Glial A2B Adenosine Receptors Modulate Abnormal Tachykininergic Responses and Prevent Enteric Inflammation Associated with High Fat Diet-Induced Obesity

 , , , ,
, , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Experiments on Animals
2.1.1. Animals and Diet
2.1.2. Recording of Colonic Contractile Activity
2.1.3. Design of Functional Experiments
2.1.4. Immunohistochemistry on Colonic Whole Mount Preparations
2.2. Experiments on Cultured Enteric Glial Cells
2.2.1. Cell Culture
2.2.2. Palmitate-Bovine Serum Albumin Complex Preparation
2.2.3. Stimulation Protocol
2.2.4. Western Blot
2.2.5. Assessment of Interleukin-1β, Substance P, and Glial Cell Derived Neurotrophic Factor Production from EGCs
2.3. Statistical Analysis
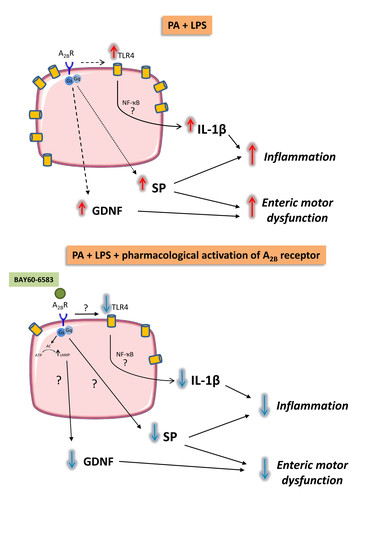
3. Results
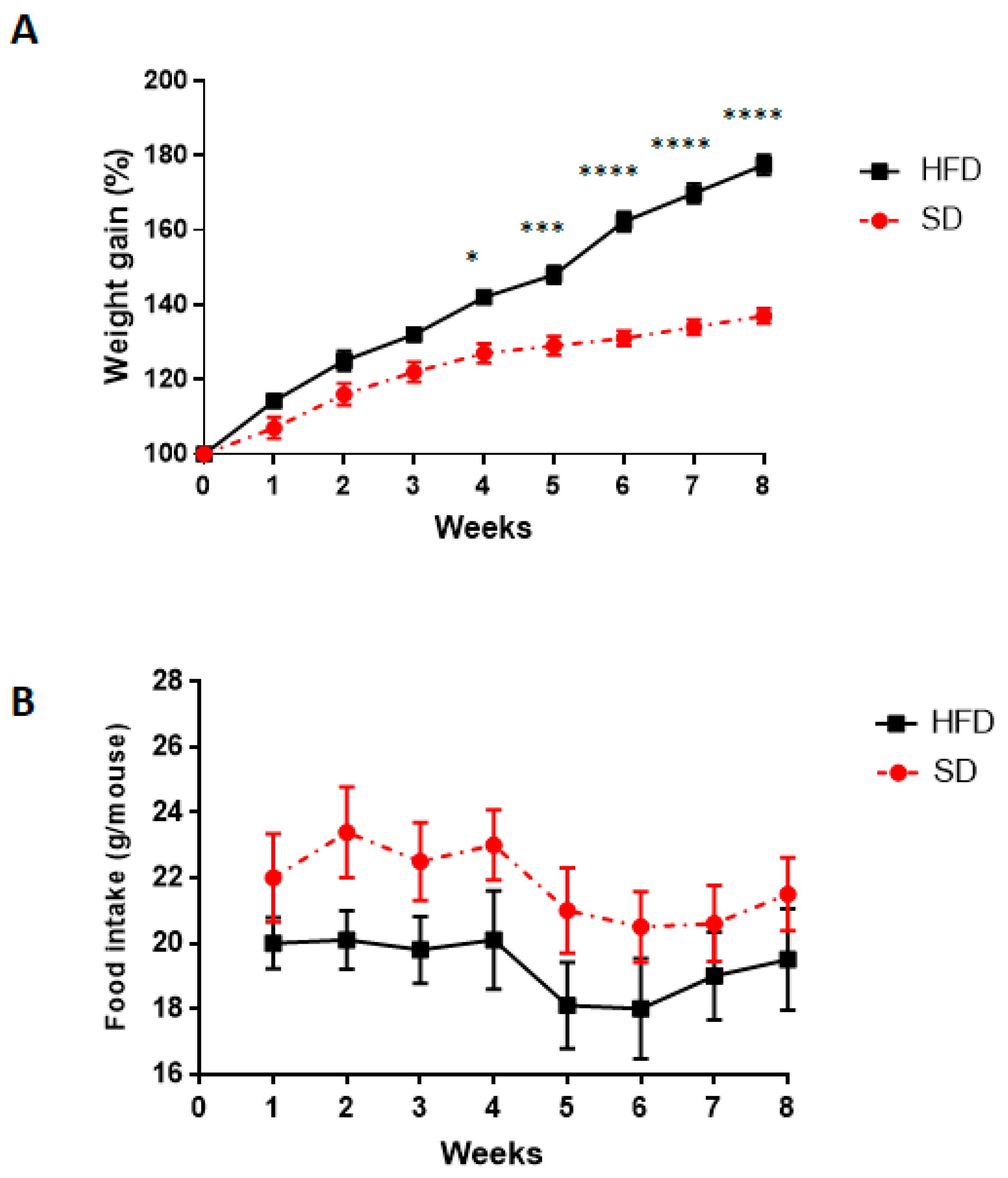
3.1. Body Weight and Food Intake
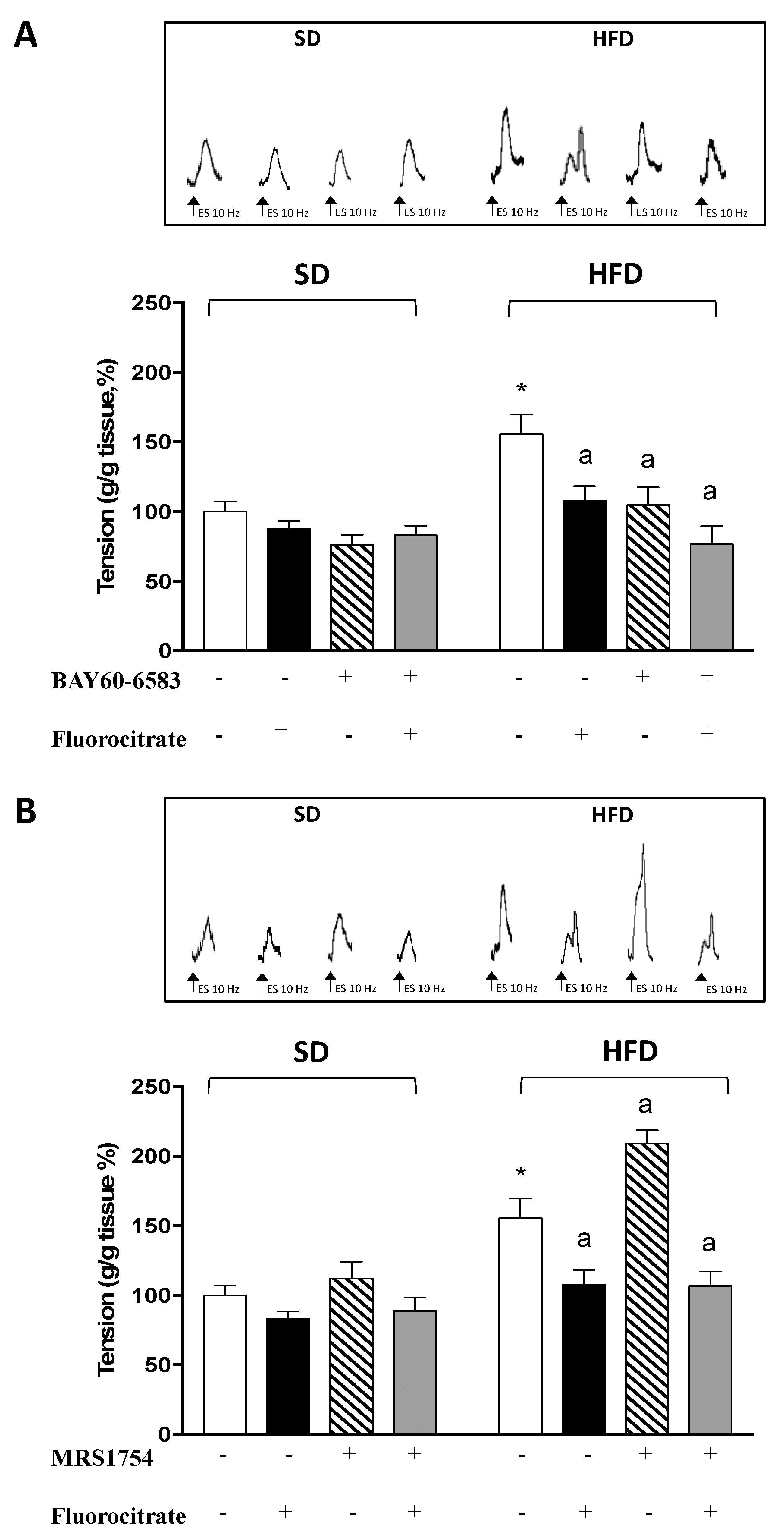
3.2. A2B Receptors on Enteric Glia Modulate Tachykininergic Contractions in HFD Mice
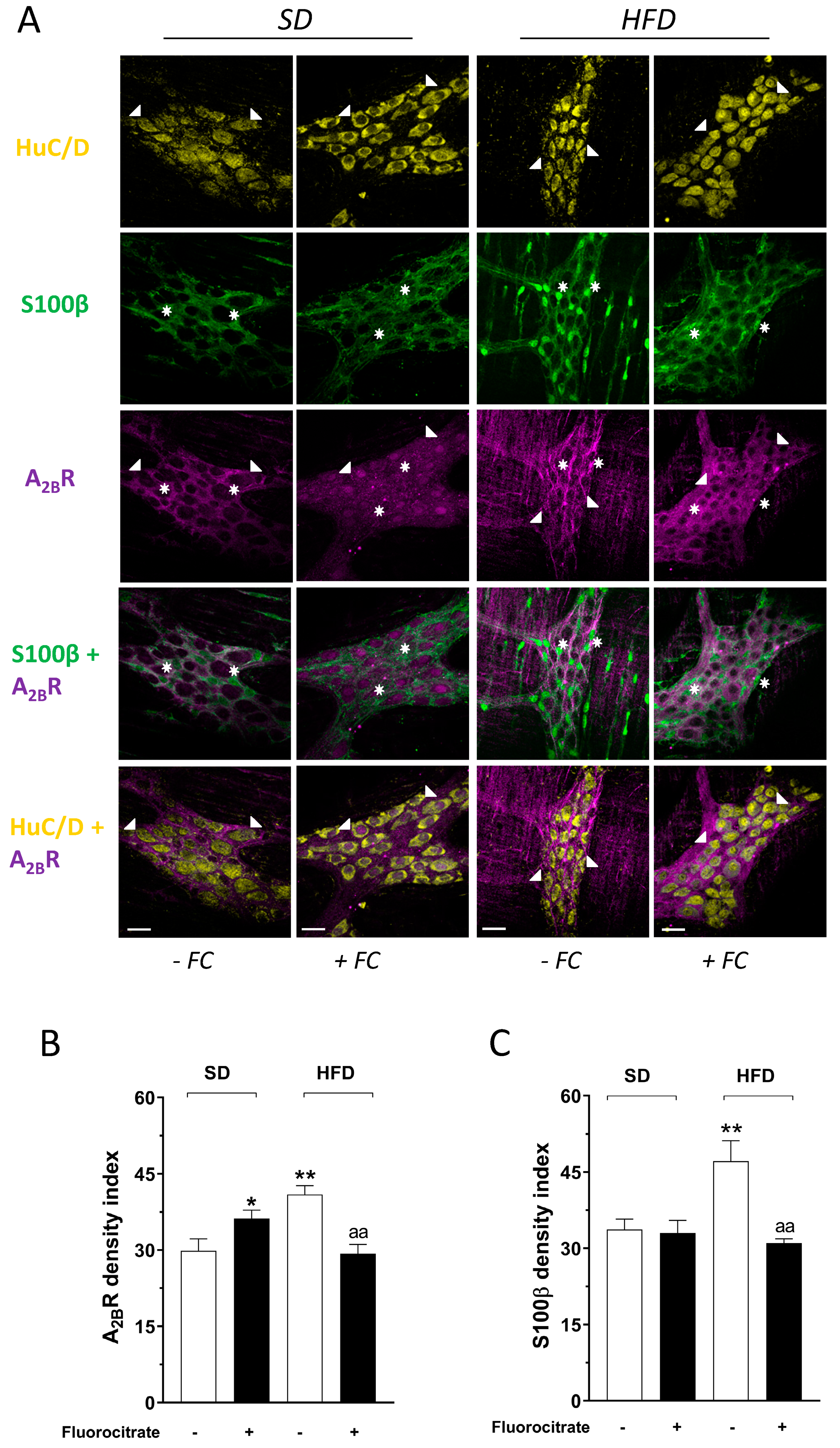
3.3. Effect of Fluorocitrate on A2B Receptor and Glial Protein S100β Distribution
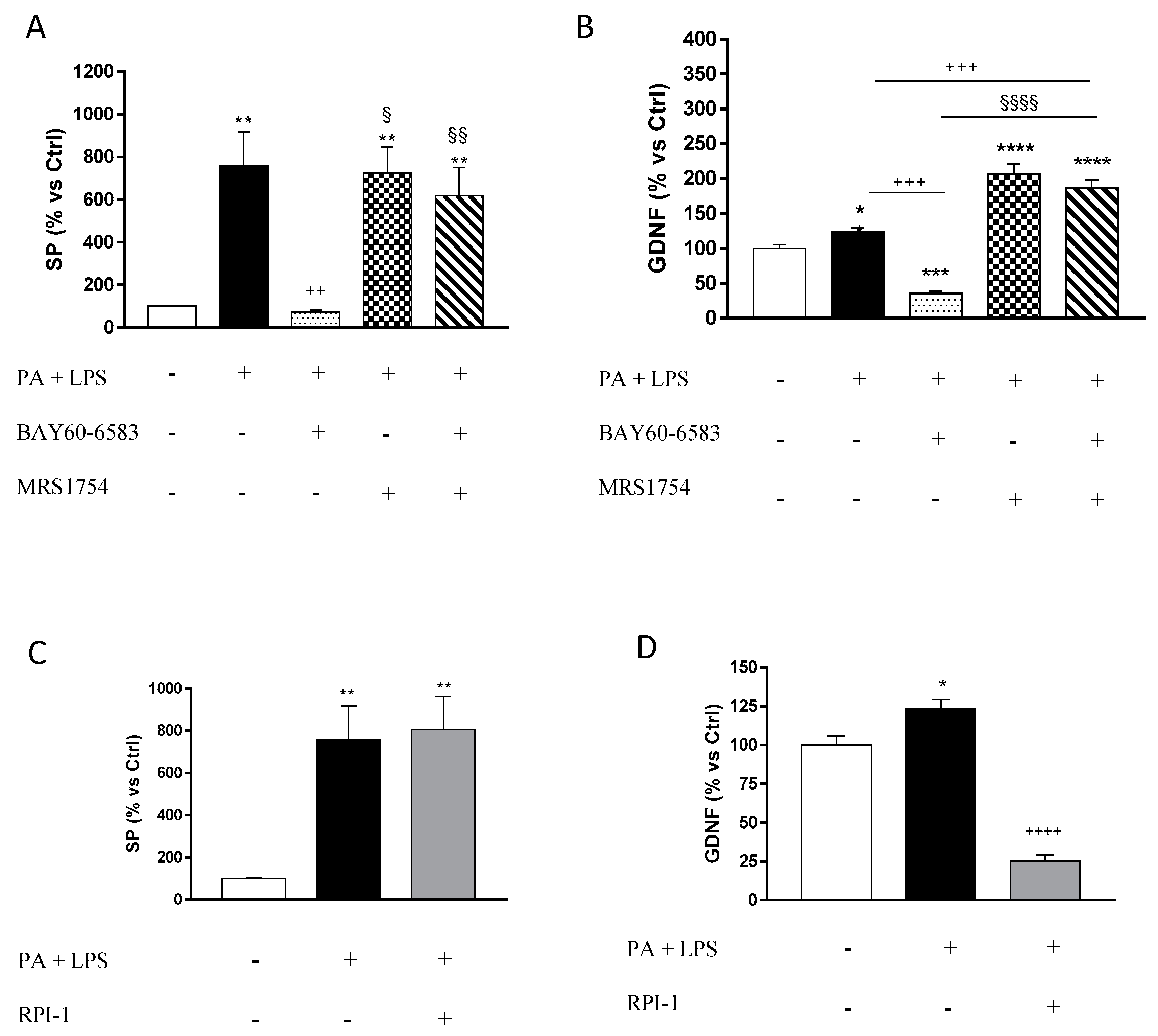
3.4. A2B Receptors Modulate the Release of Substance P and Glial Cell Derived Neurotrophic Factor from Cultured EGCs
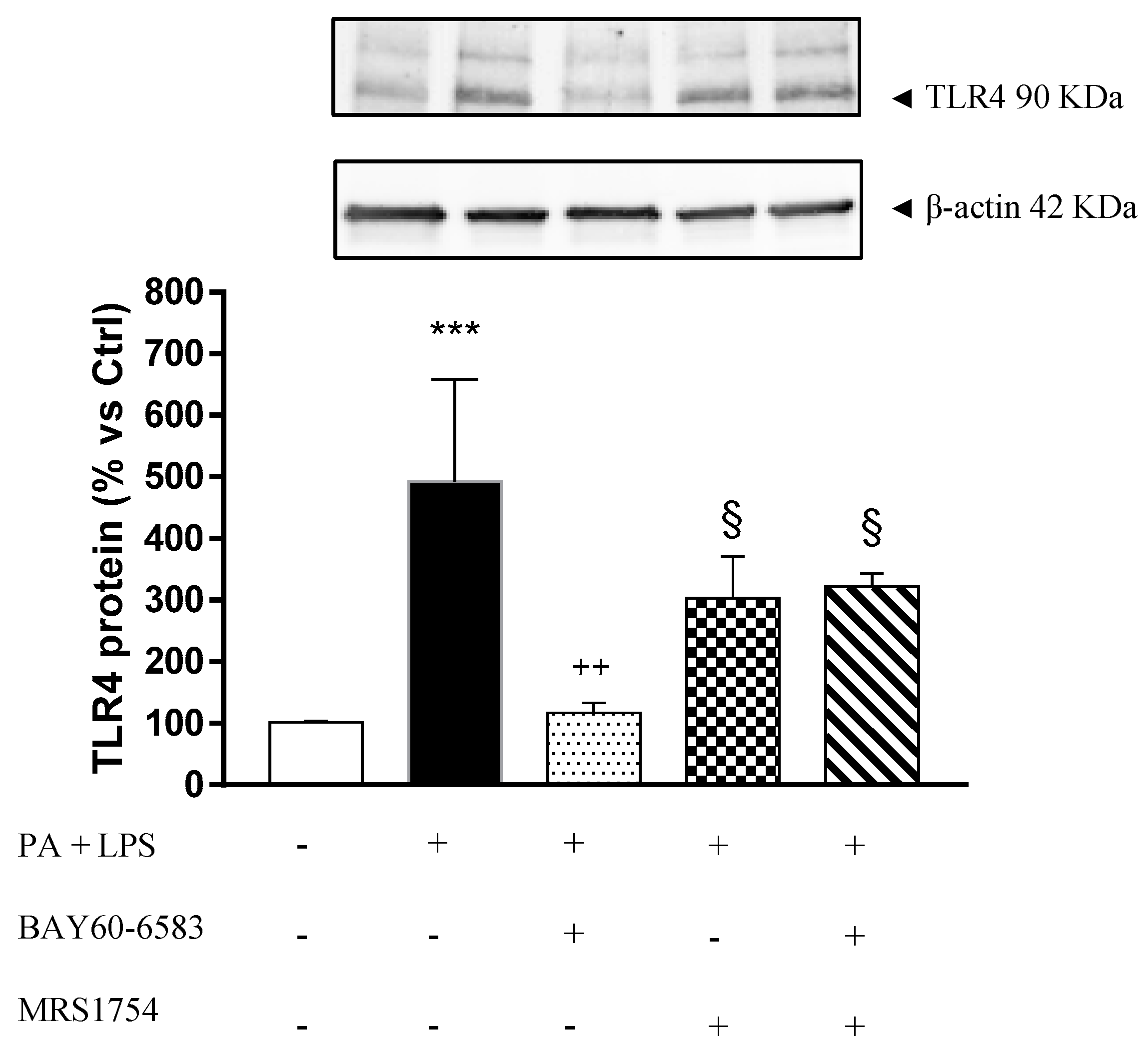
3.5. A2B Receptors Modulate TLR4 Expression in Cultured EGCs Incubated with Palmitate and Lipopolysaccharide
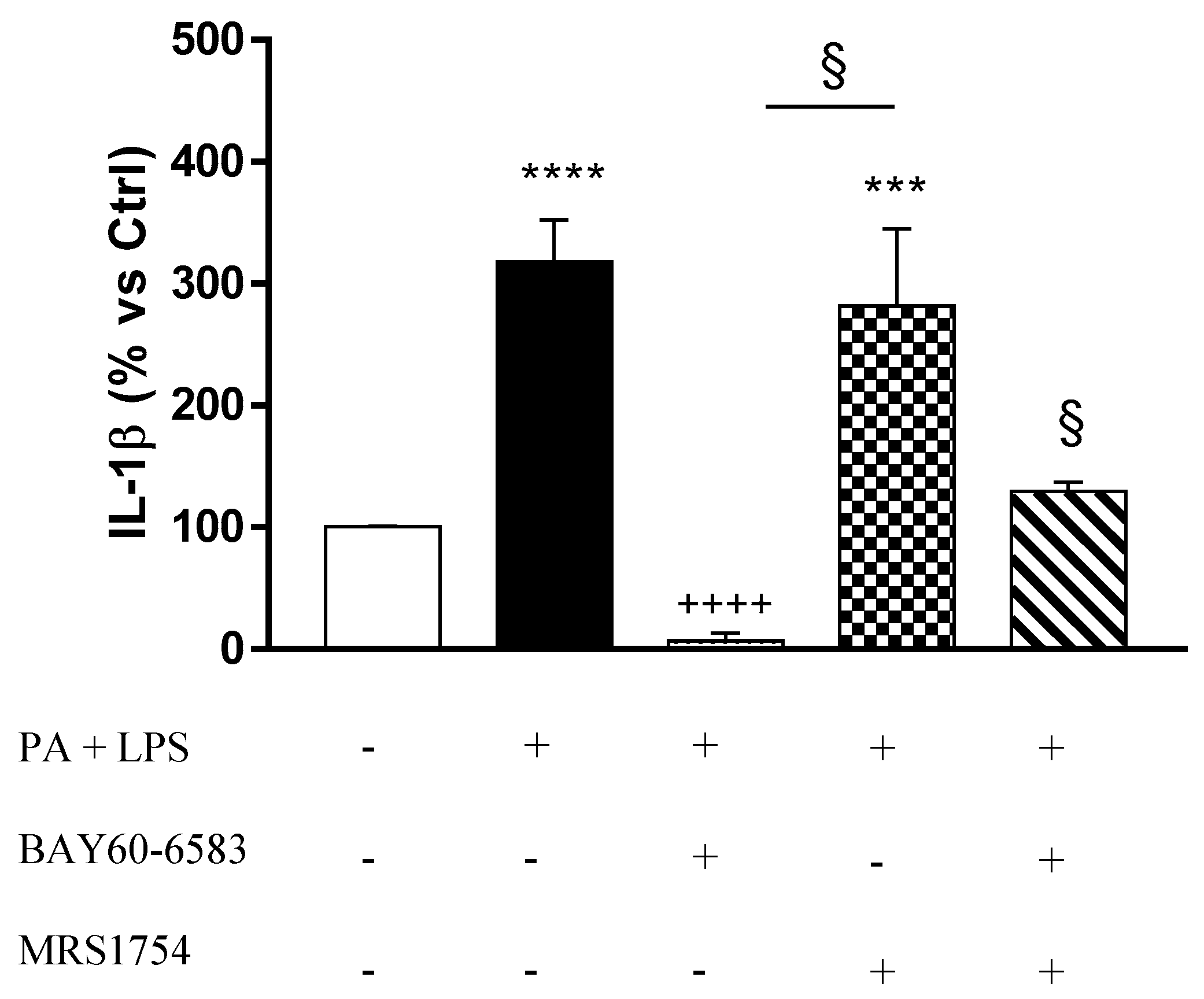
3.6. A2B Receptors Modulate Interleukin-1β Release Induced by Palmitate and Lipopolysaccharide in Cultured EGCs
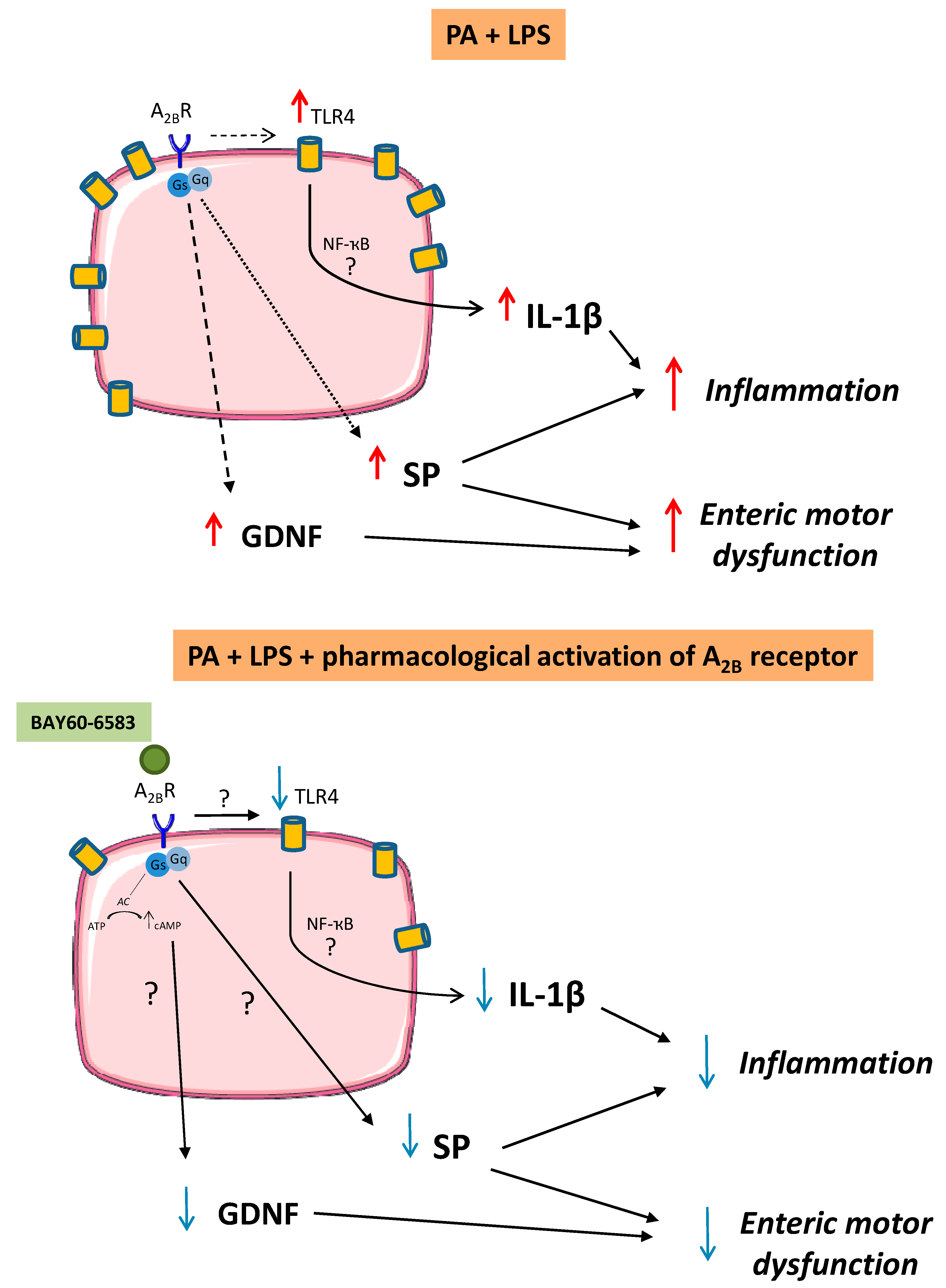
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Ethics Approval
References
- Gulbransen, B.D.; Christofi, F.L. Are We Close to Targeting Enteric Glia in Gastrointestinal Diseases and Motility Disorders? Gastroenterology 2018, 155, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, K.A. Emerging roles for enteric glia in gastrointestinal disorders. J. Clin. Investig. 2015, 125, 918–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochoa-Cortes, F.; Turco, F.; Linan-Rico, A.; Soghomonyan, S.; Whitaker, E.; Wehner, S.; Cuomo, R.; Christofi, F.L. Enteric Glial Cells: A New Frontier in Neurogastroenterology and Clinical Target for Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2016, 22, 433–449. [Google Scholar] [CrossRef] [PubMed]
