Corticosterone Induces HMGB1 Release in Primary Cultured Rat Cortical Astrocytes: Involvement of Pannexin-1 and P2X7 Receptor-Dependent Mechanisms

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Western Blotting
2.4. HMGB1 Enzyme-Linked Immunosorbent Assay (ELISA)
2.5. Real-Time PCR Analysis
2.6. Immunocytochemistry
2.7. Statistical Analysis
3. Results
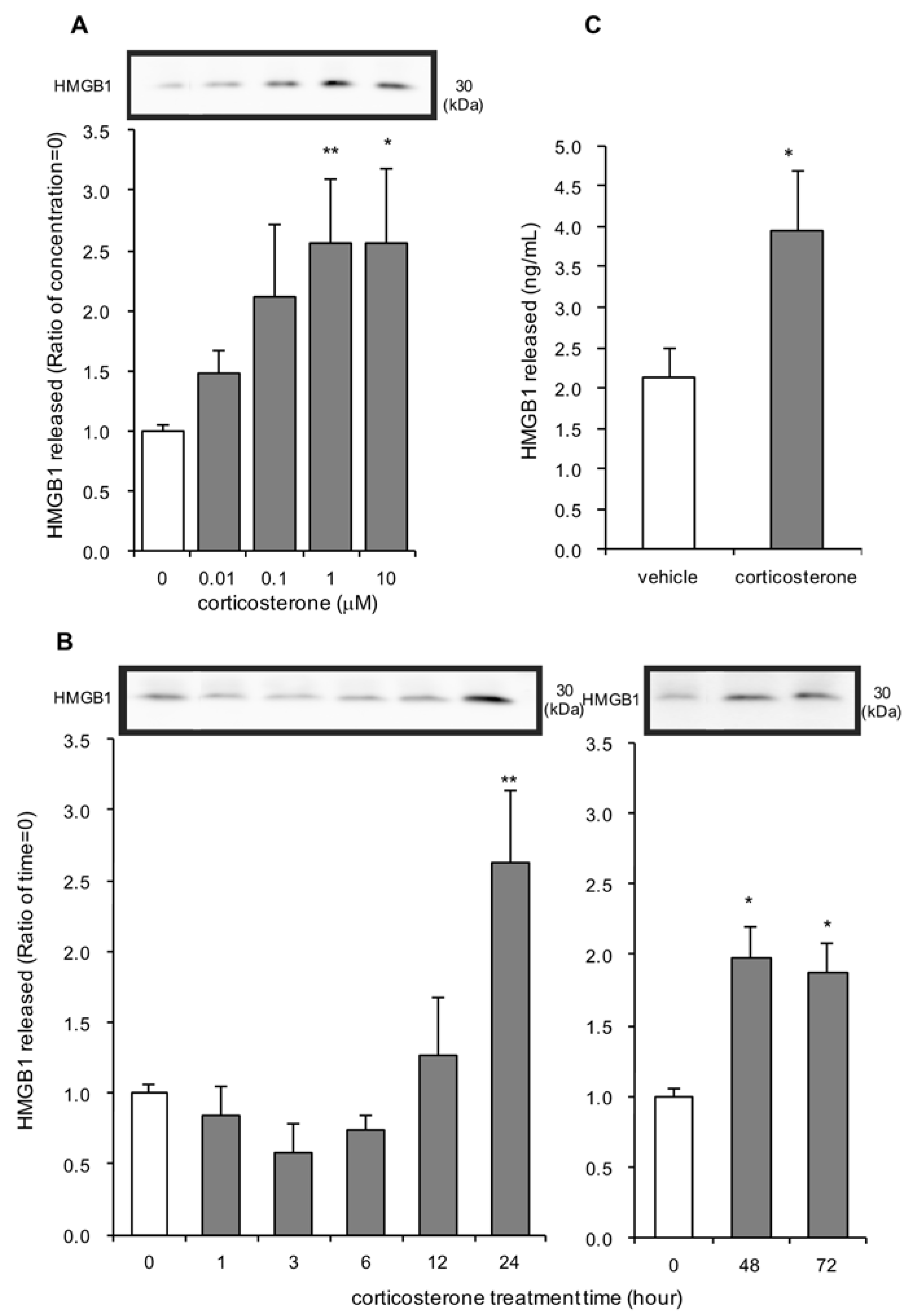
3.1. Effects of Corticosterone on HMGB1 Release in Rat Primary Cultured Cortical Astrocytes
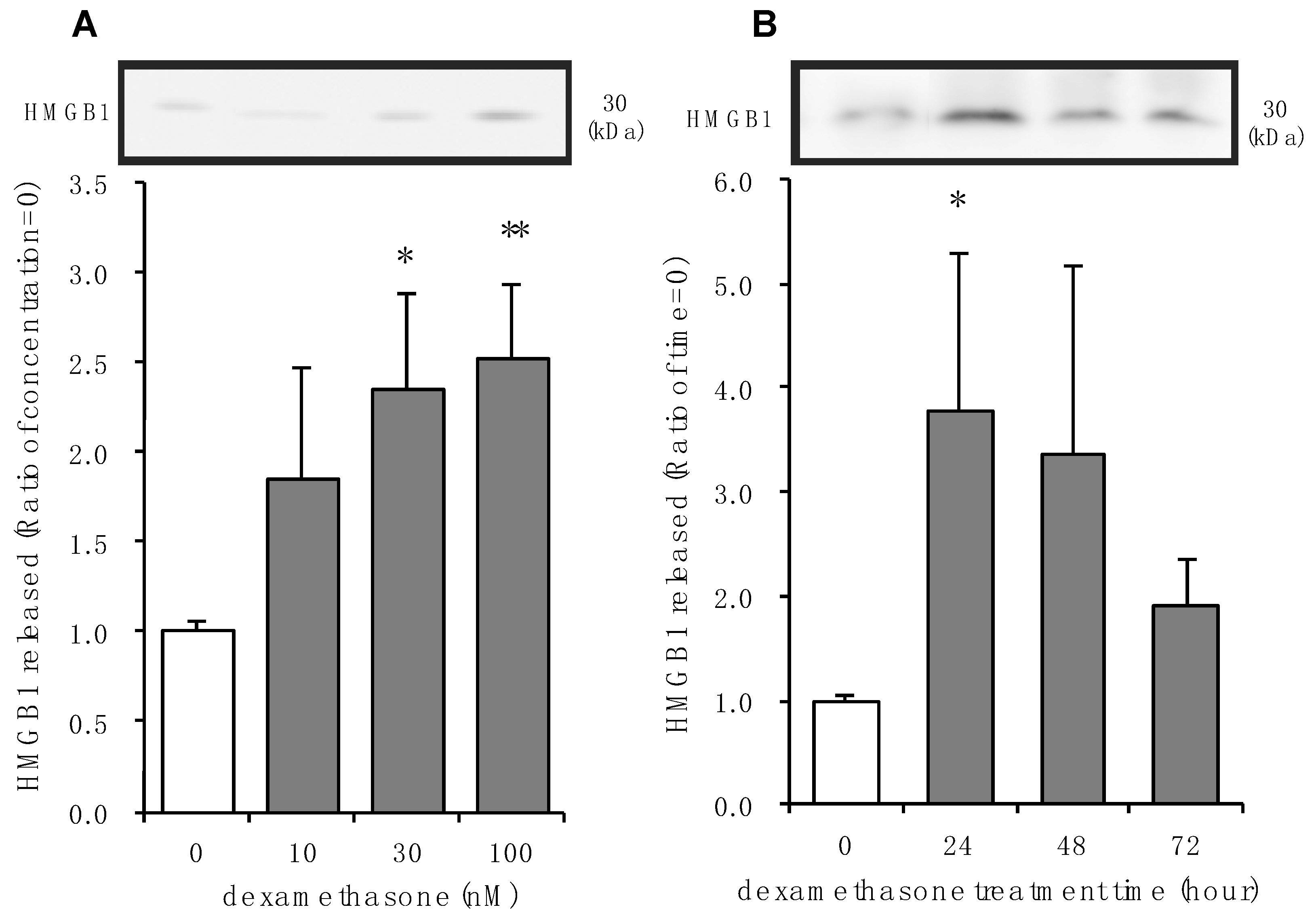
3.2. Effects of Dexamethasone on HMGB1 Release in Rat Primary Cultured Cortical Astrocytes
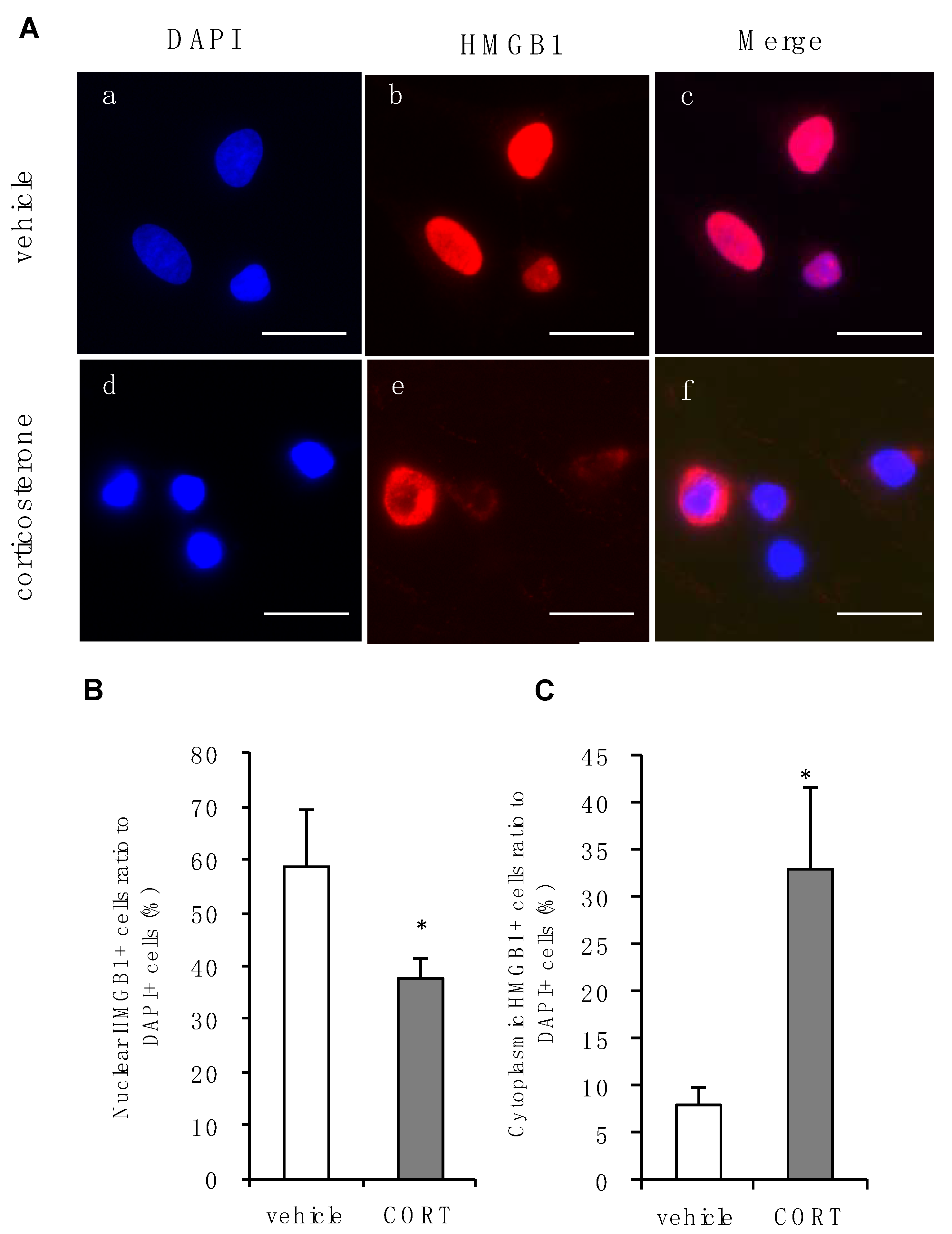
3.3. Effects of Corticosterone on Intracellular Localization of HMGB1 in Rat Primary Cultured Cortical Astrocytes
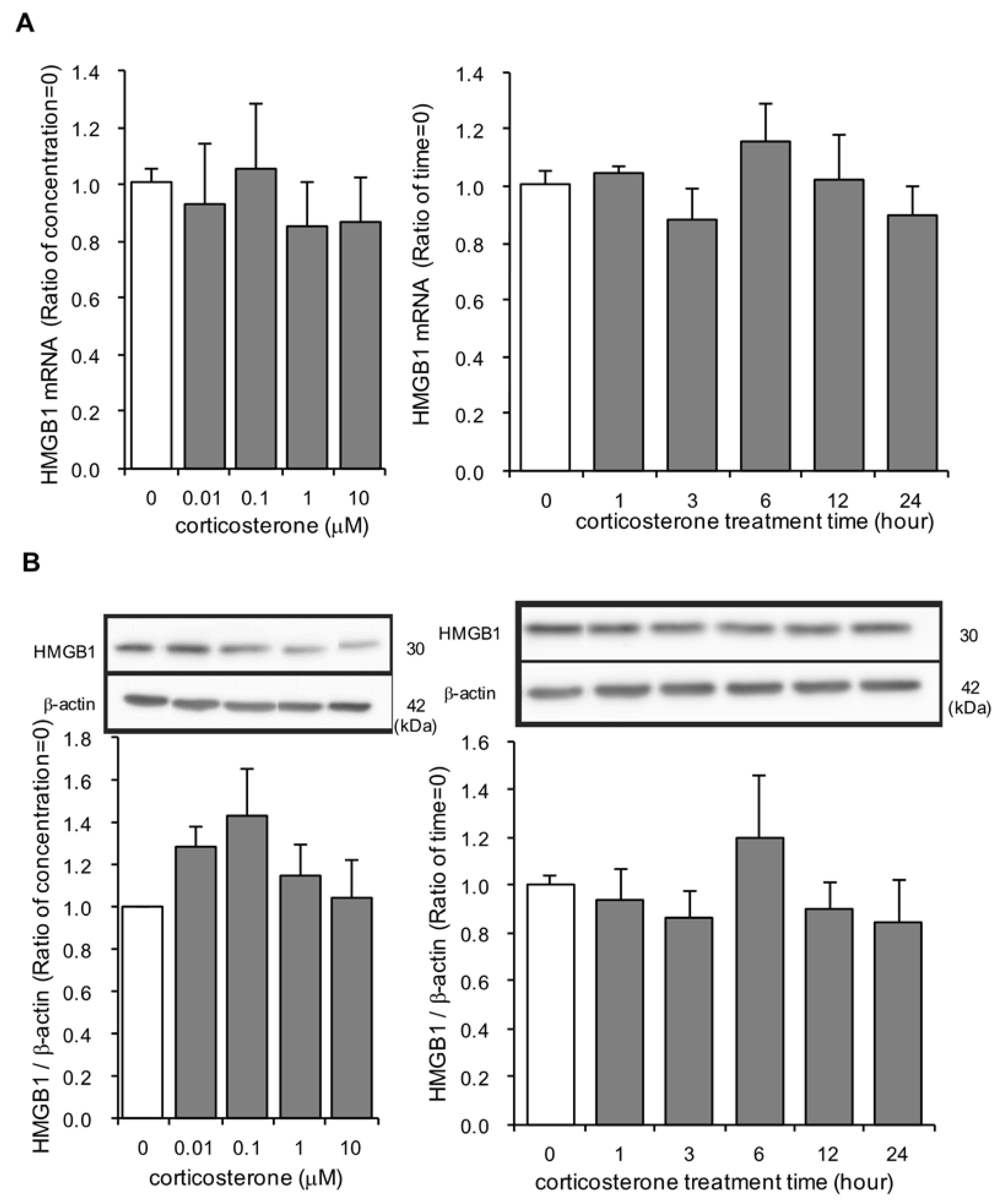
3.4. Effects of Corticosterone on HMGB1 mRNA and Intracellular HMGB1 Protein Expression in Rat Primary Cultured Cortical Astrocytes
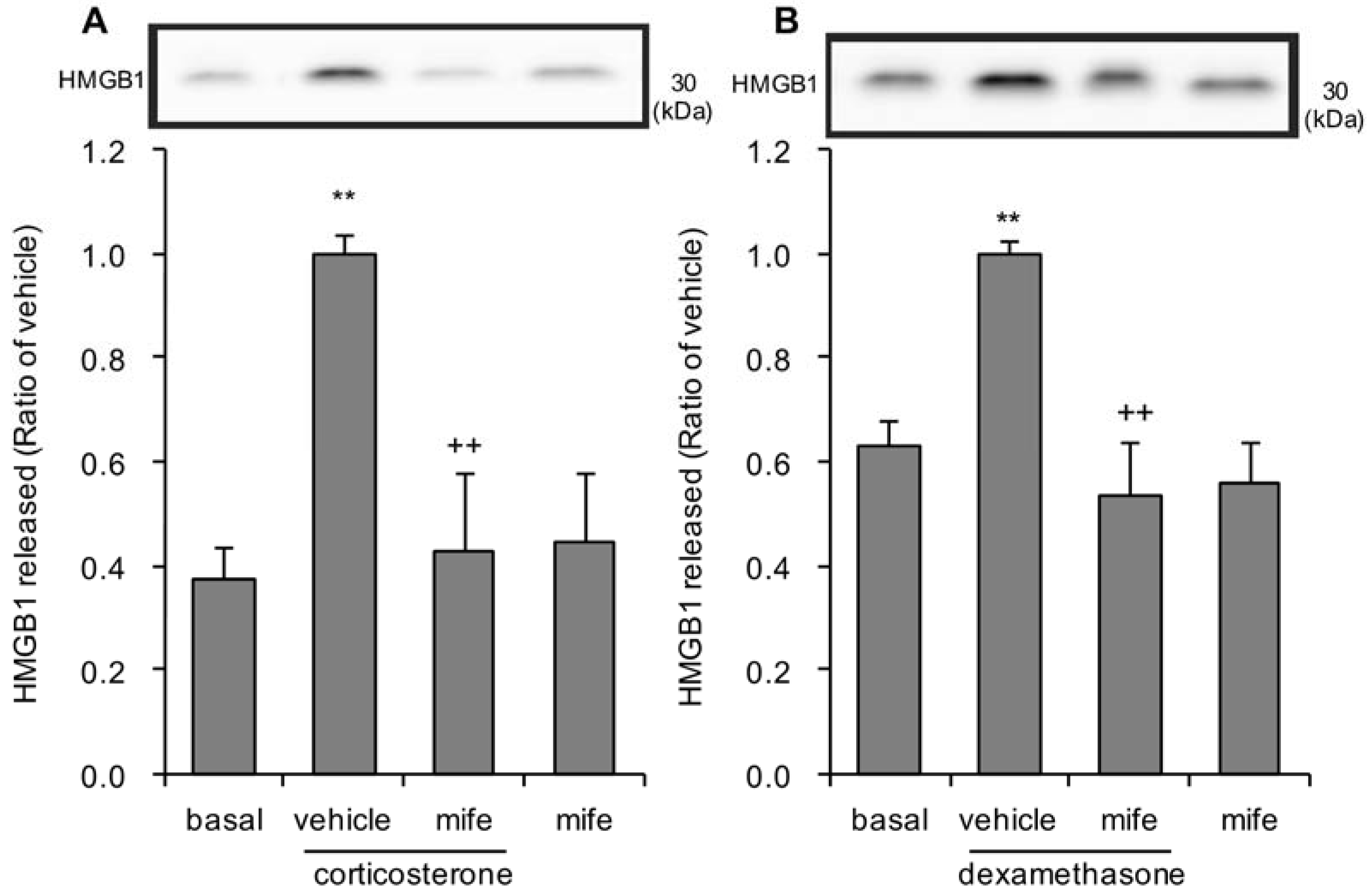
3.5. Effects of Glucocorticoid Receptor Antagonist on Corticosterone or Dexamethasone-Induced HMGB1 Release in Rat Primary Cultured Cortical Astrocytes
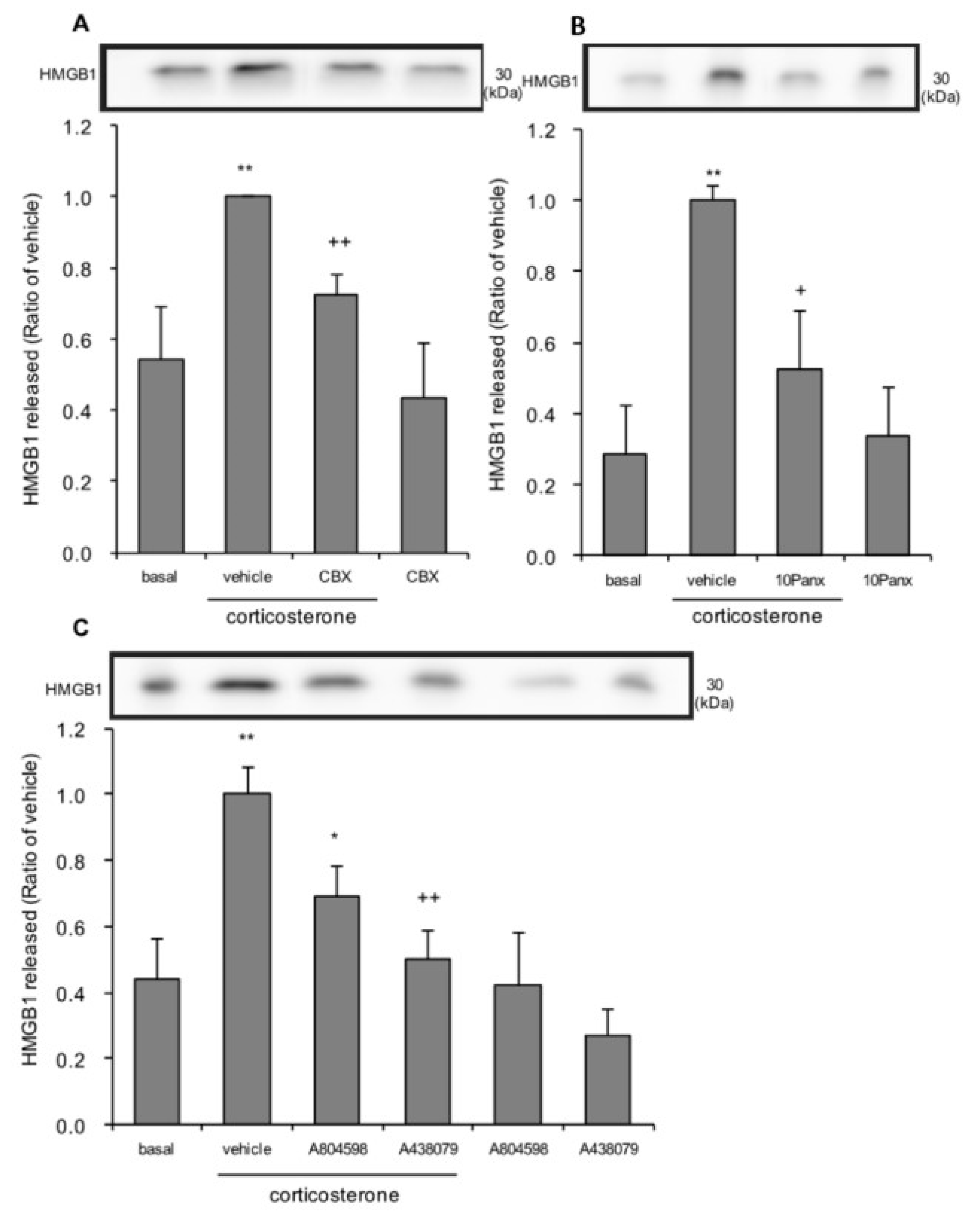
3.6. The Effects of Pannexin-1 Inhibitors and P2X7 Antagonists on Corticosterone-Induced HMGB1 Release in Rat Primary Cultured Cortical Astrocytes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Greenberg, P.E.; Fournier, A.A.; Sisitsky, T.; Pike, C.T.; Kessler, R.C. The economic burden of adults with major depressive disorder in the United States (2005 and 2010). J. Clin. Psychiatry 2015, 76, 155–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Post, R.M. Transduction of psychosocial stress into the neurobiology of recurrent affective disorder. Am. J. Psychiatry 1992, 149, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Won, E. The influence of stress on neuroinflammation and alterations in brain structure and function in major depressive disorder. Behav. Brain Res. 2017, 329, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Watkins, L.R.; Maier, S.F. Stress-induced glucocorticoids as a neuroendocrine alarm signal of danger. Brain Behav. Immun. 2013, 33, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Andersson, U.; Erlandsson-Harris, H.; Yang, H.; Tracey, K.J. HMGB1 as a DNA-binding cytokine. J. Leukoc. Biol. 2002, 72, 1084–1091. [Google Scholar]
