Suppression of Angiogenesis by Targeting Cyclin-Dependent Kinase 7 in Human Umbilical Vein Endothelial Cells and Renal Cell Carcinoma: An In Vitro and In Vivo Study

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture and siRNA Transfection
2.3. Cell Proliferation Assay
2.4. Western Blotting
2.5. Transwell Migration Assay
2.6. In Vivo Matrigel Plug Assay
2.7. Capillary Tube Formation Assay
2.8. In Vivo Xenograft Experiments
2.9. Statistical Analysis
3. Results
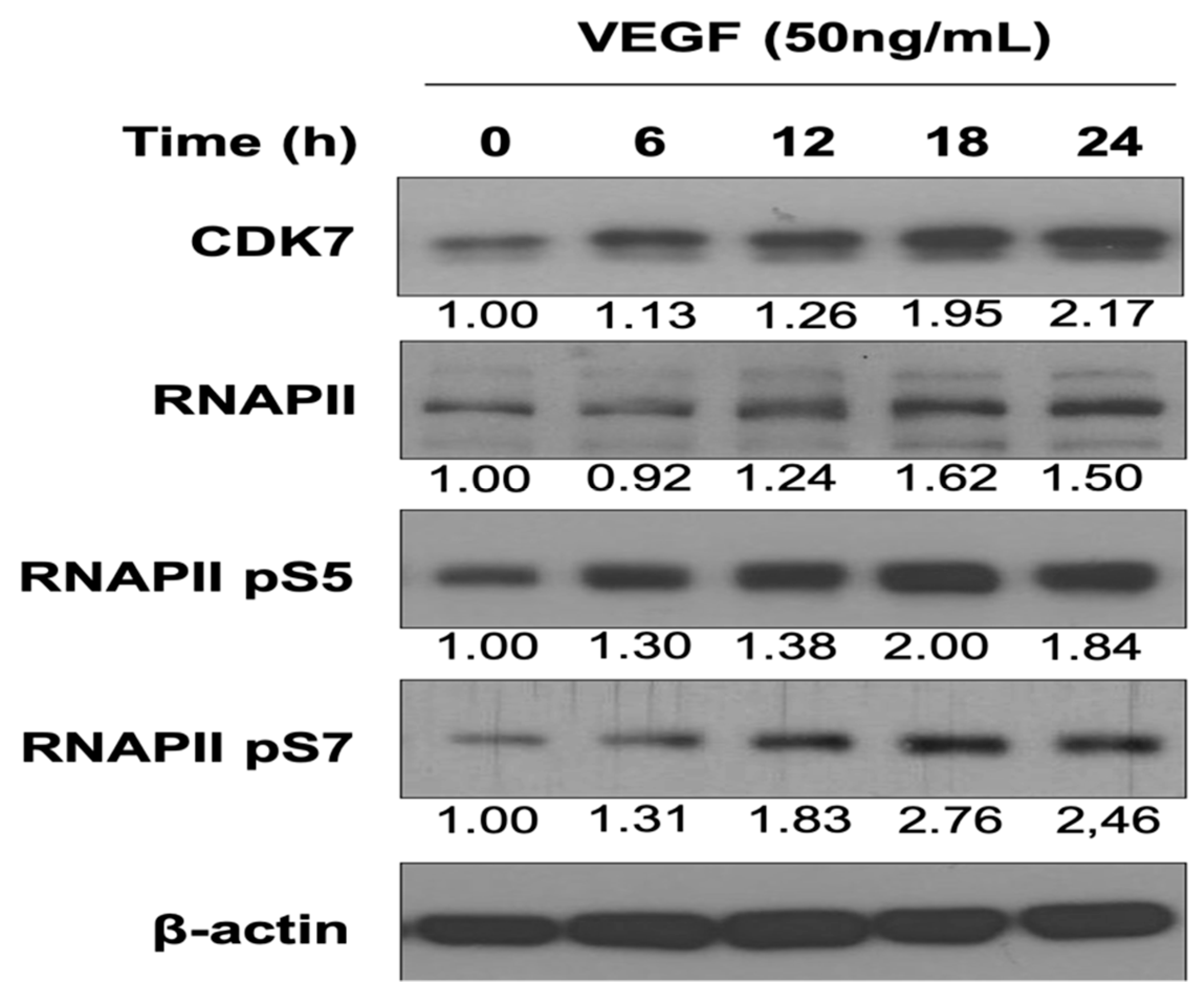
3.1. VEGF Enhances CDK7 and RNAPII Activation and Downstream RNAPII Phosphorylation at Serine 5 and 7 in HUVECs
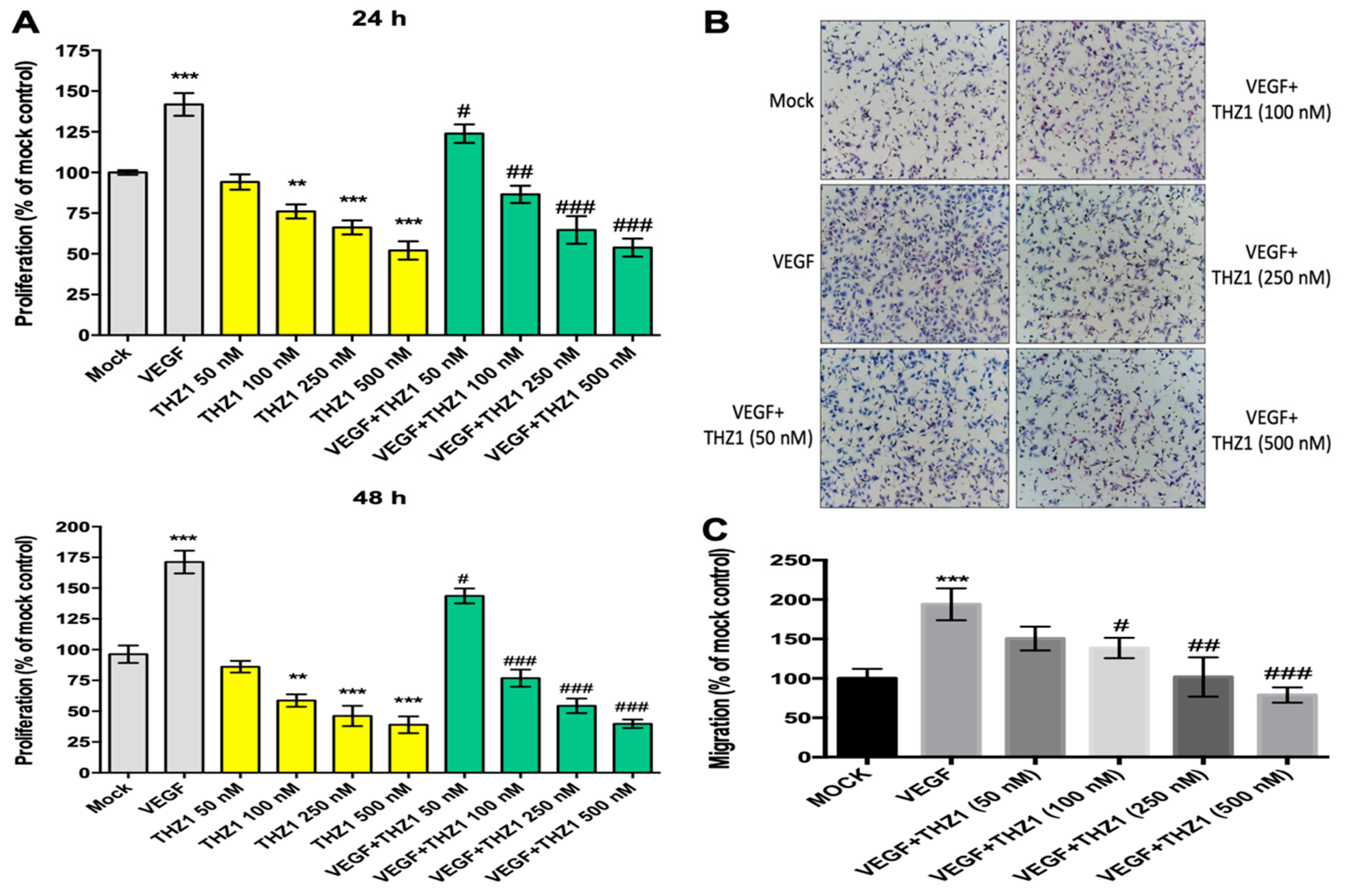
3.2. THZ1 Dose-Dependently Suppresses the Proliferation and Chemotactic Migration of HUVECs
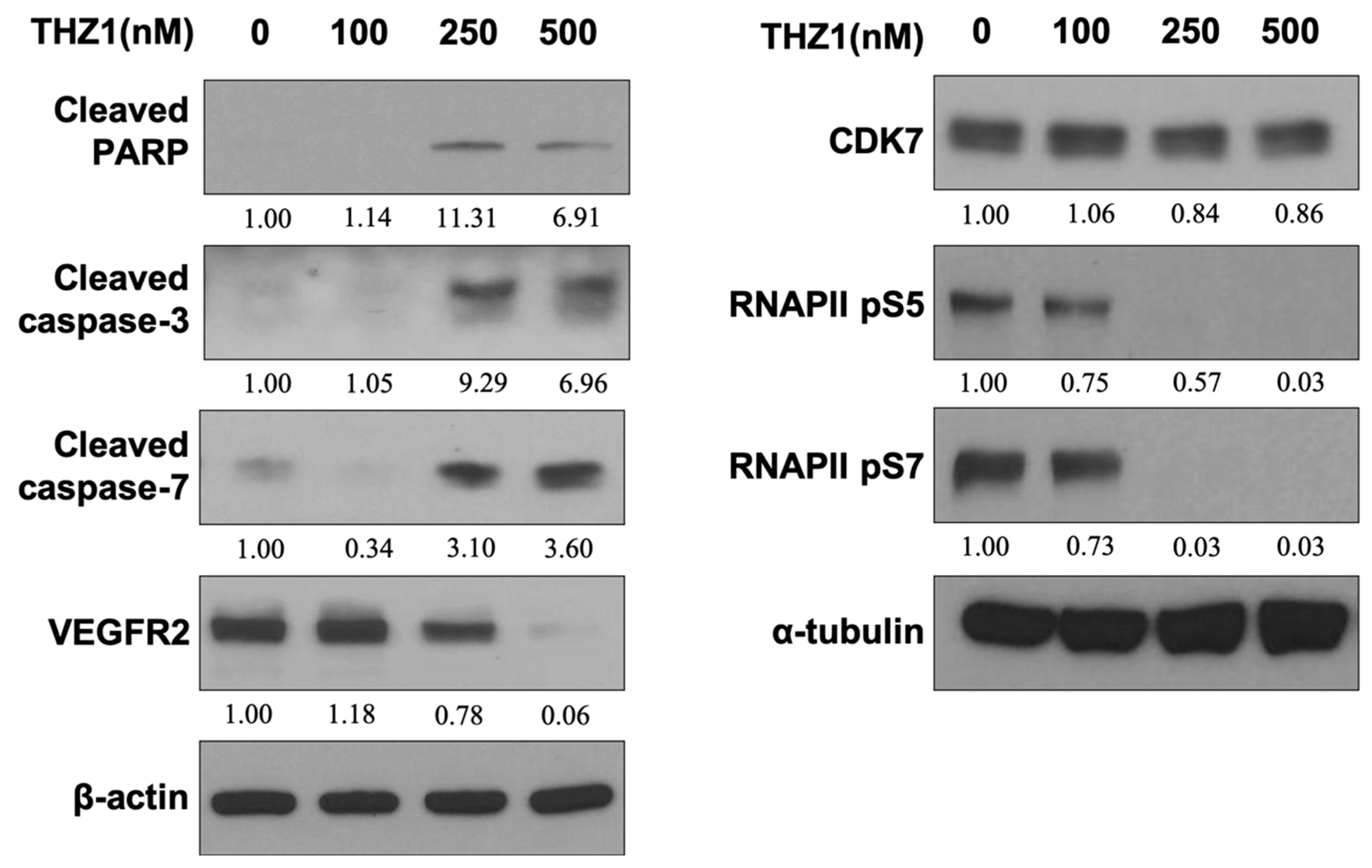
3.3. THZ1 Induces Apoptosis and Suppresses VEGFR2 Expression and RNAPII CTD Phosphorylation at Serine 5 and 7 in HUVECs
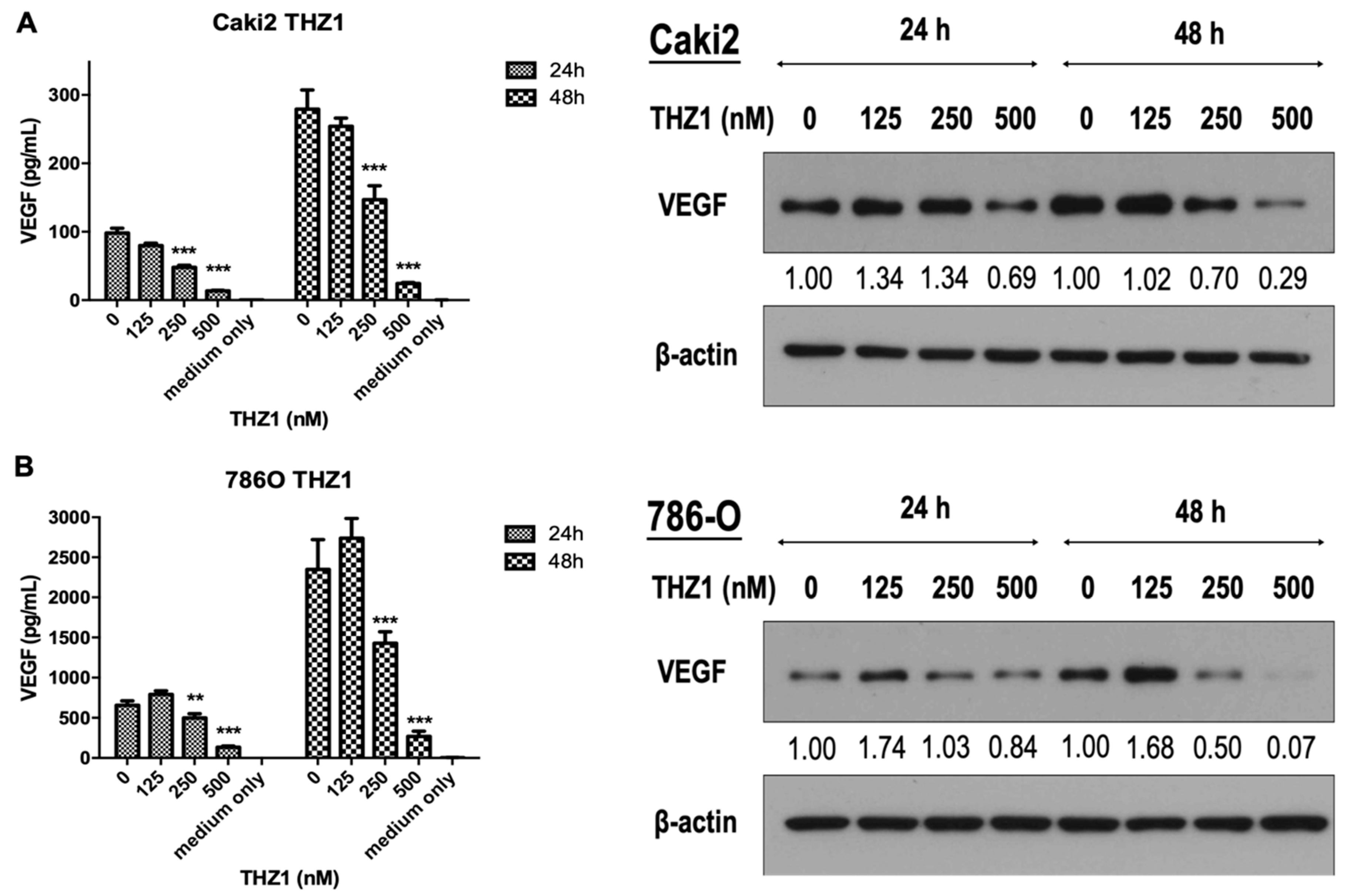
3.4. THZ1 Dose-Dependently Inhibits VEGF Secretion and Expression in Caki-2 and 786-O Cells
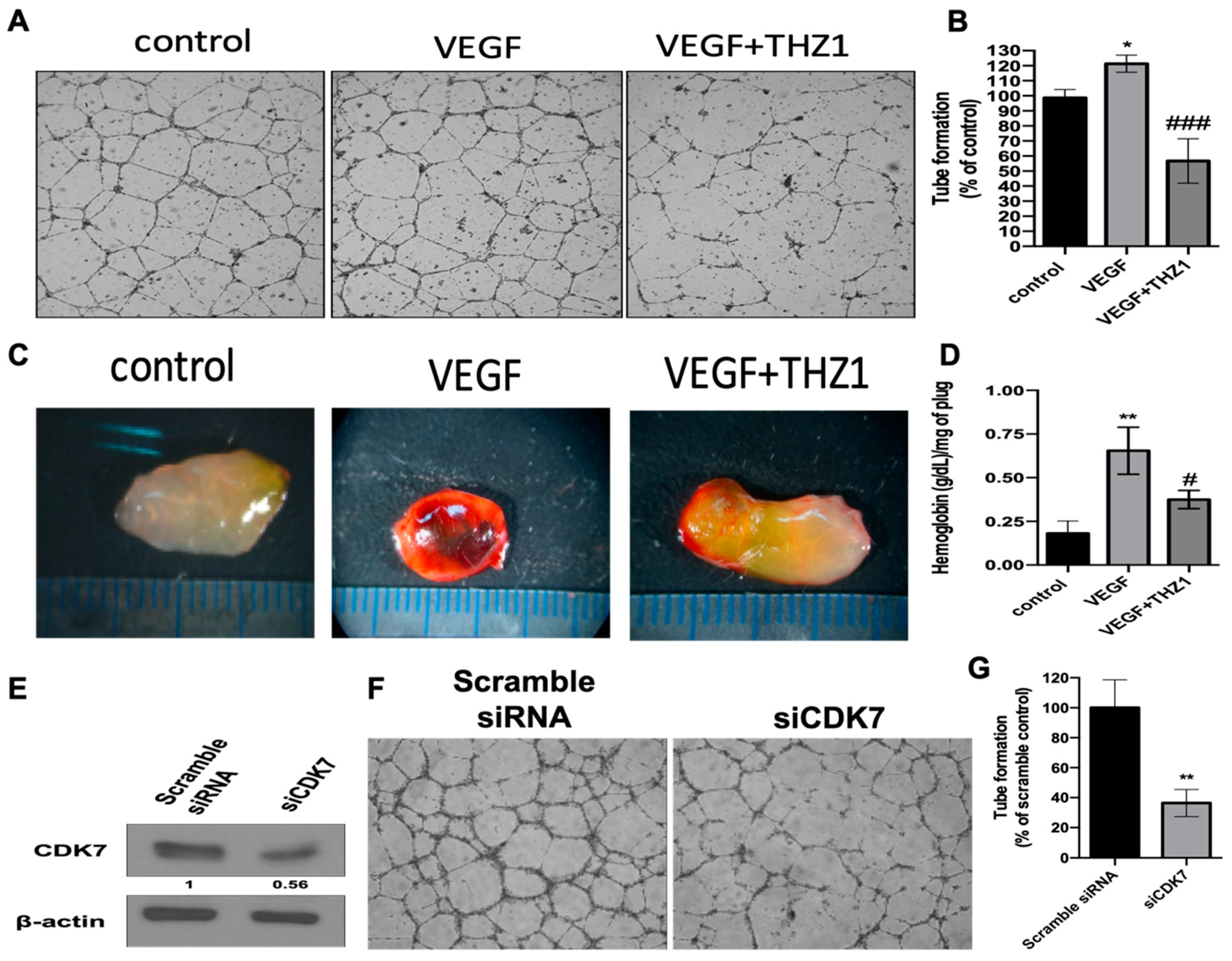
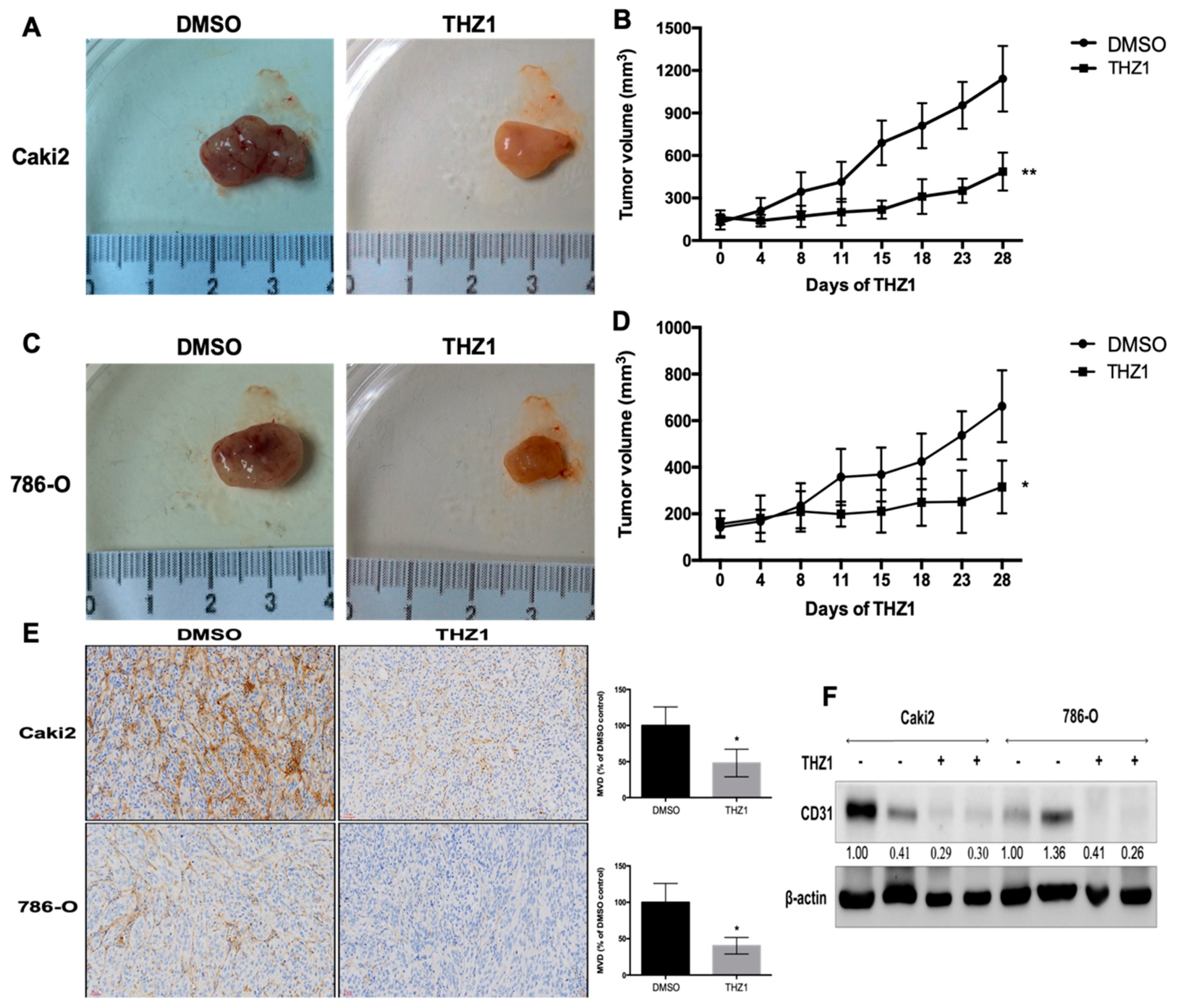
3.5. THZ1 Constrains Tumor Growth of Human RCC Cells (Caki-2 and 786-O) with the Concurrent Suppression of Angiogenesis in a Xenograft Mouse Model
