Epigenetic Regulation of Inflammatory Cytokine-Induced Epithelial-To-Mesenchymal Cell Transition and Cancer Stem Cell Generation

 ,
, 
Abstract
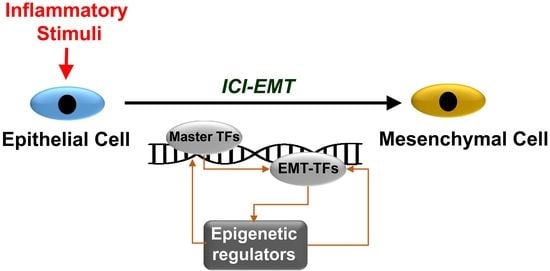
:
1. Introduction
2. Regulation of Epithelial-To-Mesenchymal Cell Transition (EMT) in Cancer, and Its Involvement in the Generation of Cancer Stem Cells (CSCs)
3. Inflammatory Molecular Regulatory Circuits Influencing EMT
3.1. TGFβ
3.2. TNFα
3.3. Interleukins
3.3.1. IL-1
3.3.2. IL-6
3.4. Converging Roles of Inflammatory Cytokines during ICI-EMT
4. Epigenetic Regulation of CSC Generation: The Role of Inflammation and the Tumor Microenvironment
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- You, J.S.; Jones, P.A. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Laird, P.W. Interplay between the cancer genome and epigenome. Cell 2013, 153, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Sarkies, P.; Sale, J.E. Cellular epigenetic stability and cancer. Trends Genet. 2012, 28, 118–127. [Google Scholar] [CrossRef]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, J.; Esteller, M. Cancer epigenomics: Beyond genomics. Curr. Opin. Genet. Dev. 2012, 22, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding rnas in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Markopoulos, G.S.; Roupakia, E.; Tokamani, M.; Chavdoula, E.; Hatziapostolou, M.; Polytarchou, C.; Marcu, K.B.; Papavassiliou, A.G.; Sandaltzopoulos, R.; Kolettas, E. A step-by-step microrna guide to cancer development and metastasis. Cell. Oncol. 2017, 40, 303–339. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding rnas. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef]
- Rodríguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330. [Google Scholar] [CrossRef] [PubMed]
- Berdasco, M.; Esteller, M. Clinical epigenetics: Seizing opportunities for translation. Nat. Rev. Genet. 2019, 20, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef]
- Brabletz, T. To differentiate or not—Routes towards metastasis. Nat. Rev. Cancer 2012, 12, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436. [Google Scholar] [CrossRef]
- Allavena, P.; Garlanda, C.; Borrello, M.G.; Sica, A.; Mantovani, A. Pathways connecting inflammation and cancer. Curr. Opin. Genet. Dev. 2008, 18, 3–10. [Google Scholar] [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef]
- López-Novoa, J.M.; Nieto, M.A. Inflammation and emt: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009, 1, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Carmona, M.; Lesage, J.; Cataldo, D.; Gilles, C. Emt and inflammation: Inseparable actors of cancer progression. Mol. Oncol. 2017, 11, 805–823. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Mantovani, A. Cancer-Related Inflammation: Common Themes and Therapeutic Opportunities. Semin. Cancer Biol. 2012, 22, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Marchesi, F.; Garlanda, C.; Mantovani, A.; Allavena, P. Inflammation-mediated promotion of invasion and metastasis. Cancer Metastasis Rev. 2010, 29, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Garlanda, C.; Allavena, P. Molecular pathways and targets in cancer-related inflammation. Ann. Med. 2010, 42, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T. Emt and met in metastasis: Where are the cancer stem cells? Cancer Cell 2012, 22, 699–701. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.Y.; Fattet, L.; Yang, J. Molecular pathways: Linking tumor microenvironment to epithelial-mesenchymal transition in metastasis. Clin. Cancer Res. 2015, 21, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell. Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Jing, Y.; Han, Z.; Zhang, S.; Liu, Y.; Wei, L. Epithelial-mesenchymal transition in tumor microenvironment. Cell Biosci. 2011, 1, 29. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Caetano, M.S.; Zhang, H.; Cumpian, A.M.; Gong, L.; Unver, N.; Ostrin, E.J.; Daliri, S.; Chang, S.H.; Ochoa, C.E.; Hanash, S.; et al. Il6 blockade reprograms the lung tumor microenvironment to limit the development and progression of k-ras–mutant lung cancer. Cancer Res. 2016, 76, 3189–3199. [Google Scholar] [CrossRef] [PubMed]
- Png, K.J.; Halberg, N.; Yoshida, M.; Tavazoie, S.F. A microrna regulon that mediates endothelial recruitment and metastasis by cancer cells. Nature 2011, 481, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.K.; Zillhardt, M.; Hua, Y.; Tiwari, P.; Murmann, A.E.; Peter, M.E.; Lengyel, E. Micrornas reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov. 2012, 2, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Shahi, P.; Werb, Z. Microrna-mediated regulation of the tumor microenvironment. Cell Cycle 2013, 12, 3262–3271. [Google Scholar] [CrossRef] [PubMed]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of emt-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [Green Version]
- Scheel, C.; Weinberg, R.A. In Cancer stem cells and epithelial–mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol. 2012, 22, 396–403. [Google Scholar] [CrossRef]
- Pencheva, N.; Tavazoie, S.F. Control of metastatic progression by microrna regulatory networks. Nat. Cell Biol. 2013, 15, 546–554. [Google Scholar] [CrossRef]
- Shimono, Y.; Zabala, M.; Cho, R.W.; Lobo, N.; Dalerba, P.; Qian, D.; Diehn, M.; Liu, H.; Panula, S.P.; Chiao, E.; et al. Downregulation of mirna-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138, 592–603. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An epigenetic switch involving nf-κb, lin28, let-7 microrna, and il6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; Agami, R. Transformation locked in a loop. Cell 2009, 139, 654–656. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Polytarchou, C.; Hatziapostolou, M.; Kottakis, F.; Maroulakou, I.G.; Struhl, K.; Tsichlis, P.N. Micrornas differentially regulated by akt isoforms control emt and stem cell renewal in cancer cells. Sci. Signal. 2009, 2, ra62. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Jaeger, S.A.; Hirsch, H.A.; Bulyk, M.L.; Struhl, K. Stat3 activation of mir-21 and mir-181b-1 via pten and cyld are part of the epigenetic switch linking inflammation to cancer. Mol. Cell 2010, 39, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Lindahl-Allen, M.; Polytarchou, C.; Hirsch, H.A.; Tsichlis, P.N.; Struhl, K. Loss of mir-200 inhibition of suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol. Cell 2010, 39, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Zahnow, C.A.; Baylin, S.B. Epigenetic networks and mirnas in stem cells and cancer. Mol. Cell 2010, 39, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Hatziapostolou, M.; Polytarchou, C.; Aggelidou, E.; Drakaki, A.; Poultsides, G.A.; Jaeger, S.A.; Ogata, H.; Karin, M.; Struhl, K.; Hadzopoulou-Cladaras, M.; et al. An hnf4alpha-mirna inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell 2011, 147, 1233–1247. [Google Scholar] [CrossRef] [PubMed]
