Targeting the Human 80S Ribosome in Cancer: From Structure to Function and Drug Design for Innovative Adjuvant Therapeutic Strategies

 , , , and
, , , and
Abstract
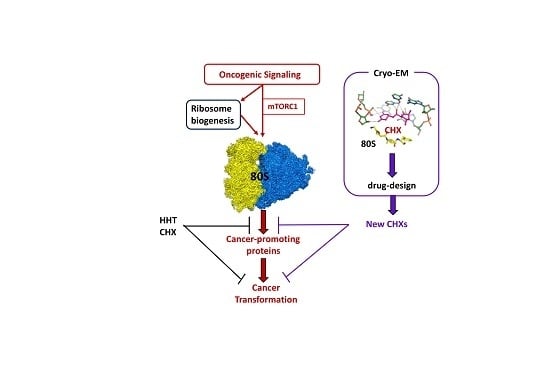
:
1. Introduction
2. The Human Ribosome
3. The Ribosome in Cancer
3.1. Ribosomal Genes Mutations in Cancer
3.2. The Ribosome as an Important Actor in Cancer
3.3. Targeting Ribosome Biogenesis
3.4. Ribosomes: Multifaceted Targets
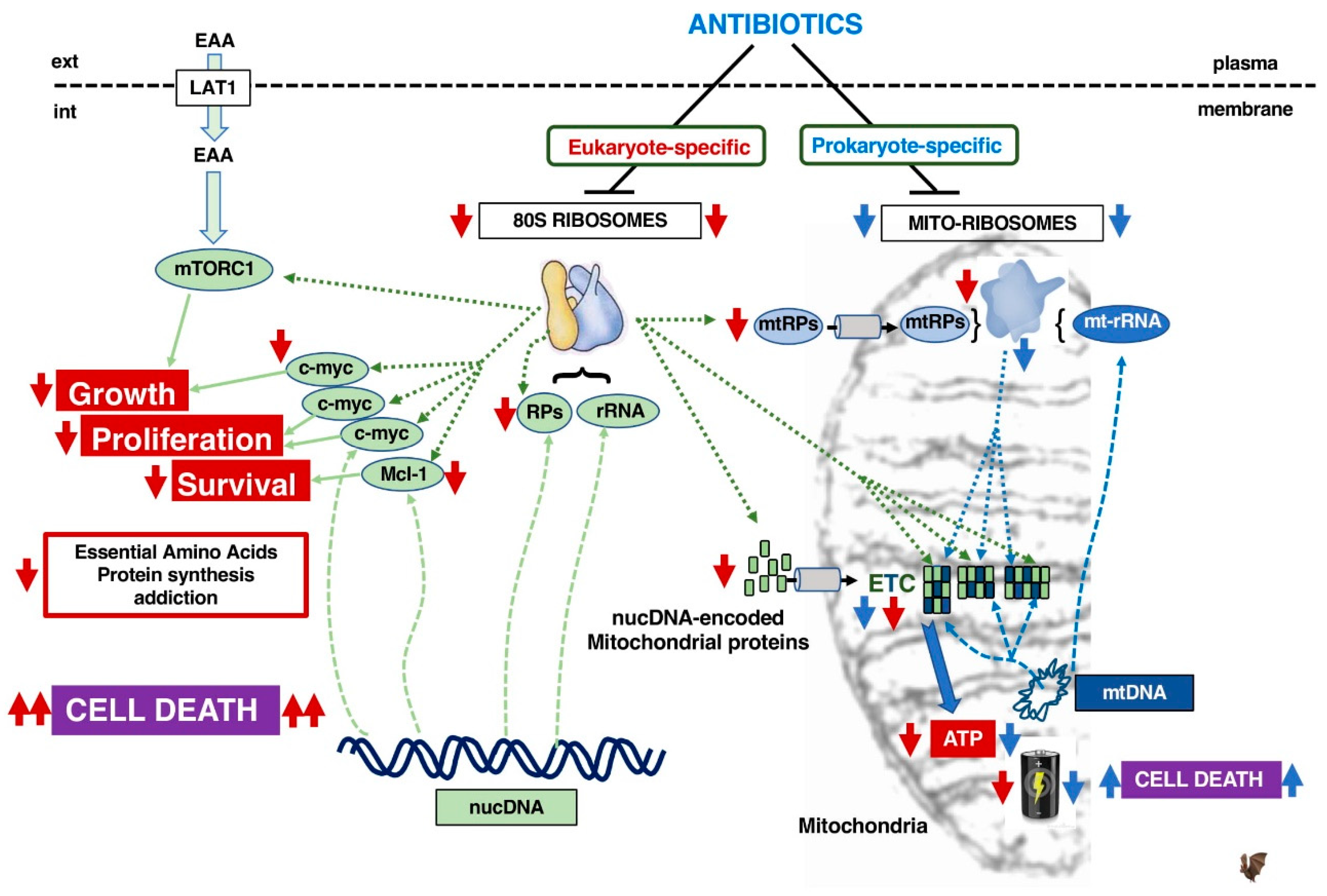
3.5. Who Is the Best Target in Cancer: 80S Ribosomes or Mitoribosomes?
4. Direct Targeting of the 80S Ribosome
4.1. Homoharringtonine, a Clinically Used Ribosome Inhibitor
4.2. Ribosome Inhibition in Hematopoietic Malignancies
4.3. Ribosome Inhibition in Various Solid Cancers
4.4. Ribosome Inhibitors and Metastasis
4.5. Ribosome Inhibitors and Immunotherapies
4.6. Combining Ribosome Inhibition with Classical Chemotherapeutic Drugs
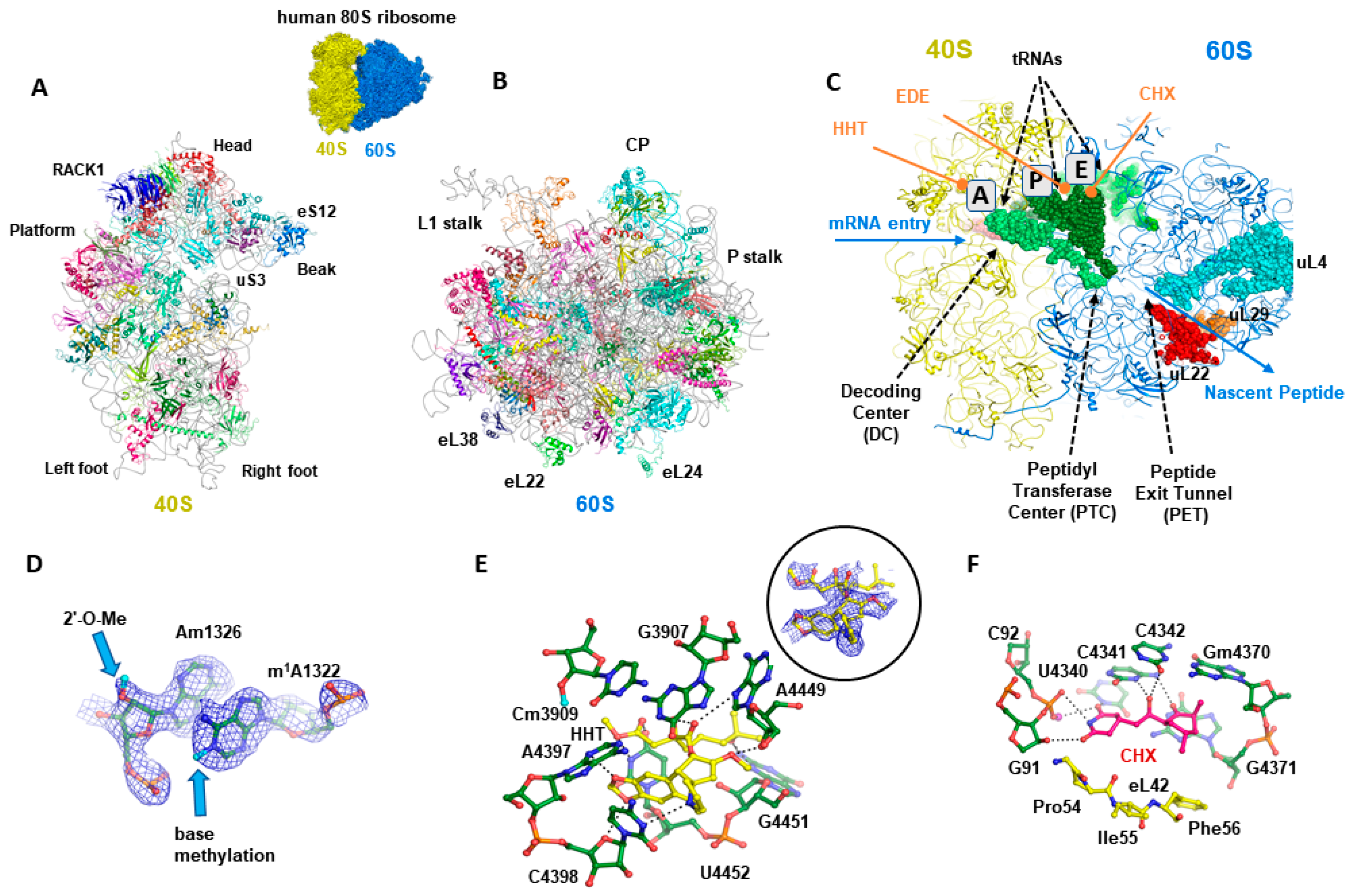
5. Advent of High-Resolution Cryo-EM Allows Structure-Guided Drug Design
Cryo-EM-Based Drug Design of Cycloheximide
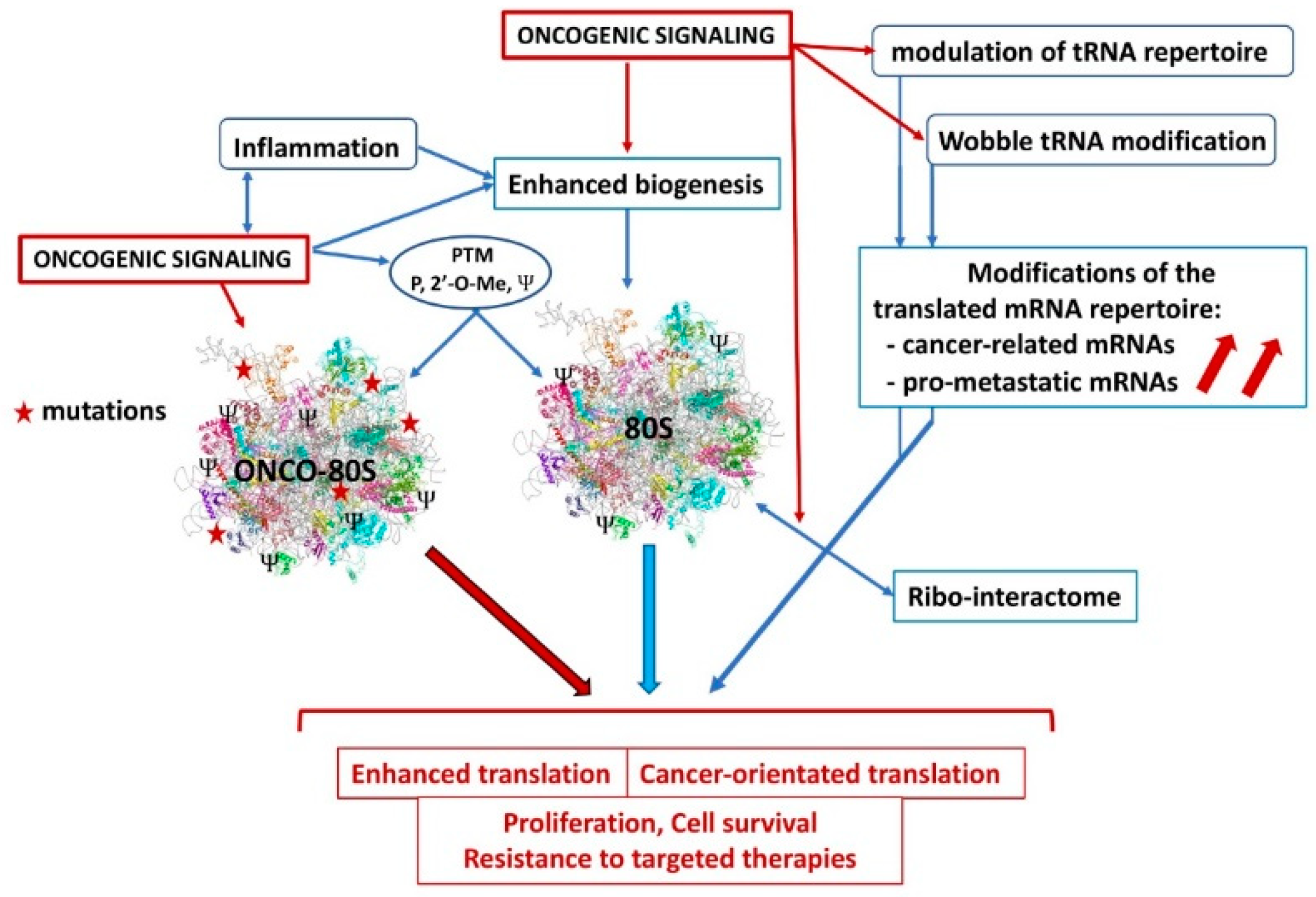
6. Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Pardee, A.B. G1 events and regulation of cell proliferation. Science 1989, 246, 603–608. [Google Scholar] [CrossRef]
- Pyronnet, S.; Sonenberg, N. Cell-cycle-dependent translational control. Curr. Opin. Genet. Dev. 2001, 11, 13–18. [Google Scholar] [CrossRef]
- Nagaraj, N.; Wisniewski, J.R.; Geiger, T.; Cox, J.; Kircher, M.; Kelso, J.; Pääbo, S.; Mann, M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 2011, 7, 548. [Google Scholar] [CrossRef]
- Truitt, M.L.; Ruggero, D. New frontiers in translational control of the cancer genome. Nat. Rev. Cancer 2016, 16, 288–304. [Google Scholar] [CrossRef] [Green Version]
- Robichaud, N.; Sonenberg, N.; Ruggero, D.; Schneider, R.J. Translational Control in Cancer. Cold Spring. Harb. Perspect. Biol. 2019, 11, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Silvera, D.; Formenti, S.C.; Schneider, R.J. Translational control in cancer. Nat. Rev. Cancer 2010, 10, 254–266. [Google Scholar] [CrossRef]
- Yusupova, G.; Yusupov, M. High-resolution structure of the eukaryotic 80S ribosome. Annu. Rev. Biochem. 2014, 83, 467–486. [Google Scholar] [CrossRef]
- Pelletier, J.; Thomas, G.; Volarević, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef]
- Khatter, H.; Myasnikov, A.G.; Natchiar, S.K.; Klaholz, B.P. Structure of the human 80S ribosome. Nature 2015, 520, 640–645. [Google Scholar] [CrossRef]
- Natchiar, S.K.; Myasnikov, A.G.; Kratzat, H.; Hazemann, I.; Klaholz, B.P. Visualization of chemical modifications in the human 80S ribosome structure. Nature 2017, 551, 472–477. [Google Scholar] [CrossRef]
- Palade, G.E. Microsomes and ribunucleoprotein particles. In Microsomal Particles and Protein Synthesis; Pergamon Press: New York, NY, USA, 1958; pp. 36–61. [Google Scholar]
- Roberts, R.B. Introduction. In Microsomal Particles and Protein Synthesis; Pergamon Press: New York, NY, USA, 1958; pp. 7–8. [Google Scholar]
- Genuth, N.R.; Barna, M. The Discovery of Ribosome Heterogeneity and Its Implications for Gene Regulation and Organismal Life. Mol. Cell 2018, 71, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Tahmasebi, S.; Khoutorsky, A.; Mathews, M.B.; Sonenberg, N. Translation deregulation in human disease. Nat. Rev. Mol. Cell. Biol. 2018, 19, 791–807. [Google Scholar] [CrossRef]
- Borovinskaya, M.A.; Shoji, S.; Fredrick, K.; Cate, J.H. Structural basis for hygromycin B inhibition of protein biosynthesis. RNA 2008, 14, 1590–1599. [Google Scholar] [CrossRef] [Green Version]
- Garreau de Loubresse, N.; Prokhorova, I.; Holtkamp, W.; Rodnina, M.V.; Yusupova, G.; Yusupov, M. Structural basis for the inhibition of the eukaryotic ribosome. Nature 2014, 513, 517–522. [Google Scholar] [CrossRef] [Green Version]
- Melnikov, S.; Ben-Shem, A.; Garreau de Loubresse, N.; Jenner, L.; Yusupova, G.; Yusupov, M. One core, two shells: Bacterial and eukaryotic ribosomes. Nat. Struct. Mol. Biol. 2012, 19, 560–567. [Google Scholar] [CrossRef]