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Folkman, J.; Merler, E.; Abernathy, C.; Williams, G. Isolation of a tumor factor responsible for angiogenesis. J. Exp. Med. 1971, 133, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb. Perspect Med. 2012, 2, a006502. [Google Scholar] [CrossRef] [PubMed]
- Haidl, F.; Pfister, D.; Heidenreich, A.; Heidegger, I. Antiangiogenic therapies in urogenital malignancies: Fiction or fact? Memo 2017, 10, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.J., 3rd; Mandal, S.S.; Reinberg, D. Recent highlights of RNA-polymerase-II-mediated transcription. Curr. Opin. Cell Biol. 2004, 16, 263–271. [Google Scholar] [CrossRef]
- Thomas, M.C.; Chiang, C.M. The general transcription machinery and general cofactors. Crit. Rev. Biochem. Mol. Biol. 2006, 41, 105–178. [Google Scholar] [CrossRef]
- Bywater, M.J.; Pearson, R.B.; McArthur, G.A.; Hannan, R.D. Dysregulation of the basal RNA polymerase transcription apparatus in cancer. Nat. Rev. Cancer 2013, 13, 299–314. [Google Scholar] [CrossRef]
- Cao, K.; Shilatifard, A. Inhibit globally, act locally: CDK7 inhibitors in cancer therapy. Cancer Cell 2014, 26, 158–159. [Google Scholar] [CrossRef]
