Angiotensin-II-Evoked Ca2+ Entry in Murine Cardiac Fibroblasts Does Not Depend on TRPC Channels

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Isolation and Primary Culture of Cardiac Fibroblasts (CFs)
2.3. RNA Isolation and qPCR Analysis
2.4. Immune Fluorescence Staining
2.5. Calcium Imaging
2.6. Agonist and Blockers
2.7. Statistical Analysis
3. Results
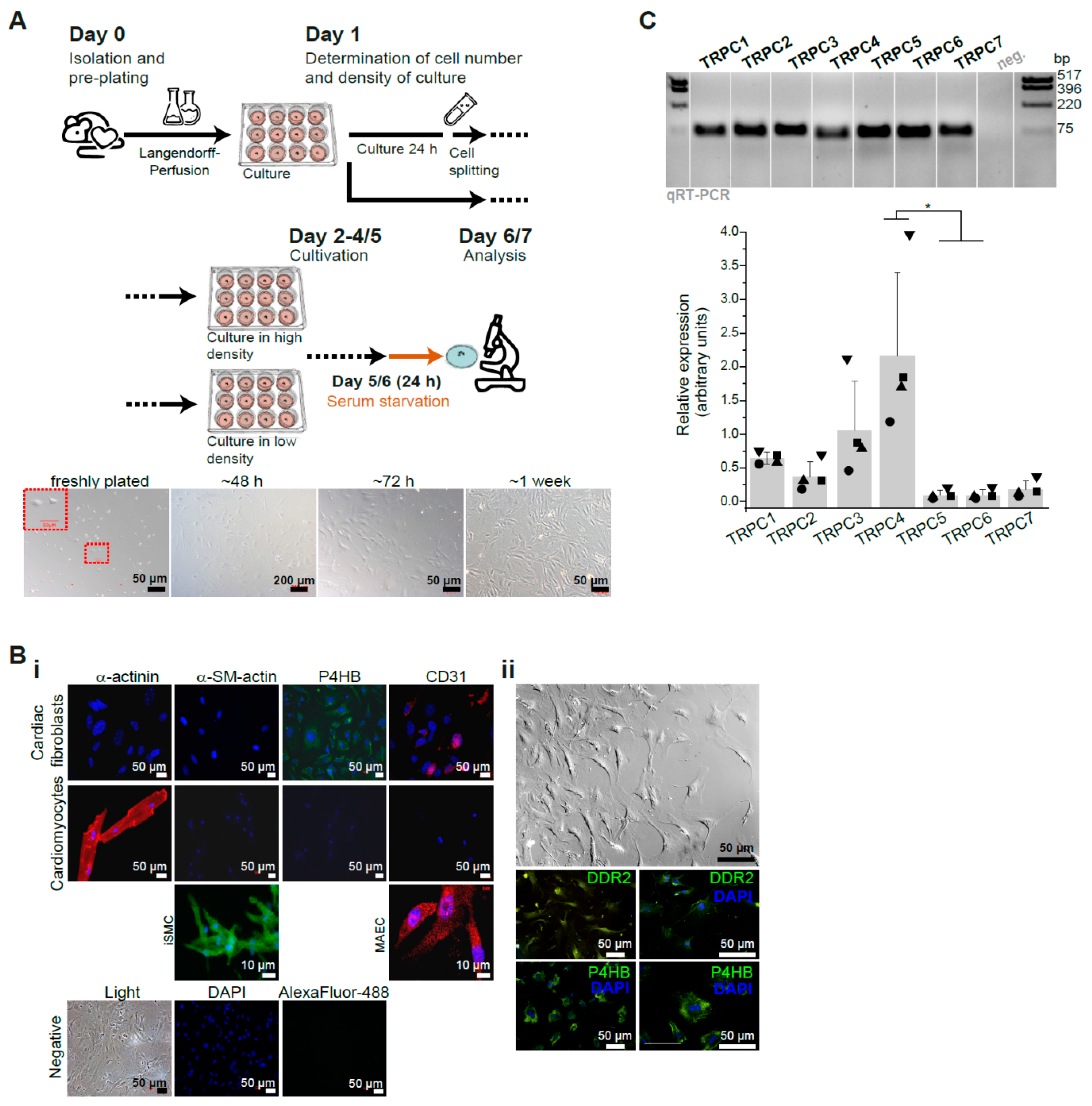
3.1. Characterization of Primary Isolated Murine Cardiac Fibroblasts (CFs)
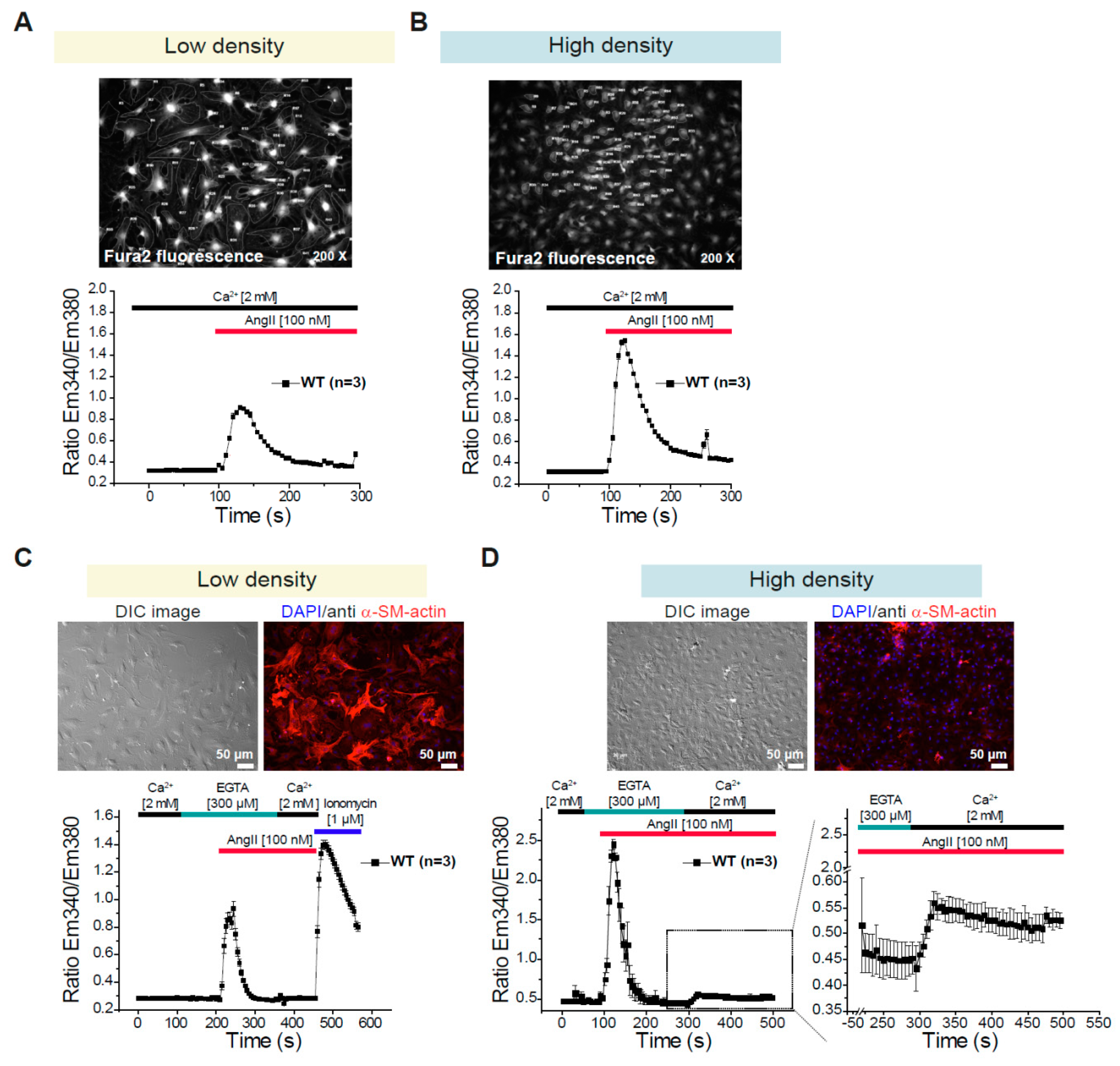
3.2. Differential Changes in Intracellular Ca2+ Concentration in CFs Depending on the Density of the Culture
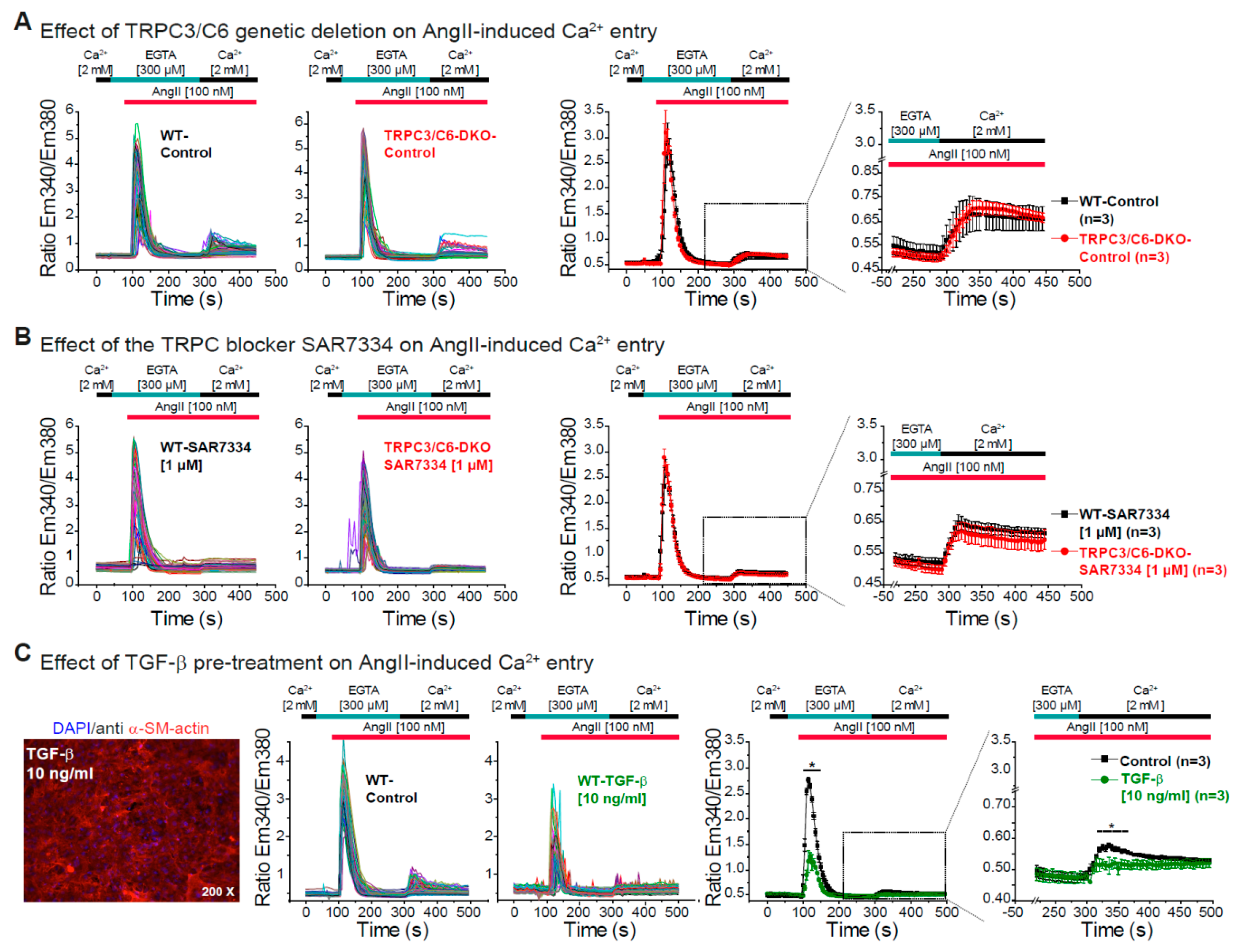
3.3. Effect of TRPC3/C6 Genetic Deletion or Its Pharmacological Inhibition with SAR7334 on the AngII-Induced Ca2+ Release and Ca2+ Entry
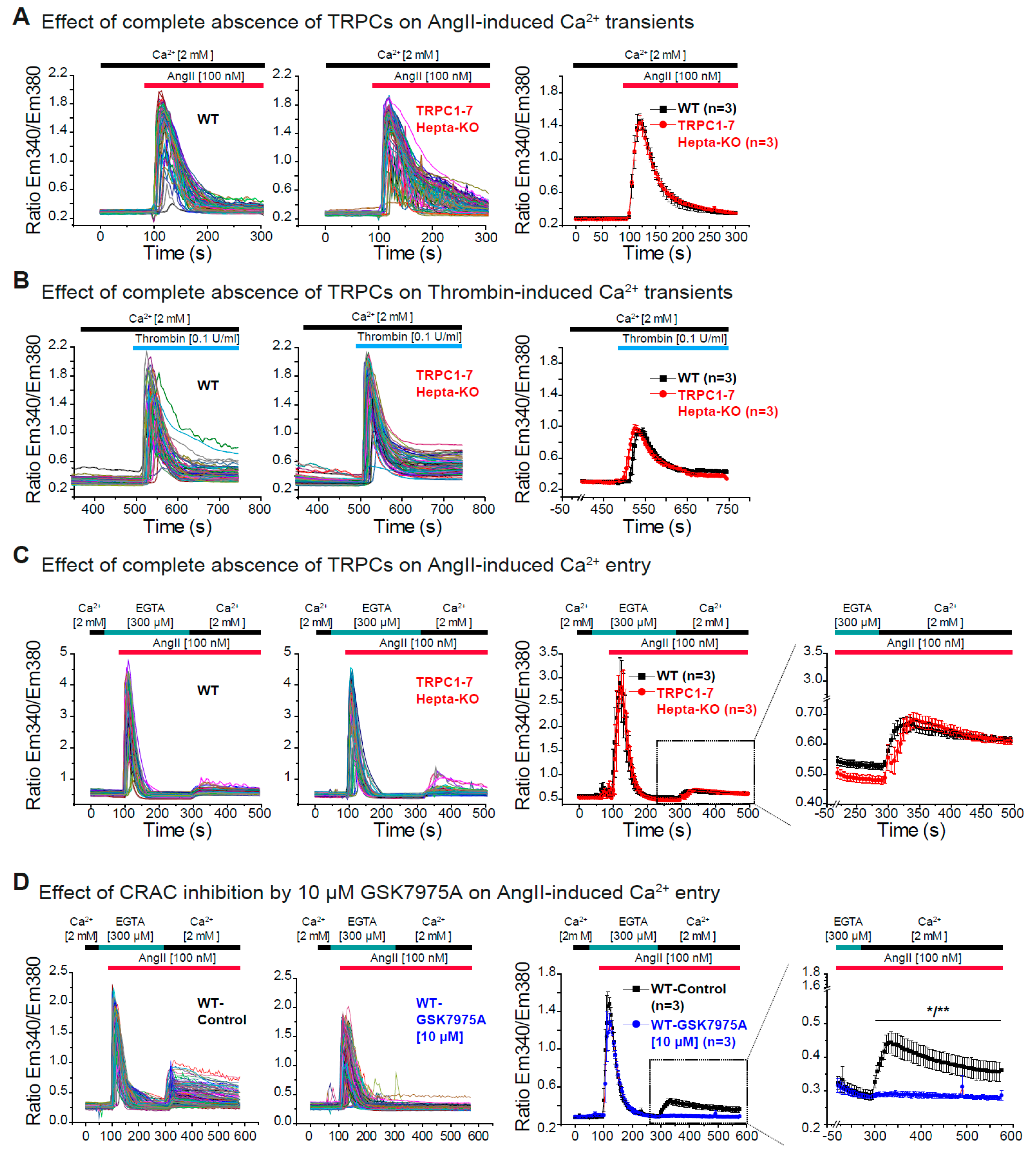
3.4. Dispensability of All TRPC Proteins for the Acute Ca2+ Release and Ca2+ Entry Induced by AngII in Cardiac Fibroblasts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humeres, C.; Frangogiannis, N.G. Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl. Sci. 2019, 4, 449–467. [Google Scholar] [CrossRef]
- Brown, R.D.; Ambler, S.K.; Mitchell, M.D.; Long, C.S. The cardiac fibroblast: Therapeutic target in myocardial remodeling and failure. Annu. Rev. Pharm. Toxicol. 2005, 45, 657–687. [Google Scholar] [CrossRef]
- Chacar, S.; Fares, N.; Bois, P.; Faivre, J.F. Basic Signaling in Cardiac Fibroblasts. J. Cell. Physiol. 2017, 232, 725–730. [Google Scholar] [CrossRef]
- Lajiness, J.D.; Conway, S.J. Origin, development, and differentiation of cardiac fibroblasts. J. Mol. Cell. Cardiol. 2014, 70, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Ivey, M.J.; Tallquist, M.D. Defining the Cardiac Fibroblast. Circ. J. 2016, 80, 2269–2276. [Google Scholar] [CrossRef] [Green Version]
- Stempien-Otero, A.; Kim, D.H.; Davis, J. Molecular networks underlying myofibroblast fate and fibrosis. J. Mol. Cell. Cardiol. 2016, 97, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Kuwabara, J.T.; Tallquist, M.D. Tracking Adventitial Fibroblast Contribution to Disease: A Review of Current Methods to Identify Resident Fibroblasts. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1598–1607. [Google Scholar] [CrossRef] [Green Version]
- Tallquist, M.D.; Molkentin, J.D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 2017, 14, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gourdie, R.G.; Dimmeler, S.; Kohl, P. Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nat. Rev. Drug Discov. 2016, 15, 620–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, Z.; Zhang, Y.; Xie, J.; Jiang, J.; Yue, L. Transient receptor potential (TRP) channels and cardiac fibrosis. Curr. Top. Med. Chem. 2013, 13, 270–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nattel, S. Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation. JACC Clin. Electrophysiol. 2017, 3, 425–435. [Google Scholar] [CrossRef]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [Green Version]
- Trebak, M.; Lemonnier, L.; Smyth, J.T.; Vazquez, G.; Putney, J.W., Jr. Phospholipase C-coupled receptors and activation of TRPC channels. Handb. Exp. Pharm. 2007, 179, 593–614. [Google Scholar] [CrossRef]
- Abramowitz, J.; Birnbaumer, L. Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J. 2009, 23, 297–328. [Google Scholar] [CrossRef] [Green Version]