- Wilson, D.N.; Doudna Cate, J.H. The structure and function of the eukaryotic ribosome. Cold Spring Harb. Perspect. Biol. 2012, 4, 1–17. [Google Scholar] [CrossRef]
- Klinge, S.; Voigts-Hoffmann, F.; Leibundgut, M.; Ban, N. Atomic structures of the eukaryotic ribosome. Trends Biochem. Sci. 2012, 37, 189–198. [Google Scholar] [CrossRef]
- Weisser, M.; Ban, N. Extensions, Extra Factors, and Extreme Complexity: Ribosomal Structures Provide Insights into Eukaryotic Translation. Cold Spring Harb. Perspect. Biol. 2019, 11, 1–20. [Google Scholar] [CrossRef]
- Sulima, S.O.; Dinman, J.D. The Expanding Riboverse. Cells 2019, 8, 1205. [Google Scholar] [CrossRef] [Green Version]
- Preiss, T. All Ribosomes Are Created Equal. Really. Trends Biochem. Sci. 2016, 41, 121–123. [Google Scholar] [CrossRef]
- Simsek, D.; Tiu, G.C.; Flynn, R.A.; Byeon, G.W.; Leppek, K.; Xu, A.F.; Chang, H.Y.; Barna, M. The Mammalian Ribo-interactome Reveals Ribosome Functional Diversity and Heterogeneity. Cell 2017, 169, 1051–1065. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Lafontaine, D.L.J. ‘View From A Bridge’: A New Perspective on Eukaryotic rRNA Base Modification. Trends Biochem. Sci. 2015, 40, 560–575. [Google Scholar] [CrossRef]
- Natchiar, S.K.; Myasnikov, A.G.; Hazemann, I.; Klaholz, B.P. Visualizing the Role of 2′-OH rRNA Methylations in the Human Ribosome Structure. Biomolecules 2018, 8, 125. [Google Scholar] [CrossRef] [Green Version]
- Montanaro, L.; Brigotti, M.; Clohessy, J.; Barbieri, S.; Ceccarelli, C.; Santini, D.; Taffurelli, M.; Calienni, M.; Teruya-Feldstein, J.; Trerè, D.; et al. Dyskerin expression influences the level of ribosomal RNA pseudo-uridylation and telomerase RNA component in human breast cancer. J. Pathol. 2006, 210, 10–18. [Google Scholar] [CrossRef]
- McMahon, M.; Contreras, A.; Ruggero, D. Small RNAs with big implications: New insights into H/ACA snoRNA function and their role in human disease. Wiley Interdiscip. Rev. RNA 2015, 6, 173–189. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.W.; Green, R. Ribosomopathies: There’s strength in numbers. Science 2017, 358. [Google Scholar] [CrossRef] [Green Version]
- Dalla Venezia, N.; Vincent, A.; Marcel, V.; Catez, F.; Diaz, J.J. Emerging Role of Eukaryote Ribosomes in Translational Control. Int. J. Mol. Sci. 2019, 20, 1226. [Google Scholar] [CrossRef] [Green Version]
- Jack, K.; Bellodi, C.; Landry, D.M.; Niederer, R.O.; Meskauskas, A.; Musalgaonkar, S.; Kopmar, N.; Krasnykh, O.; Dean, A.M.; Thompson, S.R.; et al. rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Mol. Cell 2011, 44, 660–666. [Google Scholar] [CrossRef] [Green Version]
- Marcel, V.; Ghayad, S.E.; Belin, S.; Therizols, G.; Morel, A.P.; Solano-Gonzàlez, E.; Vendrell, J.A.; Hacot, S.; Mertani, H.C.; Albaret, M.A.; et al. p53 acts as a safeguard of translational control by regulating fibrillarin and rRNA methylation in cancer. Cancer Cell 2013, 24, 318–330. [Google Scholar] [CrossRef] [Green Version]
- Krogh, N.; Jansson, M.D.; Häfner, S.J.; Tehler, D.; Birkedal, U.; Christensen-Dalsgaard, M.; Lund, A.H.; Nielsen, H. Profiling of 2′-O-Me in human rRNA reveals a subset of fractionally modified positions and provides evidence for ribosome heterogeneity. Nucleic Acids Res. 2016, 44, 7884–7895. [Google Scholar] [CrossRef] [Green Version]
- Penzo, M.; Montanaro, L. Turning Uridines around: Role of rRNA Pseudouridylation in Ribosome Biogenesis and Ribosomal Function. Biomolecules 2018, 8, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penzo, M.; Montanaro, L.; Treré, D.; Derenzini, M. The Ribosome Biogenesis-Cancer Connection. Cells 2019, 8, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Keersmaecker, K.; Sulima, S.O.; Dinman, J.D. Ribosomopathies and the paradox of cellular hypo- to hyperproliferation. Blood 2015, 125, 1377–1382. [Google Scholar] [CrossRef]
