnant women [13]. On the other hand, pre-eclampsia comprises 3–5% of hypertensive disorders in pregnancy. According to Fox et al. (2019), pre-eclampsia (PE) is defined as “new-onset hypertension after 20 weeks’ gestation, with evidence of maternal organ or uteroplacental dysfunction or proteinuria”. This disorder is a major cause of maternal morbidity and has many adverse effects associated with fetal health. There is evidence that the effects of PE go beyond fetal health and may be reflected in the long term by sequelae in the offspring’s CV system (such as hypertension and altered vascular function) [132,133].
As described above, K+-channels are extremely important for the control of HUA vascular tonus, regulating SMC membrane potential [1]. Due to its importance, it is not surprising that its molecular composition, activity, and expression are crucial parameters to know. Particularly, this issue assumes greater relevance when it comes to vascular pathologies or being associated with pregnancy, in which changes in the expression and function of these channels in SMCs have already been reported.
Starting with Kv channels, changes in the properties and functions of these channels in hypertensive vascular SMCs were reported [134,135]. These changes can lead to vasoconstriction that alters cellular mechanisms and culminates in hypertension [5]. Moreover, studies in mesenteric arteries of several hypertensive animals have shown that the expression of the α-subunit gene and the protein in these channels were elevated [134]. Additionally, studies performed in hypertensive vascular SMCs have shown that the Kv currents were lower than those in BKCa [136,137] and that this decrease had intracellular Ca2+ as the main player [134]. The same authors also demonstrated, by electrophysiological studies, that Ca2+ can be regulated by Kv channels during hypertension since an increase in protein expression seemed to be related to a decrease in functionality [136,137]. This decrease may underlie the subsequent depolarization of the membrane and, therefore, a consequent increase in vascular tonus observed in this pathology [16]. In the last year, Djokic et al. (2019) also demonstrated that during gestational hypertension, the function of the Kv channels was compromised as they observed an absence of inhibition of the functional response to pinacidil (KATP channel opener) [138]. The authors also demonstrated that 4-AP significantly decreases vasorelaxation induced by pinacidil in vascular SMCs of HUV from normotensive pregnant subjects, but not in hypertensive pregnant subjects [138]. Furthermore, the same authors reported in this year (2020) that in hypertension situations, there is a decrease in Kv1.3 channel expression and function in SM of HUV [139]. Concerning PE, studies in pregnancies complicated by uterine growth restriction have also shown that venous contractility, but not the arterial contraction of chorionic plaque arteries (CPA), was increased. The nonspecific Kv1-4 channel inhibitor (4-AP) induced contraction less effectively compared to normal pregnancies, which suggests that the activity of Kv channels is decreased in PE situations [140,141]. Indeed, studies have shown that Kv7 channels are involved in the regulation of vascular tone in CPA during normal pregnancy [142,143]. However, some authors have reported that the protein of the Kv channels (KCNQ3 and KCNE5) is significantly increased in the placenta of women with PE, while KCNQ4 and KCNQ5 appeared to be downregulated [142,144]. On the other hand, the loss of function of the Kv channels is linked to a series of lethal or debilitating channelopathies. New pharmacological approaches have been developed to selectively activate specific Kv channels [145]. In HUA, only the Kv1, 2, and 3 channels exist. However, currently, the only approved drugs are retigabine, an antiepileptic and anticonvulsant, which activates Kv7 channels in the brain, but which was discontinued in 2017 due to serious side-effects, including retention urinary, bluish skin, and retinal discoloration, and flupirtine, which also shows antiepileptic activity and acts by activating the selected Kv7 channels [146]. However, several other Kv7-specific activators are now available (see review in [146]). Regarding Kv1 channel openers, a selective activator is currently missing. However, acetazolamide, a diuretic inhibitor of carbonic anhydrase, and flupirtine, an analgesic and muscle relaxant, appear to induce the activation of Kv1 [1,147]. The Kv1 channel closers, like for the BK channels, are not specific, and the concentration determines their selectivity. The main blockers used are 4-AP (1 mM), charybdotoxin (0.1 µM), and TEA (5–10 mM) [1]. On the other hand, the inhibitor margatoxin was initially considered a selective inhibitor of the Kv1.3 channel, but recent data also show that it can inhibit Kv1.1, -1.2, and -1.7 [148]. Furthermore, 4-AP (also designated as dalfampridine) is an approved drug for improving motor function in patients with multiple sclerosis [149], and 3,4-diaminopyridine (also designated as amifampridine) is recommended as a first-line symptomatic treatment for Lambert–Eaton myasthenic syndrome [150]. However, Kv channel blockers are also approved as antiarrhythmic agents used to maintain normal sinus rhythm and cardioversion in cases of atrial fibrillation and atrial flutter (dofetilide, a Kv10 and Kv11 inhibitor) [151] and are used for the treatment of recurrent hemodynamically unstable ventricular tachycardia and recurrent ventricular fibrillation (amiodarone, a Kv1 inhibitor) [152]. In summary, due to the importance of Kv channels, namely, Kv1, and the vast knowledge that exists about them, it is clear that modulation of these channels is a promising alternative for the treatment of vascular diseases. As demonstrated, the impaired functional activity of the Kv channels contributes to hypertension. Thus, more detailed insight into the expression and function of the different Kv channels present in HUASMCs (Kv1, -2, and -3) can be an asset in understanding hypertensive diseases of pregnancy and help in the development of new strategies to reduce the outcomes in this stage of development.
Regarding the BKCa channels, studies have shown that during a stage of hypertension, there is an increase in the expression of these channels [153,154,155]. Studies performed on cerebral arteries of hypertensive rats have a decrease in BKCa activity when the expression of BKCa β1-subunit is decreased. However, changes in the BKCa α-subunit have not been found [156,157]. Thus, when the BKCa β1-subunit is deleted, there is a decrease in Ca2+ sensitization, and the channel stops responding to Ca2+ sparks. In this way, membrane depolarization begins, and intracellular Ca2+ increases, promoting vasoconstriction and increased blood pressure, which culminates in hypertension [57]. Further studies by Ledoux et al. (2006) demonstrated that changes in the channel conformation or sensitization to Ca2+ can lead them to act as pure KV [57]. Like Kv, the mechanism responsible for the currents of BKCa channels during hypertension seems to occur through negative feedback: the protein expression of the channels increases in response to an increase in vascular tonus [16,137]. Radenkovic et al. (2007) demonstrated that the role of BKCa channels is different in the force exerted by bradykinin between HUA of normotensive and hypertensive pregnancies, as demonstrated by an increase in bradykinin contractions induced by TEA in this latter case [66]. In agreement with these results, Eichhorn et al. (2007) observed an increase in Ca2+ flux and BKCa activity in SMCs of different hypertensive animals. However, these authors, contrary to others, observed an increase in the expression of both subunits (α and β) of BKCa [41]. In the same study, it was possible to observe that the inhibition of BKCa channels by specific blockers depolarizes the membrane and gives rise to vasoconstriction [41]. Recently, the regulated trafficking of BK channel subunits (including α, β1, and γ subunits) has been accepted as a functional mechanism to modulate arterial contractility. When β1 trafficking is reduced, there is a decrease in BK channel currents and vasoconstriction observed in hypertension [158,159]. In some studies on animal models of hypertension, where there was a treatment with BKCa channel blockers, exaggerated vasoconstrictor responses were observed, supporting the hypothesis that there is an increased activity of these channels in the presence of the pathology [42,48]. Joseph et al. (2013) also reported that iberiotoxin-sensitive IK (a selective blocker of BKCa channels) were elevated in hypertensive animals compared to control animals [160]. Accordingly, Djokic et al. (2019) also suggested that hypertension leads to positive regulation of BKCa as a compensatory mechanism for blood vessel hyperactivity. However, as there were no changes in the MaxiK-α subunit, this regulation does not appear to be the result of an increase in the number of channels. This regulation is likely to be the result of post-transcriptional modifications of the MaxiK-α subunits and/or changes in interaction with auxiliary subunits [138]. Concerning PE, He et al. (2017) reported that BKCa channel impairment in human CPA is potentially relevant to the development of the pathology. Specifically, the authors found that decreased expression or activation of these channels induces a pathological CPA remodeling. This process weakens vasodilation and decreases the sensitivity of the artery to vasoactive substances; consequently, there is a decrease in fetoplacental blood flow that can induce PE [161]. Also, in pre-eclampsia, NO and IKCa and SKCa channels play an important role in the vasodilation of CPA [162]. Li et al. (2017) demonstrated that these channels are in the endothelium and SM of CPA and that women with PE presented unregulated levels of expression compared to normotensive women. The expressions of endothelial (eNOS) and inducible (iNOS) oxide nitric synthase were also subregulated, which was reflected in a decrease in eNOS activity. Thus, the authors suggested that during PE, IKCa- and SKCa-mediated vasodilation was impaired and that these channels are responsible for NO-mediated vasodilation and modulation of NO synthase activity. The authors concluded that the deregulation of these channels may be involved in the pathogenesis of PE, directly promoting vascular constriction of CPAs and affecting NO functions and NOS activities [162]. As demonstrated in Section 3.2., the presence of SKCa and IKCa channels in HUA is uncertain. Thus, in the development of a new pharmacological approach, BKCa channels should be the main therapeutic target used. At the pharmacological level, BKCa channel openers can be classified based on their origin and structure in (A) endogenous BKCa channel modulators and structural analogs, (B) naturally occurring BKCa channel openers and structural analogs, and (C) synthetic BKCa channel openers. Endogenous chemicals such as arachidonic acid and the metabolites of cytochrome P450, epoxygenase, and lipoxygenase have been found to increase BKCa channel activity and be important regulators of vascular tone. Natural occurring BKCa channel activators, such as terpene derivatives (e.g., pimaric acid and maxikdiol), flavonoids (e.g., apigenine, naringenin), and phenolic derivatives (e.g., magnolol), may be found in herbs, roots, fungi, and leaves, and have been used in folk medicine for the treatment of asthma and smooth muscle disorders. Synthetic BKCa channel activators capable of increasing channel-open probabilities have been synthesized by several companies, such as benzoimidazolone derivatives (e.g., NS1619, NS 1608, and NS004), carboxylate compound CGS7181 and its analogues (e.g., CGS7184), 1,4-benzothiazine derivatives, and NS11021 [163]. Of these, NS1619 showed that it may prevent ischemia-reperfusion-induced inflammation [164], pressure overload-induced heart failure, and decreased coronary vasodilatory capacity in heart failure with preserved ejection fraction patients [165]. The BKCa channel closers are the scorpion toxins (charibdotoxin and iberiotoxin) and tetraethylammonium (TEA); however, these BKCa inhibitors are not specific to these channels, the concentration being extremely important to make them quite specific (TEA, KCa inhibitor, 5 mM; charybdotoxin (ChTx), BKCa inhibitor, 10 nM; iberiotoxin (IbTx), BKCa inhibitor, 0.1–100 nM) [1]. Thus, in the development of new pharmacological studies in HUA, the BKCa channels are, with Kv channels, the main therapeutic targets to be used in the treatment of hypertensive diseases of pregnancy.