- D’Antongiovanni, V.; Pellegrini, C.; Fornai, M.; Colucci, R.; Blandizzi, C.; Antonioli, L.; Bernardini, N. Intestinal epithelial barrier and neuromuscular compartment in health and disease. World J. Gastroenterol. 2020, 26, 1564–1579. [Google Scholar] [CrossRef] [PubMed]
- Jarolimova, J.; Tagoni, J.; Stern, T.A. Obesity: Its epidemiology, comorbidities, and management. Prim. Care Companion CNS Dis. 2013, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. AMS 2017, 13, 851–863. [Google Scholar] [CrossRef]
- Rajindrajith, S.; Devanarayana, N.M.; Benninga, M.A. Obesity and functional gastrointestinal diseases in children. J. Neurogastroenterol. Motil. 2014, 20, 414–416. [Google Scholar] [CrossRef] [Green Version]
- Le Pluart, D.; Sabate, J.M.; Bouchoucha, M.; Hercberg, S.; Benamouzig, R.; Julia, C. Functional gastrointestinal disorders in 35,447 adults and their association with body mass index. Aliment. Pharmacol. Ther. 2015, 41, 758–767. [Google Scholar] [CrossRef]
- Antonioli, L.; D’Antongiovanni, V.; Pellegrini, C.; Fornai, M.; Benvenuti, L.; di Carlo, A.; van den Wijngaard, R.; Caputi, V.; Cerantola, S.; Giron, M.C.; et al. Colonic dysmotility associated with high-fat diet-induced obesity: Role of enteric glia. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Brown, I.A.; McClain, J.L.; Watson, R.E.; Patel, B.A.; Gulbransen, B.D. Enteric glia mediate neuron death in colitis through purinergic pathways that require connexin-43 and nitric oxide. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Gulbransen, B.D.; Sharkey, K.A. Purinergic neuron-to-glia signaling in the enteric nervous system. Gastroenterology 2009, 136, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Christofi, F.L.; Zhang, H.; Yu, J.G.; Guzman, J.; Xue, J.; Kim, M.; Wang, Y.Z.; Cooke, H.J. Differential gene expression of adenosine A1, A2a, A2b, and A3 receptors in the human enteric nervous system. J. Comp. Neurol. 2001, 439, 46–64. [Google Scholar] [CrossRef] [PubMed]