- Parekh, A.B.; Penner, R. Store depletion and calcium influx. Physiol. Rev. 1997, 77, 901–930. [Google Scholar] [CrossRef]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.-H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Vig, M.; DeHaven, W.I.; Bird, G.S.; Billingsley, J.M.; Wang, H.; Rao, P.E.; Hutchings, A.B.; Jouvin, M.H.; Putney, J.W.; Kinet, J.P. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat. Immunol. 2008, 9, 89–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.L.; Yeromin, A.V.; Zhang, X.H.F.; Yu, Y.; Safrina, O.; Penna, A.; Roos, J.; Stauderman, K.A.; Cahalan, M.D. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc. Natl. Acad. Sci. USA 2006, 103, 9357–9362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, S.A.; Wissenbach, U.; Philipp, S.E.; Freichel, M.; Cavalie, A.; Flockerzi, V. Murine ORAI2 splice variants form functional Ca2+ release-activated Ca2+ (CRAC) channels. J. Biol. Chem. 2007, 282, 19375–19384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal structure of the calcium release-activated calcium channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaeth, M.; Yang, J.; Yamashita, M.; Zee, I.; Eckstein, M.; Knosp, C.; Kaufmann, U.; Karoly Jani, P.; Lacruz, R.S.; Flockerzi, V.; et al. ORAI2 modulates store-operated calcium entry and T cell-mediated immunity. Nat. Commun. 2017, 8, 14714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [Green Version]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Stathopulos, P.B.; Li, G.-Y.; Plevin, M.J.; Ames, J.B.; Ikura, M. Stored Ca2+ Depletion-induced Oligomerization of Stromal Interaction Molecule 1 (STIM1) via the EF-SAM Region: AN INITIATION MECHANISM FOR CAPACITIVE Ca2+ ENTRY. J. Biol. Chem. 2006, 281, 35855–35862. [Google Scholar] [CrossRef] [Green Version]
- Campbell, S.E.; Katwa, L.C. Angiotensin II stimulated expression of transforming growth factor-beta1 in cardiac fibroblasts and myofibroblasts. J. Mol. Cell. Cardiol. 1997, 29, 1947–1958. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.O.; Long, C.S.; Kalinyak, J.E.; Li, H.T.; Karliner, J.S. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta 1 and endothelin-1 from fibroblasts. Cardiovasc. Res. 1998, 40, 352–363. [Google Scholar] [CrossRef]
- Gao, X.; He, X.; Luo, B.; Peng, L.; Lin, J.; Zuo, Z. Angiotensin II increases collagen I expression via transforming growth factor-beta1 and extracellular signal-regulated kinase in cardiac fibroblasts. Eur. J. Pharm. 2009, 606, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.E.L.; Witt, S.A.; Glascock, B.J.; Nieman, M.L.; Reiser, P.J.; Nix, S.L.; Kimball, T.R.; Doetschman, T. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J. Clin. Investig. 2002, 109, 787–796. [Google Scholar] [CrossRef]
- Leask, A.; Abraham, D.J. TGF-beta signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Bujak, M.; Frangogiannis, N.G. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc. Res. 2007, 74, 184–195. [Google Scholar] [CrossRef] [Green Version]
- Snead, A.N.; Insel, P.A. Defining the cellular repertoire of GPCRs identifies a profibrotic role for the most highly expressed receptor, protease-activated receptor 1, in cardiac fibroblasts. FASEB J. 2012, 26, 4540–4547. [Google Scholar] [CrossRef] [Green Version]