Concerning the Kir channels, the presence of these channels in HUA is still uncertain. However, at the pharmacological level, the targeting of Kir channels could be a great strategy to induce hyperpolarization/vasodilation and to reduce the peripheral resistance and high blood pressure observed in hypertension. Watanapa et al. (2012) observed inhibition of the Kir expression by PE plasma in HUVEC cultures. The same authors also observed the expression of KCa that seems to be a compensatory mechanism for the Kir reduction. In hypertension, it has also been observed that KIR channels have their expression or function diminished [42]. Tajada et al. (2012) also demonstrated a decrease in Kir2.1, Kir4.1, Kir6.x expression, and a functional decrease in these channels [166]. These findings point out the Kir channels play an important role in the hypertensive disorders and act with a pharmacological target. As the Kir2.1 isoform is the most dominant one expressed in SMCs, pharmacological studies focusing on this subunit are of great interest. The Kir channel opener flecainide, an antiarrhythmic drug, increases Kir2.1 currents by binding to cysteine 311 and reducing polyamine-induced rectification [167]. To our knowledge, this drug is a unique drug that can activate Kir 2.1. channels [168]. Moreover, the ion Mg2+ was proposed as an activator of Kir channels in vascular SMCs [1]. In contrast, several drugs are proposed as blockers of Kir channels, namely, the antimalarial drug chloroquine [169,170], the antiarrhythmic drug quinidine [167], the diamine pentamidine analog 6 (PA-6) [171], and a more recent prototype of Kir blocker, ML133 [172]. Moreover, these channels are markedly sensitive to inorganic cations (e.g., Ba2+ and Cs+) but are insensitive to 4-AP [168]. Although the presence of Kir channels in HUA is uncertain, the next pharmacological studies focusing on the development of new targets for Kir2.1 channels can be the first promising step at the vascular level.
Concerning the KATP channels, studies have also reported their involvement in gestational pathological processes such as gestational hypertension [138]. Changes in the function and expression of these channels have already been reported in vascular SMCs of various vascular beds and different species in the presence of this pathology [7,138]. The reduction in the function of the KATP channels has been implicated in hypertension. Several studies have shown reduced relaxation of SMCs in various types of arteries when applying agonists [166,173,174,175,176,177] of the KATP channels or openers [178,179] of the same. In 2005, Gutterman et al. reported that the vascular oxidative state is regulated by physiological stimuli or pathophysiological stress and includes hypertension [180]. Indeed, the presence of chronic hypoxia or elevated oxidative stress has been demonstrated in situations of gestational hypertension and, thus, can compromise the health of the mother and fetus [181,182,183,184]. Many researchers have suggested that KATP channels are key agents in hypoxia-mediated vasodilation since hypoxia affects the ATP/ADP ratio but also because, when the activity of the channels is impaired, this process is attenuated [185,186]. Thus, it seems that the KATP channels can be excellent pharmacological targets in the treatment of pregnancy disorders such as gestational hypertension. In addition, it is described that the coexpression of both subunits of the KATP channels, in the ratio 4:4, is fundamental for their normal functional response [78]. On the other hand, the gain in Kir6.1 subunit function with the hyperactivity of these channels leads to hypotension, a phenomenon that has been confirmed in people with Cantu syndrome [187]. Djokic et al. (2019) observed in HUV that in situations of gestational hypertension, there was a decrease in the expression of the Kir6.1 pore-forming subunit, while the expression of SUR2B remained unchanged. Exposure of a too-short HUV to the hypertensive environment or the therapeutic application that maintains vascular tension during pregnancy may be possible explanations for the unchanged expression of SUR2B [138]; not least because a previous study carried out with SM of the aorta of spontaneously hypertensive rats demonstrated that as the exposure time increased, the decrease in expression observed for this subunit was increased [188]. However, Tajada et al. (2012) reported a reduced expression of SUR2B in their investigation, as well as a decrease in the function of KATP channels that impaired vascular tone [166]. In this sense, it has been suggested that hypertension can be reversed by restoring the KATP channels [178,189]. Du et al. (2019) also demonstrated that the Kir6.1 subunit was negatively regulated in the myometrium of parturient suffering from gestational hypertension. The authors suggest that since the KATP channels are responsible for uterine stillness in pregnancy, this regulation could explain the risk of premature labor associated with parturients with hypertension [190]. Thus, it seems plausible to say that in the presence of gestational hypertension, there is a decrease in the expression of KATP channels in SMCs [7,138]. However, despite the decreased expression of KATP channels observed in SMCs, Djokic et al. (2019) have found no changes in pinacidil-induced relaxation (K+ channel opener) in the presence of glibenclamide in situations of gestational hypertension in humans, and similar results have also been obtained in spontaneously hypertensive rats [191]. The fact that pinacidil completely relaxes HUV in the presence of glibenclamide supports the hypothesis that it acts partially through mechanisms independent of KATP channels, mainly in high doses, activating another type of K+ channel (such as BKCa and Kv) [138,192,193,194,195]. On the contrary, some studies have reported that there were no changes in the functions of the KATP channels [191,196,197] or the functions being improved [198,199] in hypertensive models. Recently, Tykocki et al. (2017) pointed out in their review that hypertension may be the cause of the deregulation of these channels observed in hypertensive models [7]. However, due to the fact that the information available is not fully agreed upon, further studies are needed to understand the expression and function of the KATP channels in hypertension. Furthermore, since the KATP channels are present in HUA, the knowledge about the role of these channels is crucial to improving the treatment of hypertensive disorders of pregnancy. At the pharmacological level, KATP channel openers are chemically diverse and belong to several structural classes. These include benzopyrans (levcromakalim, bimakalim), benzothiadiazines (diazoxide), cyanoguanidines (pinacidil), cyclobutenediones (WAY-151616), nicotinamides (nicorandil), pyrimidines (minoxidil), tertiary carbonoles (ZD-6169), thioformamides (aprikalim), and dihydropyridine-like structures (ZM-244085) [200]. Some of these drugs, like diazoxide, pinacidil, minoxidil, nicorandil, and cromakalim, have been used to treat hypertension, myocardial ischemia, bronchial asthma, urinary incontinence, hyperinsulinism, angina pectoris, and some forms of skeletal muscle myopathies. However, some have been withdrawn due to their adverse effects: for example, the vasodilation caused by this mechanism resulted in reflex tachycardia and fluid retention, which oppose the antihypertensive effect. In contrast, KATP channel closers stimulate insulin secretion and, therefore, pharmacologic closers like glibenclamide are one of the only oral medicines available to treat diabetes. More recently, the development of KATP channel closers called PNU compounds (PNU-37883A, a morpholinoguanidine, and PNU-99963, a cyanoguanidine) have been shown to inhibit the vasodilation and hypotension caused by traditional KATP channel openers [201]. More studies are needed to improve the knowledge of KATP channels in HUA and unravel how these channels can be a good target in the treatment of hypertensive diseases of pregnancy.
Concerning the K2P channels, the presence of these channels in HUA is uncertain, and only Martin et al. (2014) have reported the presence of mRNA from the TREK-1 and TRAAK channels. Increasing evidence shows a role for TREK channels in cardiac pathologies such as atrial fibrillation and heart failure [202,203,204]. TREK-1 channel openers include riluzole, chloroform, and diethyl ether. Moreover, these channels are opened by volatile general anesthetics such as halothane and isoflurane [103]. New synthetic compounds have been developed to modulate these channels namely, GI-530159, a new activator of the TREK1 (but not TRAAK) channels [205] and BL-1249 [206], which, in addition to directly activating TREK-1 channels, activates other channels such as TREK-2, TASK3, BKCa, KV7.2, KV7.3, and NaV1.7 channels [205]. Moreover, a dihydroacridine analog (ML67-33) has been described to selectively activate the TREK-1, TREK-2, and TRAAK channels, and has no effect on the TASK1, TASK2, and TASK3 or KCNQ2 channels [207]. Additionally, TREK-1 channels are blocked by quinidine and reversibly blocked by Gd3+ at micromolar concentrations. In contrast, these channels are resistant to TEA and 4-AP and slightly sensitive to high concentrations of Ba2+. In terms of physiological regulation, TREK-1 (but not TRAAK) channels are inhibited by activators of PKA and PKC [103]. Recently, Cunningham et al. (2018) have demonstrated that treprostinil, a stable prostacyclin analog, has a direct inhibitory effect on TREK-1 channels. These results are quite promising because treprostinil is commonly used in the therapy of pulmonary arterial hypertension to keep blood vessels open [208]. Indeed, there are relatively few selective antagonists for the TREK channels. Some examples of blockers include chlorpromazine, sipatrigine, fluoxetine, and norfluoxetine [209]. However, there is currently a highly specific and fast-acting inhibitor on TREK-1 channels, spadin. This drug does not affect the TREK-2 or TRAAK channels and acts by promoting a decrease in currents by internalizing the TREK-1 channel [210]. In clinical practice, the antiarrhythmic drug diltiazem inhibits TREK-1 (but not TRAAK) channels [211] and is used to treat hypertension and angina pectoris. Moreover, TREK channels are also modulated by other cardiac-related drugs, e.g., mibefradil [119] and quinidine [212]. Taken together, these results suggest that TREK-1 channels are promising therapeutic candidates for the development of new pharmacological studies in HUA to be used in the treatment of gestational hypertension. Concerning TRAAK channel openers, riluzole [213] and ML67-33 [207] activate these channels. On the other hand, these channels are reversibly blocked by micromolar concentrations of Gd3+, but resistant to TEA and 4-AP, and slightly sensitive to high concentrations of Ba2+ [103]. The drugs sipatrigine [214] and pimozide [215] inhibit TRAAK channels, but several other drugs tested (e.g., fluoxetine, paroxetine [216], lamotrigine [214], chlorpromazine [217], haloperidol, sulpiride [215], and diltiazem [211]) had no effect on them. Due to the fact that pharmacological knowledge of these channels is still very premature, more studies are needed to investigate the role of TRAAK channels as pharmacological targets in the treatment of hypertensive diseases of pregnancy.
In summary, the study of the physiological regulation of K+ channels present in HUA is crucial to detect potential targets for the treatment of pregnancy-related pathologies [9]. Gestational hypertension and pre-eclampsia are multifactorial disorders and are important risk factors in the development of CV complications that endanger not only the health of the mother but also the health of the fetus. As evidenced, studies on the UC vasculature and, in particular, HUA are practically scarce, even though morphological changes have been associated with these pathologies [13]. Thus, further studies are needed to understand what mechanisms underlie these pathologies, namely, with concern to the involvement of HUA K+ channels. As mentioned, there are several K+ channel activators and inhibitors that could be used in the treatment of hypertensive disorders, but some challenges remain to be overcome. One of these is the use of inhibitors that induce contraction, which is hard to reconcile with treating hypertension. However, there may exist an interaction between the various types of channels since the blocking of one type of K+ channel can activate another K+ channel, and vice versa; for example, it was observed with 4-AP that it can enhance Kv7.4 channel activity [149]. Moreover, a very important additional issue is the selectivity; most of the compounds are nonselective, and they act on a wide range of channels. Furthermore, since the K+ channels are present in many types of cells, the likelihood of adverse effects on other organs increases. Therefore, achieving specific targeting is an extremely challenging task.