- McClain, J.; Grubisic, V.; Fried, D.; Gomez-Suarez, R.A.; Leinninger, G.M.; Sevigny, J.; Parpura, V.; Gulbransen, B.D. Ca2+ responses in enteric glia are mediated by connexin-43 hemichannels and modulate colonic transit in mice. Gastroenterology 2014, 146, 497–507 e491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linan-Rico, A.; Turco, F.; Ochoa-Cortes, F.; Harzman, A.; Needleman, B.J.; Arsenescu, R.; Abdel-Rasoul, M.; Fadda, P.; Grants, I.; Whitaker, E.; et al. Molecular Signaling and Dysfunction of the Human Reactive Enteric Glial Cell Phenotype: Implications for GI Infection, IBD, POI, Neurological, Motility, and GI Disorders. Inflamm. Bowel Dis. 2016, 22, 1812–1834. [Google Scholar] [CrossRef]
- Boison, D.; Chen, J.F.; Fredholm, B.B. Adenosine signaling and function in glial cells. Cell Death Differ. 2010, 17, 1071–1082. [Google Scholar] [CrossRef] [Green Version]
- Vieira, C.; Ferreirinha, F.; Silva, I.; Duarte-Araujo, M.; Correia-de-Sa, P. Localization and function of adenosine receptor subtypes at the longitudinal muscle--myenteric plexus of the rat ileum. Neurochem. Int. 2011, 59, 1043–1055. [Google Scholar] [CrossRef]
- Antonioli, L.; Fornai, M.; Colucci, R.; Ghisu, N.; Tuccori, M.; Del Tacca, M.; Blandizzi, C. Regulation of enteric functions by adenosine: Pathophysiological and pharmacological implications. Pharmacol. Ther. 2008, 120, 233–253. [Google Scholar] [CrossRef]
- Antonioli, L.; Blandizzi, C.; Csoka, B.; Pacher, P.; Hasko, G. Adenosine signalling in diabetes mellitus—Pathophysiology and therapeutic considerations. Nat. Rev. Endocrinol. 2015, 11, 228–241. [Google Scholar] [CrossRef]
- Pardo, F.; Villalobos-Labra, R.; Chiarello, D.I.; Salsoso, R.; Toledo, F.; Gutierrez, J.; Leiva, A.; Sobrevia, L. Molecular implications of adenosine in obesity. Mol. Asp. Med. 2017, 55, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Dal Ben, D.; Antonioli, L.; Lambertucci, C.; Spinaci, A.; Fornai, M.; D’Antongiovanni, V.; Pellegrini, C.; Blandizzi, C.; Volpini, R. Approaches for designing and discovering purinergic drugs for gastrointestinal diseases. Expert Opin. Drug Discov. 2020, 1–17. [Google Scholar] [CrossRef]