- Sulima, S.O.; Kampen, K.R.; De Keersmaecker, K. Cancer Biogenesis in Ribosomopathies. Cells 2019, 8, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belver, L.; Ferrando, A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2016, 16, 494–507. [Google Scholar] [CrossRef]
- Zhou, X.; Hao, Q.; Zhang, Q.; Liao, J.M.; Ke, J.W.; Liao, P.; Cao, B.; Lu, H. Ribosomal proteins L11 and L5 activate TAp73 by overcoming MDM2 inhibition. Cell Death Differ. 2015, 22, 755–766. [Google Scholar] [CrossRef]
- Kampen, K.R.; Sulima, S.O.; Verbelen, B.; Girardi, T.; Vereecke, S.; Fancello, L.; Rinaldi, G.; Verbeeck, J.; Op de Beeck, J.; Uyttebroeck, A.; et al. Correction: The ribosomal RPL10 R98S mutation drives IRES-dependent BCL-2 translation in T-ALL. Leukemia 2019, 33, 1055–1062. [Google Scholar] [CrossRef] [Green Version]
- Ruggero, D.; Pandolfi, P.P. Does the ribosome translate cancer? Nat. Rev. Cancer 2003, 3, 179–192. [Google Scholar] [CrossRef]
- Sulima, S.O.; Hofman, I.J.F.; De Keersmaecker, K.; Dinman, J.D. How Ribosomes Translate Cancer. Cancer Discov. 2017, 7, 1069–1087. [Google Scholar] [CrossRef] [Green Version]
- Myasnikov, A.G.; Kundhavai Natchiar, S.; Nebout, M.; Hazemann, I.; Imbert, V.; Khatter, H.; Peyron, J.F.; Klaholz, B.P. Structure-function insights reveal the human ribosome as a cancer target for antibiotics. Nat. Commun. 2016, 7, 12856. [Google Scholar] [CrossRef]
- Gani, R. The nucleoli of cultured human lymphocytes. I. Nucleolar morphology in relation to transformation and the DNA cycle. Exp. Cell Res. 1976, 97, 249–258. [Google Scholar] [CrossRef]
- Derenzini, M.; Farabegoli, F.; Trerè, D. Relationship between interphase AgNOR distribution and nucleolar size in cancer cells. Histochem. J. 1992, 24, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Grummt, I. Cell cycle-dependent regulation of RNA polymerase I transcription: The nucleolar transcription factor UBF is inactive in mitosis and early G1. Proc. Natl. Acad. Sci. USA 1999, 96, 6096–6101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diakos, C.I.; Charles, K.A.; McMillan, D.C.; Clarke, S.J. Cancer-related inflammation and treatment effectiveness. Lancet Oncol. 2014, 15, e493–e503. [Google Scholar] [CrossRef]
- Brighenti, E.; Calabrese, C.; Liguori, G.; Giannone, F.A.; Trerè, D.; Montanaro, L.; Derenzini, M. Interleukin 6 downregulates p53 expression and activity by stimulating ribosome biogenesis: A new pathway connecting inflammation to cancer. Oncogene 2014, 33, 4396–4406. [Google Scholar] [CrossRef] [Green Version]
- Gingold, H.; Tehler, D.; Christoffersen, N.R.; Nielsen, M.M.; Asmar, F.; Kooistra, S.M.; Christophersen, N.S.; Christensen, L.L.; Borre, M.; Sørensen, K.D.; et al. A dual program for translation regulation in cellular proliferation and differentiation. Cell 2014, 158, 1281–1292. [Google Scholar] [CrossRef] [Green Version]
- Goodarzi, H.; Nguyen, H.C.B.; Zhang, S.; Dill, B.D.; Molina, H.; Tavazoie, S.F. Modulated Expression of Specific tRNAs Drives Gene Expression and Cancer Progression. Cell 2016, 165, 1416–1427. [Google Scholar] [CrossRef] [Green Version]
- Rapino, F.; Delaunay, S.; Rambow, F.; Zhou, Z.; Tharun, L.; De Tullio, P.; Sin, O.; Shostak, K.; Schmitz, S.; Piepers, J.; et al. Codon-specific translation reprogramming promotes resistance to targeted therapy. Nature 2018, 558, 605–609. [Google Scholar] [CrossRef]
- Ruggero, D. The role of Myc-induced protein synthesis in cancer. Cancer Res. 2009, 69, 8839–8843. [Google Scholar] [CrossRef] [Green Version]
