Recent Advances and Future Perspectives of In Vivo Targeted Delivery of Genome-Editing Reagents to Germ cells, Embryos, and Fetuses in Mice


Abstract
:
1. Introduction
1.1. Genome-Editing Technology
1.2. Developments in Genome-Edited (GE) Mice Production Technology
2. Ex Vivo Delivery of Genome-Editing Components into Zygotes/2-Cell Embryos, PGCs, and Spermatogonial Stem Cells
2.1. Microinjection Technique
2.2. EP Technique
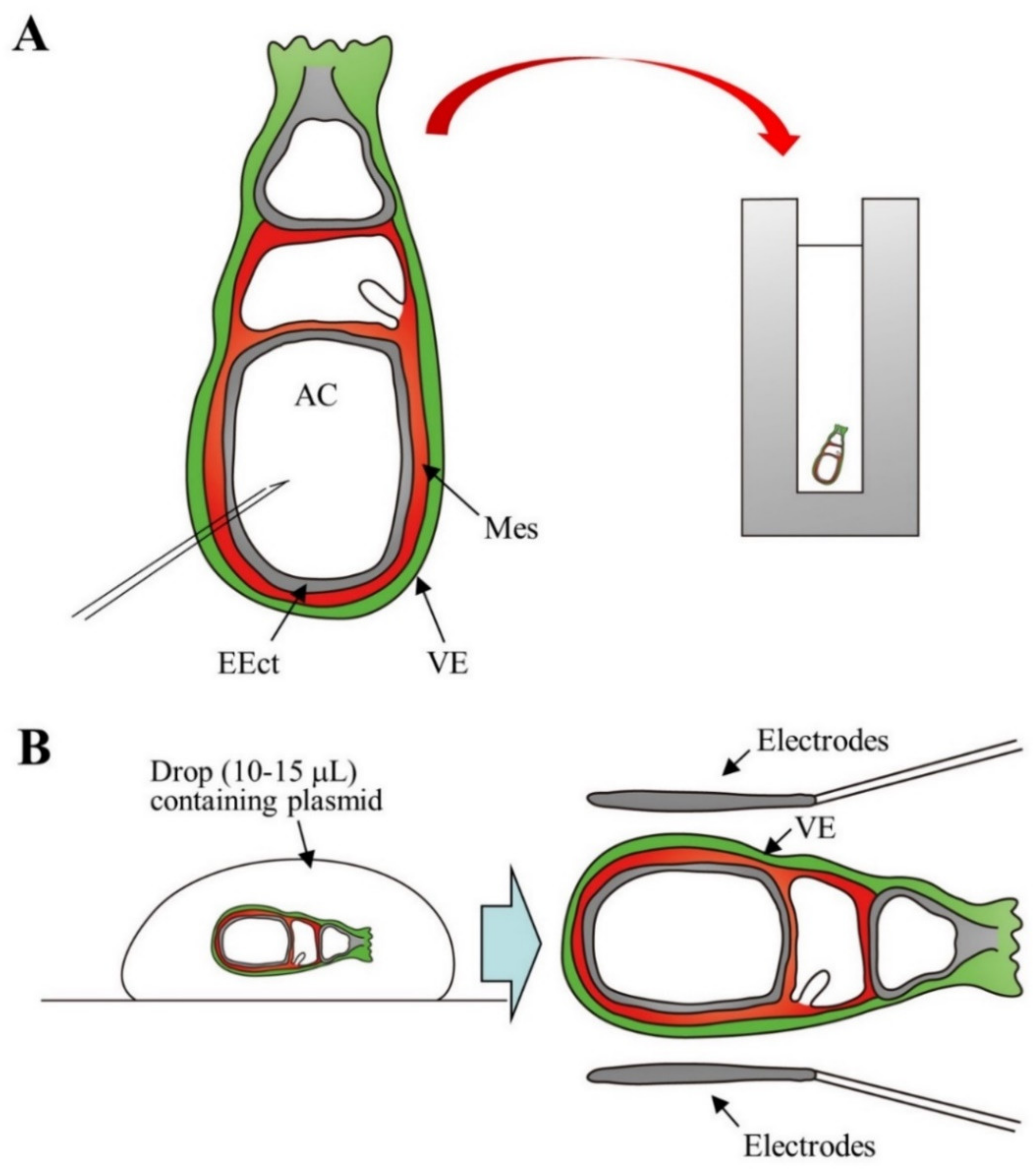
2.3. Gene Delivery to PGCs in the Genital Ridges
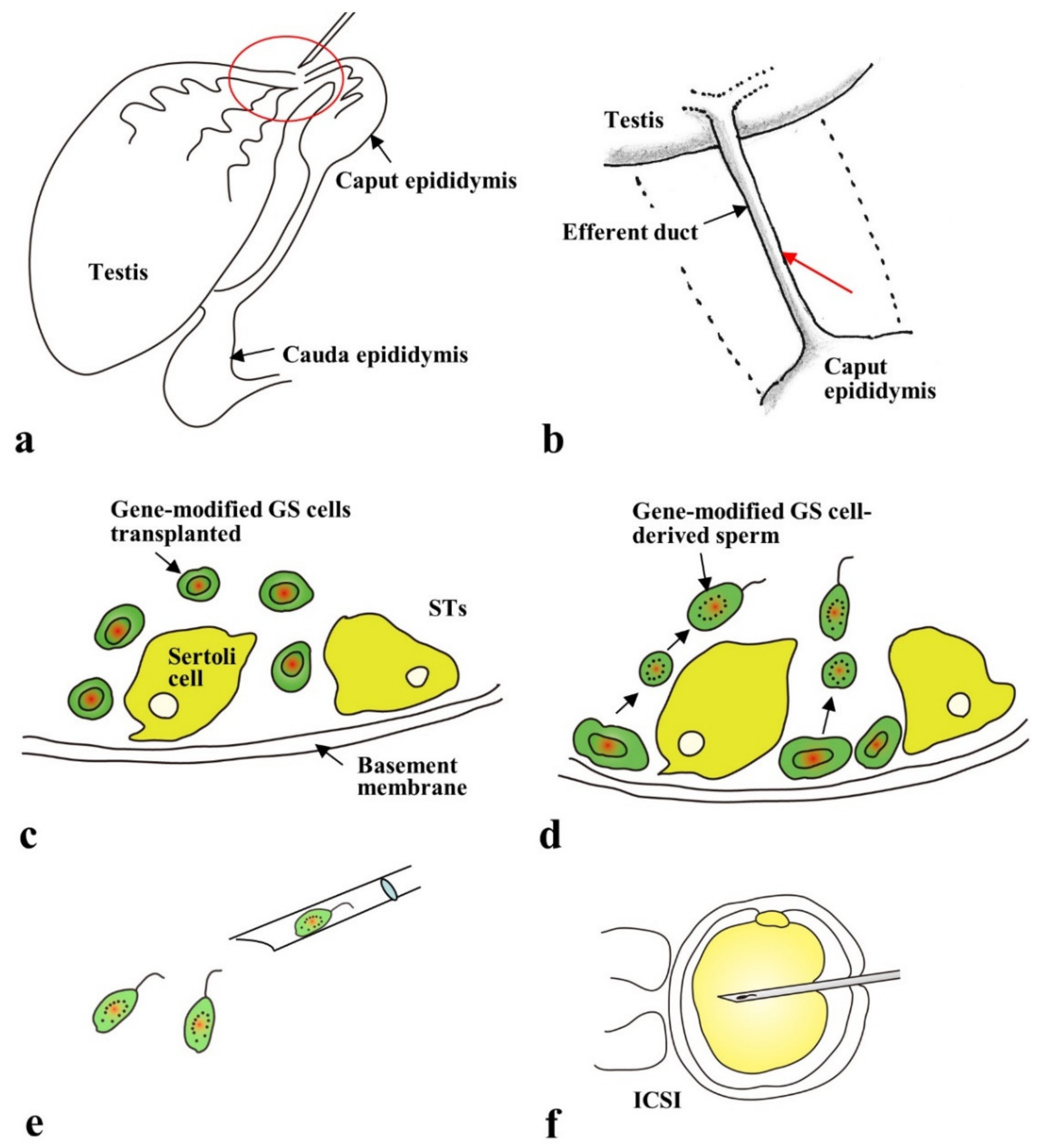
2.4. Gene Delivery to Spermatogonial Stem Cells
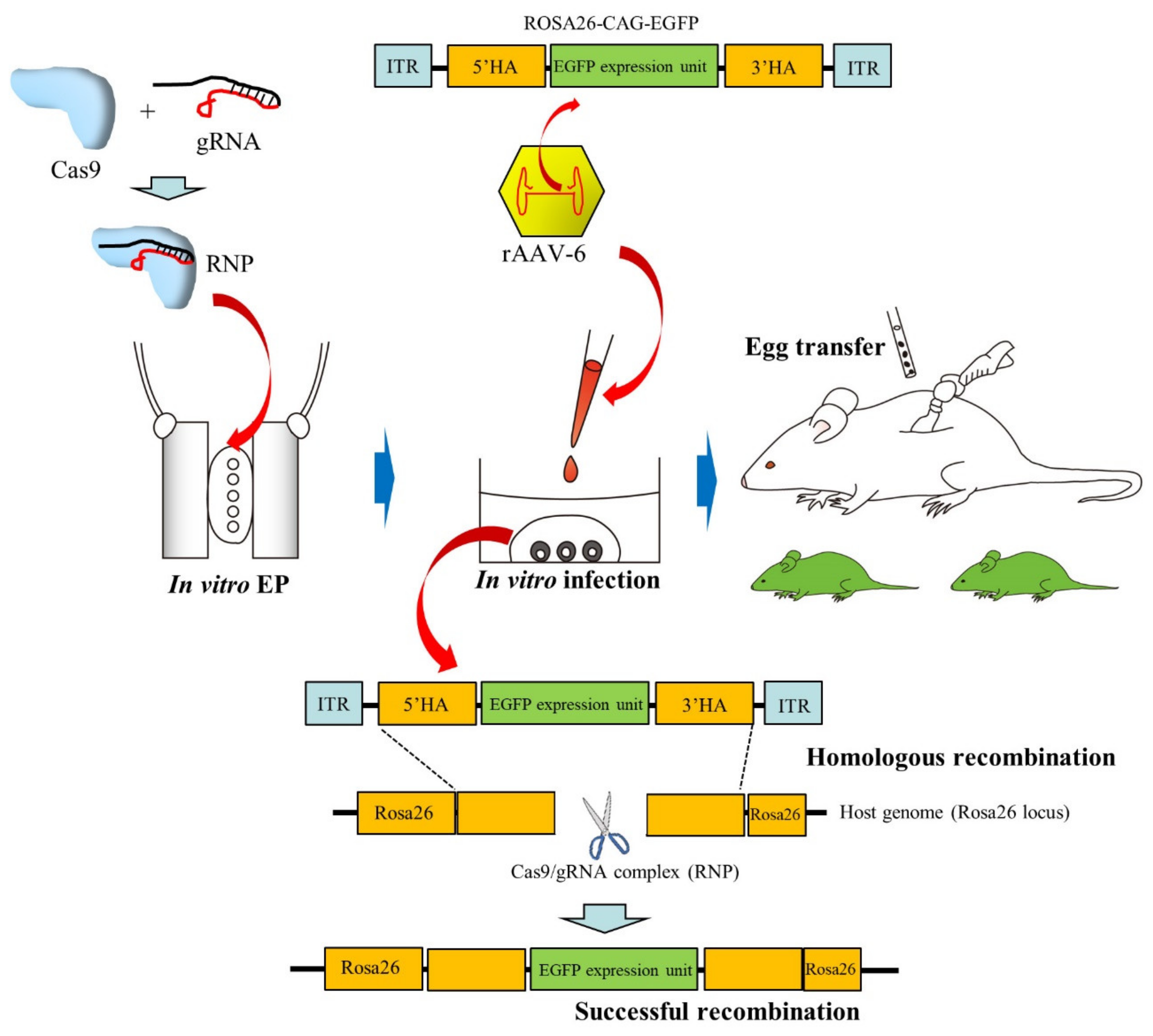
2.5. Infection of Preimplantation Embryos with AAVs
2.6. Gene Delivery to Postimplantation Embryos at Somite Stage
3. In Vivo Delivery Targeted to Zygotes, 2-Cell Embryos, and Fetuses
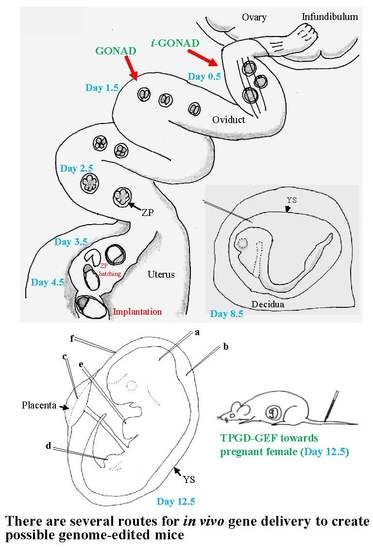
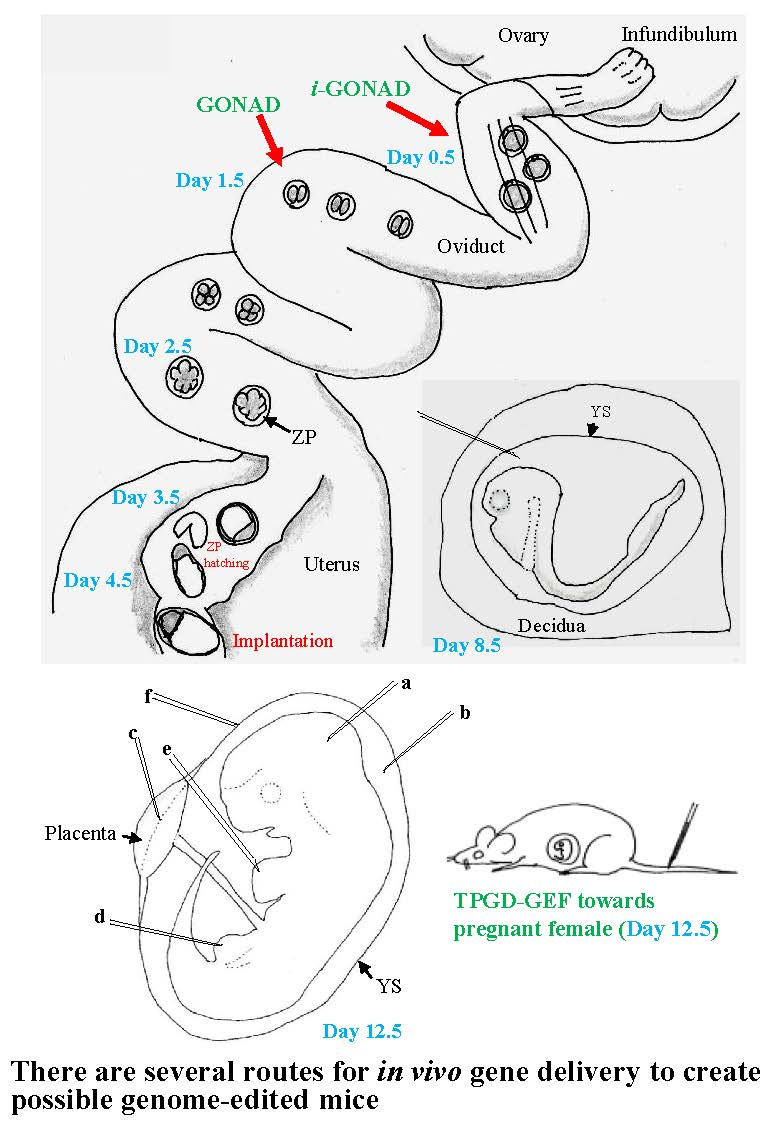
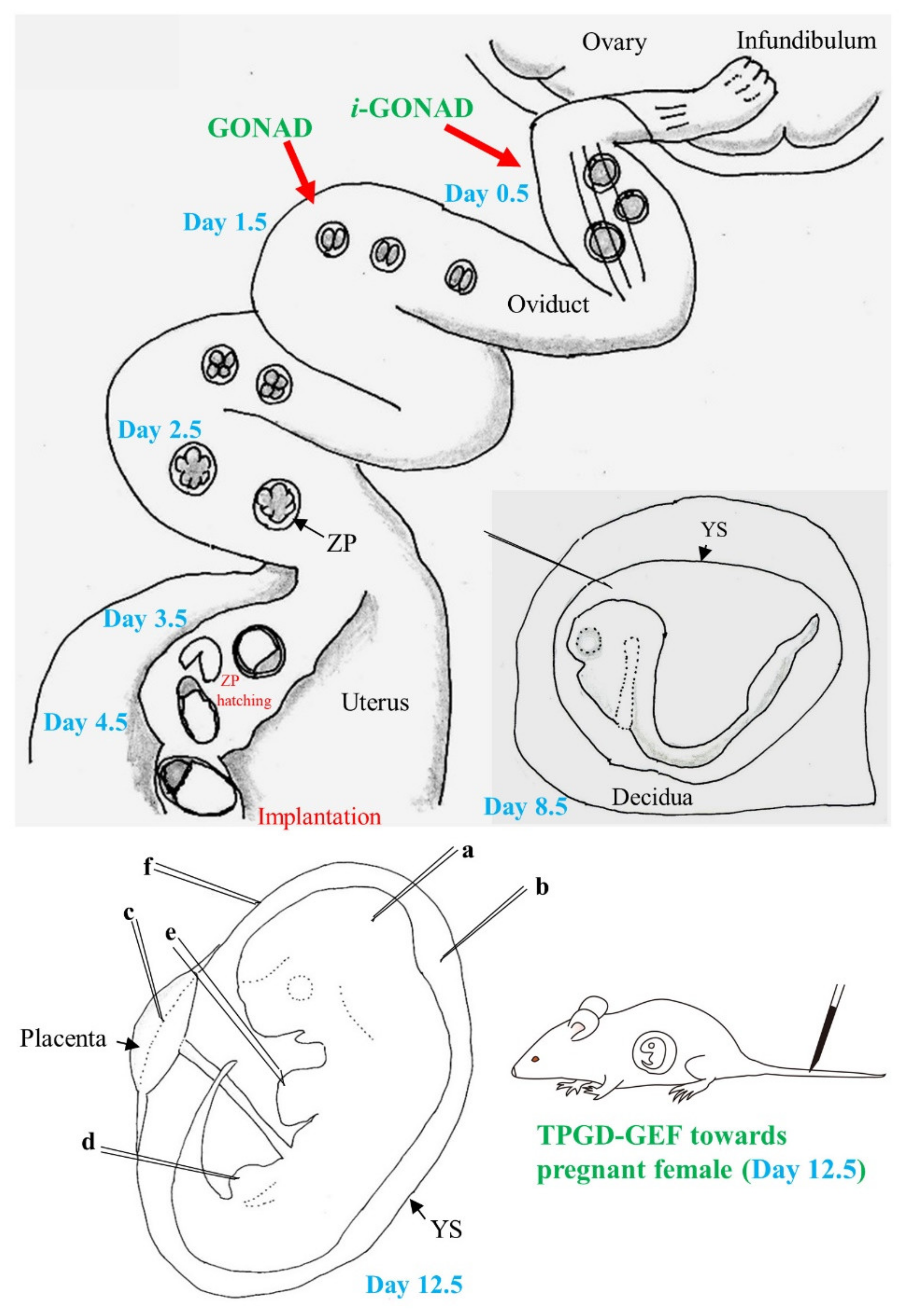
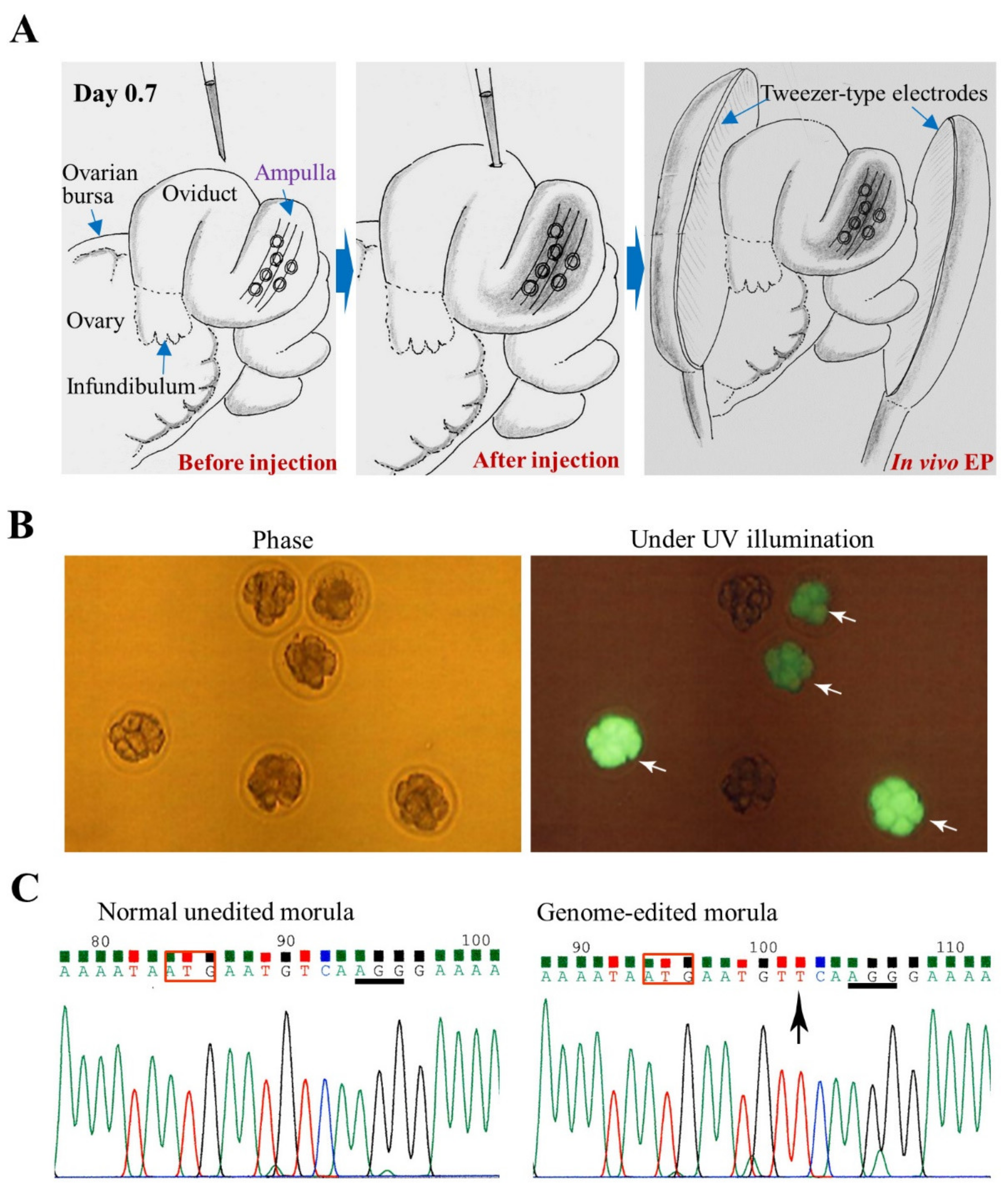
3.1. GONAD/i-GONAD for Obtaining GE Animals
3.2. TPGD-GEF Technique
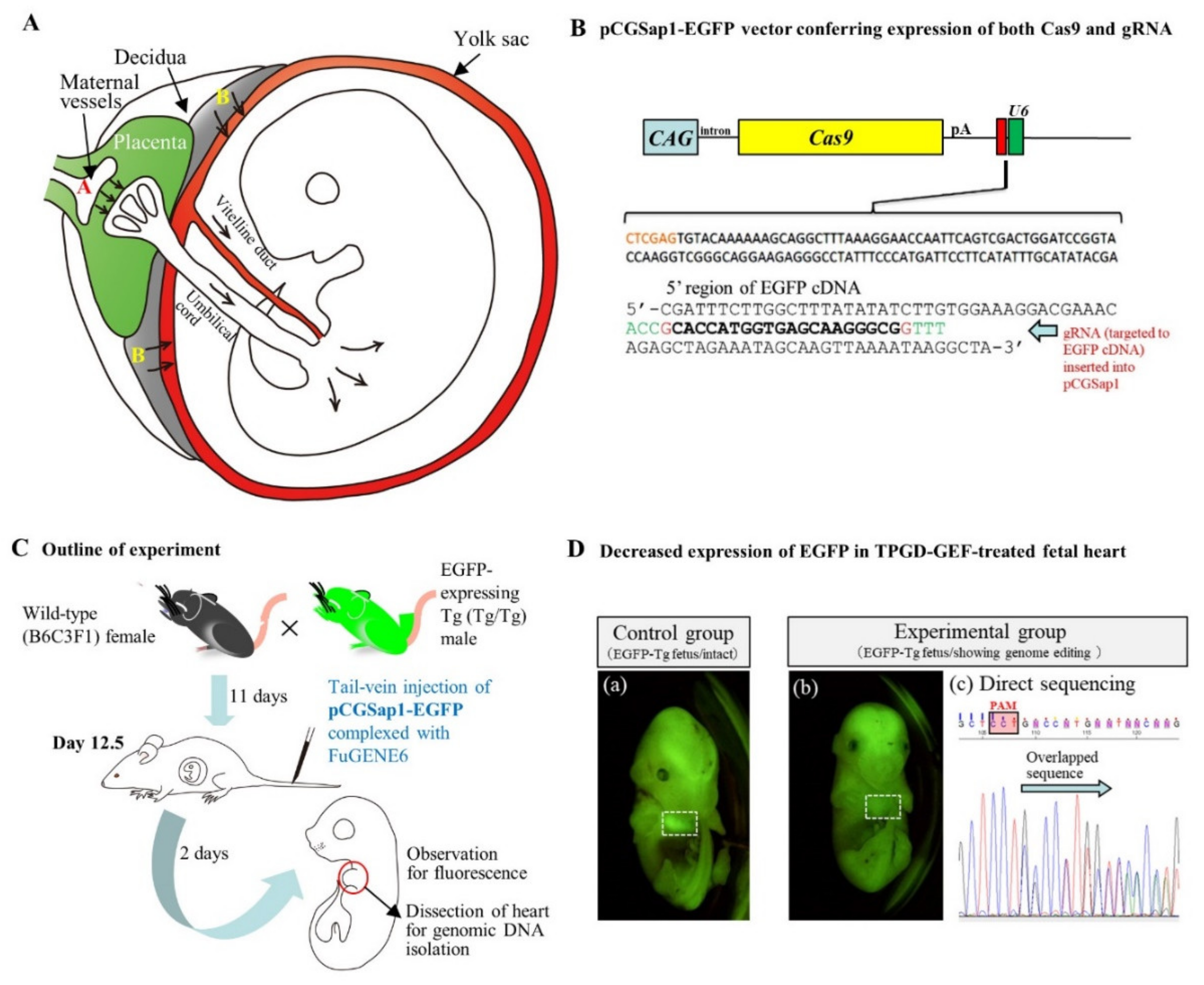
3.3. In Utero Gene Delivery
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| AAVs | Adeno-associated viruses |
| CRISPR/Cas9 | Clustered regularly interspaced palindrome repeats (CRISPR)/Caspase 9 (Cas9) |
| crRNA | CRISPR RNA |
| Crygc | Crystalline gamma C |
| DOGS | 5-carboxyspermylglycine dioctadecylamide |
| CaPO4 | Calcium phosphate |
| cDNA | Complementary DNA |
| ds | Double-stranded |
| DSBs | Double-strand breaks |
| dsRNA | Double-stranded RNA |
| EEct | Embryonic ectoderm |
| EGFP | Enhanced green fluorescent protein |
| EP | Electroporation |
| ExEct | Extra-embryonic ectoderm |
| FITC | Fluorescein isothiocyanate |
| Foxa2 | Forkhead box protein A2 |
| GE | Genome-edited |
| GFP | Green fluorescent protein |
| GONAD | Genome-editing via oviductal nucleic acids delivery |
| gRNA | Guide RNA |
| HDR | Homology-directed repair |
| HR | Homologous recombination |
| ICSI | Intracytoplasmic sperm injection |
| i-GONAD | Improved GONAD |
| IVF | In vitro fertilization |
| KI | Knock-in |
| KO | Knock-out |
| NHEJ | Nonhomologous end joining |
| ODNs | Oligodeoxynucleotides |
| Otx2 | Orthodenticle homeobox 2 |
| PAM | Protospacer adjacent motif |
| PGCs | Primordial germ cells |
| rAAVs | Recombinant AAVs |
| rAAV-6 | Recombinant AAV serotype 6 |
| RNP | Ribonucleoprotein |
| SaCas9 | Cas9 nuclease of the Staphylococcus aureus bacteria |
| sgRNA | single guide RNA |
| Sry | Sex-determining region Y |
| ss | Single-stranded |
| TALENs | Transcription activator-like effector nucleases |
| Tg | Transgenic |
| TPGD | Transplacental gene delivery |
| TPGD-GEF | Transplacental gene delivery for acquiring genome-edited fetuses |
| tracrRNA | Trans-activating CRISPR RNA |
| ZFNs | Zinc-finger nucleases |
References
- Khan, S.H. Genome-editing technologies: Concept, pros, and cons of various genome-editing techniques and bioethical concerns for clinical application. Mol. Ther. Nucleic Acids 2019, 16, 326–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, F.; Jacobson, A. Nonsense-mediated mRNA decay: Degradation of defective transcripts is only part of the story. Annu. Rev. Genet. 2015, 49, 339–366. [Google Scholar] [CrossRef] [Green Version]