- Antonioli, L.; Pellegrini, C.; Fornai, M.; Tirotta, E.; Gentile, D.; Benvenuti, L.; Giron, M.C.; Caputi, V.; Marsilio, I.; Orso, G.; et al. Colonic motor dysfunctions in a mouse model of high-fat diet-induced obesity: An involvement of A2B adenosine receptors. Purinergic Signal. 2017, 13, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Nasser, Y.; Fernandez, E.; Keenan, C.M.; Ho, W.; Oland, L.D.; Tibbles, L.A.; Schemann, M.; MacNaughton, W.K.; Ruhl, A.; Sharkey, K.A. Role of enteric glia in intestinal physiology: Effects of the gliotoxin fluorocitrate on motor and secretory function. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G912–G927. [Google Scholar] [CrossRef] [PubMed]
- McClain, J.L.; Gulbransen, B.D. The acute inhibition of enteric glial metabolism with fluoroacetate alters calcium signaling, hemichannel function, and the expression of key proteins. J. Neurophysiol. 2017, 117, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Cerantola, S.; Caputi, V.; Marsilio, I.; Ridolfi, M.; Faggin, S.; Bistoletti, M.; Giaroni, C.; Giron, M.C. Involvement of Enteric Glia in Small Intestine Neuromuscular Dysfunction of Toll-Like Receptor 4-Deficient Mice. Cells 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- Caputi, V.; Marsilio, I.; Cerantola, S.; Roozfarakh, M.; Lante, I.; Galuppini, F.; Rugge, M.; Napoli, E.; Giulivi, C.; Orso, G.; et al. Toll-Like Receptor 4 Modulates Small Intestine Neuromuscular Function through Nitrergic and Purinergic Pathways. Front. Pharmacol. 2017, 8, 350. [Google Scholar] [CrossRef] [Green Version]
- Murakami, M.; Ohta, T.; Ito, S. Lipopolysaccharides enhance the action of bradykinin in enteric neurons via secretion of interleukin-1beta from enteric glial cells. J. Neurosci. Res. 2009, 87, 2095–2104. [Google Scholar] [CrossRef]
- Voss, U.; Sand, E.; Olde, B.; Ekblad, E. Enteric neuropathy can be induced by high fat diet in vivo and palmitic acid exposure in vitro. PLoS ONE 2013, 8, e81413. [Google Scholar] [CrossRef]
- Dai, X.J.; Li, N.; Yu, L.; Chen, Z.Y.; Hua, R.; Qin, X.; Zhang, Y.M. Activation of BV2 microglia by lipopolysaccharide triggers an inflammatory reaction in PC12 cell apoptosis through a toll-like receptor 4-dependent pathway. Cell Stress Chaperones 2015, 20, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Koscso, B.; Csoka, B.; Selmeczy, Z.; Himer, L.; Pacher, P.; Virag, L.; Hasko, G. Adenosine augments IL-10 production by microglial cells through an A2B adenosine receptor-mediated process. J. Immunol. 