5. Conclusions and Future Perspectives
HUA is an excellent sample for obtaining human vascular SMCs, allowing the study of different cellular mechanisms and their functions. The study of its physiological regulation is extremely important because as HUA does not have nerve endings, the regulation of its vascular tone depends completely on other factors, where the K+ ion is included. Moreover, K+-channels are one of the main mechanisms involved in the vasorelaxation of this artery. Indeed, the K+-channels are fundamental in determining and/or modulating the membrane potential, allowing them to regulate the vascular tone of the artery. The functional presence of the Kv and BKCa channels is clearly demonstrated in HUA, and its function of regulating the contractile state is also well established. Concerning the SKCa, Kir, KATP and K2P channels, their presence is also suggested, but there are still very few studies and, therefore, much of the information about their functions remain unknown. On the other hand, regarding the IKCa channels, we found no evidence in the literature to demonstrate its presence in the artery. In this sense, new studies are needed, not only focused on the expression but also the functionality of the different K+ channels of HUA. Several vasodilators and vasoconstrictors can act as modulators of these channels, and it is crucial to understand in detail the pathways by which the different K+-channels are regulated and the interaction that exists between them. These studies include determining the structure of the channel, the role of different subunits, and the molecular basis of regulating channel activity by different intracellular pathways. On the other hand, the localization of the different types of channels also plays an important role in the understanding of each one at different vascular levels. In this line of thought, genetic and molecular studies are suggested, complemented with electrophysiological studies. It is important to note that, as demonstrated, K+ channels are clinically relevant, particularly in hypertensive diseases of pregnancy (gestational hypertension and pre-eclampsia). Thus, the identification of changes in the expression and/or functionality of the different K+ channels present in these disorders can be an asset when it is intended for developing new drugs for their treatment. The study of HUA K+-channels and the signaling pathways involved in regulating artery contractility may contribute to the development of new specific drugs that inhibit or activate these channels, which can be used as therapeutic drugs or pharmacological tools. The complexity of the different combinations of K+ channels allows the vascular response to be directed to the most appropriate pharmacological treatment, depending on the pathology, which is why the study of K+ channels is so important and promising, as it represents an open window on the progression and enrichment of the knowledge of human vascular physiology.
Author Contributions
E.C. identified the need for this review; E.C. and M.L. designed the manuscript; M.L. and N.O. wrote the manuscript; M.L., N.O., and E.C. reviewed the literature and E.C. critically reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was also supported by FEDER funds through POCI-COMPETE 2020—Operational Programme Competitiveness and Internationalisation in Axis I-Strengthening Research, Technological Development and Innovation (Project POCI-01-0145-FEDER007491), and National Funds by FCT—Foundation for Science and Technology (Project UID/Multi/00709/2019).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| (Ca2+)i | intracellular Ca2+ concentration |
| 4-AP | 4-aminopyridine |
| ANP | atrial natriuretic peptide |
| Ba2+ | barium ion |
| BKCa | large-conductance Ca2+-activated K+ channels |
| Ca2+ | calcium |
| CaM | calmodulin |
| CaM-BD | calmodulin-binding domain |
| cGMP | cyclic guanosine monophosphate |
| Cl- | chloride |
| CPA | chorionic plaque arteries |
| Cs+ | caesium ion |
| CV | cardiovascular |
| CVD | cardiovascular diseases |
| DHS-1 | dedydrosoyasaponin-1 |
| EC | endothelial cells |
| EK | K+ equilibrium potential |
| eNOS | endothelial oxide nitric synthase |
| HUA | human umbilical artery |
| HUASMC | human umbilical artery smooth muscle cells |
| HUV | human umbilical vein |
| IK | K+ currents |
| IKCa | intermediate-conductance Ca2+-activated K+ channels |
| iNOS | inducible oxide nitric synthase |
| K+ | Potassium |
| K2P | 2-pore domains K+ channels |
| KATP | ATP-sensitive K+ channels |
| KCa | Ca2+-activated K+ channels |
| Kir | inward rectifier K+ channels |
| KV | voltage-dependent K+ channels |
| MP | resting membrane potential |
| Na2S | sodium sulfide |
| NO | nitric oxide |
| NP | natriuretic peptides |
| PDE | phosphodiesteras’s |
| pGC | particulate guanylyl cyclase |
| PKA | protein kinase A |
| PKC | protein kinase C |
| PKG | protein kinase G |
| sGC | soluble guanylyl cyclase |
| SKCa | small-conductance Ca2+-activated K+ channels |
| SM | smooth muscle |
| SMC | smooth muscle cells |
| STOC | spontaneous transient outward currents |
| SUR | sulfonylurea receptor |
| TASK | acid-sensitive rectifiers K+ channels |
| TEA | tetraethylammonium |
| TWIK | lipid sensitive mechano-gated K+ channels |
| TREK | weak inward rectifiers K+ channels |
| UC | umbilical cord |
| VGCC | voltage-gated Ca2+-channels |
| Zn+2 | zinc ion |
References
- Dogan, M.F.; Yildiz, O.; Arslan, S.O.; Ulusoy, K.G. Potassium channels in vascular smooth muscle: A pathophysiological and pharmacological perspective. Fundam. Clin. Pharmacol. 2019, 33, 504–523. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.F. KV channels and the regulation of vascular smooth muscle tone. Microcirculation 2018, 25. [Google Scholar] [CrossRef]
- Hayabuchi, Y. The Action of Smooth Muscle Cell Potassium Channels in the Pathology of Pulmonary Arterial Hypertension. Pediatr. Cardiol. 2017, 38, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Burg, E.D.; Remillard, C.V.; Yuan, J.X. Potassium channels in the regulation of pulmonary artery smooth muscle cell proliferation and apoptosis: Pharmacotherapeutic implications. Br. J. Pharmacol. 2008, 153 (Suppl. 1), S99–S111. [Google Scholar] [CrossRef] [Green Version]
- Nelson, M.T.; Quayle, J.M. Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol 1995, 268, C799–C822. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y. Electrical and Mechanical Properties of Vascular Smooth Muscle. In Biology of Vascular Smooth Muscle: Vasoconstriction and Dilatation; Springer: Berlin, Germany, 2017; pp. 41–55. [Google Scholar] [CrossRef]
- Tykocki, N.R.; Boerman, E.M.; Jackson, W.F. Smooth Muscle Ion Channels and Regulation of Vascular Tone in Resistance Arteries and Arterioles. Compr. Physiol. 2017, 7, 485–581. [Google Scholar] [CrossRef] [Green Version]
- Jackson, W.F. Potassium Channels in Regulation of Vascular Smooth Muscle Contraction and Growth. Adv. Pharmacol. 2017, 78, 89–144. [Google Scholar] [CrossRef] [Green Version]
- Lorigo, M.; Mariana, M.; Feiteiro, J.; Cairrao, E. How is the human umbilical artery regulated? J. Obs. Gynaecol Res. 2018. [Google Scholar] [CrossRef] [Green Version]
- Margarida Lorigo, M.; Melissa, M.; Joana, F.; Elisa, C. Human Umbilical Artery Smooth Muscle Cells: Vascular Function and Clinical Importance. In Horizons in World Cardiovascular Research; Bennington, E.H., Ed.; Nova Science Publisher: New York, NY, USA, 2019; Volume 16. [Google Scholar]
- Meyer, W.W.; Rumpelt, H.J.; Yao, A.C.; Lind, J. Structure and closure mechanism of the human umbilical artery. Eur J. Pediatr. 1978, 128, 247–259. [Google Scholar] [CrossRef]
- Ferguson, V.L.; Dodson, R.B. Bioengineering aspects of the umbilical cord. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 144 (Suppl. 1), S108–S113. [Google Scholar] [CrossRef]
- Chillakuru, S.; Velichety, S.D.; Rajagopalan, V. Human umbilical cord and its vessels: A histomorphometric study in difference severity of hypertensive disorders of pregnancy. Anat. Cell Biol. 2020, 53, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Cairrao, E.; Santos-Silva, A.J.; Alvarez, E.; Correia, I.; Verde, I. Isolation and culture of human umbilical artery smooth muscle cells expressing functional calcium channels. Vitr. Cell. Dev. Biol. Anim. 2009, 45, 175–184. [Google Scholar] [CrossRef]
- Martin, P.; Rebolledo, A.; Palomo, A.R.; Moncada, M.; Piccinini, L.; Milesi, V. Diversity of potassium channels in human umbilical artery smooth muscle cells: A review of their roles in human umbilical artery contraction. Reprod. Sci. 2014, 21, 432–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, W.F. Ion channels and vascular tone. Hypertension 2000, 35, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Brayden, J.E. Potassium channels in vascular smooth muscle. Clin. Exp. Pharm. Physiol. 1996, 23, 1069–1076. [Google Scholar] [CrossRef]
- Milesi, V.; Raingo, J.; Rebolledo, A.; Grassi de Gende, A.O. Potassium channels in human umbilical artery cells. J. Soc. Gynecol. Investig. 2003, 10, 339–346. [Google Scholar] [CrossRef]
- Ko, E.A.; Park, W.S.; Firth, A.L.; Kim, N.; Yuan, J.X.; Han, J. Pathophysiology of voltage-gated K+ channels in vascular smooth muscle cells: Modulation by protein kinases. Prog. Biophys. Mol. Biol. 2010, 103, 95–101. [Google Scholar] [CrossRef]
- Ko, E.A.; Han, J.; Jung, I.D.; Park, W.S. Physiological roles of K+ channels in vascular smooth muscle cells. J. Smooth Muscle Res. 2008, 44, 65–81. [Google Scholar] [CrossRef] [Green Version]