- Akhtar, M.S.; Heidemann, M.; Tietjen, J.R.; Zhang, D.W.; Chapman, R.D.; Eick, D.; Ansari, A.Z. TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II. Mol. Cell 2009, 34, 387–393. [Google Scholar] [CrossRef]
- Kaldis, P.; Solomon, M.J. Analysis of CAK activities from human cells. Eur. J. Biochem. 2000, 267, 4213–4221. [Google Scholar] [CrossRef]
- Larochelle, S.; Merrick, K.A.; Terret, M.E.; Wohlbold, L.; Barboza, N.M.; Zhang, C.; Shokat, K.M.; Jallepalli, P.V.; Fisher, R.P. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol. Cell 2007, 25, 839–850. [Google Scholar] [CrossRef]
- Desai, D.; Wessling, H.C.; Fisher, R.P.; Morgan, D.O. Effects of phosphorylation by CAK on cyclin binding by CDC2 and CDK2. Mol. Cell Biol. 1995, 15, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Glover-Cutter, K.; Larochelle, S.; Erickson, B.; Zhang, C.; Shokat, K.; Fisher, R.P.; Bentley, D.L. TFIIH-associated Cdk7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing, and termination by RNA polymerase II. Mol. Cell Biol. 2009, 29, 5455–5464. [Google Scholar] [CrossRef] [PubMed]