- Hatziapostolou, M.; Iliopoulos, D. Epigenetic aberrations during oncogenesis. Cell Mol. Life Sci. 2011, 68, 1681–1702. [Google Scholar] [CrossRef]
- Kong, D.; Piao, Y.S.; Yamashita, S.; Oshima, H.; Oguma, K.; Fushida, S.; Fujimura, T.; Minamoto, T.; Seno, H.; Yamada, Y.; et al. Inflammation-induced repression of tumor suppressor mir-7 in gastric tumor cells. Oncogene 2012, 31, 3949–3960. [Google Scholar] [CrossRef]
- Tian, X.J.; Zhang, H.; Xing, J. Coupled reversible and irreversible bistable switches underlying tgfbeta-induced epithelial to mesenchymal transition. Biophys. J. 2013, 105, 1079–1089. [Google Scholar] [CrossRef]
- Zhang, J.; Tian, X.J.; Zhang, H.; Teng, Y.; Li, R.; Bai, F.; Elankumaran, S.; Xing, J. Tgf-beta-induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci. Signal. 2014, 7, ra91. [Google Scholar] [CrossRef] [PubMed]
- Polytarchou, C.; Hommes, D.W.; Palumbo, T.; Hatziapostolou, M.; Koutsioumpa, M.; Koukos, G.; van der Meulen-de Jong, A.E.; Oikonomopoulos, A.; van Deen, W.K.; Vorvis, C.; et al. Microrna214 is associated with progression of ulcerative colitis, and inhibition reduces development of colitis and colitis-associated cancer in mice. Gastroenterology 2015, 149, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between zeb1 and members of the mir-200 family promotes emt and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The emt-activator zeb1 promotes tumorigenicity by repressing stemness-inhibiting micrornas. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, S.; Brabletz, T. The zeb/mir-200 feedback loop—A motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D. Microrna circuits regulate the cancer-inflammation link. Sci. Signal. 2014, 7, pe8. [Google Scholar] [CrossRef] [PubMed]
- Carstens, J.L.; Lovisa, S.; Kalluri, R. Microenvironment-dependent cues trigger mirna-regulated feedback loop to facilitate the emt/met switch. J. Clin. Invest. 2014, 124, 1458–1460. [Google Scholar] [CrossRef] [PubMed]
- Markopoulos, G.S.; Roupakia, E.; Tokamani, M.; Alabasi, G.; Sandaltzopoulos, R.; Marcu, K.B.; Kolettas, E. Roles of nf-kappab signaling in the regulation of mirnas impacting on inflammation in cancer. Biomedicines 2018, 6, E40. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L. Immune regulation of cancer. J. Clin. Oncol. 2010, 28, 4531–4538. [Google Scholar] [CrossRef] [PubMed]
- McAllister, S.S.; Weinberg, R.A. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat. Cell Biol. 2014, 16, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I.; Katsura, A.; Matsuyama, H.; Miyazono, K. Microrna regulons in tumor microenvironment. Oncogene 2015, 34, 3085–3094. [Google Scholar] [CrossRef] [PubMed]
- Kuninty, P.R.; Schnittert, J.; Storm, G.; Prakash, J. Microrna targeting to modulate tumor microenvironment. Front. Oncol. 2016, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Rokavec, M.; Oner, M.G.; Hermeking, H. Lnflammation-induced epigenetic switches in cancer. Cell Mol. Life Sci. 2016, 73, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Kohlhapp, F.J.; Mitra, A.K.; Lengyel, E.; Peter, M.E. Micrornas as mediators and communicators between cancer cells and the tumor microenvironment. Oncogene 2015, 34, 5857–5868. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; David, J.M.; Palena, C. Epithelial-mesenchymal transition and inflammation at the site of the primary tumor. Semin. Cancer Biol. 2017, 47, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Wang, G.; Struhl, K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via il6 secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Stadler, S.C.; Allis, C.D. Linking epithelial-to-mesenchymal-transition and epigenetic modifications. Semin. Cancer Biol. 2012, 22, 404–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiesslich, T.; Pichler, M.; Neureiter, D. Epigenetic control of epithelial-mesenchymal-transition in human cancer. Mol. Clin. Oncol. 2013, 1, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shang, Y. Epigenetic control of epithelial-to-mesenchymal transition and cancer metastasis. Exp. Cell Res. 2013, 319, 160–169. [Google Scholar] [CrossRef]
- Rokavec, M.; Oner, M.G.; Li, H.; Jackstadt, R.; Jiang, L.; Lodygin, D.; Kaller, M.; Horst, D.; Ziegler, P.K.; Schwitalla, S.; et al. Il-6r/stat3/mir-34a feedback loop promotes emt-mediated colorectal cancer invasion and metastasis. J. Clin. Invest. 2014, 124, 1853–1867. [Google Scholar] [CrossRef]
- Hu, Y.; Yan, F.; Ying, L.; Xu, D. Emerging roles for epigenetic programming in the control of inflammatory signaling integration in heath and disease. Adv. Exp. Med. Biol. 2017, 1024, 63–90. [Google Scholar] [PubMed]
- He, G.; Dhar, D.; Nakagawa, H.; Font-Burgada, J.; Ogata, H.; Jiang, Y.; Shalapour, S.; Seki, E.; Yost, S.E.; Jepsen, K.; et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine il-6 signaling. Cell 2013, 155, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Rodger, E.J.; Eccles, M.R. Epigenetic drivers of tumourigenesis and cancer metastasis. Semin. Cancer Biol. 2018, 51, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Scheel, C.; Onder, T.; Karnoub, A.; Weinberg, R.A. Adaptation versus selection: The origins of metastatic behavior. Cancer Res. 2007, 67, 11476–11480. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.A. Mechanisms of malignant progression. Carcinogenesis 2008, 29, 1092–1095. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [Green Version]
- Paduch, R. The role of lymphangiogenesis and angiogenesis in tumor metastasis. Cell. Oncol. 2016, 39, 397–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. Emt: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Invest. 2009, 119, 1417–1419. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Min, C.; Eddy, S.F.; Sherr, D.H.; Sonenshein, G.E. Nf-kappab and epithelial to mesenchymal transition of cancer. J. Cell. Biochem. 2008, 104, 733–744. [Google Scholar] [CrossRef]
- Wu, Y.; Deng, J.; Rychahou, P.G.; Qiu, S.; Evers, B.M.; Zhou, B.P. Stabilization of snail by nf-kappab is required for inflammation-induced cell migration and invasion. Cancer Cell 2009, 15, 416–428. [Google Scholar] [CrossRef]