- Kleeschulte, S.; Jerrentrup, J.; Gorski, D.; Schmitt, J.; Fender, A.C. Evidence for functional PAR-4 thrombin receptor expression in cardiac fibroblasts and its regulation by high glucose: PAR-4 in cardiac fibroblasts. Int. J. Cardiol. 2018, 252, 163–166. [Google Scholar] [CrossRef]
- Geisler, T. PAR-4—The PARagon of protease-activated receptors? Int. J. Cardiol. 2018, 252, 167–168. [Google Scholar] [CrossRef]
- Ide, J.; Aoki, T.; Ishivata, S.; Glusa, E.; Strukova, S.M. Proteinase-activated receptor agonists stimulate the increase in intracellular Ca2+ in cardiomyocytes and proliferation of cardiac fibroblasts from chick embryos. Bull. Exp. Biol. Med. 2007, 144, 760–763. [Google Scholar] [CrossRef]
- Tiruppathi, C.; Ahmmed, G.U.; Vogel, S.M.; Malik, A.B. Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation 2006, 13, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Montell, C. The TRP superfamily of cation channels. Sci. Signal. 2005, 2005, re3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, S.F.; Owsianik, G.; Nilius, B. TRP channels: An overview. Cell Calcium 2005, 38, 233–252. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Szallasi, A. Transient receptor potential channels as drug targets: From the science of basic research to the art of medicine. Pharm. Rev. 2014, 66, 676–814. [Google Scholar] [CrossRef]
- Broker-Lai, J.; Kollewe, A.; Schindeldecker, B.; Pohle, J.; Chi, V.N.; Mathar, I.; Guzman, R.; Schwarz, Y.; Lai, A.; Weissgerber, P.; et al. Heteromeric channels formed by TRPC1, TRPC4 and TRPC5 define hippocampal synaptic transmission and working memory. EMBO J. 2017, 36, 2770–2789. [Google Scholar] [CrossRef]
- Freichel, M.; Berlin, M.; Schurger, A.; Mathar, I.; Bacmeister, L.; Medert, R.; Frede, W.; Marx, A.; Segin, S.; Londono, J.E.C. TRP Channels in the Heart. In Neurobiology of TRP Channels, 2nd ed.; Emir, T.L.R., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2017; pp. 149–185. [Google Scholar]
- Numaga-Tomita, T.; Oda, S.; Shimauchi, T.; Nishimura, A.; Mangmool, S.; Nishida, M. TRPC3 Channels in Cardiac Fibrosis. Front. Cardiovasc. Med. 2017, 4, 56. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Li, J. Canonical transient receptor potential 3 channels in atrial fibrillation. Eur. J. Pharm. 2018, 837, 1–7. [Google Scholar] [CrossRef]
- Zhan, L.; Li, J. The role of TRPV4 in fibrosis. Gene 2018, 642, 1–8. [Google Scholar] [CrossRef]
- Falcon, D.; Galeano-Otero, I.; Calderon-Sanchez, E.; Del Toro, R.; Martin-Bornez, M.; Rosado, J.A.; Hmadcha, A.; Smani, T. TRP Channels: Current Perspectives in the Adverse Cardiac Remodeling. Front. Physiol. 2019, 10, 159. [Google Scholar] [CrossRef]
- Feng, J.; Armillei, M.K.; Yu, A.S.; Liang, B.T.; Runnels, L.W.; Yue, L. Ca (2+) Signaling in Cardiac Fibroblasts and Fibrosis-Associated Heart Diseases. J. Cardiovasc. Dev. Dis. 2019, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Hof, T.; Chaigne, S.; Recalde, A.; Salle, L.; Brette, F.; Guinamard, R. Transient receptor potential channels in cardiac health and disease. Nat. Rev. Cardiol. 2019, 16, 344–360. [Google Scholar] [CrossRef] [PubMed]