- Agalave, N.M.; Svensson, C.I. Extracellular high-mobility group box 1 protein (HMGB1) as a mediator of persistent pain. Mol. Med. 2015, 20, 569–578. [Google Scholar] [CrossRef] [Green Version]
- Das, N.; Dewan, V.; Grace, P.M.; Gunn, R.J.; Tamura, R.; Tzarum, N.; Watkins, L.R.; Wilson, I.A.; Yin, H. HMGB1 Activates Proinflammatory Signaling via TLR5 Leading to Allodynia. Cell Rep. 2016, 17, 1128–1140. [Google Scholar] [CrossRef] [Green Version]
- Fang, P.; Schachner, M.; Shen, Y.Q. HMGB1 in development and diseases of the central nervous system. Mol. Neurobiol. 2012, 45, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Ulloa, L.; Messmer, D. High-mobility group box 1 (HMGB1) protein: Friend and foe. Cytokine Growth Factor Rev. 2006, 17, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Weber, M.D.; Watkins, L.R.; Maier, S.F. Stress sounds the alarmin: The role of the danger-associated molecular pattern HMGB1 in stress-induced neuroinflammatory priming. Brain Behav. Immun. 2015, 48, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Ding, L.; Shen, T.; Peng, D. HMGB1 involved in stress-induced depression and its neuroinflammatory priming role: A systematic review. Gen. Psychiatry 2019, 32, e100084. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Pardo, M.; Armini, R.S.; Martinez, A.; Mouhsine, H.; Zagury, J.F.; Jope, R.S.; Beurel, E. Stress-induced neuroinflammation is mediated by GSK3-dependent TLR4 signaling that promotes susceptibility to depression-like behavior. Brain Behav. Immun. 2016, 53, 207–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Lian, Y.J.; Su, W.J.; Peng, W.; Dong, X.; Liu, L.L.; Gong, H.; Zhang, T.; Jiang, C.L.; Wang, Y.X. HMGB1 mediates depressive behavior induced by chronic stress through activating the kynurenine pathway. Brain Behav. Immun. 2018, 72, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.Y.; Liu, L.; Zhang, W.; Zhang, Y.; Liu, Y.Z.; Shen, X.L.; Gong, H.; Yang, Y.Y.; Bi, X.Y.; Jiang, C.L.; et al. High-mobility group box-1 was released actively and involved in LPS induced depressive-like behavior. J. Psychiatr. Res. 2015, 64, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Lian, Y.J.; Gong, H.; Wu, T.Y.; Su, W.J.; Zhang, Y.; Yang, Y.Y.; Peng, W.; Zhang, T.; Zhou, J.R.; Jiang, C.L.; et al. Ds-HMGB1 and fr-HMGB induce depressive behavior through neuroinflammation in contrast to nonoxid-HMGB1. Brain Behav. Immun. 2017, 59, 322–332. [Google Scholar] [CrossRef]