- Sonoda, E.; Hochegger, H.; Saberi, A.; Taniguchi, Y.; Takeda, S. Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair 2006, 5, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., III. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, T. Reproductive technologies for the generation and maintenance of valuable animal strains. J. Reprod. Dev. 2018, 64, 209–215. [Google Scholar] [CrossRef]
- Mizuno, N.; Mizutani, E.; Sato, H.; Kasai, M.; Ogawa, A.; Suchy, F.; Yamaguchi, T.; Nakauchi, H. Intra-embryo gene cassette knockin by CRISPR/Cas9-mediated genome editing with adeno-associated viral vector. iScience 2018, 9, 286–297. [Google Scholar] [CrossRef] [Green Version]
- Yoon, Y.; Wang, D.; Tai, P.W.L.; Riley, J.; Gao, G.; Rivera-Pérez, J.A. Streamlined ex vivo and in vivo genome editing in mouse embryos using recombinant adeno-associated viruses. Nat. Commun. 2018, 9, 412. [Google Scholar] [CrossRef]
- Fanslow, D.A.; Wirt, S.E.; Barker, J.C.; Connelly, J.P.; Porteus, M.H.; Dann, C.T. Genome editing in mouse spermatogonial stem/progenitor cells using engineered nucleases. PLoS ONE 2014, 9, e112652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Sakuma, T.; Yokonishi, T.; Katagiri, K.; Kamimura, S.; Ogonuki, N.; Ogura, A.; Yamamoto, T.; Ogawa, T. Genome editing in mouse spermatogonial stem cell lines using TALEN and double-nicking CRISPR/Cas9. Stem Cell Rep. 2015, 5, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Zhou, H.; Fan, X.; Zhang, Y.; Zhang, M.; Wang, Y.; Xie, Z.; Bai, M.; Yin, Q.; Liang, D.; et al. Correction of a genetic disease by CRISPR-Cas9-mediated gene editing in mouse spermatogonial stem cells. Cell Res. 2015, 25, 67–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ding, Y.; Li, J. CRISPR-Cas9-mediated gene editing in mouse spermatogonial stem cells. Methods Mol. Biol. 2017, 1622, 293–305. [Google Scholar] [CrossRef]
- Li, X.; Sun, T.; Wang, X.; Tang, J.; Liu, Y. Restore natural fertility of Kitw/Kitwv mouse with nonobstructive azoospermia through gene editing on SSCs mediated by CRISPR-Cas9. Stem Cell Res. Ther. 2019, 10, 271. [Google Scholar] [CrossRef]
- Takahashi, G.; Gurumurthy, C.B.; Wada, K.; Miura, H.; Sato, M.; Ohtsuka, M. GONAD: Genome-editing via Oviductal Nucleic Acids Delivery system: A novel microinjection independent genome engineering method in mice. Sci. Rep. 2015, 5, 11406. [Google Scholar] [CrossRef] [Green Version]
- Alapati, D.; Zacharias, W.J.; Hartman, H.A.; Rossidis, A.C.; Stratigis, J.D.; Ahn, N.J.; Coons, B.; Zhou, S.; Li, H.; Singh, K.; et al. In utero gene editing for monogenic lung disease. Sci. Transl. Med. 2019, 11, eaav8375. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, S.; Ishihara, M.; Ando, N.; Watanabe, S.; Sakurai, T.; Sato, M. Transplacental delivery of genome editing components causes mutations in embryonic cardiomyocytes of mid-gestational murine fetuses. IUBMB Life 2019, 71, 835–844. [Google Scholar] [CrossRef]
