Mitochondrial Fusion Via OPA1 and MFN1 Supports Liver Tumor Cell Metabolism and Growth

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Organoids Culture
2.2. Gene Knockdown by Lentiviral Vectors
2.3. Immunofluorescence of Live Cells and Frozen Tissues
2.4. Colony Formation Assay and Alamar Blue Assay
2.5. Analysis of Cell Cycle and Cell Apoptosis
2.6. Xenograft Mouse Model in Nude Mice
2.7. RNA Isolation and Sequencing
2.8. Real-Time PCR
2.9. Oxygen Consumption and ATP Production Measurement
2.10. Measurement of Reactive Oxygen Species (ROS) Production
2.11. Glucose Consumption Measurement
2.12. Lactate Measurement
2.13. Statistical Analysis
3. Results
3.1. Activation of Mitochondrial Fusion in Liver Cancer
3.2. Mitochondrial Fusion Sustains CCA Organoid Growth
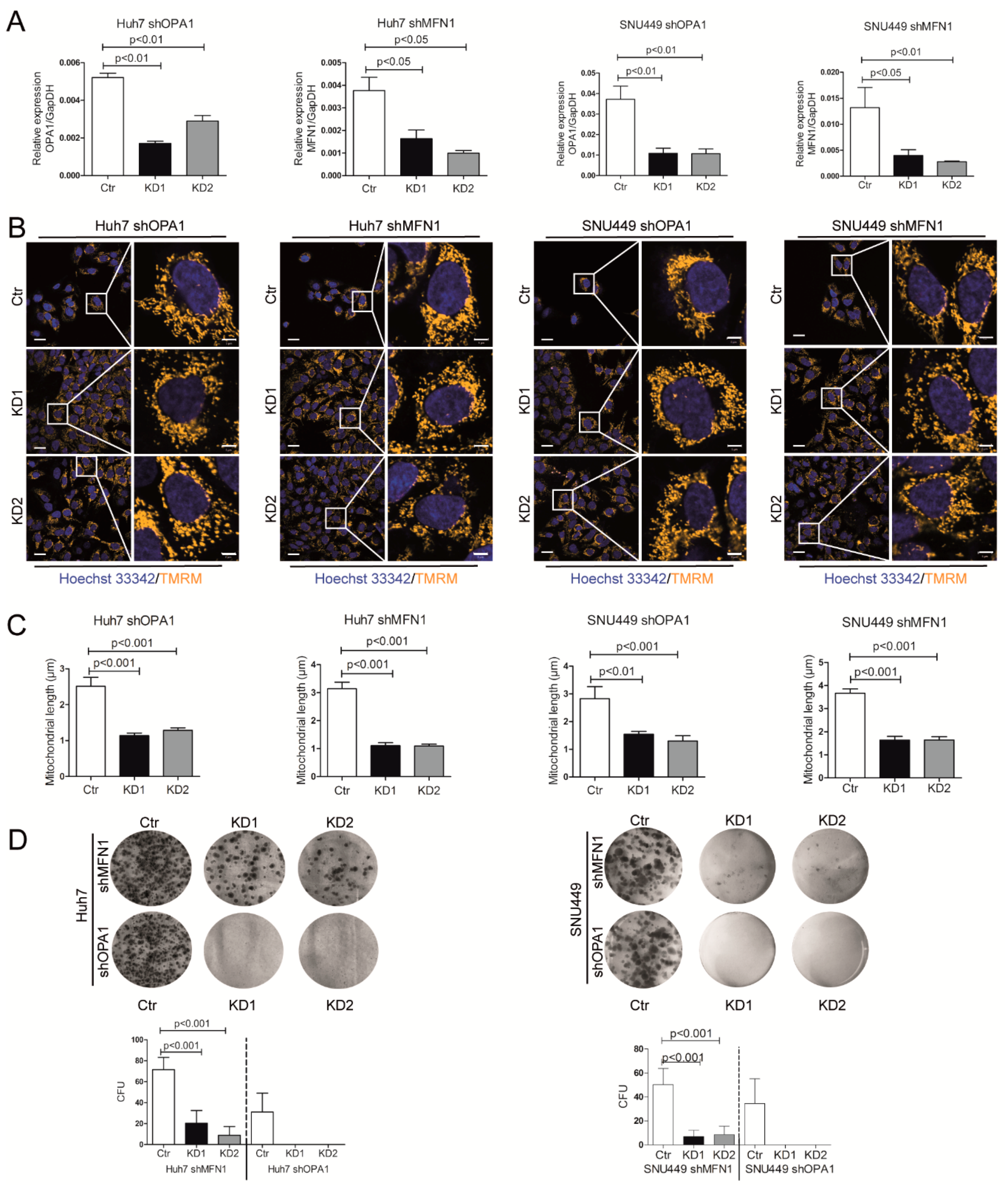
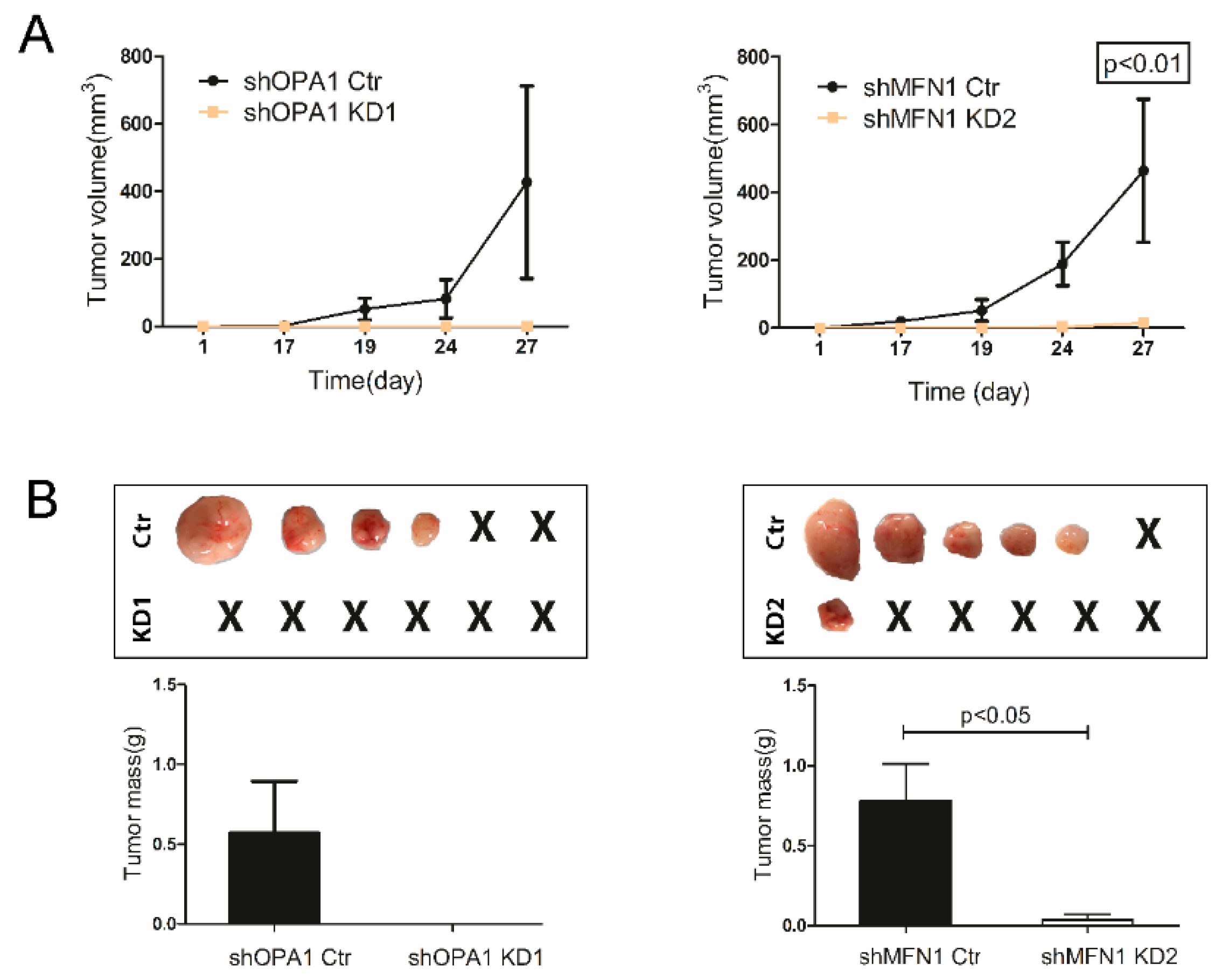
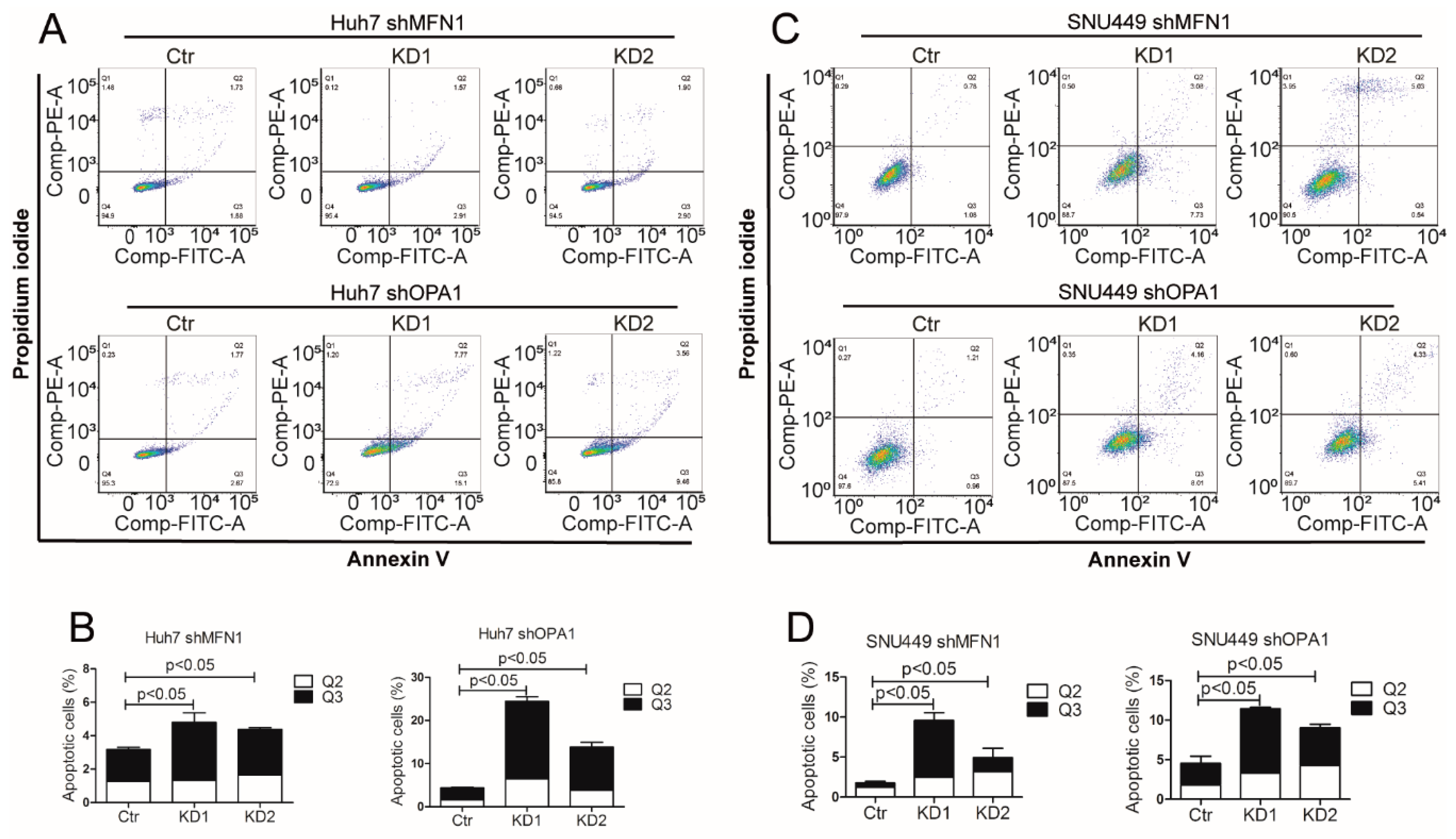
3.3. Dysfunction of Mitochondrial Fusion Inhibits Liver Cancer Cell Growth In Vitro and In Vivo
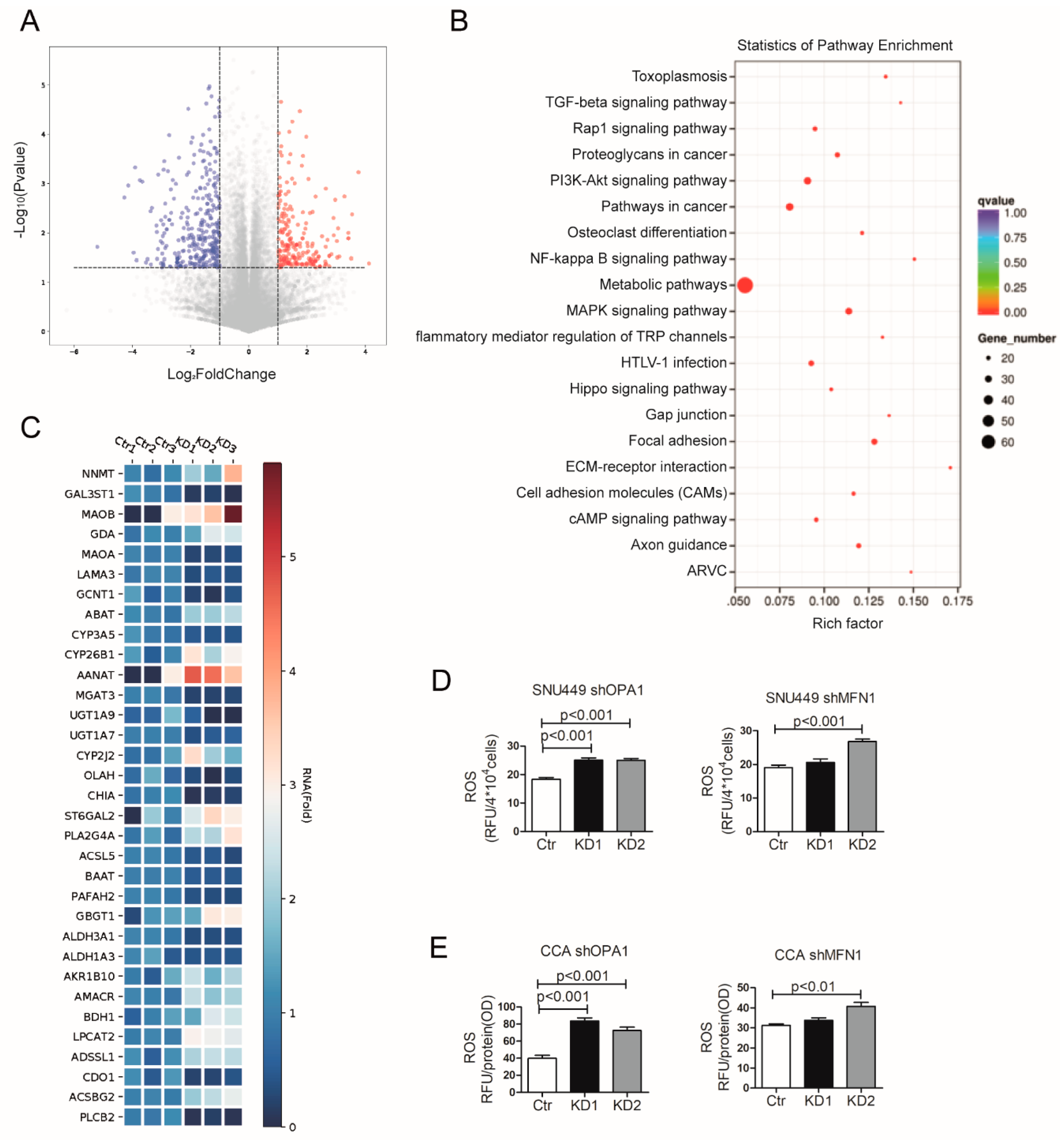
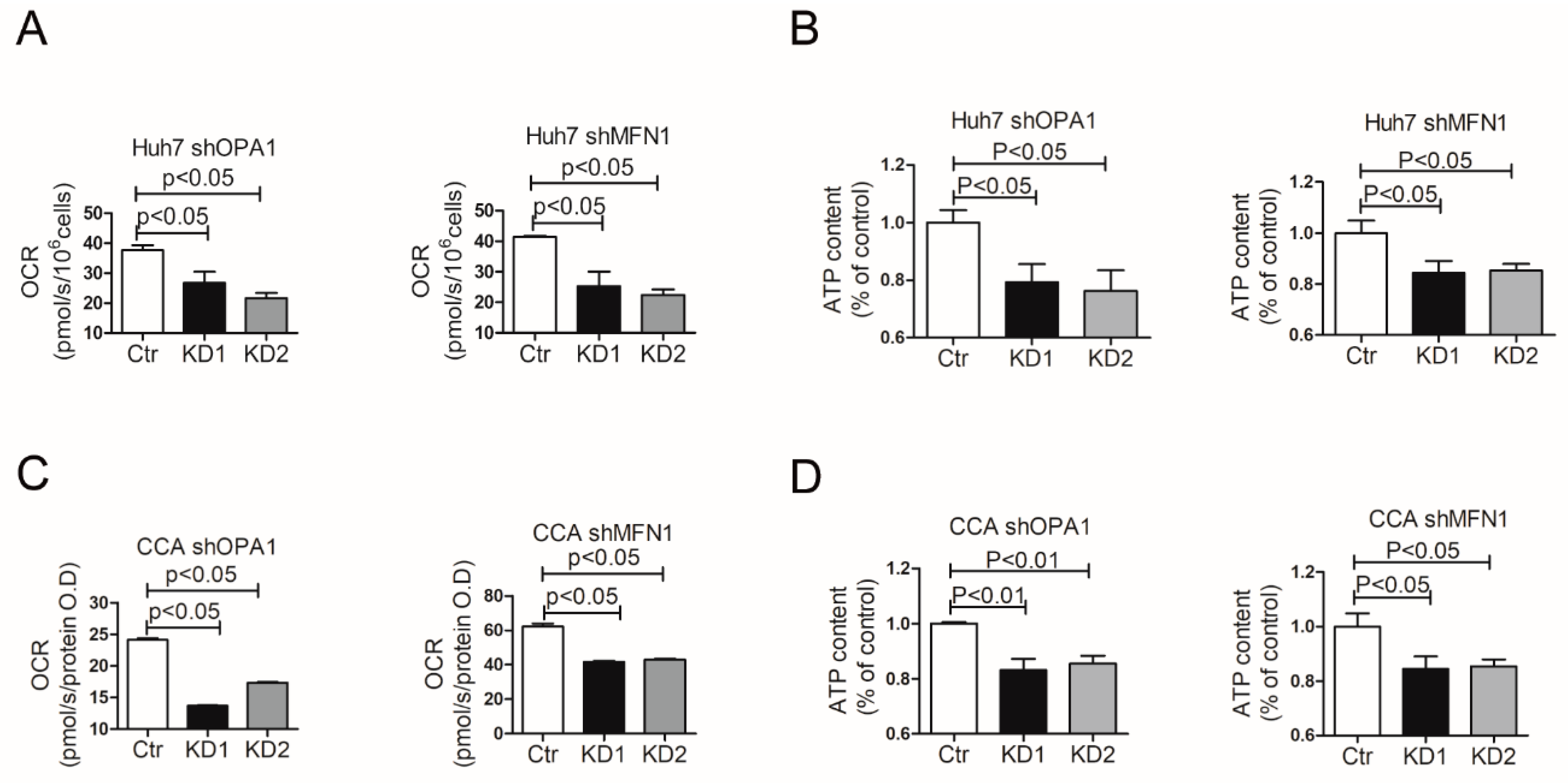
3.4. Mitochondrial Fusion Fuels Cellular Metabolism of Liver Cancer Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandel, N.S. Evolution of mitochondria as signaling organelles. Cell Metab. 2015, 22, 204–206. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Grandemange, S.; Herzig, S.; Martinou, J.C. Mitochondrial dynamics and cancer. Semin. Cancer Biol. 2009, 19, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-the cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef]