- Hisaoka-Nakashima, K.; Tomimura, Y.; Yoshii, T.; Ohata, K.; Takada, N.; Zhang, F.F.; Nakamura, Y.; Liu, K.; Wake, H.; Nishibori, M.; et al. High-mobility group box 1-mediated microglial activation induces anxiodepressive-like behaviors in mice with neuropathic pain. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 92, 347–362. [Google Scholar] [CrossRef]
- Franklin, T.C.; Wohleb, E.S.; Zhang, Y.; Fogaça, M.; Hare, B.; Duman, R.S. Persistent Increase in Microglial RAGE Contributes to Chronic Stress-Induced Priming of Depressive-like Behavior. Biol. Psychiatry 2018, 83, 50–60. [Google Scholar] [CrossRef]
- Kurosinski, P.; Götz, J. Glial cells under physiologic and pathologic conditions. Arch. Neurol. 2002, 59, 1524–1528. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Segura, L.M.; McCarthy, M.M. Minireview: Role of glia in neuroendocrine function. Endocrinology 2004, 145, 1082–1086. [Google Scholar] [CrossRef] [Green Version]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Quintana, F.J. Control of autoimmune CNS inflammation by astrocytes. Semin Immunopathol. 2015, 37, 625–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Jie, W.; Liu, J.H.; Yang, J.M.; Gao, T.M. An astroglial basis of major depressive disorder? An overview. Glia 2017, 65, 1227–1250. [Google Scholar] [CrossRef] [PubMed]
- Passalacqua, M.; Zicca, A.; Sparatore, B.; Patrone, M.; Melloni, E.; Pontremoli, S. Secretion and binding of HMG1 protein to the external surface of the membrane are required for murine erythroleukemia cell differentiation. FEBS Lett. 1997, 400, 275–279. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Arai, K.; Lo, E.H. Role of ERK map kinase and CRM1 in IL-1beta-stimulated release of HMGB1 from cortical astrocytes. Glia 2010, 58, 1007–1015. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.B.; Lim, C.M.; Yu, Y.M.; Lee, J.K. Induction and subcellular localization of high-mobility group box-1 (HMGB1) in the postischemic rat brain. J. Neurosci. Res. 2008, 86, 1125–1131. [Google Scholar] [CrossRef]