- Li, D.; Qiu, Z.; Shao, Y.; Chen, Y.; Guan, Y.; Liu, M.; Li, Y.; Gao, N.; Wang, L.; Lu, X.; et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 681–683. [Google Scholar] [CrossRef]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, H.; Shivalila, C.S.; Cheng, A.W.; Shi, L.; Jaenisch, R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 2013, 154, 1370–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashiko, D.; Fujihara, Y.; Satouh, Y.; Miyata, H.; Isotani, A.; Ikawa, M. Generation of mutant mice by pronuclear injection of circular plasmid expressing Cas9 and single guided RNA. Sci. Rep. 2013, 3, 3355. [Google Scholar] [CrossRef] [PubMed]
- Fujii, W.; Kawasaki, K.; Sugiura, K.; Naito, K. Efficient generation of large-scale genome-modified mice using gRNA and CAS9 endonuclease. Nucleic Acids Res. 2013, 41, e187. [Google Scholar] [CrossRef]
- Shen, B.; Zhang, J.; Wu, H.; Wang, J.; Ma, K.; Li, Z.; Zhang, X.; Zhang, P.; Huang, X. Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res. 2013, 23, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Horii, T.; Arai, Y.; Yamazaki, M.; Morita, S.; Kimura, M.; Itoh, M.; Abe, Y.; Hatada, I. Validation of microinjection methods for generating knockout mice by CRISPR/Cas-mediated genome engineering. Sci. Rep. 2014, 4, 4513. [Google Scholar] [CrossRef]
- Yen, S.T.; Zhang, M.; Deng, J.M.; Usman, S.J.; Smith, C.N.; Parker-Thornburg, J.; Swinton, P.G.; Martin, J.F.; Behringer, R.R. Somatic mosaicism and allele complexity induced by CRISPR/Cas9 RNA injections in mouse zygotes. Dev. Biol. 2014, 393, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Sung, Y.H.; Kim, J.M.; Kim, H.T.; Lee, J.; Jeon, J.; Jin, Y.; Choi, J.H.; Ban, Y.H.; Ha, S.J.; Kim, C.H.; et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Res. 2014, 24, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Wang, J.; Shen, B.; Chen, L.; Su, Y.; Yang, J.; Zhang, W.; Tian, X.; Huang, X. Dual sgRNAs facilitate CRISPR/Cas9-mediated mouse genome targeting. FEBS J. 2014, 281, 1717–1725. [Google Scholar] [CrossRef]
- Wu, Y.; Liang, D.; Wang, Y.; Bai, M.; Tang, W.; Bao, S.; Yan, Z.; Li, D.; Li, J. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 2013, 13, 659–662. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [Green Version]
- Chu, V.T.; Weber, T.; Graf, R.; Sommermann, T.; Petsch, K.; Sack, U.; Volchkov, P.; Rajewsky, K.; Kühn, R. Efficient generation of Rosa26 knock-in mice using CRISPR/Cas9 in C57BL/6 zygotes. BMC Biotechnol. 2016, 16, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raveux, A.; Vandormael-Pournin, S.; Cohen-Tannoudji, M. Optimization of the production of knock-in alleles by CRISPR/Cas9 microinjection into the mouse zygote. Sci. Rep. 2017, 7, 42661. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Posfai, E.; Rossant, J. Efficient generation of targeted large insertions by microinjection into two-cell-stage mouse embryos. Nat. Biotechnol. 2018, 36, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Harms, D.W.; Quadros, R.M.; Seruggia, D.; Ohtsuka, M.; Takahashi, G.; Montoliu, L.; Gurumurthy, C.B. Mouse genome editing using the CRISPR/Cas system. Curr. Protoc. Hum. Genet. 2014, 83, 15.7.1–15.7.27. [Google Scholar] [CrossRef] [Green Version]
- Jacobi, A.M.; Rettig, G.R.; Turk, R.; Collingwood, M.A.; Zeiner, S.A.; Quadros, R.M.; Harms, D.W.; Bonthuis, P.J.; Gregg, C.; Ohtsuka, M.; et al. Simplified CRISPR tools for efficient genome editing and streamlined protocols for their delivery into mammalian cells and mouse zygotes. Methods 2017, 121–122, 16–28. [Google Scholar] [CrossRef]