- Rose, R.A.; Hatano, N.; Ohya, S.; Imaizumi, Y.; Giles, W.R. C-type natriuretic peptide activates a non-selective cation current in acutely isolated rat cardiac fibroblasts via natriuretic peptide C receptor-mediated signalling. J. Physiol. 2007, 580, 255–274. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Luo, X.; Qi, X.Y.; Tadevosyan, A.; Maguy, A.; Ordog, B.; Ledoux, J.; Kato, T.; Naud, P.; Voigt, N.; et al. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation 2012, 126, 2051–2064. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, N.; Numaga-Tomita, T.; Watanabe, M.; Kuroda, T.; Nishimura, A.; Miyano, K.; Yasuda, S.; Kuwahara, K.; Sato, Y.; Ide, T.; et al. TRPC3 positively regulates reactive oxygen species driving maladaptive cardiac remodeling. Sci. Rep. 2016, 6, 37001. [Google Scholar] [CrossRef] [Green Version]
- Saliba, Y.; Jebara, V.; Hajal, J.; Maroun, R.; Chacar, S.; Smayra, V.; Abramowitz, J.; Birnbaumer, L.; Fares, N. Transient Receptor Potential Canonical 3 and Nuclear Factor of Activated T Cells C3 Signaling Pathway Critically Regulates Myocardial Fibrosis. Antioxid. Redox Signal. 2019, 30, 1851–1879. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; Onohara, N.; Sato, Y.; Suda, R.; Ogushi, M.; Tanabe, S.; Inoue, R.; Mori, Y.; Kurose, H. Galpha12/13-mediated up-regulation of TRPC6 negatively regulates endothelin-1-induced cardiac myofibroblast formation and collagen synthesis through nuclear factor of activated T cells activation. J. Biol. Chem. 2007, 282, 23117–23128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.; Burr, A.R.; Davis, G.F.; Birnbaumer, L.; Molkentin, J.D. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev. Cell 2012, 23, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Kapur, N.K.; Qiao, X.; Paruchuri, V.; Mackey, E.E.; Daly, G.H.; Ughreja, K.; Morine, K.J.; Levine, J.; Aronovitz, M.J.; Hill, N.S.; et al. Reducing endoglin activity limits calcineurin and TRPC-6 expression and improves survival in a mouse model of right ventricular pressure overload. JAHA 2014, 3. [Google Scholar] [CrossRef] [Green Version]
- Harikrishnan, V.; Titus, A.S.; Cowling, R.T.; Kailasam, S. Collagen receptor cross-talk determines alpha-smooth muscle actin-dependent collagen gene expression in angiotensin II-stimulated cardiac fibroblasts. J. Biol. Chem. 2019, 294, 19723–19739. [Google Scholar] [CrossRef]
- Ikeda, K.; Nakajima, T.; Yamamoto, Y.; Takano, N.; Tanaka, T.; Kikuchi, H.; Oguri, G.; Morita, T.; Nakamura, F.; Komuro, I. Roles of transient receptor potential canonical (TRPC) channels and reverse-mode Na+/Ca2+ exchanger on cell proliferation in human cardiac fibroblasts: Effects of transforming growth factor beta1. Cell Calcium 2013, 54, 213–225. [Google Scholar] [CrossRef]
- Lin, B.L.; Matera, D.; Doerner, J.F.; Zheng, N.; Del Camino, D.; Mishra, S.; Bian, H.; Zeveleva, S.; Zhen, X.; Blair, N.T.; et al. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc. Natl. Acad. Sci. USA 2019, 116, 10156–10161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho Londono, J.E.; Tian, Q.; Hammer, K.; Schroder, L.; Camacho Londono, J.; Reil, J.C.; He, T.; Oberhofer, M.; Mannebach, S.; Mathar, I.; et al. A background Ca2+ entry pathway mediated by TRPC1/TRPC4 is critical for development of pathological cardiac remodelling. Eur. Heart J. 2015, 36, 2257–2266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, A.; Kalwa, H.; Storch, U.; y Schnitzler, M.M.; Salanova, B.; Pinkenburg, O.; Dubrovska, G.; Essin, K.; Gollasch, M.; Birnbaumer, L.; et al. Pressure-induced and store-operated cation influx in vascular smooth muscle cells is independent of TRPC1. Pflug. Arch. 2007, 455, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Stowers, L.; Holy, T.E.; Meister, M.; Dulac, C.; Koentges, G. Loss of sex discrimination and male-male aggression in mice deficient for TRP2. Science 2002, 295, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.; Dragicevic, E.; Adelsberger, H.; Henning, H.A.; Sumser, M.; Abramowitz, J.; Blum, R.; Dietrich, A.; Freichel, M.; Flockerzi, V.; et al. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron 2008, 59, 392–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freichel, M.; Suh, S.H.; Pfeifer, A.; Schweig, U.; Trost, C.; Weissgerber, P.; Biel, M.; Philipp, S.; Freise, D.; Droogmans, G.; et al. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4-/- mice. Nat. Cell Biol. 2001, 3, 121–127. [Google Scholar] [CrossRef]
- Xue, T.; Do, M.T.; Riccio, A.; Jiang, Z.; Hsieh, J.; Wang, H.C.; Merbs, S.L.; Welsbie, D.S.; Yoshioka, T.; Weissgerber, P.; et al. Melanopsin signalling in mammalian iris and retina. Nature 2011, 479, 67–73. [Google Scholar] [CrossRef]
- Dietrich, A.; Mederos, Y.S.M.; Gollasch, M.; Gross, V.; Storch, U.; Dubrovska, G.; Obst, M.; Yildirim, E.; Salanova, B.; Kalwa, H.; et al. Increased vascular smooth muscle contractility in TRPC6-/- mice. Mol. Cell. Biol. 2005, 25, 6980–6989. [Google Scholar] [CrossRef] [Green Version]
- Perez-Leighton, C.E.; Schmidt, T.M.; Abramowitz, J.; Birnbaumer, L.; Kofuji, P. Intrinsic phototransduction persists in melanopsin-expressing ganglion cells lacking diacylglycerol-sensitive TRPC subunits. Eur. J. Neurosci. 2011, 33, 856–867. [Google Scholar] [CrossRef] [Green Version]
- Birnbaumer, L. From GTP and G proteins to TRPC channels: A personal account. J. Mol. Med. 2015, 93, 941–953. [Google Scholar] [CrossRef]
- Meissner, M.; Weissgerber, P.; Londono, J.E.; Prenen, J.; Link, S.; Ruppenthal, S.; Molkentin, J.D.; Lipp, P.; Nilius, B.; Freichel, M.; et al. Moderate calcium channel dysfunction in adult mice with inducible cardiomyocyte-specific excision of the cacnb2 gene. J. Biol. Chem. 2011, 286, 15875–15882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landeen, L.K.; Aroonsakool, N.; Haga, J.H.; Hu, B.S.; Giles, W.R. Sphingosine-1-phosphate receptor expression in cardiac fibroblasts is modulated by in vitro culture conditions. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2698–H2711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.; Follmann, M.; Hessler, G.; Kleemann, H.W.; Hachtel, S.; Fuchs, B.; Weissmann, N.; Linz, W.; Schmidt, T.; Lohn, M.; et al. Discovery and pharmacological characterization of a novel potent inhibitor of diacylglycerol-sensitive TRPC cation channels. Br. J. Pharm. 2015, 172, 3650–3660. [Google Scholar] [CrossRef]
- Zhu, Y.; Lu, Y.; Qu, C.; Miller, M.; Tian, J.; Thakur, D.P.; Zhu, J.; Deng, Z.; Hu, X.; Wu, M.; et al. Identification and optimization of 2-aminobenzimidazole derivatives as novel inhibitors of TRPC4 and TRPC5 channels. Br. J. Pharm. 2015, 172, 3495–3509. [Google Scholar] [CrossRef] [Green Version]
- Derler, I.; Schindl, R.; Fritsch, R.; Heftberger, P.; Riedl, M.C.; Begg, M.; House, D.; Romanin, C. The action of selective CRAC channel blockers is affected by the Orai pore geometry. Cell Calcium 2013, 53, 139–151. [Google Scholar] [CrossRef]