- Menke, A. Is the HPA Axis as Target for Depression Outdated, or Is There a New Hope? Front. Psychiatry 2019, 10, 101. [Google Scholar] [CrossRef]
- Frank, M.G.; Weber, M.D.; Fonken, L.K.; Hershman, S.A.; Watkins, L.R.; Maier, S.F. The redox state of the alarmin HMGB1 is a pivotal factor in neuroinflammatory and microglial priming: A role for the NLRP3 inflammasome. Brain Behav. Immun. 2016, 55, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Frank, M.G.; Annis, J.L.; Watkins, L.R.; Maier, S.F. Glucocorticoids mediate stress induction of the alarmin HMGB1 and reduction of the microglia checkpoint receptor CD200R1 in limbic brain structures. Brain Behav. Immun. 2019, 80, 678–687. [Google Scholar] [CrossRef]
- Woehrle, T.; Yip, L.; Manohar, M.; Sumi, Y.; Yao, Y.; Chen, Y.; Junger, W.G. Hypertonic stress regulates T cell function via pannexin-1 hemichannels and P2X receptors. J. Leukoc. Biol. 2010, 88, 1181–1189. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Li, J.; Sama, A.E.; Wang, H. Carbenoxolone blocks endotoxin-induced protein kinase R (PKR) activation and high mobility group box 1 (HMGB1) release. Mol. Med. 2013, 19, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Unemura, K.; Kume, T.; Kondo, M.; Maeda, Y.; Izumi, Y.; Akaike, A. Glucocorticoids decrease astrocyte numbers by reducing glucocorticoid receptor expression in vitro and in vivo. J. Pharmacol. Sci. 2012, 119, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wu, Y.; Gu, S.; Yin, Q.; Li, H.; Wang, J.; Geng, D.; Xu, Y. The P2X7 receptor (P2X7R)-specific antagonist A804598 inhibits inflammatory reaction in human fibroblast-like synoviocytes. Am. J. Transl. Res. 2020, 12, 45–53. [Google Scholar] [PubMed]
- Peña-Altamira, L.E.; Polazzi, E.; Giuliani, P.; Beraudi, A.; Massenzio, F.; Mengoni, I.; Poli, A.; Zuccarini, M.; Ciccarelli, R.; Di Iorio, P.; et al. Release of soluble and vesicular purine nucleoside phosphorylase from rat astrocytes and microglia induced by pro-inflammatory stimulation with extracellular ATP via P2X. Neurochem. Int. 2018, 115, 37–49. [Google Scholar] [CrossRef]