- Kaneko, T.; Sakuma, T.; Yamamoto, T.; Mashimo, T. Simple knockout by electroporation of engineered endonucleases into intact rat embryos. Sci. Rep. 2014, 4, 6382. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, M.; Takemoto, T. Electroporation enables the efficient mRNA delivery into the mouse zygotes and facilitates CRISPR/Cas9-based genome editing. Sci. Rep. 2015, 5, 11315. [Google Scholar] [CrossRef] [Green Version]
- Qin, W.; Dion, S.L.; Kutny, P.M.; Zhang, Y.; Cheng, A.W.; Jillette, N.L.; Malhotra, A.; Geurts, A.M.; Chen, Y.G.; Wang, H. Efficient CRISPR/Cas9-mediated genome editing in mice by zygote electroporation of nuclease. Genetics 2015, 200, 423–430. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Lee, B.; Lee, A.Y.; Modzelewski, A.J.; He, L. Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. J. Biol. Chem. 2016, 291, 14457–14467. [Google Scholar] [CrossRef] [Green Version]
- Tröder, S.E.; Ebert, L.K.; Butt, L.; Assenmacher, S.; Schermer, B.; Zevnik, B. An optimized electroporation approach for efficient CRISPR/Cas9 genome editing in murine zygotes. PLoS ONE 2018, 13, e0196891. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, M.; Py, B.F.; Bosc, C.; Laubreton, D.; Moutin, M.J.; Marvel, J.; Flamant, F.; Markossian, S. Electroporation of mice zygotes with dual guide RNA/Cas9 complexes for simple and efficient cloning-free genome editing. Sci. Rep. 2018, 8, 474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Sun, S.; Moonen, D.; Lee, C.; Lee, A.Y.; Schaffer, D.V.; He, L. CRISPR-READI: Efficient generation of knockin mice by CRISPR RNP electroporation and AAV donor infection. Cell Rep. 2019, 27, 3780–3789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, T. Genome editing in mouse and rat by electroporation. Methods Mol. Biol. 2017, 1630, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Wang, H. Delivery of CRISPR-Cas9 into mouse zygotes by electroporation. Methods Mol. Biol. 2019, 1874, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Modzelewski, A.J.; Chen, S.; Willis, B.J.; Lloyd, K.C.K.; Wood, J.A.; He, L. Efficient mouse genome engineering by CRISPR-EZ technology. Nat. Protoc. 2018, 13, 1253–1274. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, M.; Snow, M.H.; McLaren, A. Primordial germ cells in the mouse embryo during gastrulation. Development 1990, 110, 521–528. [Google Scholar]
- Tam, P.P.; Zhou, S.X. The allocation of epiblast cells to ectodermal and germ-line lineages is influenced by the position of the cells in the gastrulating mouse embryo. Dev. Biol. 1996, 178, 124–132. [Google Scholar] [CrossRef]
- Tsang, T.E.; Khoo, P.L.; Jamieson, R.V.; Zhou, S.X.; Ang, S.L.; Behringer, R.; Tam, P.P. The allocation and differentiation of mouse primordial germ cells. Int. J. Dev. Biol. 2001, 45, 549–555. [Google Scholar]
- Tam, P.P.; Snow, M.H. Proliferation and migration of primordial germ cells during compensatory growth in mouse embryos. J. Embryol. Exp. Morphol. 1981, 64, 133–147. [Google Scholar]
- Molyneaux, K.A.; Stallock, J.; Schaible, K.; Wylie, C. Time-lapse analysis of living mouse germ cell migration. Dev. Biol. 2001, 240, 488–498. [Google Scholar] [CrossRef] [Green Version]