- Storci, G.; Sansone, P.; Mari, S.; D’Uva, G.; Tavolari, S.; Guarnieri, T.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Chieco, P.; et al. Tnfalpha up-regulates slug via the nf-kappab/hif1alpha axis, which imparts breast cancer cells with a stem cell-like phenotype. J. Cell. Physiol. 2010, 225, 682–691. [Google Scholar] [CrossRef]
- D’Ignazio, L.; Batie, M.; Rocha, S. Hypoxia and inflammation in cancer, focus on hif and nf-kappab. Biomedicines 2017, 5, E21. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; Gabrielson, E.; Fujii, H.; Baylin, S.B.; Herman, J.G. Methylation patterns of the e-cadherin 5’ cpg island are unstable and reflect the dynamic, heterogeneous loss of e-cadherin expression during metastatic progression. J. Biol. Chem. 2000, 275, 2727–2732. [Google Scholar] [CrossRef] [PubMed]
- Nass, S.J.; Herman, J.G.; Gabrielson, E.; Iversen, P.W.; Parl, F.F.; Davidson, N.E.; Graff, J.R. Aberrant methylation of the estrogen receptor and e-cadherin 5’ cpg islands increases with malignant progression in human breast cancer. Cancer Res. 2000, 60, 4346–4348. [Google Scholar] [PubMed]
- Sideridou, M.; Zakopoulou, R.; Evangelou, K.; Liontos, M.; Kotsinas, A.; Rampakakis, E.; Gagos, S.; Kahata, K.; Grabusic, K.; Gkouskou, K. Cdc6 expression represses e-cadherin transcription and activates adjacent replication origins. J. Cell Biol. 2011, 195, 1123–1140. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Yu, J.; Dhanasekaran, S.M.; Kim, J.H.; Mani, R.S.; Tomlins, S.A.; Mehra, R.; Laxman, B.; Cao, X.; Yu, J.; et al. Repression of e-cadherin by the polycomb group protein ezh2 in cancer. Oncogene 2008, 27, 7274–7284. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Pasini, D.; Diaz, V.M.; Franci, C.; Gutierrez, A.; Dave, N.; Escriva, M.; Hernandez-Munoz, I.; Di Croce, L.; Helin, K.; et al. Polycomb complex 2 is required for e-cadherin repression by the snail1 transcription factor. Mol. Cell Biol. 2008, 28, 4772–4781. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Qin, L.; He, T.; Qin, J.; Hong, J.; Wong, J.; Liao, L.; Xu, J. The twist/mi2/nurd protein complex and its essential role in cancer metastasis. Cell Res. 2011, 21, 275–289. [Google Scholar] [CrossRef]
- Drakaki, A.; Iliopoulos, D. Microrna gene networks in oncogenesis. Curr. Genom. 2009, 10, 35–41. [Google Scholar] [CrossRef]
- Lin, T.; Ponn, A.; Hu, X.; Law, B.K.; Lu, J. Requirement of the histone demethylase lsd1 in snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene 2010, 29, 4896–4904. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, Y.; Li, J.; Dong, C.; Ye, X.; Chi, Y.I.; Evers, B.M.; Zhou, B.P. The snag domain of snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 2010, 29, 1803–1816. [Google Scholar] [CrossRef]
- Siemens, H.; Jackstadt, R.; Hunten, S.; Kaller, M.; Menssen, A.; Gotz, U.; Hermeking, H. Mir-34 and snail form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 2011, 10, 4256–4271. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.H.; Kim, H.S.; Li, X.Y.; Lee, I.; Choi, H.S.; Kang, S.E.; Cha, S.Y.; Ryu, J.K.; Yoon, D.; Fearon, E.R.; et al. A p53/mirna-34 axis regulates snail1-dependent cancer cell epithelial-mesenchymal transition. J. Cell Biol. 2011, 195, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Ni, T.; Li, X.Y.; Lu, N.; An, T.; Liu, Z.P.; Fu, R.; Lv, W.C.; Zhang, Y.W.; Xu, X.J.; Grant Rowe, R.; et al. Snail1-dependent p53 repression regulates expansion and activity of tumour-initiating cells in breast cancer. Nat. Cell Biol. 2016, 18, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Lohrum, M.; Stunnenberg, H.G.; Logie, C. The new frontier in cancer research: Deciphering cancer epigenetics. Int. J. Biochem. Cell Biol. 2007, 39, 1450–1461. [Google Scholar] [CrossRef] [PubMed]
- Park, P.J. Chip–seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; Rauch, T.A. In DNA methylation patterns in lung carcinomas. Semin. Cancer Biol. 2009, 19, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.T.; Otterson, G.A.; Plass, C. Unraveling the epigenetic code of cancer for therapy. Trends Genet. 2007, 23, 449–456. [Google Scholar] [CrossRef]
- Julien, S.; Puig, I.; Caretti, E.; Bonaventure, J.; Nelles, L.; van Roy, F.; Dargemont, C.; de Herreros, A.G.; Bellacosa, A.; Larue, L. Activation of nf-kappab by akt upregulates snail expression and induces epithelium mesenchyme transition. Oncogene 2007, 26, 7445–7456. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, zeb and bhlh factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- Beltran, M.; Puig, I.; Pena, C.; Garcia, J.M.; Alvarez, A.B.; Pena, R.; Bonilla, F.; de Herreros, A.G. A natural antisense transcript regulates zeb2/sip1 gene expression during snail1-induced epithelial-mesenchymal transition. Genes Dev. 2008, 22, 756–769. [Google Scholar] [CrossRef]
- Gill, J.G.; Langer, E.M.; Lindsley, R.C.; Cai, M.; Murphy, T.L.; Kyba, M.; Murphy, K.M. Snail and the microrna-200 family act in opposition to regulate epithelial-to-mesenchymal transition and germ layer fate restriction in differentiating escs. Stem Cells 2011, 29, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Kumar, B.; Datta, J.; Teknos, T.N.; Kumar, P. Il-6 promotes head and neck tumor metastasis by inducing epithelial–mesenchymal transition via the jak-stat3-snail signaling pathway. Mol. Cancer Res. 2011, 9, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog lsd1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, H.; Chen, Y.; Sun, Y.; Yang, F.; Yu, W.; Liang, J.; Sun, L.; Yang, X.; Shi, L.; et al. Lsd1 is a subunit of the nurd complex and targets the metastasis programs in breast cancer. Cell 2009, 138, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Janzer, A.; Becker, A.; Zimmer, A.; Schule, R.; Buettner, R.; Kirfel, J. Lysine-specific demethylase 1 (lsd1) is highly expressed in er-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 2010, 31, 512–520. [Google Scholar] [CrossRef]
- Dong, C.; Wu, Y.; Yao, J.; Wang, Y.; Yu, Y.; Rychahou, P.G.; Evers, B.M.; Zhou, B.P. G9a interacts with snail and is critical for snail-mediated e-cadherin repression in human breast cancer. J. Clin. Invest. 2012, 122, 1469–1486. [Google Scholar] [CrossRef]
- Dong, C.; Wu, Y.; Wang, Y.; Wang, C.; Kang, T.; Rychahou, P.G.; Chi, Y.I.; Evers, B.M.; Zhou, B.P. Interaction with suv39h1 is critical for snail-mediated e-cadherin repression in breast cancer. Oncogene 2013, 32, 1351–1362. [Google Scholar] [CrossRef]