- Hisaoka-Nakashima, K.; Taki, S.; Watanabe, S.; Nakamura, Y.; Nakata, Y.; Morioka, N. Mirtazapine increases glial cell line-derived neurotrophic factor production through lysophosphatidic acid 1 receptor-mediated extracellular signal-regulated kinase signaling in astrocytes. Eur. J. Pharmacol. 2019, 860, 172539. [Google Scholar] [CrossRef]
- Thoeringer, C.K.; Sillaber, I.; Roedel, A.; Erhardt, A.; Mueller, M.B.; Ohl, F.; Holsboer, F.; Keck, M.E. The temporal dynamics of intrahippocampal corticosterone in response to stress-related stimuli with different emotional and physical load: An in vivo microdialysis study in C57BL/6 and DBA/2 inbred mice. Psychoneuroendocrinology 2007, 32, 746–757. [Google Scholar] [CrossRef]
- Droste, S.K.; de Groote, L.; Lightman, S.L.; Reul, J.M.; Linthorst, A.C. The ultradian and circadian rhythms of free corticosterone in the brain are not affected by gender: An in vivo microdialysis study in Wistar rats. J. Neuroendocrinol. 2009, 21, 132–140. [Google Scholar] [CrossRef]
- Zhang, J.; Takahashi, H.K.; Liu, K.; Wake, H.; Liu, R.; Maruo, T.; Date, I.; Yoshino, T.; Ohtsuka, A.; Mori, S.; et al. Anti-high mobility group box-1 monoclonal antibody protects the blood-brain barrier from ischemia-induced disruption in rats. Stroke 2011, 42, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, J.; Kim, B.; Jaitpal, S.; Meng, S.S.; Adjepong, K.; Imamura, S.; Wake, H.; Nishibori, M.; Stopa, E.G.; et al. High-mobility group box-1 translocation and release after hypoxic ischemic brain injury in neonatal rats. Exp. Neurol. 2019, 311, 1–14. [Google Scholar] [CrossRef]
- Reul, J.M.; Gesing, A.; Droste, S.; Stec, I.S.; Weber, A.; Bachmann, C.; Bilang-Bleuel, A.; Holsboer, F.; Linthorst, A.C. The brain mineralocorticoid receptor: Greedy for ligand, mysterious in function. Eur. J. Pharmacol. 2000, 405, 235–249. [Google Scholar] [CrossRef]
- Sekiguchi, F.; Domoto, R.; Nakashima, K.; Yamasoba, D.; Yamanishi, H.; Tsubota, M.; Wake, H.; Nishibori, M.; Kawabata, A. Paclitaxel-induced HMGB1 release from macrophages and its implication for peripheral neuropathy in mice: Evidence for a neuroimmune crosstalk. Neuropharmacology 2018, 141, 201–213. [Google Scholar] [CrossRef]