2012, 188, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Daniele, S.; Zappelli, E.; Natali, L.; Martini, C.; Trincavelli, M.L. Modulation of A1 and A2B adenosine receptor activity: A new strategy to sensitise glioblastoma stem cells to chemotherapy. Cell Death Dis. 2014, 5, e1539. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Bolton, E.C. Glial cell line-derived neurotrophic factor induces cell proliferation in the mouse urogenital sinus. Mol. Endocrinol. 2015, 29, 289–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gale, Z.; Cooper, P.R.; Scheven, B.A. Glial cell line-derived neurotrophic factor influences proliferation of osteoblastic cells. Cytokine 2012, 57, 276–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, S.; D’Antongiovanni, V.; Martinelli, S.; Bechmann, N.; Riverso, M.; Poitz, D.M.; Pacak, K.; Eisenhofer, G.; Mannelli, M.; Rapizzi, E. Primary fibroblast co-culture stimulates growth and metabolism in Sdhb-impaired mouse pheochromocytoma MTT cells. Cell Tissue Res. 2018, 374, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Smoktunowicz, N.; Plate, M.; Stern, A.O.; D’Antongiovanni, V.; Robinson, E.; Chudasama, V.; Caddick, S.; Scotton, C.J.; Jarai, G.; Chambers, R.C. TGFbeta upregulates PAR-1 expression and signalling responses in A549 lung adenocarcinoma cells. Oncotarget 2016, 7, 65471–65484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delvalle, N.M.; Dharshika, C.; Morales-Soto, W.; Fried, D.E.; Gaudette, L.; Gulbransen, B.D. Communication Between Enteric Neurons, Glia, and Nociceptors Underlies the Effects of Tachykinins on Neuroinflammation. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 321–344. [Google Scholar] [CrossRef] [Green Version]
- Grubisic, V.; Verkhratsky, A.; Zorec, R.; Parpura, V. Enteric glia regulate gut motility in health and disease. BrainRes. Bull. 2018, 136, 109–117. [Google Scholar] [CrossRef]
- Chow, A.K.; Gulbransen, B.D. Potential roles of enteric glia in bridging neuroimmune communication in the gut. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G145–G152. [Google Scholar] [CrossRef]