- McLaren, A. Primordial germ cells in the mouse. Dev. Biol. 2003, 262, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Matsui, Y.; Zsebo, K.; Hogan, B.L.M. Derivation of pluripotent embryonic cells from murine primordial germ cells in culture. Cell 1992, 70, 841–847. [Google Scholar] [CrossRef]
- Resnick, J.L.; Bixler, L.S.; Cheng, L.; Donovan, P.J. Long term proliferation of mouse primordial germ cells in culture. Nature 1992, 359, 550–551. [Google Scholar] [CrossRef] [PubMed]
- Labosky, P.A.; Barlow, D.P.; Hogan, B.L.M. Mouse embryonic germ (EG) cell lines: Transmission through the germ line and differences in the methylation imprint of insulin-like growth factor 2 receptor (Igf2r) gene compared with embryonic stem (ES) cell lines. Development 1994, 120, 3197–3204. [Google Scholar]
- Watanabe, M.; Shirayoshi, Y.; Koshimizu, U.; Hashimoto, S.; Yonehara, S.; Eguchi, Y.; Tsujimoto, Y.; Nakatsuji, N. Gene transfection of mouse primordial germ cells in vitro and analysis of their survival and growth control. Exp. Cell Res. 1997, 230, 76–83. [Google Scholar] [CrossRef]
- De Miguel, M.P.; Cheng, L.; Holland, E.C.; Federspiel, M.J.; Donovan, P.J. Dissection of the c-Kit signaling pathway in mouse primordial germ cells by retroviral-mediated gene transfer. Proc. Natl. Acad. Sci. USA 2002, 99, 10458–10463. [Google Scholar] [CrossRef] [Green Version]
- Chuma, S.; Kanatsu-Shinohara, M.; Inoue, K.; Ogonuki, N.; Miki, H.; Toyokuni, S.; Hosokawa, M.; Nakatsuji, N.; Ogura, A.; Shinohara, T. Spermatogenesis from epiblast and primordial germ cells following transplantation into postnatal mouse testis. Development 2005, 132, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Svingen, T.; Wilhelm, D.; Combes, A.N.; Hosking, B.; Harley, V.R.; Sinclair, A.H.; Koopman, P. Ex vivo magnetofection: A novel strategy for the study of gene function in mouse organogenesis. Dev. Dyn. 2009, 238, 956–964. [Google Scholar] [CrossRef] [Green Version]
- Morohaku, K.; Tanimoto, R.; Sasaki, K.; Kawahara-Miki, R.; Kono, T.; Hayashi, K.; Hirao, Y.; Obata, Y. Complete in vitro generation of fertile oocytes from mouse primordial germ cells. Proc. Natl. Acad. Sci. USA 2016, 113, 9021–9026. [Google Scholar] [CrossRef] [Green Version]
- Morohaku, K.; Hirao, Y.; Obata, Y. Development of fertile mouse oocytes from mitotic germ cells in vitro. Nat. Protoc. 2017, 12, 1817–1829. [Google Scholar] [CrossRef]
- Meistrich, M.L.; van Beek, M.E.A.B. Spermatogonial stem cells. In Cell and Molecular Biology of the Testis; Desjardins, C., Ewing, L.L., Eds.; Oxford University Press: New York, NY, USA, 1993; pp. 266–295. [Google Scholar]
- De Rooij, D.G.; Russell, L.D. All you wanted to know about spermatogonia but were afraid to ask. J. Androl. 2000, 21, 776–798. [Google Scholar] [CrossRef] [PubMed]
- Brinster, R.L.; Zimmermann, J.W. Spermatogenesis following male germ-cell transplantation. Proc. Natl. Acad. Sci. USA 1994, 91, 11298–11302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanatsu-Shinohara, M.; Ogonuki, N.; Inoue, K.; Miki, H.; Ogura, A.; Toyokuni, S.; Shinohara, T. Long-term proliferation in culture and germline transmission of mouse male germline stem cells. Biol. Reprod. 2003, 69, 612–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, H.; Avarbock, M.R.; Brinster, R.L. Growth factors essential for self-renewal and expansion of mouse spermatogonial stem cells. Proc. Natl. Acad. Sci. USA 2004, 101, 16489–16494. [Google Scholar] [CrossRef] [Green Version]