- Alavi, M.V.; Fuhrmann, N. Dominant optic atrophy, opa1, and mitochondrial quality control: Understanding mitochondrial network dynamics. Mol. Neurodegener. 2013, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Zorzano, A.; Liesa, M.; Sebastian, D.; Segales, J.; Palacin, M. Mitochondrial fusion proteins: Dual regulators of morphology and metabolism. Semin. Cell Dev. Biol. 2010, 21, 566–574. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Hoppins, S.; Lackner, L.; Nunnari, J. The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 2007, 76, 751–780. [Google Scholar] [CrossRef]
- Patten, D.A.; Wong, J.; Khacho, M.; Soubannier, V.; Mailloux, R.J.; Pilon-Larose, K.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Trinkle-Mulcahy, L.; et al. Opa1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 2014, 33, 2676–2691. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, J.; Zhang, Z.; Wakabayashi, N.; Tamura, Y.; Fukaya, M.; Kensler, T.W.; Iijima, M.; Sesaki, H. The dynamin-related gtpase drp1 is required for embryonic and brain development in mice. J. Cell Biol. 2009, 186, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Wakabayashi, N.; Wakabayashi, J.; Tamura, Y.; Song, W.J.; Sereda, S.; Clerc, P.; Polster, B.M.; Aja, S.M.; Pletnikov, M.V.; et al. The dynamin-related gtpase opa1 is required for glucose-stimulated atp production in pancreatic beta cells. Mol. Biol. Cell 2011, 22, 2235–2245. [Google Scholar] [CrossRef]