- Serizawa, H.; Makela, T.P.; Conaway, J.W.; Conaway, R.C.; Weinberg, R.A.; Young, R.A. Association of Cdk-activating kinase subunits with transcription factor TFIIH. Nature 1995, 374, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Senderowicz, A.M. Targeting cell cycle and apoptosis for the treatment of human malignancies. Curr. Opin. Cell Biol. 2004, 16, 670–678. [Google Scholar] [CrossRef] [PubMed]
- DiPippo, A.J.; Patel, N.K.; Barnett, C.M. Cyclin-Dependent Kinase Inhibitors for the Treatment of Breast Cancer: Past, Present, and Future. Pharmacotherapy 2016. [Google Scholar] [CrossRef]
- Abadi, A.H.; Abou-Seri, S.M.; Abdel-Rahman, D.E.; Klein, C.; Lozach, O.; Meijer, L. Synthesis of 3-substituted-2-oxoindole analogues and their evaluation as kinase inhibitors, anticancer and antiangiogenic agents. Eur. J. Med. Chem. 2006, 41, 296–305. [Google Scholar] [CrossRef]
- Maggiorella, L.; Aubel, C.; Haton, C.; Milliat, F.; Connault, E.; Opolon, P.; Deutsch, E.; Bourhis, J. Cooperative effect of roscovitine and irradiation targets angiogenesis and induces vascular destabilization in human breast carcinoma. Cell Prolif. 2009, 42, 38–48. [Google Scholar] [CrossRef]
- Zahler, S.; Liebl, J.; Furst, R.; Vollmar, A.M. Anti-angiogenic potential of small molecular inhibitors of cyclin dependent kinases in vitro. Angiogenesis 2010, 13, 239–249. [Google Scholar] [CrossRef]