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. Snail mediates e-cadherin repression by the recruitment of the sin3a/histone deacetylase 1 (hdac1)/hdac2 complex. Mol. Cell Biol. 2004, 24, 306–319. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Cano, A.; Nieto, M.A. Non-coding rnas take centre stage in epithelial-to-mesenchymal transition. Trends Cell Biol. 2008, 18, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The mir-200 family determines the epithelial phenotype of cancer cells by targeting the e-cadherin repressors zeb1 and zeb2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.N.; Yin, J.J.; Abou-Kheir, W.; Hynes, P.G.; Casey, O.M.; Fang, L.; Yi, M.; Stephens, R.M.; Seng, V.; Sheppard-Tillman, H.; et al. Mir-1 and mir-200 inhibit emt via slug-dependent and tumorigenesis via slug-independent mechanisms. Oncogene 2013, 32, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Perdigao-Henriques, R.; Petrocca, F.; Altschuler, G.; Thomas, M.P.; Le, M.T.; Tan, S.M.; Hide, W.; Lieberman, J. Mir-200 promotes the mesenchymal to epithelial transition by suppressing multiple members of the zeb2 and snail1 transcriptional repressor complexes. Oncogene 2016, 35, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, Y.; Wang, K.; Wang, Y.; Yin, W.; Li, L. P38/nf-kappab/snail pathway is involved in caffeic acid-induced inhibition of cancer stem cells-like properties and migratory capacity in malignant human keratinocyte. PLoS ONE 2013, 8, e58915. [Google Scholar]
- Yamakuchi, M.; Lowenstein, C.J. Mir-34, sirt1, and p53: The feedback loop. Cell Cycle 2009, 8, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H. The mir-34 family in cancer and apoptosis. Cell Death Differ. 2010, 17, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Rokavec, M.; Li, H.; Jiang, L.; Hermeking, H. The p53/mir-34 axis in development and disease. J. Mol. Cell Biol. 2014, 6, 214–230. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H. Micrornas in the p53 network: Micromanagement of tumour suppression. Nat. Rev. Cancer 2012, 12, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Chao, C.H.; Xia, W.; Yang, J.Y.; Xiong, Y.; Li, C.W.; Yu, W.H.; Rehman, S.K.; Hsu, J.L.; Lee, H.H.; et al. P53 regulates epithelial-mesenchymal transition and stem cell properties through modulating mirnas. Nat. Cell Biol. 2011, 13, 317–323. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. P53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Lodygin, D.; Tarasov, V.; Epanchintsev, A.; Berking, C.; Knyazeva, T.; Korner, H.; Knyazev, P.; Diebold, J.; Hermeking, H. Inactivation of mir-34a by aberrant cpg methylation in multiple types of cancer. Cell Cycle 2008, 7, 2591–2600. [Google Scholar] [CrossRef] [PubMed]
- Granados Lopez, A.J.; Lopez, J.A. Multistep model of cervical cancer: Participation of mirnas and coding genes. Int. J. Mol. Sci. 2014, 15, 15700–15733. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, R.; Saini, N. Micrornas in cancer stem cells: Current status and future directions. Tumour Biol. 2014, 35, 8395–8405. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Croce, C.M. Role of micrornas in maintaining cancer stem cells. Adv. Drug Deliv. Rev. 2015, 81, 53–61. [Google Scholar] [CrossRef]
- Ma, X.; Becker Buscaglia, L.E.; Barker, J.R.; Li, Y. Micrornas in nf-kappab signaling. J. Mol. Cell Biol. 2011, 3, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Boldin, M.P.; Baltimore, D. Micrornas, new effectors and regulators of nf-kappab. Immunol. Rev. 2012, 246, 205–220. [Google Scholar] [CrossRef]
- Mann, M.; Mehta, A.; Zhao, J.L.; Lee, K.; Marinov, G.K.; Garcia-Flores, Y.; Baltimore, D. An nf-kappab-microrna regulatory network tunes macrophage inflammatory responses. Nat. Commun. 2017, 8, 851. [Google Scholar] [CrossRef]
- Romeo, E.; Caserta, C.A.; Rumio, C.; Marcucci, F. The vicious cross-talk between tumor cells with an emt phenotype and cells of the immune system. Cells 2019, 8, 460. [Google Scholar] [CrossRef]
- Natoli, G.; Ghisletti, S.; Barozzi, I. The genomic landscapes of inflammation. Genes Dev. 2011, 25, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Massague, J. Tgfbeta signalling in context. Nat. Rev. Mol. Cell. Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Brandl, M.; Seidler, B.; Haller, F.; Adamski, J.; Schmid, R.M.; Saur, D.; Schneider, G. Ikk(alpha) controls canonical tgf(ss)-smad signaling to regulate genes expressing snail and slug during emt in panc1 cells. J. Cell Sci. 2010, 123, 4231–4239. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.N.; Hilyard, A.C.; Lagna, G.; Hata, A. Smad proteins control drosha-mediated microrna maturation. Nature 2008, 454, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. Tgfbeta in cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. Tgf-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Tian, L.; Han, Y.; Vogelbaum, M.; Stark, G.R. Dose-dependent cross-talk between the transforming growth factor-beta and interleukin-1 signaling pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 4365–4370. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.D.; Parvani, J.G.; Schiemann, W.P. The relevance of the tgf-beta paradox to emt-met programs. Cancer Lett. 2013, 341, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Thillainadesan, G.; Chitilian, J.M.; Isovic, M.; Ablack, J.N.; Mymryk, J.S.; Tini, M.; Torchia, J. Tgf-beta-dependent active demethylation and expression of the p15ink4b tumor suppressor are impaired by the znf217/corest complex. Mol. Cell 2012, 46, 636–649. [Google Scholar] [CrossRef]
- Gomis, R.R.; Alarcon, C.; Nadal, C.; Van Poznak, C.; Massague, J. C/ebpbeta at the core of the tgfbeta cytostatic response and its evasion in metastatic breast cancer cells. Cancer Cell 2006, 10, 203–214. [Google Scholar] [CrossRef]
- Seoane, J.; Pouponnot, C.; Staller, P.; Schader, M.; Eilers, M.; Massague, J. Tgfbeta influences myc, miz-1 and smad to control the cdk inhibitor p15ink4b. Nat. Cell Biol. 2001, 3, 400–408. [Google Scholar] [CrossRef]
- Staller, P.; Peukert, K.; Kiermaier, A.; Seoane, J.; Lukas, J.; Karsunky, H.; Moroy, T.; Bartek, J.; Massague, J.; Hanel, F.; et al. Repression of p15ink4b expression by myc through association with miz-1. Nat. Cell Biol. 2001, 3, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Padua, D.; Massague, J. Roles of tgfbeta in metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Muthusamy, B.P.; Saeteurn, K.Y. Signaling pathway cooperation in tgf-beta-induced epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 2014, 31, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Massague, J. Transforming growth factor-beta signaling in immunity and cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Owens, P.; Moses, H.L. Tgf-beta, bone morphogenetic protein, and activin signaling and the tumor microenvironment. Cold Spring Harb. Perspect. Biol. 2017, 9, a022285. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Massague, J. Contextual determinants of tgfbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell. Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Ammanamanchi, S.; Howell, G.M. Epigenetic targeting of transforming growth factor beta receptor ii and implications for cancer therapy. Mol. Cell Pharmacol. 2009, 1, 57–70. [Google Scholar] [CrossRef]