- Zhao, X.; Tian, C.; Puszyk, W.M.; Ogunwobi, O.O.; Cao, M.; Wang, T.; Cabrera, R.; Nelson, D.R.; Liu, C. Opa1 downregulation is involved in sorafenib-induced apoptosis in hepatocellular carcinoma. Lab. Investig. 2013, 93, 8–19. [Google Scholar] [CrossRef]
- Liu, P.; Cao, W.; Ma, B.; Li, M.; Chen, K.; Sideras, K.; Duitman, J.W.; Sprengers, D.; Khe Tran, T.C.; Ijzermans, J.N.M.; et al. Action and clinical significance of ccaat/enhancer-binding protein delta in hepatocellular carcinoma. Carcinogenesis 2019, 40, 155–163. [Google Scholar] [CrossRef]
- Broutier, L.; Andersson-Rolf, A.; Hindley, C.J.; Boj, S.F.; Clevers, H.; Koo, B.K.; Huch, M. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3d organoids and their genetic manipulation. Nat. Protoc. 2016, 11, 1724–1743. [Google Scholar] [CrossRef]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.; Francies, H.E.; Gavarro, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef]
- Vicent, S.; Lieshout, R.; Saborowski, A.; Verstegen, M.M.A.; Raggi, C.; Recalcati, S.; Invernizzi, P.; van der Laan, L.J.W.; Alvaro, D.; Calvisi, D.F.; et al. Experimental models to unravel the molecular pathogenesis, cell of origin and stem cell properties of cholangiocarcinoma. Liver Int. 2019, 39, 79–97. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Zhou, X.; Wang, W.; Wang, Y.; Yin, Y.; Laan, L.J.; Sprengers, D.; Metselaar, H.J.; Peppelenbosch, M.P.; Pan, Q. Ifn regulatory factor 1 restricts hepatitis e virus replication by activating stat1 to induce antiviral ifn-stimulated genes. FASEB J. 2016, 30, 3352–3367. [Google Scholar] [CrossRef] [Green Version]
- Drost, J.; Artegiani, B.; Clevers, H. The generation of organoids for studying wnt signaling. Methods Mol. Biol. 2016, 1481, 141–159. [Google Scholar]
- Petrat, F.; Pindiur, S.; Kirsch, M.; de Groot, H. “Mitochondrial” photochemical drugs do not release toxic amounts of 1o(2) within the mitochondrial matrix space. Arch. Biochem. Biophys. 2003, 412, 207–215. [Google Scholar] [CrossRef]
- Cao, W.; Chen, K.; Bolkestein, M.; Yin, Y.; Verstegen, M.M.A.; Bijvelds, M.J.C.; Wang, W.; Tuysuz, N.; Ten Berge, D.; Sprengers, D.; et al. Dynamics of proliferative and quiescent stem cells in liver homeostasis and injury. Gastroenterology 2017, 153, 1133–1147. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Cao, W.; Li, J.; Sprengers, D.; Y Hernanda, P.; Kong, X.; Jw van der Laan, L.; Man, K.; Kwekkeboom, J.; J Metselaar, H.; et al. Differential sensitivities of fast- and slow-cycling cancer cells to inosine monophosphate dehydrogenase 2 inhibition by mycophenolic acid. Mol. Med. 2016, 21, 792–802. [Google Scholar] [CrossRef]