- Van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Lohrum, M.A.; Ludwig, R.L.; Kubbutat, M.H.; Hanlon, M.; Vousden, K.H. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 2003, 3, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lu, H. Signaling to p53: Ribosomal proteins find their way. Cancer Cell 2009, 16, 369–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catez, F.; Dalla Venezia, N.; Marcel, V.; Zorbas, C.; Lafontaine, D.L.J.; Diaz, J.J. Ribosome biogenesis: An emerging druggable pathway for cancer therapeutics. Biochem. Pharmacol. 2019, 159, 74–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, K.; Mühl, B.; Harasim, T.; Rohrmoser, M.; Malamoussi, A.; Orban, M.; Kellner, M.; Gruber-Eber, A.; Kremmer, E.; Hölzel, M.; et al. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J. Biol. Chem. 2010, 285, 12416–12425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, P.M.; Liu, Y.; Park, G.Y.; Murai, J.; Koch, C.E.; Eisen, T.J.; Pritchard, J.R.; Pommier, Y.; Lippard, S.J.; Hemann, M.T. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 2017, 23, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Volarevic, S.; Stewart, M.J.; Ledermann, B.; Zilberman, F.; Terracciano, L.; Montini, E.; Grompe, M.; Kozma, S.C.; Thomas, G. Proliferation, but not growth, blocked by conditional deletion of 40S ribosomal protein S6. Science 2000, 288, 2045–2047. [Google Scholar] [CrossRef]
- Barna, M.; Pusic, A.; Zollo, O.; Costa, M.; Kondrashov, N.; Rego, E.; Rao, P.H.; Ruggero, D. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 2008, 456, 971–975. [Google Scholar] [CrossRef]
- Rodnina, M.V. The ribosome in action: Tuning of translational efficiency and protein folding. Protein Sci. 2016, 25, 1390–1406. [Google Scholar] [CrossRef] [Green Version]
- Wilson, D.N. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 2014, 12, 35–48. [Google Scholar] [CrossRef]
- Park, M.H.; Wolff, E.C. Hypusine, a polyamine-derived amino acid critical for eukaryotic translation. J. Biol. Chem. 2018, 293, 18710–18718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubas, M.; Harder, L.M.; Kumsta, C.; Tiessen, I.; Hansen, M.; Andersen, J.S.; Lund, A.H.; Frankel, L.B. eIF5A is required for autophagy by mediating ATG3 translation. EMBO Rep. 2018, 19, e46072. [Google Scholar] [CrossRef] [PubMed]
- Killock, D. Drug therapy: Can the mitochondrial adverse effects of antibiotics be exploited to target cancer metabolism. Nat. Rev. Clin. Oncol. 2015, 12, 190. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584. [Google Scholar] [CrossRef] [Green Version]
- Cuyàs, E.; Martin-Castillo, B.; Corominas-Faja, B.; Massaguer, A.; Bosch-Barrera, J.; Menendez, J.A. Anti-protozoal and anti-bacterial antibiotics that inhibit protein synthesis kill cancer subtypes enriched for stem cell-like properties. Cell Cycle 2015, 14, 3527–3532. [Google Scholar]
- Skrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Guo, Y. Inhibition of mitochondrial translation as a therapeutic strategy for human ovarian cancer to overcome chemoresistance. Biochem. Biophys. Res. Commun. 2019, 509, 373–378. [Google Scholar] [CrossRef]
- Wang, B.; Ao, J.; Yu, D.; Rao, T.; Ruan, Y.; Yao, X. Inhibition of mitochondrial translation effectively sensitizes renal cell carcinoma to chemotherapy. Biochem. Biophys. Res. Commun. 2017, 490, 767–773. [Google Scholar] [CrossRef]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [Green Version]
- Jhas, B.; Sriskanthadevan, S.; Skrtic, M.; Sukhai, M.A.; Voisin, V.; Jitkova, Y.; Gronda, M.; Hurren, R.; Laister, R.C.; Bader, G.D.; et al. Metabolic adaptation to chronic inhibition of mitochondrial protein synthesis in acute myeloid leukemia cells. PLoS ONE 2013, 8, e58367. [Google Scholar] [CrossRef]
- Gray, M.W. Mosaic nature of the mitochondrial proteome: Implications for the origin and evolution of mitochondria. Proc. Natl. Acad. Sci. USA 2015, 112, 10133–10138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amunts, A.; Brown, A.; Bai, X.C.; Llácer, J.L.; Hussain, T.; Emsley, P.; Long, F.; Murshudov, G.; Scheres, S.H.W.; Ramakrishnan, V. Structure of the yeast mitochondrial large ribosomal subunit. Science 2014, 343, 1485–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greber, B.J.; Bieri, P.; Leibundgut, M.; Leitner, A.; Aebersold, R.; Boehringer, D.; Ban, N. The complete structure of the 55S mammalian mitochondrial ribosome. Science 2015, 348, 303–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrov, A.S.; Wood, E.C.; Bernier, C.R.; Norris, A.M.; Brown, A.; Amunts, A. Structural Patching Fosters Divergence of Mitochondrial Ribosomes. Mol. Biol. Evol. 2019, 36, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Manning, B.D. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 2013, 15, 555–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosilio, C.; Nebout, M.; Imbert, V.; Griessinger, E.; Neffati, Z.; Benadiba, J.; Hagenbeek, T.; Spits, H.; Reverso, J.; Ambrosetti, D.; et al. L-type amino-acid transporter 1 (LAT1): A therapeutic target supporting growth and survival of T-cell lymphoblastic lymphoma/T-cell acute lymphoblastic leukemia. Leukemia 2015, 29, 1253–1266. [Google Scholar] [CrossRef]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6, 282–300. [Google Scholar] [CrossRef] [Green Version]
- Trendowski, M. Recent Advances in the Development of Antineoplastic Agents Derived from Natural Products. Drugs 2015, 75, 1993–2016. [Google Scholar] [CrossRef] [Green Version]
- Powell, R.G.; Weisleder, D.; Smith, C.R.; Rohwedder, W.K. Structures of harringtonine, isoharringtonine, and homoharringtonine. Tetrahedron Lett. 1970, 815–818. [Google Scholar] [CrossRef]
- Ma, G.Z.; Li, P.F.; Liu, L.; Li, W.Z.; Chen, L. Diastereoselective Synthesis of Cephalotaxus Esters via Asymmetric Mukaiyama Aldol Reaction. Org. Lett. 2017, 19, 2250–2253. [Google Scholar] [CrossRef]
- Ju, X.; Beaudry, C.M. Total Synthesis of (-)-Cephalotaxine and (-)-Homoharringtonine via Furan Oxidation-Transannular Mannich Cyclization. Angew. Chem. Int. Ed. Engl. 2019, 58, 6752–6755. [Google Scholar] [CrossRef] [PubMed]
- Tujebajeva, R.M.; Graifer, D.M.; Karpova, G.G.; Ajtkhozhina, N.A. Alkaloid homoharringtonine inhibits polypeptide chain elongation on human ribosomes on the step of peptide bond formation. FEBS Lett. 1989, 257, 254–256. [Google Scholar] [CrossRef] [Green Version]
- Gürel, G.; Blaha, G.; Steitz, T.A.; Moore, P.B. Structures of triacetyloleandomycin and mycalamide A bind to the large ribosomal subunit of Haloarcula marismortui. Antimicrob. Agents Chemother. 2009, 53, 5010–5014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantarjian, H.M.; Talpaz, M.; Santini, V.; Murgo, A.; Cheson, B.; O’Brien, S.M. Homoharringtonine: History, current research, and future direction. Cancer 2001, 92, 1591–1605. [Google Scholar] [CrossRef]
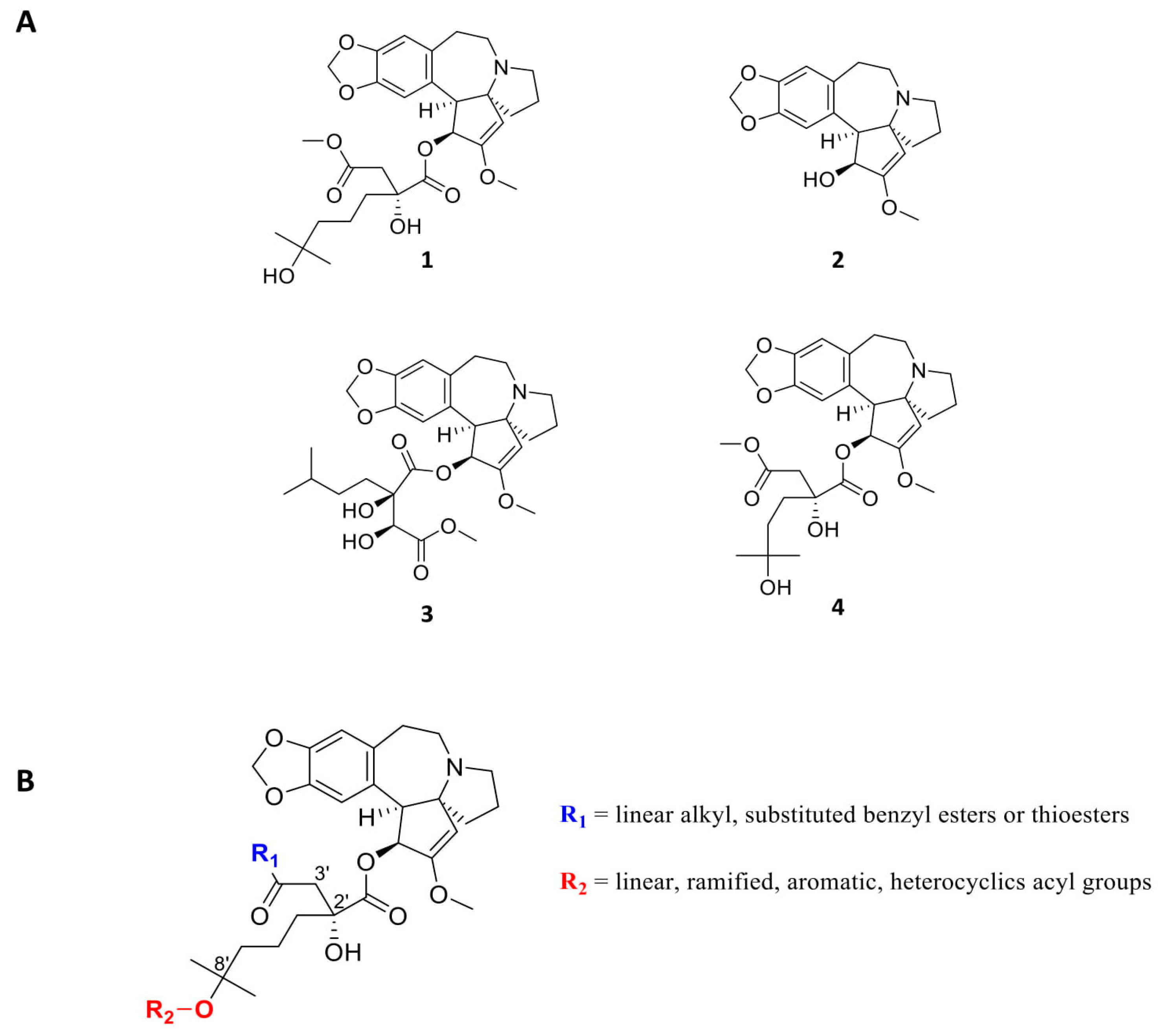
- Chang, Y.; Meng, F.C.; Wang, R.; Wang, C.M.; Lu, X.Y.; Zhang, Q.W. Chemistry, Bioactivity, and the Structure-Activity Relationship of Cephalotaxine-Type Alkaloids from Cephalotaxus sp. Stud. Nat. Prod. Chem. 2017, 53, 339–373. [Google Scholar]
- Robin, J.P.; Dhal, R.; Drouye, F.; Marie, J.P.; Radosevic, N.; Robin, J.; Souchaud, K.; Bataille, P. New Cephalotaxanes, Their Method of Preparation and Their Use in Treatment of Cancers, Leukemias, Parasites Including Thus Resistant to Usual Chemotherapeutic Agents and as Reversal Agents. Patent #WO2002032904, 25 April 2002. [Google Scholar]
- Abdelkafi, H.; Nay, B. Natural products from Cephalotaxus sp.: Chemical diversity and synthetic aspects. Nat. Prod. Rep. 2012, 29, 845–869. [Google Scholar] [CrossRef]
- Chen, R.; Gandhi, V.; Plunkett, W. A sequential blockade strategy for the design of combination therapies to overcome oncogene addiction in chronic myelogenous leukemia. Cancer Res. 2006, 66, 10959–10966. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, V.; Plunkett, W.; Cortes, J.E. Omacetaxine: A protein translation inhibitor for treatment of chronic myelogenous leukemia. Clin. Cancer Res. 2014, 20, 1735–1740. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.T.; Zeng, Q.C.; Zhao, W.H.; Tan, Y. Homoharringtonine contributes to imatinib sensitivity by blocking the EphB4/RhoA pathway in chronic myeloid leukemia cell lines. Med. Oncol. 2014, 31, 836. [Google Scholar] [CrossRef]