- McDonald, O.G.; Wu, H.; Timp, W.; Doi, A.; Feinberg, A.P. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat. Struct. Mol. Biol. 2011, 18, 867–874. [Google Scholar] [CrossRef]
- Villarejo, A.; Cortes-Cabrera, A.; Molina-Ortiz, P.; Portillo, F.; Cano, A. Differential role of snail1 and snail2 zinc fingers in e-cadherin repression and epithelial to mesenchymal transition. J. Biol. Chem. 2014, 289, 930–941. [Google Scholar] [CrossRef]
- Kim, D.; Nam, H.J.; Lee, W.; Yim, H.Y.; Ahn, J.Y.; Park, S.W.; Shin, H.R.; Yu, R.; Won, K.J.; Bae, J.S.; et al. Pkcalpha-lsd1-nf-kappab-signaling cascade is crucial for epigenetic control of the inflammatory response. Mol. Cell 2018, 69, 398–411. [Google Scholar] [CrossRef]
- Ramadoss, S.; Chen, X.; Wang, C.Y. Histone demethylase kdm6b promotes epithelial-mesenchymal transition. J. Biol. Chem. 2012, 287, 44508–44517. [Google Scholar] [CrossRef] [PubMed]
- Vega, S.; Morales, A.V.; Ocana, O.H.; Valdes, F.; Fabregat, I.; Nieto, M.A. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004, 18, 1131–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Sung, I.J.; Lee, S.W.; Kim, K.W.; Kim, Y.S.; Yoo, M.A. The zinc-finger transcription factor snail downregulates proliferating cell nuclear antigen expression in colorectal carcinoma cells. Int. J. Oncol. 2005, 26, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.A. Opposing functions of zeb proteins in the regulation of the tgfbeta/bmp signaling pathway. EMBO J. 2003, 22, 2443–2452. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.A.; Depp, J.L.; Taylor, J.J.; Kroll, K.L. Regulation of smad signaling through a differential recruitment of coactivators and corepressors by zeb proteins. EMBO J. 2003, 22, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Mejlvang, J.; Kriajevska, M.; Vandewalle, C.; Chernova, T.; Sayan, A.E.; Berx, G.; Mellon, J.K.; Tulchinsky, E. Direct repression of cyclin d1 by sip1 attenuates cell cycle progression in cells undergoing an epithelial mesenchymal transition. Mol. Biol. Cell 2007, 18, 4615–4624. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, D.L.; Lin, W.; Creighton, C.J.; Rizvi, Z.H.; Gregory, P.A.; Goodall, G.J.; Thilaganathan, N.; Du, L.; Zhang, Y.; Pertsemlidis, A. Contextual extracellular cues promote tumor cell emt and metastasis by regulating mir-200 family expression. Genes Dev. 2009, 23, 2140–2151. [Google Scholar] [CrossRef] [PubMed]
- Eades, G.; Yao, Y.; Yang, M.; Zhang, Y.; Chumsri, S.; Zhou, Q. Mir-200a regulates sirt1 expression and epithelial to mesenchymal transition (emt)-like transformation in mammary epithelial cells. J. Biol. Chem. 2011, 286, 25992–26002. [Google Scholar] [CrossRef]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Gupta, S.C.; Kim, J.H. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 2012, 119, 651–665. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef]
- Perkins, N.D. Integrating cell-signalling pathways with nf-κb and ikk function. Nat. Rev. Mol. Cell. Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef]
- Perkins, N.D. The diverse and complex roles of nf-κb subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef]
- Karin, M. Nf-kappab as a critical link between inflammation and cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a000141. [Google Scholar] [CrossRef]
- DiDonato, J.A.; Mercurio, F.; Karin, M. Nf-κb and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef]
- Li, C.W.; Xia, W.; Huo, L.; Lim, S.O.; Wu, Y.; Hsu, J.L.; Chao, C.H.; Yamaguchi, H.; Yang, N.K.; Ding, Q.; et al. Epithelial-mesenchymal transition induced by tnf-alpha requires nf-kappab-mediated transcriptional upregulation of twist1. Cancer Res. 2012, 72, 1290–1300. [Google Scholar] [CrossRef]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of e-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef]
- Chua, H.L.; Bhat-Nakshatri, P.; Clare, S.E.; Morimiya, A.; Badve, S.; Nakshatri, H. Nf-kappab represses e-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: Potential involvement of zeb-1 and zeb-2. Oncogene 2007, 26, 711–724. [Google Scholar] [CrossRef]
- Sullivan, D.E.; Ferris, M.; Nguyen, H.; Abboud, E.; Brody, A.R. Tnf-alpha induces tgf-beta1 expression in lung fibroblasts at the transcriptional level via ap-1 activation. J. Cell. Mol. Med. 2009, 13, 1866–1876. [Google Scholar] [CrossRef]
- Bates, R.C.; Mercurio, A.M. Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol. Biol. Cell 2003, 14, 1790–1800. [Google Scholar] [CrossRef]
- Maier, H.J.; Schmidt-Strassburger, U.; Huber, M.A.; Wiedemann, E.M.; Beug, H.; Wirth, T. Nf-kappab promotes epithelial-mesenchymal transition, migration and invasion of pancreatic carcinoma cells. Cancer Lett. 2010, 295, 214–228. [Google Scholar] [CrossRef]
- Kumar, M.; Allison, D.F.; Baranova, N.N.; Wamsley, J.J.; Katz, A.J.; Bekiranov, S.; Jones, D.R.; Mayo, M.W. Nf-kappab regulates mesenchymal transition for the induction of non-small cell lung cancer initiating cells. PLoS ONE 2013, 8, e68597. [Google Scholar] [CrossRef]
- Cottonham, C.L.; Kaneko, S.; Xu, L. Mir-21 and mir-31 converge on tiam1 to regulate migration and invasion of colon carcinoma cells. J. Biol. Chem. 2010, 285, 35293–35302. [Google Scholar] [CrossRef]
- Saito, Y.; Liang, G.; Egger, G.; Friedman, J.M.; Chuang, J.C.; Coetzee, G.A.; Jones, P.A. Specific activation of microrna-127 with downregulation of the proto-oncogene bcl6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006, 9, 435–443. [Google Scholar] [CrossRef]
- Liu, Y.; Mayo, M.W.; Nagji, A.S.; Smith, P.W.; Ramsey, C.S.; Li, D.; Jones, D.R. Phosphorylation of rela/p65 promotes dnmt-1 recruitment to chromatin and represses transcription of the tumor metastasis suppressor gene brms1. Oncogene 2012, 31, 1143–1154. [Google Scholar] [CrossRef]
- Wang, Y.; Hou, N.; Cheng, X.; Zhang, J.; Tan, X.; Zhang, C.; Tang, Y.; Teng, Y.; Yang, X. Ezh2 acts as a tumor suppressor in kras-driven lung adenocarcinoma. Int. J. Biol. Sci. 2017, 13, 652–659. [Google Scholar] [CrossRef]