- Kinoshita, M.; Hirayama, Y.; Fujishita, K.; Shibata, K.; Shinozaki, Y.; Shigetomi, E.; Takeda, A.; Le, H.P.N.; Hayashi, H.; Hiasa, M.; et al. Anti-Depressant Fluoxetine Reveals its Therapeutic Effect Via Astrocytes. EBioMedicine 2018, 32, 72–83. [Google Scholar] [CrossRef]
- Mauborgne, A.; Polienor, H.; Hamon, M.; Cesselin, F.; Bourgoin, S. Adenosine receptor-mediated control of in vitro release of pain-related neuropeptides from the rat spinal cord. Eur. J. Pharmacol. 2002, 441, 47–55. [Google Scholar] [CrossRef]
- Santicioli, P.; Del Bianco, E.; Tramontana, M.; Maggi, C.A. Adenosine inhibits action potential-dependent release of calcitonin gene-related peptide- and substance P-like immunoreactivities from primary afferents in rat spinal cord. Neurosci. Lett. 1992, 144, 211–214. [Google Scholar] [CrossRef]
- Cabarrocas, J.; Savidge, T.C.; Liblau, R.S. Role of enteric glial cells in inflammatory bowel disease. Glia 2003, 41, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Duarte Azevedo, M.; Sander, S.; Tenenbaum, L. GDNF, A Neuron-Derived Factor Upregulated in Glial Cells during Disease. J. Clin. Med. 2020, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.K.; He, F.Q.; Li, T.K.; Pang, X.H.; Cui, D.J.; Xie, Q.; Huang, X.L.; Gan, H.T. Glial-derived neurotrophic factor regulates intestinal epithelial barrier function and inflammation and is therapeutic for murine colitis. J. Pathol. 2010, 222, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Mwangi, S.M.; Nezami, B.G.; Obukwelu, B.; Anitha, M.; Marri, S.; Fu, P.; Epperson, M.F.; Le, N.A.; Shanmugam, M.; Sitaraman, S.V.; et al. Glial cell line-derived neurotrophic factor protects against high-fat diet-induced obesity. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G515–G525. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Hughes, I.; Planer, W.; Parsadanian, A.; Grider, J.R.; Vohra, B.P.; Keller-Peck, C.; Heuckeroth, R.O. The timing and location of glial cell line-derived neurotrophic factor expression determine enteric nervous system structure and function. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 1523–1538. [Google Scholar] [CrossRef] [Green Version]