- Cairrao, E.; Alvarez, E.; Santos-Silva, A.J.; Verde, I. Potassium channels are involved in testosterone-induced vasorelaxation of human umbilical artery. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2008, 376, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Santos-Silva, A.J.; Cairrao, E.; Verde, I. Study of the mechanisms regulating human umbilical artery contractility. Health 2010, 2, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Cairrao, E.; Santos-Silva, A.J.; Verde, I. PKG is involved in testosterone-induced vasorelaxation of human umbilical artery. Eur. J. Pharmacol. 2010, 640, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Feiteiro, J.; Santos-Silva, A.J.; Verde, I.; Cairrao, E. Testosterone and atrial natriuretic Peptide share the same pathway to induce vasorelaxation of human umbilical artery. J. Cardiovasc. Pharmacol. 2014, 63, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Feiteiro, J.; Verde, I.; Cairrao, E. Cyclic guanosine monophosphate compartmentation in human vascular smooth muscle cells. Cell. Signal. 2016, 28, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Park, W.S.; Firth, A.L.; Han, J.; Ko, E.A. Patho-, physiological roles of voltage-dependent K+ channels in pulmonary arterial smooth muscle cells. J. Smooth Muscle Res. 2010, 46, 89–105. [Google Scholar] [CrossRef] [Green Version]
- Wulff, H.; Castle, N.A.; Pardo, L.A. Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov 2009, 8, 982–1001. [Google Scholar] [CrossRef] [Green Version]
- Long, S.B.; Campbell, E.B.; Mackinnon, R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 2005, 309, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Long, S.B.; Campbell, E.B.; Mackinnon, R. Voltage sensor of Kv1.2: Structural basis of electromechanical coupling. Science 2005, 309, 903–908. [Google Scholar] [CrossRef] [Green Version]
- Hasan, R.; Jaggar, J.H. KV channel trafficking and control of vascular tone. Microcirculation 2018, 25. [Google Scholar] [CrossRef]
- Lovren, F.; Triggle, C. Nitric oxide and sodium nitroprusside-induced relaxation of the human umbilical artery. Br. J. Pharmacol. 2000, 131, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Yildiz, O.; Nacitarhan, C.; Seyrek, M. Potassium channels in the vasodilating action of levosimendan on the human umbilical artery. J. Soc. Gynecol. Investig. 2006, 13, 312–315. [Google Scholar] [CrossRef]
- Mathew John, C.; Khaddaj Mallat, R.; George, G.; Kim, T.; Mishra, R.C.; Braun, A.P. Pharmacologic targeting of endothelial Ca(2+)-activated K(+) channels: A strategy to improve cardiovascular function. Channels 2018, 12, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.M.; Shim, H.; Christophersen, P.; Wulff, H. Pharmacology of Small- and Intermediate-Conductance Calcium-Activated Potassium Channels. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 219–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, D.L.; Bai, Y.L.; Cai, B.Z. Calcium-Activated Potassium Channels: Potential Target for Cardiovascular Diseases. Adv. Protein Chem. Struct. Biol. 2016, 104, 233–261. [Google Scholar] [CrossRef] [PubMed]
- Marty, A. Ca-dependent K channels with large unitary conductance in chromaffin cell membranes. Nature 1981, 291, 497–500. [Google Scholar] [CrossRef]
- Gardos, G. The function of calcium in the potassium permeability of human erythrocytes. Biochim. Biophys. Acta 1958, 30, 653–654. [Google Scholar] [CrossRef]
- Blatz, A.L.; Magleby, K.L. Single apamin-blocked Ca-activated K+ channels of small conductance in cultured rat skeletal muscle. Nature 1986, 323, 718–720. [Google Scholar] [CrossRef]
- Ghatta, S.; Nimmagadda, D.; Xu, X.; O’Rourke, S.T. Large-conductance, calcium-activated potassium channels: Structural and functional implications. Pharmacology 2006, 110, 103–116. [Google Scholar] [CrossRef]
- Brenner, R.; Perez, G.J.; Bonev, A.D.; Eckman, D.M.; Kosek, J.C.; Wiler, S.W.; Patterson, A.J.; Nelson, M.T.; Aldrich, R.W. Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature 2000, 407, 870–876. [Google Scholar] [CrossRef]
- Eichhorn, B.; Dobrev, D. Vascular large conductance calcium-activated potassium channels: Functional role and therapeutic potential. Naunyn Schmiedebergs Arch. Pharm. 2007, 376, 145–155. [Google Scholar] [CrossRef]
- Sobey, C.G. Potassium channel function in vascular disease. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Lee, U.S.; Cui, J. BK channel activation: Structural and functional insights. Trends Neurosci. 2010, 33, 415–423. [Google Scholar] [CrossRef] [Green Version]
- Kohler, R. Single-nucleotide polymorphisms in vascular Ca2+-activated K+-channel genes and cardiovascular disease. Pflug. Arch. Eur. J. Physiol. 2010, 460, 343–351. [Google Scholar] [CrossRef]
- Gollasch, M.; Lohn, M.; Furstenau, M.; Nelson, M.T.; Luft, F.C.; Haller, H. Ca2+ channels, ‘quantized’ Ca2+ release, and differentiation of myocytes in the cardiovascular system. J. Hypertens. 2000, 18, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.T.; Cheng, H.; Rubart, M.; Santana, L.F.; Bonev, A.D.; Knot, H.J.; Lederer, W.J. Relaxation of arterial smooth muscle by calcium sparks. Science 1995, 270, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Perez, G.J.; Bonev, A.D.; Patlak, J.B.; Nelson, M.T. Functional coupling of ryanodine receptors to KCa channels in smooth muscle cells from rat cerebral arteries. J. Gen. Physiol. 1999, 113, 229–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.Q.; Zhang, L. Function and regulation of large conductance Ca(2+)-activated K+ channel in vascular smooth muscle cells. Drug Discov. Today 2012, 17, 974–987. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Alioua, A.; Kumar, Y.; Eghbali, M.; Stefani, E.; Toro, L. MaxiK channel partners: Physiological impact. J. Physiol. 2006, 570, 65–72. [Google Scholar] [CrossRef]
- Alioua, A.; Mahajan, A.; Nishimaru, K.; Zarei, M.M.; Stefani, E.; Toro, L. Coupling of c-Src to large conductance voltage- and Ca2+-activated K+ channels as a new mechanism of agonist-induced vasoconstriction. Proc. Natl. Acad. Sci. USA 2002, 99, 14560–14565. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.A.; Yang, Y.; Ella, S.R.; Davis, M.J.; Braun, A.P. Large conductance, Ca2+-activated K+ channels (BKCa) and arteriolar myogenic signaling. FEBS Lett. 2010, 584, 2033–2042. [Google Scholar] [CrossRef] [Green Version]
- Kaczmarek, L.K.; Aldrich, R.W.; Chandy, K.G.; Grissmer, S.; Wei, A.D.; Wulff, H. International Union of Basic and Clinical Pharmacology. C. Nomenclature and Properties of Calcium-Activated and Sodium-Activated Potassium Channels. Pharmacol. Rev. 2017, 69, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Clements, R.T.; Terentyev, D.; Sellke, F.W. Ca(2+)-activated K(+) channels as therapeutic targets for myocardial and vascular protection. Circ. J. Off. J. Jpn. Circ. Soc. 2015, 79, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Sigworth, F.J. Structure of the BK potassium channel in a lipid membrane from electron cryomicroscopy. Nature 2009, 461, 292–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, P.; Leonetti, M.D.; Hsiung, Y.; MacKinnon, R. Open structure of the Ca2+ gating ring in the high-conductance Ca2+-activated K+ channel. Nature 2011, 481, 94–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrighton, D.C.; Muench, S.P.; Lippiat, J.D. Mechanism of inhibition of mouse Slo3 (KCa 5.1) potassium channels by quinine, quinidine and barium. Br. J. Pharmacol. 2015, 172, 4355–4363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledoux, J.; Werner, M.E.; Brayden, J.E.; Nelson, M.T. Calcium-activated potassium channels and the regulation of vascular tone. Physiol. (Bethesda) 2006, 21, 69–78. [Google Scholar] [CrossRef]
- Cui, J.; Yang, H.; Lee, U.S. Molecular mechanisms of BK channel activation. Cell. Mol. Life Sci. CMLS 2009, 66, 852–875. [Google Scholar] [CrossRef] [Green Version]
- Standen, N.B.; Quayle, J.M. K+ channel modulation in arterial smooth muscle. Acta Physiol. Scand. 1998, 164, 549–557. [Google Scholar] [CrossRef]
- Waldron, G.J.; Cole, W.C. Activation of vascular smooth muscle K+ channels by endothelium-derived relaxing factors. Clin. Exp. Pharm. Physiol. 1999, 26, 180–184. [Google Scholar] [CrossRef]
- Wallner, M.; Meera, P.; Ottolia, M.; Kaczorowski, G.J.; Latorre, R.; Garcia, M.L.; Stefani, E.; Toro, L. Characterization of and modulation by a beta-subunit of a human maxi KCa channel cloned from myometrium. Recept. Channels 1995, 3, 185–199. [Google Scholar]
- Trombetta-Lima, M.; Krabbendam, I.E.; Dolga, A.M. Calcium-activated potassium channels: Implications for aging and age-related neurodegeneration. Int. J. Biochem. Cell Biol. 2020, 123, 105748. [Google Scholar] [CrossRef]
- Tanaka, Y.; Koike, K.; Toro, L. MaxiK channel roles in blood vessel relaxations induced by endothelium-derived relaxing factors and their molecular mechanisms. J. Smooth Muscle Res. 2004, 40, 125–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Wallner, M.; Meera, P.; Toro, L. Human and rodent MaxiK channel beta-subunit genes: Cloning and characterization. Genomics 1999, 55, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Aldrich, R.W. BK potassium channel modulation by leucine-rich repeat-containing proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 7917–7922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radenkovic, M.; Grbovic, L.; Radunovic, N.; Momcilov, P. Pharmacological evaluation of bradykinin effect on human umbilical artery in normal, hypertensive and diabetic pregnancy. Pharm. Rep. 2007, 59, 64–73. [Google Scholar]
- Saldanha, P.A.; Cairrao, E.; Maia, C.J.; Verde, I. Long- and short-term effects of androgens in human umbilical artery smooth muscle. Clin. Exp. Pharmacol. Physiol. 2013, 40, 181–189. [Google Scholar] [CrossRef]
- Evaristo Rodrigues da Silva, R.; de Alencar Silva, A.; Pereira-de-Morais, L.; de Sousa Almeida, N.; Iriti, M.; Kerntopf, M.R.; Menezes, I.R.A.d.; Coutinho, H.D.M.; Barbosa, R. Relaxant Effect of Monoterpene (−)-Carveol on Isolated Human Umbilical Cord Arteries and the Involvement of Ion Channels. Molecules 2020, 25, 2681. [Google Scholar] [CrossRef]
- Rebolledo, A.; Raingo, J.; Rinaldi, G.; Grassi, A.; Milesi, V. 05 Cyclic GMP activates the big Ca-sensitive K channel in smooth muscle cells of human umbilical artery. J. Mol. Cell. Cardiol. 2002, 34, A3. [Google Scholar] [CrossRef]