- Liebl, J.; Krystof, V.; Vereb, G.; Takacs, L.; Strnad, M.; Pechan, P.; Havlicek, L.; Zatloukal, M.; Furst, R.; Vollmar, A.M.; et al. Anti-angiogenic effects of purine inhibitors of cyclin dependent kinases. Angiogenesis 2011, 14, 281–291. [Google Scholar] [CrossRef]
- Liebl, J.; Weitensteiner, S.B.; Vereb, G.; Takacs, L.; Furst, R.; Vollmar, A.M.; Zahler, S. Cyclin-dependent kinase 5 regulates endothelial cell migration and angiogenesis. J. Biol. Chem. 2010, 285, 35932–35943. [Google Scholar] [CrossRef]
- Jonasch, E.; Futreal, P.A.; Davis, I.J.; Bailey, S.T.; Kim, W.Y.; Brugarolas, J.; Giaccia, A.J.; Kurban, G.; Pause, A.; Frydman, J.; et al. State of the science: An update on renal cell carcinoma. Mol. Cancer Res. 2012, 10, 859–880. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Rusan, M.; Li, K.; Li, Y.; Christensen, C.L.; Abraham, B.J.; Kwiatkowski, N.; Buczkowski, K.A.; Bockorny, B.; Chen, T.; Li, S.; et al. Suppression of Adaptive Responses to Targeted Cancer Therapy by Transcriptional Repression. Cancer Discov. 2018, 8, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef] [PubMed]
- Christensen, C.L.; Kwiatkowski, N.; Abraham, B.J.; Carretero, J.; Al-Shahrour, F.; Zhang, T.; Chipumuro, E.; Herter-Sprie, G.S.; Akbay, E.A.; Altabef, A.; et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 2014, 26, 909–922. [Google Scholar] [CrossRef]