- Kanatsu-Shinohara, M.; Toyokuni, S.; Shinohara, T. Genetic selection of mouse male germline stem cells in vitro: Offspring from single stem cells. Biol. Reprod. 2005, 72, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Iwamori, N.; Iwamori, T.; Matzuk, M.M. Characterization of spermatogonial stem cells lacking intercellular bridges and genetic replacement of a mutation in spermatogonial stem cells. PLoS ONE 2012, 7, e38914. [Google Scholar] [CrossRef] [Green Version]
- Kanatsu-Shinohara, M.; Ikawa, M.; Takehashi, M.; Ogonuki, N.; Miki, H.; Inoue, K.; Kazuki, Y.; Lee, J.; Toyokuni, S.; Oshimura, M.; et al. Production of knockout mice by random or targeted mutagenesis in spermatogonial stem cells. Proc. Natl. Acad. Sci. USA 2006, 103, 8018–8023. [Google Scholar] [CrossRef] [Green Version]
- Kanatsu-Shinohara, M.; Lee, J.; Inoue, K.; Ogonuki, N.; Miki, H.; Toyokuni, S.; Ikawa, M.; Nakamura, T.; Ogura, A.; Shinohara, T. Pluripotency of a single spermatogonial stem cell in mice. Biol. Reprod. 2008, 78, 681–687. [Google Scholar] [CrossRef] [Green Version]
- Lois, C.; Hong, E.J.; Pease, S.; Brown, E.J.; Baltimore, D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 2002, 295, 868–872. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, A.; Ikawa, M.; Dayn, Y.; Verma, I.M. Transgenesis by lentiviral vectors: Lack of gene silencing in mammalian embryonic stem cells and preimplantation embryos. Proc. Natl. Acad. Sci. USA 2002, 99, 2140–2145. [Google Scholar] [CrossRef] [Green Version]
- Osumi-Yamashita, N.; Kuratani, S.; Ninomiya, Y.; Aoki, K.; Iseki, S.; Chareonvit, S.; Doi, H.; Fujiwara, M.; Watanabe, T.; Eto, K. Cranial anomaly of homozygous rSey rat is associated with a defect in the migration pathway of midbrain crest cells. Dev. Growth Differ. 1997, 39, 53–67. [Google Scholar] [CrossRef]
- Moore-Scott, B.A.; Gordon, J.; Blackburn, C.C.; Condie, B.G.; Manley, N.R. New serum-free in vitro culture technique for midgestation mouse embryos. Genesis 2003, 35, 164–168. [Google Scholar] [CrossRef]
- Kinder, S.J.; Tan, S.S.; Tam, P.P.L. Cell grafting and fate mapping of early-somite-stage mouse embryo. Methods Mol. Biol. 2000, 135, 425–437. [Google Scholar] [CrossRef]
- Osumi, N.; Inoue, T. Gene transfer into cultured mammalian embryos by electroporation. Methods 2001, 24, 35–42. [Google Scholar] [CrossRef]
- Mellitzer, G.; Hallonet, M.; Chen, L.; Ang, S.L. Spatial and temporal ‘knock down’ of gene expression by electroporation of double-stranded RNA and morpholinos into early postimplantation mouse embryos. Mech. Dev. 2002, 118, 57–63. [Google Scholar] [CrossRef]
- Davidson, B.P.; Tsang, T.E.; Khoo, P.L.; Gad, J.M.; Tam, P.P. Introduction of cell markers into germ layer tissues of the mouse gastrula by whole embryo electroporation. Genesis 2003, 35, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Pierreux, C.E.; Poll, A.V.; Jacquemin, P.; Lemaigre, F.P.; Rousseau, G.G. Gene transfer into mouse prepancreatic endoderm by whole embryo electroporation. JOP 2005, 6, 128–135. [Google Scholar] [PubMed]