- Liu, Y.; Peng, J.; Sun, T.; Li, N.; Zhang, L.; Ren, J.; Yuan, H.; Kan, S.; Pan, Q.; Li, X.; et al. Epithelial ezh2 serves as an epigenetic determinant in experimental colitis by inhibiting tnfalpha-mediated inflammation and apoptosis. Proc. Natl. Acad. Sci. USA 2017, 114, E3796–E3805. [Google Scholar] [CrossRef]
- Yee, D.; Shah, K.M.; Coles, M.C.; Sharp, T.V.; Lagos, D. Microrna-155 induction via tnf-alpha and ifn-gamma suppresses expression of programmed death ligand-1 (pd-l1) in human primary cells. J. Biol. Chem. 2017, 292, 20683–20693. [Google Scholar] [CrossRef]
- Shen, Z.; Zhou, R.; Liu, C.; Wang, Y.; Zhan, W.; Shao, Z.; Liu, J.; Zhang, F.; Xu, L.; Zhou, X.; et al. Microrna-105 is involved in tnf-alpha-related tumor microenvironment enhanced colorectal cancer progression. Cell Death Dis. 2017, 8, 3213. [Google Scholar] [CrossRef]
- Mantovani, A.; Barajon, I.; Garlanda, C. Il-1 and il-1 regulatory pathways in cancer progression and therapy. Immunol. Rev. 2018, 281, 57–61. [Google Scholar] [CrossRef]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and related cytokines in the regulation of inflammation and immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef]
- Kaneko, N.; Kurata, M.; Yamamoto, T.; Morikawa, S.; Masumoto, J. The role of interleukin-1 in general pathology. Inflamm. Regen. 2019, 39, 12. [Google Scholar] [CrossRef] [Green Version]
- Baker, K.J.; Houston, A.; Brint, E. Il-1 family members in cancer; two sides to every story. Front. Immunol. 2019, 10, 1197. [Google Scholar] [CrossRef]
- Boraschi, D.; Italiani, P.; Weil, S.; Martin, M.U. The family of the interleukin-1 receptors. Immunol. Rev. 2018, 281, 197–232. [Google Scholar] [CrossRef]
- Voronov, E.; Dinarello, C.A.; Apte, R.N. Interleukin-1alpha as an intracellular alarmin in cancer biology. Semin. Immunol. 2018, 38, 3–14. [Google Scholar] [CrossRef]
- Malik, A.; Kanneganti, T.D. Function and regulation of il-1alpha in inflammatory diseases and cancer. Immunol. Rev. 2018, 281, 124–137. [Google Scholar] [CrossRef]
- Soria, G.; Ofri-Shahak, M.; Haas, I.; Yaal-Hahoshen, N.; Leider-Trejo, L.; Leibovich-Rivkin, T.; Weitzenfeld, P.; Meshel, T.; Shabtai, E.; Gutman, M.; et al. Inflammatory mediators in breast cancer: Coordinated expression of tnfalpha & il-1beta with ccl2 & ccl5 and effects on epithelial-to-mesenchymal transition. BMC Cancer 2011, 11, 130. [Google Scholar]
- Li, H.J.; Reinhardt, F.; Herschman, H.R.; Weinberg, R.A. Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin e2 signaling. Cancer Discov. 2012, 2, 840–855. [Google Scholar] [CrossRef]
- Cataisson, C.; Salcedo, R.; Hakim, S.; Moffitt, B.A.; Wright, L.; Yi, M.; Stephens, R.; Dai, R.M.; Lyakh, L.; Schenten, D.; et al. Il-1r-myd88 signaling in keratinocyte transformation and carcinogenesis. J. Exp. Med. 2012, 209, 1689–1702. [Google Scholar] [CrossRef]
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. Krasg12d-induced ikk2/beta/nf-kappab activation by il-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 105–120. [Google Scholar] [CrossRef]
- Niu, J.; Li, Z.; Peng, B.; Chiao, P.J. Identification of an autoregulatory feedback pathway involving interleukin-1alpha in induction of constitutive nf-kappab activation in pancreatic cancer cells. J. Biol. Chem. 2004, 279, 16452–16462. [Google Scholar] [CrossRef]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. Il-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef]
- Krelin, Y.; Voronov, E.; Dotan, S.; Elkabets, M.; Reich, E.; Fogel, M.; Huszar, M.; Iwakura, Y.; Segal, S.; Dinarello, C.A.; et al. Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res. 2007, 67, 1062–1071. [Google Scholar] [CrossRef]
- Charuworn, B.; Dohadwala, M.; Krysan, K.; Sharma, S.; Escuadro, B.; Dubinett, S. Inflammation-mediated promotion of emt in nsclc: Il-1b mediates a mek/erk-and jnk/sapk-dependent downregulation of e-cadherin. Proc. Am. Thorac. Soc. 2006, 3, D96. [Google Scholar]
- Liu, X. Inflammatory cytokines augments tgf-β1-induced epithelial-mesenchymal transition in a549 cells by up-regulating tβr-i. Cell Motil. Cytoskelet. 2008, 65, 935–944. [Google Scholar] [CrossRef]
- Petrella, B.L.; Armstrong, D.A.; Vincenti, M.P. Interleukin-1 beta and transforming growth factor-beta 3 cooperate to activate matrix metalloproteinase expression and invasiveness in a549 lung adenocarcinoma cells. Cancer Lett. 2012, 325, 220–226. [Google Scholar] [CrossRef]
- Jiménez-Garduño, A.M.; Mendoza-Rodríguez, M.G.; Urrutia-Cabrera, D.; Domínguez-Robles, M.C.; Pérez-Yépez, E.A.; Ayala-Sumuano, J.T.; Meza, I. Il-1β induced methylation of the estrogen receptor erα gene correlates with emt and chemoresistance in breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 490, 780–785. [Google Scholar] [CrossRef]
- Perez-Yepez, E.A.; Ayala-Sumuano, J.T.; Lezama, R.; Meza, I. A novel beta-catenin signaling pathway activated by il-1beta leads to the onset of epithelial-mesenchymal transition in breast cancer cells. Cancer Lett. 2014, 354, 164–171. [Google Scholar] [CrossRef]
- Mendoza-Rodriguez, M.; Arevalo Romero, H.; Fuentes-Panana, E.M.; Ayala-Sumuano, J.T.; Meza, I. Il-1beta induces up-regulation of birc3, a gene involved in chemoresistance to doxorubicin in breast cancer cells. Cancer Lett. 2017, 390, 39–44. [Google Scholar] [CrossRef]
- Li, Y.; Wang, L.; Pappan, L.; Galliher-Beckley, A.; Shi, J. Il-1beta promotes stemness and invasiveness of colon cancer cells through zeb1 activation. Mol. Cancer 2012, 11, 87. [Google Scholar] [CrossRef]
- Caradonna, F.; Cruciata, I.; Schifano, I.; La Rosa, C.; Naselli, F.; Chiarelli, R.; Perrone, A.; Gentile, C. Methylation of cytokines gene promoters in il-1beta-treated human intestinal epithelial cells. Inflamm. Res. 2018, 67, 327–337. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting hdac2 to specifically repress il-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef]
- Ko, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Thompson, E.C.; Hastie, R.; Tsangaratou, A.; Rajewsky, K.; Koralov, S.B.; Rao, A. Ten-eleven-translocation 2 (tet2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 14566–14571. [Google Scholar] [CrossRef]
- Pan, W.; Zhu, S.; Qu, K.; Meeth, K.; Cheng, J.; He, K.; Ma, H.; Liao, Y.; Wen, X.; Roden, C.; et al. The DNA methylcytosine dioxygenase tet2 sustains immunosuppressive function of tumor-infiltrating myeloid cells to promote melanoma progression. Immunity 2017, 47, 284–297. [Google Scholar] [CrossRef]
- Ma, J.; Liu, J.; Wang, Z.; Gu, X.; Fan, Y.; Zhang, W.; Xu, L.; Zhang, J.; Cai, D. Nf-kappab-dependent microrna-425 upregulation promotes gastric cancer cell growth by targeting pten upon il-1β induction. Mol. Cancer 2014, 13, 40. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, L.F.; Wu, J.; Xu, S.J.; Xu, Y.Y.; Li, D.; Lou, J.T.; Liu, M.F. Il-1beta-mediated repression of microrna-101 is crucial for inflammation-promoted lung tumorigenesis. Cancer Res. 2014, 74, 4720–4730. [Google Scholar] [CrossRef]