- Grider, J.R.; Heuckeroth, R.O.; Kuemmerle, J.F.; Murthy, K.S. Augmentation of the ascending component of the peristaltic reflex and substance P release by glial cell line-derived neurotrophic factor. Neurogastroenterol. Motil. Off. J. Eur. Gastrointest. Motil. Soc. 2010, 22, 779–786. [Google Scholar] [CrossRef] [Green Version]
- Nicholas, D.A.; Zhang, K.; Hung, C.; Glasgow, S.; Aruni, A.W.; Unternaehrer, J.; Payne, K.J.; Langridge, W.H.R.; De Leon, M. Palmitic acid is a toll-like receptor 4 ligand that induces human dendritic cell secretion of IL-1beta. PLoS ONE 2017, 12, e0176793. [Google Scholar] [CrossRef]
- Speaker, K.J.; Fleshner, M. Interleukin-1 beta: A potential link between stress and the development of visceral obesity. BMC Physiol. 2012, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Bing, C. Is interleukin-1beta a culprit in macrophage-adipocyte crosstalk in obesity? Adipocyte 2015, 4, 149–152. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.; Leonard, F.; Ernens, I.; Rodius, S.; Vausort, M.; Rolland-Turner, M.; Devaux, Y.; Wagner, D.R. Adenosine reduces cell surface expression of toll-like receptor 4 and inflammation in response to lipopolysaccharide and matrix products. J. Cardiovasc. Transl. Res. 2011, 4, 790–800. [Google Scholar] [CrossRef]
- Hasko, G.; Szabo, C.; Nemeth, Z.H.; Kvetan, V.; Pastores, S.M.; Vizi, E.S. Adenosine receptor agonists differentially regulate IL-10, TNF-alpha, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. J. Immunol. 1996, 157, 4634–4640. [Google Scholar]
- Nemeth, Z.H.; Lutz, C.S.; Csoka, B.; Deitch, E.A.; Leibovich, S.J.; Gause, W.C.; Tone, M.; Pacher, P.; Vizi, E.S.; Hasko, G. Adenosine augments IL-10 production by macrophages through an A2B receptor-mediated posttranscriptional mechanism. J. Immunol. 2005, 175, 8260–8270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasko, G.; Kuhel, D.G.; Chen, J.F.; Schwarzschild, M.A.; Deitch, E.A.; Mabley, J.G.; Marton, A.; Szabo, C. Adenosine inhibits IL-12 and TNF-[alpha] production via adenosine A2a receptor-dependent and independent mechanisms. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2000, 14, 2065–2074. [Google Scholar] [CrossRef] [Green Version]