- Ma, B.; Chen, K.; Liu, P.; Li, M.; Liu, J.; Sideras, K.; Sprengers, D.; Biermann, K.; Wang, W.; Ijzermans, J.N.M.; et al. Dichotomal functions of phosphorylated and unphosphorylated stat1 in hepatocellular carcinoma. J. Mol. Med. (Berl.) 2019, 97, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Santel, A.; Frank, S.; Gaume, B.; Herrler, M.; Youle, R.J.; Fuller, M.T. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J. Cell Sci. 2003, 116, 2763–2774. [Google Scholar] [CrossRef] [Green Version]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Ortmayr, K.; Dubuis, S.; Zampieri, M. Metabolic profiling of cancer cells reveals genome-wide crosstalk between transcriptional regulators and metabolism. Nat. Commun. 2019, 10, 1841. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis b virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis c virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [Green Version]
- Ferreira-da-Silva, A.; Valacca, C.; Rios, E.; Populo, H.; Soares, P.; Sobrinho-Simoes, M.; Scorrano, L.; Maximo, V.; Campello, S. Mitochondrial dynamics protein drp1 is overexpressed in oncocytic thyroid tumors and regulates cancer cell migration. PLoS ONE 2015, 10, e0122308. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.Y.; Yun, S.H.; Yun, J. Downregulation of drp1, a fission regulator, is associated with human lung and colon cancers. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Maycotte, P.; Marin-Hernandez, A.; Goyri-Aguirre, M.; Anaya-Ruiz, M.; Reyes-Leyva, J.; Cortes-Hernandez, P. Mitochondrial dynamics and cancer. Tumour Biol. 2017, 39, 1010428317698391. [Google Scholar] [CrossRef] [Green Version]