- Cortes, J.; Lipton, J.H.; Rea, D.; Digumarti, R.; Chuah, C.; Nanda, N.; Benichou, A.C.; Craig, A.R.; Michallet, M.; Nicolini, F.E.; et al. Phase 2 study of subcutaneous omacetaxine mepesuccinate after TKI failure in patients with chronic-phase CML with T315I mutation. Blood 2012, 120, 2573–2580. [Google Scholar] [CrossRef] [Green Version]
- Muselli, F.; Peyron, J.F.; Mary, D. Druggable Biochemical Pathways and Potential Therapeutic Alternatives to Target Leukemic Stem Cells and Eliminate the Residual Disease in Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2019, 20, 5616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, J.; Kamitsuji, Y.; Kimura, S.; Ashihara, E.; Kawata, E.; Nakagawa, Y.; Takeuichi, M.; Murotani, Y.; Yokota, A.; Tanaka, R.; et al. Anti-myeloma effect of homoharringtonine with concomitant targeting of the myeloma-promoting molecules, Mcl-1, XIAP, and beta-catenin. Int. J. Hematol. 2008, 87, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.Y.; Man, C.H.; Kwong, Y.L. FLT3 inhibition: A moving and evolving target in acute myeloid leukaemia. Leukemia 2013, 27, 260–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, S.S.; Ho, E.S.; He, B.L.; Wong, W.W.; Cher, C.Y.; Ng, N.K.; Man, C.H.; Gill, H.; Cheung, A.M.; Ip, H.W.; et al. Homoharringtonine (omacetaxine mepesuccinate) as an adjunct for FLT3-ITD acute myeloid leukemia. Sci. Transl. Med. 2016, 8, 359. [Google Scholar] [CrossRef]
- Cao, W.; Liu, Y.; Zhang, R.; Zhang, B.; Wang, T.; Zhu, X.; Mei, L.; Chen, H.; Zhang, H.; Ming, P.; et al. Homoharringtonine induces apoptosis and inhibits STAT3 via IL-6/JAK1/STAT3 signal pathway in Gefitinib-resistant lung cancer cells. Sci. Rep. 2015, 5, 8477. [Google Scholar] [CrossRef]
- Yakhni, M.; Briat, A.; El Guerrab, A.; Furtado, L.; Kwiatkowski, F.; Miot-Noirault, E.; Cachin, F.; Penault-Llorca, F.; Radosevic-Robin, N. Homoharringtonine, an approved anti-leukemia drug, suppresses triple negative breast cancer growth through a rapid reduction of anti-apoptotic protein abundance. Am. J. Cancer Res. 2019, 9, 1043–1060. [Google Scholar]
- Paterson, E.K.; Courtneidge, S.A. Invadosomes are coming: New insights into function and disease relevance. FEBS J. 2018, 285, 8–27. [Google Scholar] [CrossRef]
- Ezzoukhry, Z.; Henriet, E.; Cordelières, F.P.; Dupuy, J.W.; Maître, M.; Gay, N.; Di-Tommaso, S.; Mercier, L.; Goetz, J.G.; Peter, M.; et al. Combining laser capture microdissection and proteomics reveals an active translation machinery controlling invadosome formation. Nat. Commun. 2018, 9, 2031. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Kingwell, K. Translational target for checkpoint inhibitors. Nat. Rev. Immunol. 2018, 18, 728–729. [Google Scholar] [CrossRef]
- Cerezo, M.; Guemiri, R.; Druillennec, S.; Girault, I.; Malka-Mahieu, H.; Shen, S.; Allard, D.; Martineau, S.; Welsch, C.; Agoussi, S.; et al. Translational control of tumor immune escape via the eIF4F-STAT1-PD-L1 axis in melanoma. Nat. Med. 2018, 24, 1877–1886. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Poggio, M.; Jin, H.Y.; Shi, Z.; Forester, C.M.; Wang, Y.; Stumpf, C.R.; Xue, L.; Devericks, E.; So, L.; et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat. Med. 2019, 25, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Weng, T.Y.; Wu, H.F.; Li, C.Y.; Hung, Y.H.; Chang, Y.W.; Chen, Y.L.; Hsu, H.P.; Chen, Y.H.; Wang, C.Y.; Chang, J.Y.; et al. Homoharringtonine induced immune alteration for an Efficient Anti-tumor Response in Mouse Models of Non-small Cell Lung Adenocarcinoma Expressing Kras Mutation. Sci. Rep. 2018, 8, 8216. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.H.; Jiang, H.; Jiang, Q.; Jia, J.S.; Qin, Y.Z.; Huang, X.J. Homoharringtonine, aclarubicin and cytarabine (HAA) regimen as the first course of induction therapy is highly effective for acute myeloid leukemia with t (8;21). Leuk. Res. 2016, 44, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jiang, M.; Borthakur, G.; Luan, S.; Huang, X.; Tang, G.; Xu, Q.; Ji, D.; Boyer, A.D.; Li, F.; et al. Acute myeloid leukemia with a novel CPSF6-RARG variant is sensitive to homoharringtonine and cytarabine chemotherapy. Am. J. Hematol. 2019, in press. [Google Scholar] [CrossRef]