- Wang, Q.; Li, M.; Zhang, X.; Huang, H.; Huang, J.; Ke, J.; Ding, H.; Xiao, J.; Shan, X.; Liu, Q.; et al. Upregulation of CDK7 in gastric cancer cell promotes tumor cell proliferation and predicts poor prognosis. Exp. Mol. Pathol. 2016, 100, 514–521. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef]
- Kuo, K.L.; Ho, I.L.; Shi, C.S.; Wu, J.T.; Lin, W.C.; Tsai, Y.C.; Chang, H.C.; Chou, C.T.; Hsu, C.H.; Hsieh, J.T.; et al. MLN4924, a novel protein neddylation inhibitor, suppresses proliferation and migration of human urothelial carcinoma: In vitro and in vivo studies. Cancer Lett. 2015, 363, 127–136. [Google Scholar] [CrossRef]
- Malinda, K.M. In vivo matrigel migration and angiogenesis assay. Methods Mol. Biol. 2009, 467, 287–294. [Google Scholar] [CrossRef]
- Heltberg, M.L.; Krishna, S.; Jensen, M.H. On chaotic dynamics in transcription factors and the associated effects in differential gene regulation. Nat. Commun. 2019, 10, 71. [Google Scholar] [CrossRef]
- Chen, J.; Fu, Y.; Day, D.S.; Sun, Y.; Wang, S.; Liang, X.; Gu, F.; Zhang, F.; Stevens, S.M.; Zhou, P.; et al. VEGF amplifies transcription through ETS1 acetylation to enable angiogenesis. Nat. Commun. 2017, 8, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Tahergorabi, Z.; Khazaei, M. A review on angiogenesis and its assays. Iran. J. Basic Med. Sci. 2012, 15, 1110–1126. [Google Scholar]
- Singh, B.; Wu, P.J. Regulation of the program of DNA replication by CDK: New findings and perspectives. Curr. Genet. 2019, 65, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Lampropoulou, E.; Logoviti, I.; Koutsioumpa, M.; Hatziapostolou, M.; Polytarchou, C.; Skandalis, S.S.; Hellman, U.; Fousteris, M.; Nikolaropoulos, S.; Choleva, E.; et al. Cyclin-dependent kinase 5 mediates pleiotrophin-induced endothelial cell migration. Sci. Rep. 2018, 8, 5893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Zhang, Y.; Zhang, R.; Zhao, Z.; Zhang, H.; Wu, J.; Shen, W.; Zhong, M. Cyclin-dependent kinase 1 disruption inhibits angiogenesis by inducing cell cycle arrest and apoptosis. Exp. Med. 2019, 18, 3062–3070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenleaf, A.L. Human CDK12 and CDK13, multi-tasking CTD kinases for the new millenium. Transcription 2019, 10, 91–110. [Google Scholar] [CrossRef] [Green Version]