- von Eyss, B.; Jaenicke, L.A.; Kortlever, R.M.; Royla, N.; Wiese, K.E.; Letschert, S.; McDuffus, L.A.; Sauer, M.; Rosenwald, A.; Evan, G.I.; et al. A myc-driven change in mitochondrial dynamics limits yap/taz function in mammary epithelial cells and breast cancer. Cancer Cell 2015, 28, 743–757. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.; Mak, T.W. Mitochondrial cell death effectors. Curr. Opin. Cell Biol. 2009, 21, 871–877. [Google Scholar] [CrossRef]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [Green Version]
- Frezza, C.; Cipolat, S.; De Brito, O.M.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. Opa1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Graves, J.A.; Wang, Y.; Sims-Lucas, S.; Cherok, E.; Rothermund, K.; Branca, M.F.; Elster, J.; Beer-Stolz, D.; Van Houten, B.; Vockley, J.; et al. Mitochondrial structure, function and dynamics are temporally controlled by c-myc. PLoS ONE 2012, 7, e37699. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. Myc, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef] [Green Version]
- Serasinghe, M.N.; Wieder, S.Y.; Renault, T.T.; Elkholi, R.; Asciolla, J.J.; Yao, J.L.; Jabado, O.; Hoehn, K.; Kageyama, Y.; Sesaki, H.; et al. Mitochondrial division is requisite to ras-induced transformation and targeted by oncogenic mapk pathway inhibitors. Mol. Cell 2015, 57, 521–536. [Google Scholar] [CrossRef] [Green Version]