- Martin, P.; Enrique, N.; Palomo, A.R.; Rebolledo, A.; Milesi, V. Bupivacaine inhibits large conductance, voltage- and Ca2+- activated K+ channels in human umbilical artery smooth muscle cells. Channels 2012, 6, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Bernsteiner, H.; Zangerl-Plessl, E.M.; Chen, X.; Stary-Weinzinger, A. Conduction through a narrow inward-rectifier K(+) channel pore. J. Gen. Physiol. 2019, 151, 1231–1246. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Swale, D.R. Inwardly Rectifying Potassium (Kir) Channels Represent a Critical Ion Conductance Pathway in the Nervous Systems of Insects. Sci. Rep. 2018, 8, 1617. [Google Scholar] [CrossRef]
- Nichols, C.G.; Lopatin, A.N. Inward rectifier potassium channels. Annu. Rev. Physiol. 1997, 59, 171–191. [Google Scholar] [CrossRef]
- Lu, Z. Mechanism of rectification in inward-rectifier K+ channels. Annu. Rev. Physiol. 2004, 66, 103–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logothetis, D.E.; Jin, T.; Lupyan, D.; Rosenhouse-Dantsker, A. Phosphoinositide-mediated gating of inwardly rectifying K(+) channels. Pflug. Arch. Eur. J. Physiol. 2007, 455, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Park, W.S.; Han, J.; Earm, Y.E. Physiological role of inward rectifier K(+) channels in vascular smooth muscle cells. Pflug. Arch. Eur. J. Physiol. 2008, 457, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Doyle, D.A.; Morais Cabral, J.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, K.A.; He, S.; Jin, R.; Miller, D.M.; Bolla, J.R.; Clarke, O.B.; Johnson, P.; Windley, M.; Burns, C.J.; Hill, A.P.; et al. A constricted opening in Kir channels does not impede potassium conduction. Nat. Commun. 2020, 11, 3024. [Google Scholar] [CrossRef]
- Wu, B.N.; Luykenaar, K.D.; Brayden, J.E.; Giles, W.R.; Corteling, R.L.; Wiehler, W.B.; Welsh, D.G. Hyposmotic challenge inhibits inward rectifying K+ channels in cerebral arterial smooth muscle cells. Am. J. Physiology. Heart Circ. Physiol. 2007, 292, H1085–H1094. [Google Scholar] [CrossRef] [Green Version]
- Tennant, B.P.; Cui, Y.; Tinker, A.; Clapp, L.H. Functional expression of inward rectifier potassium channels in cultured human pulmonary smooth muscle cells: Evidence for a major role of Kir2.4 subunits. J. Membr. Biol. 2006, 213, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Longden, T.A.; Nelson, M.T. Vascular inward rectifier K+ channels as external K+ sensors in the control of cerebral blood flow. Microcirculation 2015, 22, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of potassium channels. Cell. Mol. Life Sci. CMLS 2015, 72, 3677–3693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Tang, J.; Zhou, X.; Xiang, S.; Zhu, X.; Li, N.; Shi, R.; Zhong, Y.; Zhang, L.; Sun, M.; et al. Roles of ion channels in regulation of acetylcholine-mediated vasoconstrictions in umbilical cords of rabbit/rats. Reprod. Toxicol. 2016, 65, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Noma, A. ATP-regulated K+ channels in cardiac muscle. Nature 1983, 305, 147–148. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hu, D.; Huang, C.; Nichols, C.G. Genetic Discovery of ATP-Sensitive K(+) Channels in Cardiovascular Diseases. Circulation. Arrhythmia Electrophysiol. 2019, 12, e007322. [Google Scholar] [CrossRef] [PubMed]
- Cole, W.C.; Clement-Chomienne, O. ATP-sensitive K+ channels of vascular smooth muscle cells. J. Cardiovasc. Electrophysiol. 2003, 14, 94–103. [Google Scholar] [CrossRef]
- Li, H.; Shin, S.E.; Seo, M.S.; An, J.R.; Ha, K.S.; Han, E.T.; Hong, S.H.; Kim, J.; Yim, M.J.; Lee, J.M.; et al. Alterations of ATP-sensitive K(+) channels in human umbilical arterial smooth muscle during gestational diabetes mellitus. Pflug. Arch. Eur. J. Physiol. 2018, 470, 1325–1333. [Google Scholar] [CrossRef]
- Shi, X.; Zhen, L.; Ding, H.; Chen, J.; Zhang, S.; Fu, Y. Role of ATP-sensitive potassium channels and inflammatory response of basilar artery smooth muscle cells in subarachnoid hemorrhage of rabbit and immune-modulation by shikonin. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2019, 134, 110804. [Google Scholar] [CrossRef]
- Szeto, V.; Chen, N.H.; Sun, H.S.; Feng, Z.P. The role of KATP channels in cerebral ischemic stroke and diabetes. Acta Pharmacol. Sin. 2018, 39, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Miki, T.; Seino, S. Roles of KATP channels as metabolic sensors in acute metabolic changes. J. Mol. Cell Cardiol. 2005, 38, 917–925. [Google Scholar] [CrossRef]
- Seino, S. Physiology and pathophysiology of K(ATP) channels in the pancreas and cardiovascular system: A review. J. Diabetes Complic. 2003, 17, 2–5. [Google Scholar] [CrossRef]
- Brayden, J.E. Functional roles of KATP channels in vascular smooth muscle. Clin. Exp. Pharm. Physiol. 2002, 29, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Pipatpolkai, T.; Usher, S.; Stansfeld, P.J.; Ashcroft, F.M. New insights into KATP channel gene mutations and neonatal diabetes mellitus. Nat. Rev. Endocrinol. 2020, 16, 378–393. [Google Scholar] [CrossRef]
- Rubaiy, H.N. The therapeutic agents that target ATP-sensitive potassium channels. Acta Pharm. 2016, 66, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, R.; Provitera, L.; Cavallaro, G.; Lattuada, D.; Ercoli, G.; Mosca, F.; Villamor, E. Vasomotor effects of hydrogen sulfide in human umbilical vessels. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2017, 68, 737–747. [Google Scholar]
- Bai, X.J.; Tian, H.Y.; Wang, T.Z.; Du, Y.; Xi, Y.T.; Wu, Y.; Gao, J.; Ma, A.Q. Oleic acid inhibits the K(ATP) channel subunit Kir6.1 and the K(ATP) current in human umbilical artery smooth muscle cells. Am. J. Med. Sci. 2013, 346, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.J.; Honore, E.; Maingret, F.; Lesage, F.; Fink, M.; Duprat, F.; Lazdunski, M. A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J. 1998, 17, 4283–4290. [Google Scholar] [CrossRef] [PubMed]
- Ketchum, K.A.; Joiner, W.J.; Sellers, A.J.; Kaczmarek, L.K.; Goldstein, S.A. A new family of outwardly rectifying potassium channel proteins with two pore domains in tandem. Nature 1995, 376, 690–695. [Google Scholar] [CrossRef] [Green Version]
- Hughes, S.; Foster, R.G.; Peirson, S.N.; Hankins, M.W. Expression and localisation of two-pore domain (K2P) background leak potassium ion channels in the mouse retina. Sci. Rep. 2017, 7, 46085. [Google Scholar] [CrossRef]
- Ben Soussia, I.; El Mouridi, S.; Kang, D.; Leclercq-Blondel, A.; Khoubza, L.; Tardy, P.; Zariohi, N.; Gendrel, M.; Lesage, F.; Kim, E.J.; et al. Mutation of a single residue promotes gating of vertebrate and invertebrate two-pore domain potassium channels. Nat. Commun. 2019, 10, 787. [Google Scholar] [CrossRef]
- Wiedmann, F.; Rinne, S.; Donner, B.; Decher, N.; Katus, H.A.; Schmidt, C. Mechanosensitive TREK-1 two-pore-domain potassium (K2P) channels in the cardiovascular system. Prog. Biophys. Mol. Biol. 2020. [Google Scholar] [CrossRef]
- Lesage, F.; Lazdunski, M. Molecular and functional properties of two-pore-domain potassium channels. Am. J. Physiology. Ren. Physiol. 2000, 279, F793–F801. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.J.; Honoré, E. Properties and modulation of mammalian 2P domain K+ channels. Trends Neurosci. 2001, 24, 339–346. [Google Scholar] [CrossRef]
- Renigunta, V.; Schlichthorl, G.; Daut, J. Much more than a leak: Structure and function of K(2)p-channels. Pflug. Arch. Eur. J. Physiol. 2015, 467, 867–894. [Google Scholar] [CrossRef]
- Lotshaw, D.P. Biophysical, pharmacological, and functional characteristics of cloned and native mammalian two-pore domain K+ channels. Cell Biochem. Biophys. 2007, 47, 209–256. [Google Scholar] [CrossRef]
- Kim, D. Physiology and pharmacology of two-pore domain potassium channels. Curr. Pharm. Des. 2005, 11, 2717–2736. [Google Scholar] [CrossRef]
- Lesage, F. Pharmacology of neuronal background potassium channels. Neuropharmacology 2003, 44, 1–7. [Google Scholar] [CrossRef]
- Goldstein, S.A.; Bockenhauer, D.; O’Kelly, I.; Zilberberg, N. Potassium leak channels and the KCNK family of two-P-domain subunits. Nat. Rev. Neurosci. 2001, 2, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Pathan, A.R.; Rusch, N.J. Two-pore domain K(+) channels: Evidence for TWIK-2 in blood pressure regulation. Hypertension 2011, 58, 539–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, M.; Ledoux, J. K+ channels in biological processes: Vascular K+ channels in the regulation of blood pressure. J. Recept. Ligand Channel Res. 2014, 51. [Google Scholar] [CrossRef] [Green Version]
- Pandit, L.M.; Lloyd, E.E.; Reynolds, J.O.; Lawrence, W.S.; Reynolds, C.; Wehrens, X.H.; Bryan, R.M. TWIK-2 channel deficiency leads to pulmonary hypertension through a rho-kinase-mediated process. Hypertension 2014, 64, 1260–1265. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, E.E.; Crossland, R.F.; Phillips, S.C.; Marrelli, S.P.; Reddy, A.K.; Taffet, G.E.; Hartley, C.J.; Bryan, R.M., Jr. Disruption of K(2P)6.1 produces vascular dysfunction and hypertension in mice. Hypertension 2011, 58, 672–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurney, A.M.; Osipenko, O.N.; MacMillan, D.; McFarlane, K.M.; Tate, R.J.; Kempsill, F.E. Two-pore domain K channel, TASK-1, in pulmonary artery smooth muscle cells. Circ. Res. 2003, 93, 957–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardener, M.J.; Johnson, I.T.; Burnham, M.P.; Edwards, G.; Heagerty, A.M.; Weston, A.H. Functional evidence of a role for two-pore domain potassium channels in rat mesenteric and pulmonary arteries. Br. J. Pharmacol. 2004, 142, 192–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olschewski, A.; Veale, E.L.; Nagy, B.M.; Nagaraj, C.; Kwapiszewska, G.; Antigny, F.; Lambert, M.; Humbert, M.; Czirjak, G.; Enyedi, P. TASK-1 (KCNK3) channels in the lung: From cell biology to clinical implications. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef]
- Antigny, F.; Hautefort, A.; Meloche, J.; Belacel-Ouari, M.; Manoury, B.; Rucker-Martin, C.; Pechoux, C.; Potus, F.; Nadeau, V.; Tremblay, E.; et al. Potassium Channel Subfamily K Member 3 (KCNK3) Contributes to the Development of Pulmonary Arterial Hypertension. Circulation 2016, 133, 1371–1385. [Google Scholar] [CrossRef]