- Geng, S.; Yao, H.; Weng, J.; Tong, J.; Huang, X.; Wu, P.; Deng, C.; Li, M.; Lu, Z.; Du, X. Effects of the combination of decitabine and homoharringtonine in SKM-1 and Kg-1a cells. Leuk. Res. 2016, 44, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Pan, J.; Jin, J.; Li, C.; Li, X.; Huang, J.; Huang, X.; Yan, X.; Li, F.; Yu, M.; et al. Abivertinib, a novel BTK inhibitor: Anti-Leukemia effects and synergistic efficacy with homoharringtonine in acute myeloid leukemia. Cancer Lett. 2019, 461, 132–143. [Google Scholar] [CrossRef]
- Wu, Z.; Zhuang, H.; Yu, Q.; Zhang, X.; Jiang, X.; Lu, X.; Xu, Y.; Yang, L.; Wu, B.; Ma, A.; et al. Homoharringtonine Combined with the Heat Shock Protein 90 Inhibitor IPI504 in the Treatment of FLT3-ITD Acute Myeloid Leukemia. Transl. Oncol. 2019, 12, 801–809. [Google Scholar] [CrossRef]
- Zhang, J.; Geng, H.; Liu, L.; Zhang, H. Synergistic cytotoxicity of homoharringtonine and etoposide in acute myeloid leukemia cells involves disrupted antioxidant defense. Cancer Manag. Res. 2019, 11, 1023–1032. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Lu, Y.; Zhen, T.; Chen, X.; Zhang, M.; Liu, P.; Weng, X.; Chen, B.; Wang, Y. Homoharringtonine synergy with oridonin in treatment of t(8; 21) acute myeloid leukemia. Front. Med. 2019, 13, 388–397. [Google Scholar] [CrossRef]
- Cao, H.; Cheng, Y.; You, L.; Qian, J.; Qian, W. Homoharringtonine and SAHA synergistically enhance apoptosis in human acute myeloid leukemia cells through upregulation of TRAIL and death receptors. Mol. Med. Rep. 2013, 7, 1838–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.; Zhang, Q.; Yuan, X.; Chen, Y.; Wu, Y. Synergistic killing effects of homoharringtonine and arsenic trioxide on acute myeloid leukemia stem cells and the underlying mechanisms. J. Exp. Clin. Cancer Res. 2019, 38, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Parker, R.; Zhang, Y.; Hawkins, E.; Kmieciak, M.; Craun, W.; Grant, S. Homoharringtonine interacts synergistically with bortezomib in NHL cells through MCL-1 and NOXA-dependent mechanisms. BMC Cancer 2018, 18, 1129. [Google Scholar] [CrossRef] [PubMed]
- Klanova, M.; Andera, L.; Brazina, J.; Svadlenka, J.; Benesova, S.; Soukup, J.; Prukova, D.; Vejmelkova, D.; Jaksa, R.; Helman, K.; et al. Targeting of BCL2 Family Proteins with ABT-199 and Homoharringtonine Reveals BCL2- and MCL1-Dependent Subgroups of Diffuse Large B-Cell Lymphoma. Clin. Cancer Res. 2016, 22, 1138–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.T.; Dargeon, H.W.; Burchenal, J.H. The effect of actinomycin D on cancer in childhood. Pediatrics 1959, 24, 544–561. [Google Scholar] [PubMed]
- Hollstein, U. Actinomycin. Chemistry and mechanism of action. Chem. Rev. 1974, 625–652. [Google Scholar] [CrossRef]
- Kühlbrandt, W. Cryo-EM enters a new era. Elife 2014, 3, e03678. [Google Scholar]
- Shalev-Benami, M.; Zhang, Y.; Rozenberg, H.; Nobe, Y.; Taoka, M.; Matzov, D.; Zimmerman, E.; Bashan, A.; Isobe, T.; Jaffe, C.L.; et al. Atomic resolution snapshot of Leishmania ribosome inhibition by the aminoglycoside paromomycin. Nat. Commun. 2017, 8, 1589. [Google Scholar] [CrossRef] [Green Version]
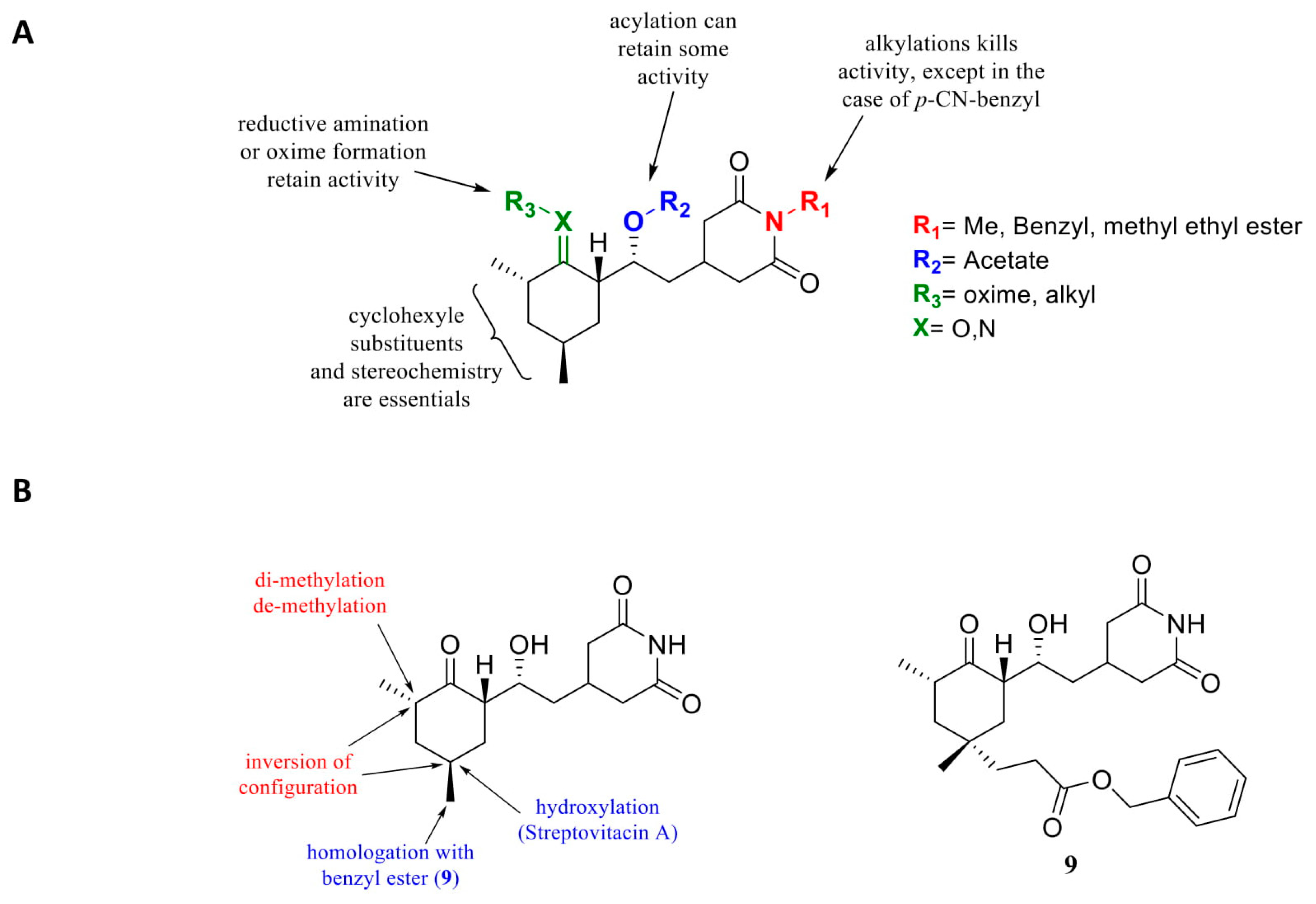
- Schneider-Poetsch, T.; Ju, J.; Eyler, D.E.; Dang, Y.; Bhat, S.; Merrick, W.C.; Green, R.; Shen, B.; Liu, J.O. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat. Chem. Biol. 2010, 6, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Piatak, D.M.; Tang, P.F.; Yen, C.C. Cycloheximide analogues as potential anticonvulsants. J. Med. Chem. 1986, 29, 50–54. [Google Scholar] [CrossRef]
- Godchaux, W.; Adamson, S.D.; Herbert, E. Effects of cycloheximide on polyribosome function in reticulocytes. J. Mol. Biol. 1967, 27, 57–72. [Google Scholar] [CrossRef]
- McKeehan, W.; Hardesty, B. The mechanism of cycloheximide inhibition of protein synthesis in rabbit reticulocytes. Biochem. Biophys. Res. Commun. 1969, 36, 625–630. [Google Scholar] [CrossRef]