- Bajzikova, M.; Kovarova, J.; Coelho, A.R.; Boukalova, S.; Oh, S.; Rohlenova, K.; Svec, D.; Hubackova, S.; Endaya, B.; Judasova, K.; et al. Reactivation of dihydroorotate dehydrogenase-driven pyrimidine biosynthesis restores tumor growth of respiration-deficient cancer cells. Cell Metab. 2019, 29, 399–416.e10. [Google Scholar] [CrossRef] [Green Version]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Huang, Q.; Zhan, L.; Cao, H.; Li, J.; Lyu, Y.; Guo, X.; Zhang, J.; Ji, L.; Ren, T.; An, J.; et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ros-modulated coordinated regulation of the nfkb and tp53 pathways. Autophagy 2016, 12, 999–1014. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, Y.; Jiang, X.; Zhang, H.; Gao, Z.; Li, Y.; Fu, R.; Li, L.; Li, J.; Cui, H.; et al. Ros-mediated activation and mitochondrial translocation of camkii contributes to drp1-dependent mitochondrial fission and apoptosis in triple-negative breast cancer cells by isorhamnetin and chloroquine. J. Exp. Clin. Cancer Res. 2019, 38, 225. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | HCC Patient (n = 13) | CCA Patient (n = 1) |
|---|---|---|
| Age at surgery (years) | ||
| Mean ± SD | 64.9 ± 13.4 | 60 |
| Median (range) | 70 (37–79) | - |
| sex (%) | ||
| Male | 8 (62) | 1 (100) |
| Female | 5 (38) | - |
| Etiology (%) | ||
| Unknown liver disease | 6 (46) | - |
| Alcohol abuse | 1 (8) | 1 (100) |