- Feliciangeli, S.; Chatelain, F.C.; Bichet, D.; Lesage, F. The family of K2P channels: Salient structural and functional properties. J. Physiol. 2015, 593, 2587–2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chemin, J.; Patel, A.; Duprat, F.; Zanzouri, M.; Lazdunski, M.; Honore, E. Lysophosphatidic acid-operated K+ channels. J. Biol. Chem. 2005, 280, 4415–4421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathie, A. Neuronal two-pore-domain potassium channels and their regulation by G protein-coupled receptors. J. Physiol. 2007, 578, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Rinne, S.; Renigunta, V.; Schlichthorl, G.; Zuzarte, M.; Bittner, S.; Meuth, S.G.; Decher, N.; Daut, J.; Preisig-Muller, R. A splice variant of the two-pore domain potassium channel TREK-1 with only one pore domain reduces the surface expression of full-length TREK-1 channels. Pflug. Arch. Eur. J. Physiol. 2014, 466, 1559–1570. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kang, D.; Kim, D. Functional properties of four splice variants of a human pancreatic tandem-pore K+ channel, TALK-1. Am. J. Physiology. Cell Physiol. 2003, 285, C529–C538. [Google Scholar] [CrossRef]
- Gu, W.; Schlichthorl, G.; Hirsch, J.R.; Engels, H.; Karschin, C.; Karschin, A.; Derst, C.; Steinlein, O.K.; Daut, J. Expression pattern and functional characteristics of two novel splice variants of the two-pore-domain potassium channel TREK-2. J. Physiol. 2002, 539, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Xian Tao, L.; Dyachenko, V.; Zuzarte, M.; Putzke, C.; Preisig-Muller, R.; Isenberg, G.; Daut, J. The stretch-activated potassium channel TREK-1 in rat cardiac ventricular muscle. Cardiovasc Res. 2006, 69, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.; Plant, L.D.; Wilkens, C.M.; McCrossan, Z.A.; Goldstein, S.A. Alternative translation initiation in rat brain yields K2P2.1 potassium channels permeable to sodium. Neuron 2008, 58, 859–870. [Google Scholar] [CrossRef] [Green Version]
- Levitz, J.; Royal, P.; Comoglio, Y.; Wdziekonski, B.; Schaub, S.; Clemens, D.M.; Isacoff, E.Y.; Sandoz, G. Heterodimerization within the TREK channel subfamily produces a diverse family of highly regulated potassium channels. Proc. Natl. Acad. Sci. USA 2016, 113, 4194–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czirjak, G.; Enyedi, P. Formation of functional heterodimers between the TASK-1 and TASK-3 two-pore domain potassium channel subunits. J. Biol. Chem. 2002, 277, 5426–5432. [Google Scholar] [CrossRef] [Green Version]
- Hwang, E.M.; Kim, E.; Yarishkin, O.; Woo, D.H.; Han, K.S.; Park, N.; Bae, Y.; Woo, J.; Kim, D.; Park, M.; et al. A disulphide-linked heterodimer of TWIK-1 and TREK-1 mediates passive conductance in astrocytes. Nat. Commun. 2014, 5, 3227. [Google Scholar] [CrossRef] [Green Version]
- Wareing, M.; Bai, X.; Seghier, F.; Turner, C.M.; Greenwood, S.L.; Baker, P.N.; Taggart, M.J.; Fyfe, G.K. Expression and function of potassium channels in the human placental vasculature. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R437–R446. [Google Scholar] [CrossRef] [Green Version]
- Naderi, S.; Tsai, S.A.; Khandelwal, A. Hypertensive Disorders of Pregnancy. Curr. Atheroscler Rep. 2017, 19, 15. [Google Scholar] [CrossRef]
- Folk, D.M. Hypertensive Disorders of Pregnancy: Overview and Current Recommendations. J. Midwifery Womens Health 2018, 63, 289–300. [Google Scholar] [CrossRef]
- Fox, R.; Kitt, J.; Leeson, P.; Aye, C.Y.L.; Lewandowski, A.J. Preeclampsia: Risk Factors, Diagnosis, Management, and the Cardiovascular Impact on the Offspring. J. Clin. Med. 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Peres, G.M.; Mariana, M.; Cairrao, E. Pre-Eclampsia and Eclampsia: An Update on the Pharmacological Treatment Applied in Portugal. J. Cardiovasc. Dev. Dis. 2018, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, R.H. Molecular determinants of voltage-gated potassium currents in vascular smooth muscle. Cell Biochem. Biophys. 2005, 42, 167–195. [Google Scholar] [CrossRef]
- Cox, R.H.; Folander, K.; Swanson, R. Differential Expression of Voltage-Gated K+ Channel Genes in Arteries From Spontaneously Hypertensive and Wistar-Kyoto Rats. Hypertension 2001, 37, 1315–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, R.H. Changes in the expression and function of arterial potassium channels during hypertension. Vasc. Pharm. 2002, 38, 13–23. [Google Scholar] [CrossRef]
- Pinterova, M.; Kunes, J.; Zicha, J. Altered neural and vascular mechanisms in hypertension. Physiol Res. 2011, 60, 381–402. [Google Scholar] [CrossRef]
- Djokic, V.; Jankovic-Raznatovic, S.; Novakovic, R.; Kostic, M.; Rajkovic, J.; Labudovic-Borovic, M.; Rakocevic, J.; Stanisic, J.; Djuric, M.; Gojkovic-Bukarica, L. Effect of gestational diabetes mellitus and pregnancy-induced hypertension on human umbilical vein smooth muscle KATP channels. Exp. Mol. Pathol. 2019, 111, 104323. [Google Scholar] [CrossRef]
- Djokic, V.; Jankovic, S.; Labudovic-Borovic, M.; Rakocevic, J.; Stanisic, J.; Rajkovic, J.; Novakovic, R.; Kostic, M.; Djuric, M.; Gostimirovic, M.; et al. Pregnancy-induced hypertension decreases Kv1.3 potassium channel expression and function in human umbilical vein smooth muscle. Eur. J. Pharm. 2020, 173281. [Google Scholar] [CrossRef]
- Wareing, M.; Greenwood, S.L.; Fyfe, G.K.; Baker, P.N. Reactivity of human placental chorionic plate vessels from pregnancies complicated by intrauterine growth restriction (IUGR). Biol. Reprod. 2006, 75, 518–523. [Google Scholar] [CrossRef]
- Wareing, M. Oxygen sensitivity, potassium channels, and regulation of placental vascular tone. Microcirculation 2014, 21, 58–66. [Google Scholar] [CrossRef]
- Wei, X.; Zhang, Y.; Yin, B.; Wen, J.; Cheng, J.; Fu, X. The expression and function of KCNQ potassium channels in human chorionic plate arteries from women with normal pregnancies and pre-eclampsia. PLoS ONE 2018, 13, e0192122. [Google Scholar] [CrossRef] [Green Version]
- Mills, T.A.; Greenwood, S.L.; Devlin, G.; Shweikh, Y.; Robinson, M.; Cowley, E.; Hayward, C.E.; Cottrell, E.C.; Tropea, T.; Brereton, M.F. Activation of KV7 channels stimulates vasodilatation of human placental chorionic plate arteries. Placenta 2015, 36, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.D.; McCallum, L.A.; Kurlak, L.O.; Greenwood, I.A.; Broughton Pipkin, F.; Tribe, R.M. Novel expression and regulation of voltage-dependent potassium channels in placentas from women with preeclampsia. Hypertension 2011, 58, 497–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manville, R.W.; Abbott, G.W. Isoform-Selective KCNA1 Potassium Channel Openers Built from Glycine. J. Pharm. Exp. 2020, 373, 391–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jepps, T.A.; Olesen, S.P.; Greenwood, I.A. One man’s side effect is another man’s therapeutic opportunity: Targeting Kv7 channels in smooth muscle disorders. Br. J. Pharmacol. 2013, 168, 19–27. [Google Scholar] [CrossRef]
- Lawson, K.; McKay, N.G. Modulation of potassium channels as a therapeutic approach. Curr. Pharm. Des. 2006, 12, 459–470. [Google Scholar] [CrossRef]
- Bartok, A.; Toth, A.; Somodi, S.; Szanto, T.G.; Hajdu, P.; Panyi, G.; Varga, Z. Margatoxin is a non-selective inhibitor of human Kv1.3 K+ channels. Toxicon Off. J. Int. Soc. Toxinol. 2014, 87, 6–16. [Google Scholar] [CrossRef] [Green Version]
- Khammy, M.M.; Kim, S.; Bentzen, B.H.; Lee, S.; Choi, I.; Aalkjaer, C.; Jepps, T.A. 4-Aminopyridine: A pan voltage-gated potassium channel inhibitor that enhances Kv 7.4 currents and inhibits noradrenaline-mediated contraction of rat mesenteric small arteries. Br. J. Pharmacol. 2018, 175, 501–516. [Google Scholar] [CrossRef] [Green Version]
- Lindquist, S.; Stangel, M. Update on treatment options for Lambert-Eaton myasthenic syndrome: Focus on use of amifampridine. Neuropsychiatr. Dis. Treat. 2011, 7, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.C.; Trager, L.; Anzia, L.E.; Saliba, W.; Bassiouny, M.; Bhargava, M.; Chung, M.; Desai, M.; Garberich, R.; Lever, H.; et al. Dofetilide for suppression of atrial fibrillation in hypertrophic cardiomyopathy: A case series and literature review. Pacing Clin. Electrophysiol. Pace 2018, 41, 396–401. [Google Scholar] [CrossRef]
- Colunga Biancatelli, R.M.; Congedo, V.; Calvosa, L.; Ciacciarelli, M.; Polidoro, A.; Iuliano, L. Adverse reactions of Amiodarone. J. Geriatr. Cardiol. JGC 2019, 16, 552–566. [Google Scholar] [CrossRef]
- Faraci, F.M.; Heistad, D.D. Regulation of the cerebral circulation: Role of endothelium and potassium channels. Physiol. Rev. 1998, 78, 53–97. [Google Scholar] [CrossRef] [PubMed]
- Rusch, N.J.; Liu, Y. Potassium channels in hypertension: Homeostatic pathways to buffer arterial contraction. J. Lab. Clin. Med. 1997, 130, 245–251. [Google Scholar] [CrossRef]
- Wang, R.; Wu, L. The chemical modification of KCa channels by carbon monoxide in vascular smooth muscle cells. J. Biol. Chem. 1997, 272, 8222–8226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amberg, G.C.; Bonev, A.D.; Rossow, C.F.; Nelson, M.T.; Santana, L.F. Modulation of the molecular composition of large conductance, Ca2+ activated K+ channels in vascular smooth muscle during hypertension. J. Clin. Investig. 2003, 112, 717–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amberg, G.C.; Santana, L.F. Downregulation of the BK channel beta1 subunit in genetic hypertension. Circ. Res. 2003, 93, 965–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, X.; Leo, M.D.; Jaggar, J.H. Endothelin-1 Stimulates Vasoconstriction Through Rab11A Serine 177 Phosphorylation. Circ. Res. 2017, 121, 650–661. [Google Scholar] [CrossRef]
- Leo, M.D.; Zhai, X.; Yin, W.; Jaggar, J.H. Impaired Trafficking of beta1 Subunits Inhibits BK Channels in Cerebral Arteries of Hypertensive Rats. Hypertension 2018, 72, 765–775. [Google Scholar] [CrossRef]