- Gerasimenko, J.V.; Gryshchenko, O.; Ferdek, P.E.; Stapleton, E.; Hebert, T.O.; Bychkova, S.; Peng, S.; Begg, M.; Gerasimenko, O.V.; Petersen, O.H. Ca2+ release-activated Ca2+ channel blockade as a potential tool in antipancreatitis therapy. Proc. Natl. Acad. Sci. USA 2013, 110, 13186–13191. [Google Scholar] [CrossRef] [Green Version]
- Rice, L.V.; Bax, H.J.; Russell, L.J.; Barrett, V.J.; Walton, S.E.; Deakin, A.M.; Thomson, S.A.; Lucas, F.; Solari, R.; House, D.; et al. Characterization of selective Calcium-Release Activated Calcium channel blockers in mast cells and T-cells from human, rat, mouse and guinea-pig preparations. Eur. J. Pharm. 2013, 704, 49–57. [Google Scholar] [CrossRef]
- Jaffre, F.; Bonnin, P.; Callebert, J.; Debbabi, H.; Setola, V.; Doly, S.; Monassier, L.; Mettauer, B.; Blaxall, B.C.; Launay, J.M.; et al. Serotonin and angiotensin receptors in cardiac fibroblasts coregulate adrenergic-dependent cardiac hypertrophy. Circ. Res. 2009, 104, 113–123. [Google Scholar] [CrossRef] [Green Version]
- Graziani, A.; Poteser, M.; Heupel, W.M.; Schleifer, H.; Krenn, M.; Drenckhahn, D.; Romanin, C.; Baumgartner, W.; Groschner, K. Cell-cell contact formation governs Ca2+ signaling by TRPC4 in the vascular endothelium: Evidence for a regulatory TRPC4-beta-catenin interaction. J. Biol. Chem. 2010, 285, 4213–4223. [Google Scholar] [CrossRef] [Green Version]
- Eichelbaum, K.; Winter, M.; Berriel Diaz, M.; Herzig, S.; Krijgsveld, J. Selective enrichment of newly synthesized proteins for quantitative secretome analysis. Nat. Biotechnol. 2012, 30, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Kashpur, O.; LaPointe, D.; Ambady, S.; Ryder, E.F.; Dominko, T. FGF2-induced effects on transcriptome associated with regeneration competence in adult human fibroblasts. BMC Genom. 2013, 14, 656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schleifer, H.; Doleschal, B.; Lichtenegger, M.; Oppenrieder, R.; Derler, I.; Frischauf, I.; Glasnov, T.N.; Kappe, C.O.; Romanin, C.; Groschner, K. Novel pyrazole compounds for pharmacological discrimination between receptor-operated and store-operated Ca (2+) entry pathways. Br. J. Pharm. 2012, 167, 1712–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Jiang, J.; Yue, Z.; Liu, S.; Ma, Y.; Yu, N.; Gao, Y.; Sun, S.; Chen, S.; Liu, P. Store-Operated Ca (2+) Entry (SOCE) contributes to angiotensin II-induced cardiac fibrosis in cardiac fibroblasts. J. Pharm. Sci. 2016, 132, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [CrossRef]
- Hofmann, K.; Fiedler, S.; Vierkotten, S.; Weber, J.; Klee, S.; Jia, J.; Zwickenpflug, W.; Flockerzi, V.; Storch, U.; Yildirim, A.O.; et al. Classical transient receptor potential 6 (TRPC6) channels support myofibroblast differentiation and development of experimental pulmonary fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 560–568. [Google Scholar] [CrossRef]
- Du, J.; Xie, J.; Zhang, Z.; Tsujikawa, H.; Fusco, D.; Silverman, D.; Liang, B.; Yue, L. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ. Res. 2010, 106, 992–1003. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Tadross, M.R.; Tsien, R.W. Sequential ionic and conformational signaling by calcium channels drives neuronal gene expression. Science 2016, 351, 863–867. [Google Scholar] [CrossRef] [Green Version]