| Chronic HBV | 3 (23) | - |
| Hemochromatosis | 1 (8) | - |
| Hemochromatosis and alcohol abuse | 2 (15) | - |
| Cirrhosis (%) | ||
| Yes | 2 (15) | - |
| No | 11 (85) | 1 (100) |
| Tumor differentiation (%) | ||
| Well | 1 (8) | - |
| Moderate | 9 (69) | - |
| Poor | 2 (15) | - |
| Unknown | 1 (8) | 1 (100) |
| Vascular invasion (%) | ||
| Micro-invasion | 6 (46) | - |
| Vaso-invasion | 2 (15) | - |
| Severe vaso-invasion | 2 (15) | 1 (100) |
| No | 3 (23) | - |
| AFP (pre-operative) (µg/L) | ||
| Mean ± SD | 4076.2 ± 12607.2 | 3.8 |
| Median (range) | 16 (3-45803) | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Wang, L.; Wang, Y.; Zhang, S.; Zhou, G.; Lieshout, R.; Ma, B.; Liu, J.; Qu, C.; Verstegen, M.M.A.; et al. Mitochondrial Fusion Via OPA1 and MFN1 Supports Liver Tumor Cell Metabolism and Growth. Cells 2020, 9, 121. https://doi.org/10.3390/cells9010121
Li M, Wang L, Wang Y, Zhang S, Zhou G, Lieshout R, Ma B, Liu J, Qu C, Verstegen MMA, et al. Mitochondrial Fusion Via OPA1 and MFN1 Supports Liver Tumor Cell Metabolism and Growth. Cells. 2020; 9(1):121. https://doi.org/10.3390/cells9010121
Chicago/Turabian StyleLi, Meng, Ling Wang, Yijin Wang, Shaoshi Zhang, Guoying Zhou, Ruby Lieshout, Buyun Ma, Jiaye Liu, Changbo Qu, Monique M. A. Verstegen, and et al. 2020. "Mitochondrial Fusion Via OPA1 and MFN1 Supports Liver Tumor Cell Metabolism and Growth" Cells 9, no. 1: 121. https://doi.org/10.3390/cells9010121