- Arts, N.; Cane, S.; Hennequart, M.; Lamy, J.; Bommer, G.; Van den Eynde, B.; De Plaen, E. Microrna-155, induced by interleukin-1ss, represses the expression of microphthalmia-associated transcription factor (mitf-m) in melanoma cells. PLoS ONE 2015, 10, e0122517. [Google Scholar] [CrossRef]
- Hai Ping, P.; Feng Bo, T.; Li, L.; Nan Hui, Y.; Hong, Z. Il-1beta/nf-kb signaling promotes colorectal cancer cell growth through mir-181a/pten axis. Arch. Biochem. Biophys. 2016, 604, 20–26. [Google Scholar] [CrossRef]
- Hu, M.; Yuan, X.; Liu, Y.; Tang, S.; Miao, J.; Zhou, Q.; Chen, S. Il-1beta-induced nf-kappab activation down-regulates mir-506 expression to promotes osteosarcoma cell growth through jag1. Biomed. Pharm. 2017, 95, 1147–1155. [Google Scholar] [CrossRef]
- Fisher, D.T.; Appenheimer, M.M.; Evans, S.S. The two faces of il-6 in the tumor microenvironment. Semin. Immunol. 2014, 26, 38–47. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. Il-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin. Immunol. 2014, 26, 54–74. [Google Scholar] [CrossRef]
- Chang, Q.; Daly, L.; Bromberg, J. The il-6 feed-forward loop: A driver of tumorigenesis. Semin. Immunol. 2014, 26, 48–53. [Google Scholar] [CrossRef]
- Unver, N.; McAllister, F. Il-6 family cytokines: Key inflammatory mediators as biomarkers and potential therapeutic targets. Cytokine Growth Factor Rev. 2018, 41, 10–17. [Google Scholar] [CrossRef]
- Taher, M.Y.; Davies, D.M.; Maher, J. The role of the interleukin (il)-6/il-6 receptor axis in cancer. Biochem. Soc. Trans. 2018, 46, 1449–1462. [Google Scholar] [CrossRef]
- Kang, S.; Tanaka, T.; Narazaki, M.; Kishimoto, T. Targeting interleukin-6 signaling in clinic. Immunity 2019, 50, 1007–1023. [Google Scholar] [CrossRef]
- Heink, S.; Yogev, N.; Garbers, C.; Herwerth, M.; Aly, L.; Gasperi, C.; Husterer, V.; Croxford, A.L.; Moller-Hackbarth, K.; Bartsch, H.S.; et al. Trans-presentation of il-6 by dendritic cells is required for the priming of pathogenic th17 cells. Nat. Immunol. 2017, 18, 74–85. [Google Scholar] [CrossRef]
- Scheller, J.; Garbers, C.; Rose-John, S. Interleukin-6: From basic biology to selective blockade of pro-inflammatory activities. Semin. Immunol. 2014, 26, 2–12. [Google Scholar] [CrossRef]
- Ancrile, B.; Lim, K.H.; Counter, C.M. Oncogenic ras-induced secretion of il6 is required for tumorigenesis. Genes Dev. 2007, 21, 1714–1719. [Google Scholar] [CrossRef]
- Leslie, K.; Gao, S.P.; Berishaj, M.; Podsypanina, K.; Ho, H.; Ivashkiv, L.; Bromberg, J. Differential interleukin-6/stat3 signaling as a function of cellular context mediates ras-induced transformation. Breast Cancer Res. 2010, 12, R80. [Google Scholar] [CrossRef]
- Chou, C.H.; Wei, L.H.; Kuo, M.L.; Huang, Y.J.; Lai, K.P.; Chen, C.A.; Hsieh, C.Y. Up-regulation of interleukin-6 in human ovarian cancer cell via a gi/pi3k-akt/nf-kappab pathway by lysophosphatidic acid, an ovarian cancer-activating factor. Carcinogenesis 2005, 26, 45–52. [Google Scholar] [CrossRef]
- Dagia, N.M.; Agarwal, G.; Kamath, D.V.; Chetrapal-Kunwar, A.; Gupte, R.D.; Jadhav, M.G.; Dadarkar, S.S.; Trivedi, J.; Kulkarni-Almeida, A.A.; Kharas, F.; et al. A preferential p110alpha/gamma pi3k inhibitor attenuates experimental inflammation by suppressing the production of proinflammatory mediators in a nf-kappab-dependent manner. Am. J. Physiol. Cell Physiol. 2010, 298, C929–C941. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef]
- Naugler, W.E.; Karin, M. The wolf in sheep’s clothing: The role of interleukin-6 in immunity, inflammation and cancer. Trends Mol. Med. 2008, 14, 109–119. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an nf-kappab-dependent manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef]
- Hirano, T.; Ishihara, K.; Hibi, M. Roles of stat3 in mediating the cell growth, differentiation and survival signals relayed through the il-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556. [Google Scholar] [CrossRef]
- Gritsko, T.; Williams, A.; Turkson, J.; Kaneko, S.; Bowman, T.; Huang, M.; Nam, S.; Eweis, I.; Diaz, N.; Sullivan, D.; et al. Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin. Cancer Res. 2006, 12, 11–19. [Google Scholar] [CrossRef]
- Liu, C.C.; Lin, J.H.; Hsu, T.W.; Su, K.; Li, A.F.; Hsu, H.S.; Hung, S.C. Il-6 enriched lung cancer stem-like cell population by inhibition of cell cycle regulators via dnmt1 upregulation. Int. J. Cancer 2015, 136, 547–559. [Google Scholar]
- Hodge, D.R.; Cho, E.; Copeland, T.D.; Guszczynski, T.; Yang, E.; Seth, A.K.; Farrar, W.L. Il-6 enhances the nuclear translocation of DNA cytosine-5-methyltransferase 1 (dnmt1) via phosphorylation of the nuclear localization sequence by the akt kinase. Cancer Genom. Proteom. 2007, 4, 387–398. [Google Scholar]
- Hodge, D.R.; Xiao, W.; Peng, B.; Cherry, J.C.; Munroe, D.J.; Farrar, W.L. Enforced expression of superoxide dismutase 2/manganese superoxide dismutase disrupts autocrine interleukin-6 stimulation in human multiple myeloma cells and enhances dexamethasone-induced apoptosis. Cancer Res. 2005, 65, 6255–6263. [Google Scholar] [CrossRef]
- D’Anello, L.; Sansone, P.; Storci, G.; Mitrugno, V.; D’Uva, G.; Chieco, P.; Bonafe, M. Epigenetic control of the basal-like gene expression profile via interleukin-6 in breast cancer cells. Mol. Cancer 2010, 9, 300. [Google Scholar] [CrossRef]
- Angelo, L.S.; Talpaz, M.; Kurzrock, R. Autocrine interleukin-6 production in renal cell carcinoma: Evidence for the involvement of p53. Cancer Res. 2002, 62, 932–940. [Google Scholar]
- Esteve, P.O.; Chin, H.G.; Pradhan, S. Human maintenance DNA (cytosine-5)-methyltransferase and p53 modulate expression of p53-repressed promoters. Proc. Natl. Acad. Sci. USA 2005, 102, 1000–1005. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Zhang, P.; Herrmann, A.; Yang, C.; Xin, H.; Wang, Z.; Hoon, D.S.; Forman, S.J.; Jove, R.; Riggs, A.D.; et al. Acetylated stat3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. USA 2012, 109, 7765–7769. [Google Scholar] [CrossRef]
- Gasche, J.A.; Hoffmann, J.; Boland, C.R.; Goel, A. Interleukin-6 promotes tumorigenesis by altering DNA methylation in oral cancer cells. Int. J. Cancer 2011, 129, 1053–1063. [Google Scholar] [CrossRef]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef]