- Farmer, L.K.; Rollason, R.; Whitcomb, D.J.; Ni, L.; Goodliff, A.; Lay, A.C.; Birnbaumer, L.; Heesom, K.J.; Xu, S.Z.; Saleem, M.A.; et al. TRPC6 Binds to and Activates Calpain, Independent of Its Channel Activity, and Regulates Podocyte Cytoskeleton, Cell Adhesion, and Motility. J. Am. Soc. Nephrol. 2019, 30, 1910–1924. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, Y.; Li, D.; Zhang, Y.; Tang, B.; Li, G.; Yang, Y.; Yang, D. Transgenic overexpression of transient receptor potential vanilloid subtype 1 attenuates isoproterenol-induced myocardial fibrosis in mice. Int J. Mol. Med. 2016, 38, 601–609. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Ma, S.; Li, D.; Zhang, Y.; Tang, B.; Qiu, C.; Yang, Y.; Yang, D. Dietary capsaicin ameliorates pressure overload-induced cardiac hypertrophy and fibrosis through the transient receptor potential vanilloid type 1. Am. J. Hypertens. 2014, 27, 1521–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Qi, H.; Mingyao, E.; Shi, P.; Zhang, Q.; Li, S.; Wang, Y.; Cao, Y.; Chen, Y.; Ba, L.; et al. Transient receptor potential vanilloid-3 (TRPV3) activation plays a central role in cardiac fibrosis induced by pressure overload in rats via TGF-beta1 pathway. Naunyn Schmiedeberg’s Arch. Pharm. 2018, 391, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, S.O.; Grove, L.M.; Paruchuri, S.; Southern, B.D.; Abraham, S.; Niese, K.A.; Scheraga, R.G.; Ghosh, S.; Thodeti, C.K.; Zhang, D.X.; et al. TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J. Clin. Investig. 2014, 124, 5225–5238. [Google Scholar] [CrossRef] [PubMed]
- Adapala, R.K.; Thoppil, R.J.; Luther, D.J.; Paruchuri, S.; Meszaros, J.G.; Chilian, W.M.; Thodeti, C.K. TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J. Mol. Cell. Cardiol. 2013, 54, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Solis-Lopez, A.; Kriebs, U.; Marx, A.; Mannebach, S.; Liedtke, W.B.; Caterina, M.J.; Freichel, M.; Tsvilovskyy, V.V. Analysis of TRPV channel activation by stimulation of FCepsilonRI and MRGPR receptors in mouse peritoneal mast cells. PLoS ONE 2017, 12, e0171366. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Sakamoto, K.; Kimura, J. Hypoxic stress induces transient receptor potential melastatin 2 (TRPM2) channel expression in adult rat cardiac fibroblasts. J. Pharm. Sci. 2012, 118, 186–197. [Google Scholar] [CrossRef] [Green Version]
- Runnels, L.W.; Yue, L.; Clapham, D.E. The TRPM7 channel is inactivated by PIP (2) hydrolysis. Nat. Cell Biol. 2002, 4, 329–336. [Google Scholar] [CrossRef]
- Qin, X.; Yue, Z.; Sun, B.; Yang, W.; Xie, J.; Ni, E.; Feng, Y.; Mahmood, R.; Zhang, Y.; Yue, L. Sphingosine and FTY720 are potent inhibitors of the transient receptor potential melastatin 7 (TRPM7) channels. Br. J. Pharm. 2013, 168, 1294–1312. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Liu, Y.; Pan, Y.; Lu, C.; Xu, H.; Wang, X.; Liu, T.; Feng, K.; Tang, Y. MicroRNA-135a inhibits cardiac fibrosis induced by isoproterenol via TRPM7 channel. Biomed. Pharm. 2018, 104, 252–260. [Google Scholar] [CrossRef]
- Lu, J.; Wang, Q.Y.; Zhou, Y.; Lu, X.C.; Liu, Y.H.; Wu, Y.; Guo, Q.; Ma, Y.T.; Tang, Y.Q. Astragaloside against cardiac fibrosis by inhibiting TRPM7 channel. Phytomedicine 2017, 30, 10–17. [Google Scholar] [CrossRef]