- Otrock, Z.K.; Mahfouz, R.A.; Makarem, J.A.; Shamseddine, A.I. Understanding the biology of angiogenesis: Review of the most important molecular mechanisms. Blood Cells Mol. Dis. 2007, 39, 212–220. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, C.-S.; Kuo, K.-L.; Chen, M.-S.; Chow, P.-M.; Liu, S.-H.; Chang, Y.-W.; Lin, W.-C.; Liao, S.-M.; Hsu, C.-H.; Hsu, F.-S.; et al. Suppression of Angiogenesis by Targeting Cyclin-Dependent Kinase 7 in Human Umbilical Vein Endothelial Cells and Renal Cell Carcinoma: An In Vitro and In Vivo Study. Cells 2019, 8, 1469. https://doi.org/10.3390/cells8111469
Shi C-S, Kuo K-L, Chen M-S, Chow P-M, Liu S-H, Chang Y-W, Lin W-C, Liao S-M, Hsu C-H, Hsu F-S, et al. Suppression of Angiogenesis by Targeting Cyclin-Dependent Kinase 7 in Human Umbilical Vein Endothelial Cells and Renal Cell Carcinoma: An In Vitro and In Vivo Study. Cells. 2019; 8(11):1469. https://doi.org/10.3390/cells8111469
Chicago/Turabian StyleShi, Chung-Sheng, Kuan-Lin Kuo, Mei-Sin Chen, Po-Ming Chow, Shing-Hwa Liu, Yu-Wei Chang, Wei-Chou Lin, Shih-Ming Liao, Chen-Hsun Hsu, Fu-Shun Hsu, and et al. 2019. "Suppression of Angiogenesis by Targeting Cyclin-Dependent Kinase 7 in Human Umbilical Vein Endothelial Cells and Renal Cell Carcinoma: An In Vitro and In Vivo Study" Cells 8, no. 11: 1469. https://doi.org/10.3390/cells8111469