- Koyanagi, S.; Kusunose, N.; Taniguchi, M.; Akamine, T.; Kanado, Y.; Ozono, Y.; Masuda, T.; Kohro, Y.; Matsunaga, N.; Tsuda, M.; et al. Glucocorticoid regulation of ATP release from spinal astrocytes underlies diurnal exacerbation of neuropathic mechanical allodynia. Nat. Commun. 2016, 7, 13102. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, R.; Barbe, M.T.; Jakob, N.J.; Monyer, H. Pharmacological properties of homomeric and heteromeric pannexin hemichannels expressed in Xenopus oocytes. J. Neurochem. 2005, 92, 1033–1043. [Google Scholar] [CrossRef]
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.G.; Yan, Z.; et al. HMGB1 in health and disease. Mol. Asp. Med. 2014, 40, 1–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauvala, H.; Rouhiainen, A. RAGE as a receptor of HMGB1 (Amphoterin): Roles in health and disease. Curr. Mol. Med. 2007, 7, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Nishimura, M.; Wang, Y.; Sims, J.R.; Qiu, S.; Savitz, S.I.; Salomone, S.; Moskowitz, M.A. Early release of HMGB-1 from neurons after the onset of brain ischemia. J. Cereb. Blood Flow Metab. 2008, 28, 927–938. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef]
- Klune, J.R.; Dhupar, R.; Cardinal, J.; Billiar, T.R.; Tsung, A. HMGB1: Endogenous danger signaling. Mol. Med. 2008, 14, 476–484. [Google Scholar] [CrossRef]
- Deng, M.; Scott, M.J.; Fan, J.; Billiar, T.R. Location is the key to function: HMGB1 in sepsis and trauma-induced inflammation. J. Leukoc. Biol. 2019, 106, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Venereau, E.; De Leo, F.; Mezzapelle, R.; Careccia, G.; Musco, G.; Bianchi, M.E. HMGB1 as biomarker and drug target. Pharmacol. Res. 2016, 111, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Venereau, E.; Casalgrandi, M.; Schiraldi, M.; Antoine, D.J.; Cattaneo, A.; De Marchis, F.; Liu, J.; Antonelli, A.; Preti, A.; Raeli, L.; et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 2012, 209, 1519–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohn, M.C.; Howard, E.; Vielkind, U.; Krozowski, Z. Glial cells express both mineralocorticoid and glucocorticoid receptors. J. Steroid Biochem. Mol. Biol. 1991, 40, 105–111. [Google Scholar] [CrossRef]
- Nicolaides, N.C.; Pavlaki, A.N.; Alexandra, M.A.M.; Chrousos, G.P. Endotext. In Glucocorticoid Therapy and Adrenal Suppression; Feingold, K.R., Anawalt, B., Boyce, A., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2018. [Google Scholar]