- Joseph, B.K.; Thakali, K.M.; Moore, C.L.; Rhee, S.W. Ion channel remodeling in vascular smooth muscle during hypertension: Implications for novel therapeutic approaches. Pharmacol. Res. 2013, 70, 126–138. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Li, F.; Yang, M.; Fan, Y.; Beejadhursing, R.; Xie, Y.; Zhou, Y.; Deng, D. Impairment of BKca channels in human placental chorionic plate arteries is potentially relevant to the development of preeclampsia. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2018, 41, 126–134. [Google Scholar] [CrossRef]
- Li, F.F.; He, M.Z.; Xie, Y.; Wu, Y.Y.; Yang, M.T.; Fan, Y.; Qiao, F.Y.; Deng, D.R. Involvement of dysregulated IKCa and SKCa channels in preeclampsia. Placenta 2017, 58, 9–16. [Google Scholar] [CrossRef]
- Bentzen, B.H.; Olesen, S.P.; Ronn, L.C.; Grunnet, M. BK channel activators and their therapeutic perspectives. Front. Physiol. 2014, 5, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, H.; Wang, M.; Patel, P.N.; Kalogeris, T.; Liu, Y.; Durante, W.; Korthuis, R.J. Preconditioning with the BKCa channel activator NS-1619 prevents ischemia-reperfusion-induced inflammation and mucosal barrier dysfunction: Roles for ROS and heme oxygenase-1. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H988–H999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olver, T.D.; Edwards, J.C.; Ferguson, B.S.; Hiemstra, J.A.; Thorne, P.K.; Hill, M.A.; Laughlin, M.H.; Emter, C.A. Chronic interval exercise training prevents BKCa channel-mediated coronary vascular dysfunction in aortic-banded miniswine. J. Appl. Physiol. 2018, 125, 86–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajada, S.; Cidad, P.; Moreno-Dominguez, A.; Perez-Garcia, M.T.; Lopez-Lopez, J.R. High blood pressure associates with the remodelling of inward rectifier K+ channels in mice mesenteric vascular smooth muscle cells. J. Physiol. 2012, 590, 6075–6091. [Google Scholar] [CrossRef] [Green Version]
- Caballero, R.; Dolz-Gaiton, P.; Gomez, R.; Amoros, I.; Barana, A.; Gonzalez de la Fuente, M.; Osuna, L.; Duarte, J.; Lopez-Izquierdo, A.; Moraleda, I.; et al. Flecainide increases Kir2.1 currents by interacting with cysteine 311, decreasing the polyamine-induced rectification. Proc. Natl. Acad. Sci. USA 2010, 107, 15631–15636. [Google Scholar] [CrossRef] [Green Version]
- Walsh, K.B. Screening Technologies for Inward Rectifier Potassium Channels: Discovery of New Blockers and Activators. Slas Discov. Adv. Life Sci. R D 2020, 25, 420–433. [Google Scholar] [CrossRef]
- Rodriguez-Menchaca, A.A.; Navarro-Polanco, R.A.; Ferrer-Villada, T.; Rupp, J.; Sachse, F.B.; Tristani-Firouzi, M.; Sanchez-Chapula, J.A. The molecular basis of chloroquine block of the inward rectifier Kir2.1 channel. Proc. Natl. Acad. Sci. USA 2008, 105, 1364–1368. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Chapula, J.; Salinas-Stefanon, E.; Torres-Jácome, J.; Benavides-Haro, D.; Navarro-Polanco, R. Blockade of currents by the antimalarial drug chloroquine in feline ventricular myocytes. J. Pharmacol. Exp. Ther. 2001, 297, 437–445. [Google Scholar]
- Takanari, H.; Nalos, L.; Stary-Weinzinger, A.; de Git, K.C.; Varkevisser, R.; Linder, T.; Houtman, M.J.; Peschar, M.; de Boer, T.P.; Tidwell, R.R.; et al. Efficient and specific cardiac IK(1) inhibition by a new pentamidine analogue. Cardiovasc Res. 2013, 99, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.R.; Wu, M.; Yu, H.; Long, S.; Stevens, A.; Engers, D.W.; Sackin, H.; Daniels, J.S.; Dawson, E.S.; Hopkins, C.R.; et al. Selective inhibition of the K(ir)2 family of inward rectifier potassium channels by a small molecule probe: The discovery, SAR, and pharmacological characterization of ML133. ACS Chem. Biol. 2011, 6, 845–856. [Google Scholar] [CrossRef] [Green Version]
- Van de Voorde, J.; Vanheel, B.; Leusen, I. Endothelium-dependent relaxation and hyperpolarization in aorta from control and renal hypertensive rats. Circ. Res. 1992, 70, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitazono, T.; Heistad, D.D.; Faraci, F.M. ATP-sensitive potassium channels in the basilar artery during chronic hypertension. Hypertension 1993, 22, 677–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kam, K.L.; Pfaffendorf, M.; van Zwieten, P.A. Drug-induced endothelium-dependent and -independent relaxations in isolated resistance vessels taken from simultaneously hypertensive and streptozotocin-diabetic rats. Blood Press. 1994, 3, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Hanna, S.T.; Wang, R.; McNeill, J.R. Altered vascular reactivity and KATP channel currents in vascular smooth muscle cells from deoxycorticosterone acetate (DOCA)-salt hypertensive rats. J. Cardiovasc. Pharmacol. 2004, 44, 525–531. [Google Scholar] [CrossRef]
- Kalliovalkama, J. Arterial function in nitric oxide-deficient hypertension: Influence of long-term angiotensin II receptor antagonism. Cardiovasc. Res. 1999, 42, 773–782. [Google Scholar] [CrossRef]
- Takaba, H.; Nagao, T.; Ibayashi, S.; Kitazono, T.; Fujii, K.; Fujishima, M. Altered cerebrovascular response to a potassium channel opener in hypertensive rats. Hypertension 1996, 28, 143–146. [Google Scholar] [CrossRef]
- Kawata, T.; Mimuro, T.; Onuki, T.; Tsuchiya, K.; Nihei, H.; Koike, T. The K(ATP) channel opener nicorandil: Effect on renal hemodynamics in spontaneously hypertensive and Wistar Kyoto rats. Kidney Int. Suppl. 1998, 67, S231–S233. [Google Scholar] [CrossRef] [Green Version]
- Gutterman, D.D.; Miura, H.; Liu, Y. Redox modulation of vascular tone: Focus of potassium channel mechanisms of dilation. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 671–678. [Google Scholar] [CrossRef]
- Bisseling, T.M.; Versteegen, M.G.; van der Wal, S.; Copius Peereboom-Stegeman, J.J.; Borggreven, J.M.; Steegers, E.A.; van der Laak, J.A.; Russel, F.G.; Smits, P. Impaired KATP channel function in the fetoplacental circulation of patients with type 1 diabetes mellitus. Am. J. Obstet. Gynecol. 2005, 192, 973–979. [Google Scholar] [CrossRef]
- Taricco, E.; Radaelli, T.; Rossi, G.; Nobile de Santis, M.S.; Bulfamante, G.P.; Avagliano, L.; Cetin, I. Effects of gestational diabetes on fetal oxygen and glucose levels in vivo. BJOG Int. J. Obstet. Gynaecol. 2009, 116, 1729–1735. [Google Scholar] [CrossRef]
- Toljic, M.; Egic, A.; Munjas, J.; Karadzov Orlic, N.; Milovanovic, Z.; Radenkovic, A.; Vuceljic, J.; Joksic, I. Increased oxidative stress and cytokinesis-block micronucleus cytome assay parameters in pregnant women with gestational diabetes mellitus and gestational arterial hypertension. Reprod. Toxicol. 2017, 71, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Tang, P.L.; Liu, P.Y.; Huang, W.C.; Chen, Y.Y.; Wang, H.P.; Chang, J.T.; Lin, L.T. Maternal pregnancy-induced hypertension increases subsequent neonatal necrotizing enterocolitis risk: A nationwide population-based retrospective cohort study in Taiwan. Medicine 2018, 97, e11739. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, C.M.; Braunstein, T.H.; Holstein-Rathlou, N.H.; Salomonsson, M. Role of vascular potassium channels in the regulation of renal hemodynamics. Am. J. Physiol. Ren. Physiol. 2012, 302, F505–F518. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Xiao, D.; Zhang, L. Potassium channels and uterine vascular adaptation to pregnancy and chronic hypoxia. Curr. Vasc. Pharmacol. 2013, 11, 737–747. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Knutsen, R.H.; Zhang, H.; Osei-Owusu, P.; Moreno-Dominguez, A.; Harter, T.M.; Uchida, K.; Remedi, M.S.; Dietrich, H.H.; Bernal-Mizrachi, C.; et al. Hypotension due to Kir6.1 gain-of-function in vascular smooth muscle. J. Am. Heart Assoc. 2013, 2, e000365. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Duan, P.; Hu, X.; Li, R.; Zhu, Q. Altered KATP Channel Subunits Expression and Vascular Reactivity in Spontaneously Hypertensive Rats With Age. J. Cardiovasc. Pharmacol. 2016, 68, 143–149. [Google Scholar] [CrossRef]
- Ohya, Y.; Setoguchi, M.; Fujii, K.; Nagao, T.; Abe, I.; Fujishima, M. Impaired action of levcromakalim on ATP-sensitive K+ channels in mesenteric artery cells from spontaneously hypertensive rats. Hypertension 1996, 27, 1234–1239. [Google Scholar] [CrossRef]
- Du, Q.; Jovanovic, S.; Tulic, L.; Tulic, I.; Jovanovic, A. Pregnancy-induced hypertension is associated with down-regulation of Kir6.1 in human myometrium. Pregnancy Hypertens. 2019, 18, 96–98. [Google Scholar] [CrossRef]
- Blanco-Rivero, J.; Gamallo, C.; Aras-Lopez, R.; Cobeno, L.; Cogolludo, A.; Perez-Vizcaino, F.; Ferrer, M.; Balfagon, G. Decreased expression of aortic KIR6.1 and SUR2B in hypertension does not correlate with changes in the functional role of K(ATP) channels. Eur J. Pharm. 2008, 587, 204–208. [Google Scholar] [CrossRef]
- Thorne, G.D.; Conforti, L.; Paul, R.J. Hypoxic vasorelaxation inhibition by organ culture correlates with loss of Kv channels but not Ca(2+) channels. Am. J. Physiology. Heart Circ. Physiol. 2002, 283, H247–H253. [Google Scholar] [CrossRef] [Green Version]
- Khan, R.N.; Morrison, J.J.; Smith, S.K.; Ashford, M.L.J. Activation of large-conductance potassium channels in pregnant human myometrium by pinacidil. Am. J. Obstet. Gynecol. 1998, 178, 1027–1034. [Google Scholar] [CrossRef]
- Novakovic, A.; Pavlovic, M.; Milojevic, P.; Stojanovic, I.; Nenezic, D.; Jovic, M.; Ugresic, N.; Kanjuh, V.; Yang, Q.; He, G.W. Different potassium channels are involved in relaxation of rat renal artery induced by P1075. Basic Clin. Pharmacol. Toxicol. 2012, 111, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Stockbridge, N.; Zhang, H.; Weir, B. Effects of K+ channel agonists cromakalim and pinacidil on rat basilar artery smooth muscle cells are mediated by Ca++ activated K+ channels. Biochem. Biophys. Res. Commun. 1991, 181, 172–178. [Google Scholar] [CrossRef]
- Kolias, T.J.; Chai, S.; Webb, R.C. Potassium channel antagonists and vascular reactivity in stroke-prone spontaneously hypertensive rats. Am. J. Hypertens. 1993, 6, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Hutri-Kahonen, N.; Kahonen, M.; Wu, X.; Sand, J.; Nordback, I.; Taurio, J.; Porsti, I. Control of vascular tone in isolated mesenteric arterial segments from hypertensive patients. Br. J. Pharmacol. 1999, 127, 1735–1743. [Google Scholar] [CrossRef] [Green Version]