- Sakai, D.; Trainor, P.A. Gene transfer techniques in whole embryo cultured post-implantation mouse embryos. Methods Mol. Biol. 2014, 1092, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Mazari, E.; Zhao, X.; Migeotte, I.; Collignon, J.; Gosse, C.; Perea-Gomez, A. A microdevice to locally electroporate embryos with high efficiency and reduced cell damage. Development 2014, 141, 2349–2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtsuka, M.; Sato, M.; Miura, H.; Takabayashi, S.; Matsuyama, M.; Koyano, T.; Arifin, N.; Nakamura, S.; Wada, K.; Gurumurthy, C.B. i-GONAD: A robust method for in situ germline genome engineering using CRISPR nucleases. Genome Biol. 2018, 19, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, T.; Namba, M.; Koyano, T.; Fukushima, M.; Sato, M.; Ohtsuka, M.; Matsuyama, M. Successful production of genome-edited rats by the rGONAD method. BMC Biotechnol. 2018, 18, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takabayashi, S.; Aoshima, T.; Kabashima, K.; Aoto, K.; Ohtsuka, M.; Sato, M. i-GONAD (improved genome-editing via oviductal nucleic acids delivery), a convenient in vivo tool to produce genome-edited rats. Sci. Rep. 2018, 8, 12059. [Google Scholar] [CrossRef]
- Gurumurthy, C.B.; Takahashi, G.; Wada, K.; Miura, H.; Sato, M.; Ohtsuka, M. GONAD: A novel CRISPR/Cas9 genome editing method that does not require ex vivo handling of embryos. Curr. Protoc. Hum. Genet. 2016, 88, 15.8.1–15.8.12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurumurthy, C.B.; Sato, M.; Nakamura, A.; Inui, M.; Kawano, N.; Islam, M.; Ogiwara, S.; Takabayashi, S.; Matsuyama, M.; Nakagawa, S.; et al. Creation of CRISPR-based germline-genome-engineered mice without ex vivo handling of zygotes by i-GONAD. Nat. Protoc. 2019, 14, 2452–2482. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, M.; Sato, M. i-GONAD: A method for generating genome-edited animals without ex vivo handling of embryos. Dev. Growth Differ. 2019, 61, 306–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukamoto, M.; Ochiya, T.; Yoshida, S.; Sugimura, T.; Terada, M. Gene transfer and expression in progeny after intravenous DNA injection into pregnant mice. Nat. Genet. 1995, 9, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Watanabe, S.; Ando, N.; Ishihara, M.; Sato, M. Transplacental gene delivery (TPGD) as a noninvasive tool for fetal gene manipulation in mice. Int. J. Mol. Sci. 2019, 20, 5926. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, N.; Nakamura, S.; Ohtsuka, M.; Kimura, M.; Sato, M. Possible mechanism of gene transfer into early to mid-gestational mouse fetuses by tail vein injection. Gene Ther. 2002, 9, 1529–1541. [Google Scholar] [CrossRef]
- Sheehy, N.T.; Cordes, K.R.; White, M.P.; Ivey, K.N.; Srivastava, D. The neural crest-enriched microRNA miR-452 regulates epithelial-mesenchymal signaling in the first pharyngeal arch. Development 2010, 137, 4307–4316. [Google Scholar] [CrossRef] [Green Version]
- Tsunekawa, Y.; Terhune, R.K.; Fujita, I.; Shitamukai, A.; Suetsugu, T.; Matsuzaki, F. Developing a de novo targeted knock-in method based on in utero electroporation into the mammalian brain. Development 2016, 143, 3216–3222. [Google Scholar] [CrossRef] [Green Version]