- Ennis, H.L.; Lubin, M. Cyclohehimide: Aspects of inhibition of protein synthesis in mammalian cells. Science 1964, 146, 1474–1476. [Google Scholar] [CrossRef] [PubMed]
- Piatak, M.; Yen, C.C.; Kennedy Jr, R.V. Pyrazole and pyrimidine derivatives of dehydrocycloheximide analogs. J. Med. Chem. 1970, 13, 770. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Oritani, T.; Yamashita, K. Synthesis and biological activities of demethylcycloheximides. Agric. Biol. Chem. 1988, 129–133. [Google Scholar]
- Paoletti, F.; Ainger, K.; Donati, I.; Scardigli, R.; Vetere, A.; Cattaneo, A.; Campa, C. Novel fluorescent cycloheximide derivatives for the imaging of protein synthesis. Biochem. Biophys. Res. Commun. 2010, 396, 258–264. [Google Scholar] [CrossRef]
- Christner, C.; Wyrwa, R.; Marsch, S.; Küllertz, G.; Thiericke, R.; Grabley, S.; Schumann, D.; Fischer, G. Synthesis and cytotoxic evaluation of cycloheximide derivatives as potential inhibitors of FKBP12 with neuroregenerative properties. J. Med. Chem. 1999, 42, 3615–3622. [Google Scholar] [CrossRef]
- Ji, X.Y.; Zhong, Z.J.; Xue, S.T.; Meng, S.; He, W.Y.; Gao, R.M.; Li, Y.H.; Li, Z.R. Synthesis and antiviral activities of synthetic glutarimide derivatives. Chem. Pharm. Bull. (Tokyo) 2010, 58, 1436–1441. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Koga, Y.; Su, C.; Waterbury, A.L.; Johnny, C.L.; Liau, B.B. Versatile Synthetic Route to Cycloheximide and Analogues That Potently Inhibit Translation Elongation. Angew. Chem. Int. 2019, 58, 5387–5391. [Google Scholar] [CrossRef]
- Shi, Z.; Fujii, K.; Kovary, K.M.; Genuth, N.R.; Röst, H.L.; Teruel, M.N.; Barna, M. Heterogeneous Ribosomes Preferentially Translate Distinct Subpools of mRNAs Genome-wide. Mol. Cell 2017, 67, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.; Warner, J.R. Ribosome-omics of the human ribosome. RNA 2014, 20, 1004–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauro, V.P.; Edelman, G.M. The ribosome filter hypothesis. Proc. Natl. Acad. Sci. USA 2002, 99, 12031–12036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guimaraes, J.C.; Zavolan, M. Patterns of ribosomal protein expression specify normal and malignant human cells. Genome Biol. 2016, 17, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Drug | Target | Reference |
|---|---|---|---|
| AML | AraC | anti-metabolite | [113] |
| AML | aclarubicin | anthracyclin | [107] |
| AML | decitabine | methylation inhibitor | [109] |
| AML | abivertinib | BTK inhibitor | [110] |
| AML | IPI504 | HSP90 inhibitor | [111] |
| AML | etoposide | DNA topoisomerase II | [113] |
| t(8, 21)AML | oridonin | cKIT, MDR1, MRP-1 | [113] |
| AML | SAHA | HDAC inhibitor | [114] |
| AML | AsO3 | apoptosis induction | [115] |
| DLBCL | bortezomib | proteasome inhibitor | [116] |
| BCL-2+ DLBCL | ABT-199 | BCL-2 | [117] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gilles, A.; Frechin, L.; Natchiar, K.; Biondani, G.; Loeffelholz, O.v.; Holvec, S.; Malaval, J.-L.; Winum, J.-Y.; Klaholz, B.P.; Peyron, J.-F. Targeting the Human 80S Ribosome in Cancer: From Structure to Function and Drug Design for Innovative Adjuvant Therapeutic Strategies. Cells 2020, 9, 629. https://doi.org/10.3390/cells9030629
Gilles A, Frechin L, Natchiar K, Biondani G, Loeffelholz Ov, Holvec S, Malaval J-L, Winum J-Y, Klaholz BP, Peyron J-F. Targeting the Human 80S Ribosome in Cancer: From Structure to Function and Drug Design for Innovative Adjuvant Therapeutic Strategies. Cells. 2020; 9(3):629. https://doi.org/10.3390/cells9030629
Chicago/Turabian StyleGilles, Arnaud, Léo Frechin, Kundhavai Natchiar, Giulia Biondani, Ottilie von Loeffelholz, Samuel Holvec, Julie-Lisa Malaval, Jean-Yves Winum, Bruno P. Klaholz, and Jean-François Peyron. 2020. "Targeting the Human 80S Ribosome in Cancer: From Structure to Function and Drug Design for Innovative Adjuvant Therapeutic Strategies" Cells 9, no. 3: 629. https://doi.org/10.3390/cells9030629