- Bonaldi, T.; Talamo, F.; Scaffidi, P.; Ferrera, D.; Porto, A.; Bachi, A.; Rubartelli, A.; Agresti, A.; Bianchi, M.E. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003, 22, 5551–5560. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, K.; Hashiguchi, T.; Kikuchi, K.; Tancharoen, S.; Miura, N.; Ito, T.; Oyama, Y.; Nawa, Y.; Biswas, K.K.; Meng, X.; et al. Induction of high mobility group box 1 release from serotonin-stimulated human umbilical vein endothelial cells. Int. J. Mol. Med. 2008, 22, 639–644. [Google Scholar]
- Feghali, K.; Iwasaki, K.; Tanaka, K.; Komaki, M.; Machigashira, M.; Ishikawa, I.; Izumi, Y. Human gingival fibroblasts release high-mobility group box-1 protein through active and passive pathways. Oral Microbiol. Immunol. 2009, 24, 292–298. [Google Scholar] [CrossRef]
- Ishii, T.; Warabi, E.; Mann, G.E. Circadian control of BDNF-mediated Nrf2 activation in astrocytes protects dopaminergic neurons from ferroptosis. Free Radic. Biol. Med. 2019, 133, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Simard, M.; Couldwell, W.T.; Zhang, W.; Song, H.; Liu, S.; Cotrina, M.L.; Goldman, S.; Nedergaard, M. Glucocorticoids-potent modulators of astrocytic calcium signaling. Glia 1999, 28, 1–12. [Google Scholar] [CrossRef]
- Guthrie, P.B.; Knappenberger, J.; Segal, M.; Bennett, M.V.; Charles, A.C.; Kater, S.B. ATP released from astrocytes mediates glial calcium waves. J. Neurosci. 1999, 19, 520–528. [Google Scholar] [CrossRef]
- Iwata, M.; Ota, K.T.; Li, X.Y.; Sakaue, F.; Li, N.; Dutheil, S.; Banasr, M.; Duric, V.; Yamanashi, T.; Kaneko, K.; et al. Psychological Stress Activates the Inflammasome via Release of Adenosine Triphosphate and Stimulation of the Purinergic Type 2X7 Receptor. Biol. Psychiatry 2016, 80, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Yue, N.; Huang, H.; Zhu, X.; Han, Q.; Wang, Y.; Li, B.; Liu, Q.; Wu, G.; Zhang, Y.; Yu, J. Activation of P2X7 receptor and NLRP3 inflammasome assembly in hippocampal glial cells mediates chronic stress-induced depressive-like behaviors. J. Neuroinflamm. 2017, 14, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamkanfi, M.; Sarkar, A.; Vande Walle, L.; Vitari, A.C.; Amer, A.O.; Wewers, M.D.; Tracey, K.J.; Kanneganti, T.D.; Dixit, V.M. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 2010, 185, 4385–4392. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Zhao, Y.; Yang, T.; Song, M.; Wang, C.; Yao, Y.; Fan, H. Glucocorticoid-Driven NLRP3 Inflammasome Activation in Hippocampal Microglia Mediates Chronic Stress-Induced Depressive-Like Behaviors. Front. Mol. Neurosci. 2019, 12, 210. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Watkins, L.R.; Maier, S.F. The permissive role of glucocorticoids in neuroinflammatory priming: Mechanisms and insights. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 300–305. [Google Scholar] [CrossRef] [Green Version]
- Rosciszewski, G.; Cadena, V.; Auzmendi, J.; Cieri, M.B.; Lukin, J.; Rossi, A.R.; Murta, V.; Villarreal, A.; Reinés, A.; Gomes, F.C.A.; et al. Detrimental Effects of HMGB-1 Require Microglial-Astroglial Interaction: Implications for the Status Epilepticus -Induced Neuroinflammation. Front. Cell Neurosci. 2019, 13, 380. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Xu, J.; Zheng, Y.; Wei, Y.; Zhu, X.; Lo, E.H.; Moskowitz, M.A.; Sims, J.R. High-mobility group box 1 promotes metalloproteinase-9 upregulation through Toll-like receptor 4 after cerebral ischemia. Stroke 2010, 41, 2077–2082. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hisaoka-Nakashima, K.; Azuma, H.; Ishikawa, F.; Nakamura, Y.; Wang, D.; Liu, K.; Wake, H.; Nishibori, M.; Nakata, Y.; Morioka, N. Corticosterone Induces HMGB1 Release in Primary Cultured Rat Cortical Astrocytes: Involvement of Pannexin-1 and P2X7 Receptor-Dependent Mechanisms. Cells 2020, 9, 1068. https://doi.org/10.3390/cells9051068
Hisaoka-Nakashima K, Azuma H, Ishikawa F, Nakamura Y, Wang D, Liu K, Wake H, Nishibori M, Nakata Y, Morioka N. Corticosterone Induces HMGB1 Release in Primary Cultured Rat Cortical Astrocytes: Involvement of Pannexin-1 and P2X7 Receptor-Dependent Mechanisms. Cells. 2020; 9(5):1068. https://doi.org/10.3390/cells9051068
Chicago/Turabian StyleHisaoka-Nakashima, Kazue, Honami Azuma, Fumina Ishikawa, Yoki Nakamura, Dengli Wang, Keyue Liu, Hidenori Wake, Masahiro Nishibori, Yoshihiro Nakata, and Norimitsu Morioka. 2020. "Corticosterone Induces HMGB1 Release in Primary Cultured Rat Cortical Astrocytes: Involvement of Pannexin-1 and P2X7 Receptor-Dependent Mechanisms" Cells 9, no. 5: 1068. https://doi.org/10.3390/cells9051068