- Uemura, T.; Mori, T.; Kurihara, T.; Kawase, S.; Koike, R.; Satoga, M.; Cao, X.; Li, X.; Yanagawa, T.; Sakurai, T.; et al. Fluorescent protein tagging of endogenous protein in brain neurons using CRISPR/Cas9-mediated knock-in and in utero electroporation techniques. Sci. Rep. 2016, 6, 35861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricciardi, A.S.; Bahal, R.; Farrelly, J.S.; Quijano, E.; Bianchi, A.H.; Luks, V.L.; Putman, R.; López-Giráldez, F.; Coşkun, S.; Song, E.; et al. In utero nanoparticle delivery for site-specific genome editing. Nat. Commun. 2018, 9, 2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossidis, A.C.; Stratigis, J.D.; Chadwick, A.C.; Hartman, H.A.; Ahn, N.J.; Li, H.; Singh, K.; Coons, B.E.; Li, L.; Lv, W.; et al. In utero CRISPR-mediated therapeutic editing of metabolic genes. Nat. Med. 2018, 24, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Chu, A.H.Y.; Bao, S.; Hoang, D.A.; Kebede, F.T.; Xiong, W.; Ji, M.; Shi, J.; Zheng, Z. Rationally engineered Staphylococcus aureus Cas9 nucleases with high genome-wide specificity. Proc. Natl. Acad. Sci. USA 2019, 116, 20969–20976. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Delivery mode(s) | Method(s) | Target Embryos, Fetuses and Cells | Equipment | Notes | References |
|---|---|---|---|---|---|
| Ex vivo | Microinjection | Zygotes 2-Cell embryos | Requires micromanipulator system | Requires egg transfer to allow further development of the treated embryos | [19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35] |
| Ex vivo | In vitro EP | Zygotes Germ stem cells | Requires electroporator and micromanipulator system | Requires ICSI and egg transfer to allow further development of the treated embryos | [12,13,14,15,36,37,38,39,40,41,42,43,44,45] |
| Ex vivo | Transduction by viral vectors | Zygotes | No special equipment | Requires egg transfer to allow further development of the treated embryos | [9,10] |
| In vivo | GONAD/i-GONAD/AAV -based GONAD | Zygotes 2-cell embryos | Requires electroporator but no special equipment in the case of AAV-based GONAD | No recipients are required | [10,16,81,84,85,86] |
| In vivo | TPGD/TPGD-GEF | Mid-gestational fetuses | No special equipment | No recipients are required | [18,88] |
| In vivo | In utero gene delivery using viral vectors or in vivo EP | Mid-gestational fetuses | Requires electroporator in the case of using nonviral vectors | No recipients are required | [91,92,93,94] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, M.; Takabayashi, S.; Akasaka, E.; Nakamura, S. Recent Advances and Future Perspectives of In Vivo Targeted Delivery of Genome-Editing Reagents to Germ cells, Embryos, and Fetuses in Mice. Cells 2020, 9, 799. https://doi.org/10.3390/cells9040799
Sato M, Takabayashi S, Akasaka E, Nakamura S. Recent Advances and Future Perspectives of In Vivo Targeted Delivery of Genome-Editing Reagents to Germ cells, Embryos, and Fetuses in Mice. Cells. 2020; 9(4):799. https://doi.org/10.3390/cells9040799
Chicago/Turabian StyleSato, Masahiro, Shuji Takabayashi, Eri Akasaka, and Shingo Nakamura. 2020. "Recent Advances and Future Perspectives of In Vivo Targeted Delivery of Genome-Editing Reagents to Germ cells, Embryos, and Fetuses in Mice" Cells 9, no. 4: 799. https://doi.org/10.3390/cells9040799