- Csoka, B.; Nemeth, Z.H.; Virag, L.; Gergely, P.; Leibovich, S.J.; Pacher, P.; Sun, C.X.; Blackburn, M.R.; Vizi, E.S.; Deitch, E.A.; et al. A2A adenosine receptors and C/EBPbeta are crucially required for IL-10 production by macrophages exposed to Escherichia coli. Blood 2007, 110, 2685–2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csoka, B.; Selmeczy, Z.; Koscso, B.; Nemeth, Z.H.; Pacher, P.; Murray, P.J.; Kepka-Lenhart, D.; Morris, S.M., Jr.; Gause, W.C.; Leibovich, S.J.; et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 376–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csoka, B.; Koscso, B.; Toro, G.; Kokai, E.; Virag, L.; Nemeth, Z.H.; Pacher, P.; Bai, P.; Hasko, G. A2B adenosine receptors prevent insulin resistance by inhibiting adipose tissue inflammation via maintaining alternative macrophage activation. Diabetes 2014, 63, 850–866. [Google Scholar] [CrossRef] [Green Version]
- Mashaghi, A.; Marmalidou, A.; Tehrani, M.; Grace, P.M.; Pothoulakis, C.; Dana, R. Neuropeptide substance P and the immune response. Cell. Mol. Life Sci. CMLS 2016, 73, 4249–4264. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Antongiovanni, V.; Benvenuti, L.; Fornai, M.; Pellegrini, C.; van den Wijngaard, R.; Cerantola, S.; Giron, M.C.; Caputi, V.; Colucci, R.; Haskó, G.; et al. Glial A2B Adenosine Receptors Modulate Abnormal Tachykininergic Responses and Prevent Enteric Inflammation Associated with High Fat Diet-Induced Obesity. Cells 2020, 9, 1245. https://doi.org/10.3390/cells9051245
D’Antongiovanni V, Benvenuti L, Fornai M, Pellegrini C, van den Wijngaard R, Cerantola S, Giron MC, Caputi V, Colucci R, Haskó G, et al. Glial A2B Adenosine Receptors Modulate Abnormal Tachykininergic Responses and Prevent Enteric Inflammation Associated with High Fat Diet-Induced Obesity. Cells. 2020; 9(5):1245. https://doi.org/10.3390/cells9051245
Chicago/Turabian StyleD’Antongiovanni, Vanessa, Laura Benvenuti, Matteo Fornai, Carolina Pellegrini, Renè van den Wijngaard, Silvia Cerantola, Maria Cecilia Giron, Valentina Caputi, Rocchina Colucci, Gyorgy Haskó, and et al. 2020. "Glial A2B Adenosine Receptors Modulate Abnormal Tachykininergic Responses and Prevent Enteric Inflammation Associated with High Fat Diet-Induced Obesity" Cells 9, no. 5: 1245. https://doi.org/10.3390/cells9051245