- Miyata, N.; Tsuchida, K.; Otomo, S. Functional changes in potassium channels in carotid arteries from stroke-prone spontaneously hypertensive rats. Eur. J. Pharmacol. 1990, 182, 209–210. [Google Scholar] [CrossRef]
- Furspan, P.B.; Webb, R.C. Decreased ATP sensitivity of a K+ channel and enhanced vascular smooth muscle relaxation in genetically hypertensive rats. J. Hypertens. 1993, 11, 1067–1072. [Google Scholar] [CrossRef]
- Jahangir, A.; Terzic, A. K(ATP) channel therapeutics at the bedside. J. Mol. Cell Cardiol. 2005, 39, 99–112. [Google Scholar] [CrossRef] [Green Version]
- Roy Chowdhury, U.; Dosa, P.I.; Fautsch, M.P. ATP sensitive potassium channel openers: A new class of ocular hypotensive agents. Exp. Eye Res. 2017, 158, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Wiedmann, F.; Schmidt, C.; Lugenbiel, P.; Staudacher, I.; Rahm, A.K.; Seyler, C.; Schweizer, P.A.; Katus, H.A.; Thomas, D. Therapeutic targeting of two-pore-domain potassium (K(2P)) channels in the cardiovascular system. Clin. Sci. 2016, 130, 643–650. [Google Scholar] [CrossRef]
- Abraham, D.M.; Lee, T.E.; Watson, L.J.; Mao, L.; Chandok, G.; Wang, H.G.; Frangakis, S.; Pitt, G.S.; Shah, S.H.; Wolf, M.J. The two-pore domain potassium channel TREK-1 mediates cardiac fibrosis and diastolic dysfunction. J. Clin. Investig. 2018, 128, 4843–4855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamas, J.A.; Fernandez-Fernandez, D. Tandem pore TWIK-related potassium channels and neuroprotection. Neural Regen. Res. 2019, 14, 1293–1308. [Google Scholar] [CrossRef] [PubMed]
- Loucif, A.J.C.; Saintot, P.P.; Liu, J.; Antonio, B.M.; Zellmer, S.G.; Yoger, K.; Veale, E.L.; Wilbrey, A.; Omoto, K.; Cao, L. GI-530159, a novel, selective, mechanosensitive two-pore-domain potassium (K2P ) channel opener, reduces rat dorsal root ganglion neuron excitability. Br. J. Pharmacol. 2018, 175, 2272–2283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veale, E.L.; Al-Moubarak, E.; Bajaria, N.; Omoto, K.; Cao, L.; Tucker, S.J.; Stevens, E.B.; Mathie, A. Influence of the N terminus on the biophysical properties and pharmacology of TREK1 potassium channels. Mol. Pharmacol. 2014, 85, 671–681. [Google Scholar] [CrossRef]
- Bagriantsev, S.N.; Ang, K.H.; Gallardo-Godoy, A.; Clark, K.A.; Arkin, M.R.; Renslo, A.R.; Minor, D.L., Jr. A high-throughput functional screen identifies small molecule regulators of temperature- and mechano-sensitive K2P channels. ACS Chem. Biol. 2013, 8, 1841–1851. [Google Scholar] [CrossRef]
- Cunningham, K.; Veale, E.; Clapp, L.; Mathie, A. The Role of the K2P Channels TASK-1, TREK-1 and TREK-2 in the Use of Treprostinil Therapy in Pulmonary Arterial Hypertension. FASEB J. 2018, 32. [Google Scholar] [CrossRef]
- Mathie, A.; Veale, E.L.; Cunningham, K.P.; Holden, R.G.; Wright, P.D. Two-Pore Domain Potassium Channels as Drug Targets: Anesthesia and Beyond. Annu. Rev. Pharmacol. Toxicol. 2020, 61. [Google Scholar] [CrossRef]
- Mazella, J.; Petrault, O.; Lucas, G.; Deval, E.; Beraud-Dufour, S.; Gandin, C.; El-Yacoubi, M.; Widmann, C.; Guyon, A.; Chevet, E. Spadin, a sortilin-derived peptide, targeting rodent TREK-1 channels: A new concept in the antidepressant drug design. Plos Biol. 2010, 8, e1000355. [Google Scholar] [CrossRef] [Green Version]
- Takahira, M.; Sakurai, M.; Sakurada, N.; Sugiyama, K. Fenamates and diltiazem modulate lipid-sensitive mechano-gated 2P domain K(+) channels. Pflug. Arch. Eur. J. Physiol. 2005, 451, 474–478. [Google Scholar] [CrossRef]
- Bodnar, M.; Schlichthorl, G.; Daut, J. The potassium current carried by TREK-1 channels in rat cardiac ventricular muscle. Pflug. Arch. Eur. J. Physiol. 2015, 467, 1069–1079. [Google Scholar] [CrossRef]
- Fink, M.; Lesage, F.; Duprat, F.; Heurteaux, C.; Reyes, R.; Fosset, M.; Lazdunski, M. A neuronal two P domain K+ channel stimulated by arachidonic acid and polyunsaturated fatty acids. EMBO J. 1998, 17, 3297–3308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meadows, H.J.; Chapman, C.G.; Duckworth, D.M.; Kelsell, R.E.; Murdock, P.R.; Nasir, S.; Rennie, G.; Randall, A.D. The neuroprotective agent sipatrigine (BW619C89) potently inhibits the human tandem pore-domain K+ channels TREK-1 and TRAAK. Brain Res. 2001, 892, 94–101. [Google Scholar] [CrossRef]
- Thummler, S.; Duprat, F.; Lazdunski, M. Antipsychotics inhibit TREK but not TRAAK channels. Biochem. Biophys. Res. Commun. 2007, 354, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Lee, D.K.; Hong, S.G.; Han, J.; Kang, D. Activation of TREK-1, but Not TREK-2, Channel by Mood Stabilizers. Int. J. Mol. Sci. 2017, 18, 2460. [Google Scholar] [CrossRef] [Green Version]
- Maingret, F.; Patel, A.J.; Lesage, F.; Lazdunski, M.; Honore, E. Lysophospholipids open the two-pore domain mechano-gated K(+) channels TREK-1 and TRAAK. J. Biol. Chem. 2000, 275, 10128–10133. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Schematic representation of the K+ channels present in human umbilical artery.

Figure 2.
Schematic representation of the mechanisms implicated in activation of Kv and BKCa channels in human umbilical artery smooth muscle cells (HUASMCs). LEGEND: ![Cells 09 01956 i001]() —G-protein coupled receptor;
—G-protein coupled receptor; ![Cells 09 01956 i002]() —peptide receptor;
—peptide receptor; ![Cells 09 01956 i003]() /green arrows—stimulation;
/green arrows—stimulation; ![Cells 09 01956 i004]() /red arrows—inhibition; ?—unknown mechanism; [Ca2+]I—intracellular Ca2+ concentration; BKCa—large-conductance Ca2+-activated K+ channels; Ca2+—calcium; cAMP—cyclic adenosine monophosphate; cGMP—cyclic guanosine monophosphate; eNOS—endothelial nitric oxide synthase; K+—potassium; Kv—voltage-gated K+ channels; NO—nitric oxide; PDE—phosphodiesterase; PKA—protein kinase A; PKG—protein kinase G; SKCa—small-conductance Ca2+-activated K+ channels; VGCC—voltage-gated Ca2+ channels.
/red arrows—inhibition; ?—unknown mechanism; [Ca2+]I—intracellular Ca2+ concentration; BKCa—large-conductance Ca2+-activated K+ channels; Ca2+—calcium; cAMP—cyclic adenosine monophosphate; cGMP—cyclic guanosine monophosphate; eNOS—endothelial nitric oxide synthase; K+—potassium; Kv—voltage-gated K+ channels; NO—nitric oxide; PDE—phosphodiesterase; PKA—protein kinase A; PKG—protein kinase G; SKCa—small-conductance Ca2+-activated K+ channels; VGCC—voltage-gated Ca2+ channels.
 —G-protein coupled receptor;
—G-protein coupled receptor;  —peptide receptor;
—peptide receptor;  /green arrows—stimulation;
/green arrows—stimulation;  /red arrows—inhibition; ?—unknown mechanism; [Ca2+]I—intracellular Ca2+ concentration; BKCa—large-conductance Ca2+-activated K+ channels; Ca2+—calcium; cAMP—cyclic adenosine monophosphate; cGMP—cyclic guanosine monophosphate; eNOS—endothelial nitric oxide synthase; K+—potassium; Kv—voltage-gated K+ channels; NO—nitric oxide; PDE—phosphodiesterase; PKA—protein kinase A; PKG—protein kinase G; SKCa—small-conductance Ca2+-activated K+ channels; VGCC—voltage-gated Ca2+ channels.
/red arrows—inhibition; ?—unknown mechanism; [Ca2+]I—intracellular Ca2+ concentration; BKCa—large-conductance Ca2+-activated K+ channels; Ca2+—calcium; cAMP—cyclic adenosine monophosphate; cGMP—cyclic guanosine monophosphate; eNOS—endothelial nitric oxide synthase; K+—potassium; Kv—voltage-gated K+ channels; NO—nitric oxide; PDE—phosphodiesterase; PKA—protein kinase A; PKG—protein kinase G; SKCa—small-conductance Ca2+-activated K+ channels; VGCC—voltage-gated Ca2+ channels.
Figure 2.
Schematic representation of the mechanisms implicated in activation of Kv and BKCa channels in human umbilical artery smooth muscle cells (HUASMCs). LEGEND: ![Cells 09 01956 i001]() —G-protein coupled receptor;
—G-protein coupled receptor; ![Cells 09 01956 i002]() —peptide receptor;
—peptide receptor; ![Cells 09 01956 i003]() /green arrows—stimulation;
/green arrows—stimulation; ![Cells 09 01956 i004]() /red arrows—inhibition; ?—unknown mechanism; [Ca2+]I—intracellular Ca2+ concentration; BKCa—large-conductance Ca2+-activated K+ channels; Ca2+—calcium; cAMP—cyclic adenosine monophosphate; cGMP—cyclic guanosine monophosphate; eNOS—endothelial nitric oxide synthase; K+—potassium; Kv—voltage-gated K+ channels; NO—nitric oxide; PDE—phosphodiesterase; PKA—protein kinase A; PKG—protein kinase G; SKCa—small-conductance Ca2+-activated K+ channels; VGCC—voltage-gated Ca2+ channels.
/red arrows—inhibition; ?—unknown mechanism; [Ca2+]I—intracellular Ca2+ concentration; BKCa—large-conductance Ca2+-activated K+ channels; Ca2+—calcium; cAMP—cyclic adenosine monophosphate; cGMP—cyclic guanosine monophosphate; eNOS—endothelial nitric oxide synthase; K+—potassium; Kv—voltage-gated K+ channels; NO—nitric oxide; PDE—phosphodiesterase; PKA—protein kinase A; PKG—protein kinase G; SKCa—small-conductance Ca2+-activated K+ channels; VGCC—voltage-gated Ca2+ channels.
—G-protein coupled receptor; —peptide receptor; /green arrows—stimulation; /red arrows—inhibition; ?—unknown mechanism; [Ca2+]I—intracellular Ca2+ concentration; BKCa—large-conductance Ca2+-activated K+ channels; Ca2+—calcium; cAMP—cyclic adenosine monophosphate; cGMP—cyclic guanosine monophosphate; eNOS—endothelial nitric oxide synthase; K+—potassium; Kv—voltage-gated K+ channels; NO—nitric oxide; PDE—phosphodiesterase; PKA—protein kinase A; PKG—protein kinase G; SKCa—small-conductance Ca2+-activated K+ channels; VGCC—voltage-gated Ca2+ channels.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lorigo, M.; Oliveira, N.; Cairrao, E. Clinical Importance of the Human Umbilical Artery Potassium Channels. Cells 2020, 9, 1956. https://doi.org/10.3390/cells9091956
AMA Style
Lorigo M, Oliveira N, Cairrao E. Clinical Importance of the Human Umbilical Artery Potassium Channels. Cells. 2020; 9(9):1956. https://doi.org/10.3390/cells9091956
Chicago/Turabian StyleLorigo, Margarida, Nelson Oliveira, and Elisa Cairrao. 2020. "Clinical Importance of the Human Umbilical Artery Potassium Channels" Cells 9, no. 9: 1956. https://doi.org/10.3390/cells9091956
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.