- Guo, J.L.; Yu, Y.; Jia, Y.Y.; Ma, Y.Z.; Zhang, B.Y.; Liu, P.Q.; Chen, S.R.; Jiang, J.M. Transient receptor potential melastatin 7 (TRPM7) contributes to H2O2-induced cardiac fibrosis via mediating Ca(2+) influx and extracellular signal-regulated kinase 1/2 (ERK1/2) activation in cardiac fibroblasts. J. Pharm. Sci. 2014, 125, 184–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rios, F.J.; Zou, Z.G.; Harvey, A.P.; Harvey, K.Y.; Nosalski, R.; Anyfanti, P.; Camargo, L.L.; Lacchini, S.; Ryazanov, A.G.; Ryazanova, L.; et al. Chanzyme TRPM7 protects against cardiovascular inflammation and fibrosis. Cardiovasc. Res. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oguri, G.; Nakajima, T.; Yamamoto, Y.; Takano, N.; Tanaka, T.; Kikuchi, H.; Morita, T.; Nakamura, F.; Yamasoba, T.; Komuro, I. Effects of methylglyoxal on human cardiac fibroblast: Roles of transient receptor potential ankyrin 1 (TRPA1) channels. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1339–H1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Sun, X.; Wu, H.; Yu, P.; Wang, X.; Jiang, Z.; Gao, E.; Chen, J.; Li, D.; Qiu, C.; et al. TRPA1 Promotes Cardiac Myofibroblast Transdifferentiation after Myocardial Infarction Injury via the Calcineurin-NFAT-DYRK1A Signaling Pathway. Oxidative Med. Cell. Longev. 2019, 2019, 6408352. [Google Scholar] [CrossRef] [Green Version]
- Rubaiy, H.N.; Ludlow, M.J.; Henrot, M.; Gaunt, H.J.; Miteva, K.; Cheung, S.Y.; Tanahashi, Y.; Hamzah, N.; Musialowski, K.E.; Blythe, N.M.; et al. Picomolar, selective, and subtype-specific small-molecule inhibition of TRPC1/4/5 channels. J. Biol. Chem. 2017, 292, 8158–8173. [Google Scholar] [CrossRef] [Green Version]
- Minard, A.; Bauer, C.C.; Wright, D.J.; Rubaiy, H.N.; Muraki, K.; Beech, D.J.; Bon, R.S. Remarkable Progress with Small-Molecule Modulation of TRPC1/4/5 Channels: Implications for Understanding the Channels in Health and Disease. Cells 2018, 7, 52. [Google Scholar] [CrossRef] [Green Version]
- Rubaiy, H.N.; Seitz, T.; Hahn, S.; Choidas, A.; Habenberger, P.; Klebl, B.; Dinkel, K.; Nussbaumer, P.; Waldmann, H.; Christmann, M.; et al. Identification of an (-)-englerin A analogue, which antagonizes (-)-englerin A at TRPC1/4/5 channels. Br. J. Pharm. 2018, 175, 830–839. [Google Scholar] [CrossRef]
- Minard, A.; Bauer, C.C.; Chuntharpursat-Bon, E.; Pickles, I.B.; Wright, D.J.; Ludlow, M.J.; Burnham, M.P.; Warriner, S.L.; Beech, D.J.; Muraki, K.; et al. Potent, selective, and subunit-dependent activation of TRPC5 channels by a xanthine derivative. Br. J. Pharm. 2019, 176, 3924–3938. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camacho Londoño, J.E.; Marx, A.; Kraft, A.E.; Schürger, A.; Richter, C.; Dietrich, A.; Lipp, P.; Birnbaumer, L.; Freichel, M. Angiotensin-II-Evoked Ca2+ Entry in Murine Cardiac Fibroblasts Does Not Depend on TRPC Channels. Cells 2020, 9, 322. https://doi.org/10.3390/cells9020322
Camacho Londoño JE, Marx A, Kraft AE, Schürger A, Richter C, Dietrich A, Lipp P, Birnbaumer L, Freichel M. Angiotensin-II-Evoked Ca2+ Entry in Murine Cardiac Fibroblasts Does Not Depend on TRPC Channels. Cells. 2020; 9(2):322. https://doi.org/10.3390/cells9020322
Chicago/Turabian StyleCamacho Londoño, Juan E., André Marx, Axel E. Kraft, Alexander Schürger, Christin Richter, Alexander Dietrich, Peter Lipp, Lutz Birnbaumer, and Marc Freichel. 2020. "Angiotensin-II-Evoked Ca2+ Entry in Murine Cardiac Fibroblasts Does Not Depend on TRPC Channels" Cells 9, no. 2: 322. https://doi.org/10.3390/cells9020322