- Al-Ismaeel, Q.; Neal, C.P.; Al-Mahmoodi, H.; Almutairi, Z.; Al-Shamarti, I.; Straatman, K.; Jaunbocus, N.; Irvine, A.; Issa, E.; Moreman, C.; et al. Zeb1 and il-6/11-stat3 signalling cooperate to define invasive potential of pancreatic cancer cells via differential regulation of the expression of s100 proteins. Br. J. Cancer 2019, 121, 65–75. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Huerta-Yepez, S.; Law, I.; Baay-Guzman, G.J.; Tirado-Rodriguez, B.; Hoffman, J.M.; Iliopoulos, D.; Hommes, D.W.; Verspaget, H.W.; Chang, L. Diminished expression of crhr2 in human colon cancer promotes tumor growth and emt via persistent il-6/stat3 signaling. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 610–630. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Lo, H.W. Stat3 target genes relevant to human cancers. Cancers (Basel) 2014, 6, 897–925. [Google Scholar] [CrossRef]
- Pires, B.R.; Mencalha, A.L.; Ferreira, G.M.; de Souza, W.F.; Morgado-Diaz, J.A.; Maia, A.M.; Correa, S.; Abdelhay, E.S. Nf-kappab is involved in the regulation of emt genes in breast cancer cells. PLoS ONE 2017, 12, e0169622. [Google Scholar] [CrossRef]
- Huang, C.; Yang, G.; Jiang, T.; Zhu, G.; Li, H.; Qiu, Z. The effects and mechanisms of blockage of stat3 signaling pathway on il-6 inducing emt in human pancreatic cancer cells in vitro. Neoplasma 2011, 58, 396. [Google Scholar] [CrossRef]
- Dehai, C.; Bo, P.; Qiang, T.; Lihua, S.; Fang, L.; Shi, J.; Jingyan, C.; Yan, Y.; Guangbin, W.; Zhenjun, Y. Enhanced invasion of lung adenocarcinoma cells after co-culture with thp-1-derived macrophages via the induction of emt by il-6. Immunol. Lett. 2014, 160, 1–10. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, H.; Liu, Y.; Song, Y.; Lai, L.; Han, Q.; Cao, X.; Wang, Q. Inducible microrna-223 down-regulation promotes tlr-triggered il-6 and il-1β production in macrophages by targeting stat3. PLoS ONE 2012, 7, e42971. [Google Scholar] [CrossRef]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. Micrornas bind to toll-like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 2012, 109, E2110–E2116. [Google Scholar] [CrossRef]
- Xue, X.; Xia, W.; Wenzhong, H. A modeled dynamic regulatory network of nf-kappab and il-6 mediated by mirna. Biosystems 2013, 114, 214–218. [Google Scholar] [CrossRef]
- Xiang, M.; Birkbak, N.J.; Vafaizadeh, V.; Walker, S.R.; Yeh, J.E.; Liu, S.; Kroll, Y.; Boldin, M.; Taganov, K.; Groner, B. Stat3 induction of mir-146b forms a feedback loop to inhibit the nf-κb to il-6 signaling axis and stat3-driven cancer phenotypes. Sci. Signal. 2014, 7, ra11. [Google Scholar] [CrossRef]
- Kumar, M.S.; Erkeland, S.J.; Pester, R.E.; Chen, C.Y.; Ebert, M.S.; Sharp, P.A.; Jacks, T. Suppression of non-small cell lung tumor development by the let-7 microrna family. Proc. Natl. Acad. Sci. USA 2008, 105, 3903–3908. [Google Scholar] [CrossRef]
- Jiang, S.; Yan, W.; Wang, S.E.; Baltimore, D. Dual mechanisms of posttranscriptional regulation of tet2 by let-7 microrna in macrophages. Proc. Natl. Acad. Sci. USA 2019, 116, 12416–12421. [Google Scholar] [CrossRef]
- Fabbri, M. Tlrs as mirna receptors. Cancer Res. 2012, 72, 6333–6337. [Google Scholar] [CrossRef]
- Gupta, P.B.; Chaffer, C.L.; Weinberg, R.A. Cancer stem cells: Mirage or reality? Nat. Med. 2009, 15, 1010–1012. [Google Scholar] [CrossRef]
- Katsuno, Y.; Qin, J.; Oses-Prieto, J.; Wang, H.; Jackson-Weaver, O.; Zhang, T.; Lamouille, S.; Wu, J.; Burlingame, A.; Xu, J.; et al. Arginine methylation of smad7 by prmt1 in tgf-beta-induced epithelial-mesenchymal transition and epithelial stem-cell generation. J. Biol. Chem. 2018, 293, 13059–13072. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.A.; Sossey-Alaoui, K.; Thompson, C.L.; Danielpour, D.; Schiemann, W.P. Tgf-beta upregulates mir-181a expression to promote breast cancer metastasis. J. Clin. Invest. 2013, 123, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Hsu, S.H.; Majumder, S.; Kutay, H.; Huang, W.; Jacob, S.T.; Ghoshal, K. Tgfbeta-mediated upregulation of hepatic mir-181b promotes hepatocarcinogenesis by targeting timp3. Oncogene 2010, 29, 1787–1797. [Google Scholar] [CrossRef] [PubMed]
- Keklikoglou, I.; Koerner, C.; Schmidt, C.; Zhang, J.; Heckmann, D.; Shavinskaya, A.; Allgayer, H.; Gückel, B.; Fehm, T.; Schneeweiss, A. Microrna-520/373 family functions as a tumor suppressor in estrogen receptor negative breast cancer by targeting nf-κb and tgf-β signaling pathways. Oncogene 2012, 31, 4150. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Storci, G.; Tavolari, S.; Guarnieri, T.; Giovannini, C.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Paterini, P.; Marcu, K.B.; et al. Il-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Invest. 2007, 117, 3988–4002. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Kim, G.I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K.; et al. Activation of an il6 inflammatory loop mediates trastuzumab resistance in her2+ breast cancer by expanding the cancer stem cell population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Cho, H.J.; Cho, S.M.; Jo, K.; Park, J.A.; Kim, N.H.; Amidon, G.L.; Kim, J.S.; Shin, H.C. Blockade of interleukin-6 receptor suppresses the proliferation of h460 lung cancer stem cells. Int. J. Oncol. 2012, 41, 310–316. [Google Scholar]
- Studebaker, A.W.; Storci, G.; Werbeck, J.L.; Sansone, P.; Sasser, A.K.; Tavolari, S.; Huang, T.; Chan, M.W.; Marini, F.C.; Rosol, T.J.; et al. Fibroblasts isolated from common sites of breast cancer metastasis enhance cancer cell growth rates and invasiveness in an interleukin-6-dependent manner. Cancer Res. 2008, 68, 9087–9095. [Google Scholar] [CrossRef]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The jak2/stat3 signaling pathway is required for growth of cd44(+)cd24(-) stem cell-like breast cancer cells in human tumors. J. Clin. Invest. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kang, J.W.; Song, X.; Kim, B.K.; Yoo, Y.D.; Kwon, Y.T.; Lee, Y.J. Role of the il-6-jak1-stat3-oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell Signal. 2013, 25, 961–969. [Google Scholar] [CrossRef]
- Kim, M.Y.; Oskarsson, T.; Acharyya, S.; Nguyen, D.X.; Zhang, X.H.; Norton, L.; Massague, J. Tumor self-seeding by circulating cancer cells. Cell 2009, 139, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Ouzounova, M.; Quraishi, A.A.; Davis, A.; Tawakkol, N.; Clouthier, S.G.; Malik, F.; Paulson, A.K.; D’Angelo, R.C.; Korkaya, S.; et al. Socs3-mediated regulation of inflammatory cytokines in pten and p53 inactivated triple negative breast cancer model. Oncogene 2015, 34, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Hodge, D.R.; Peng, B.; Cherry, J.C.; Hurt, E.M.; Fox, S.D.; Kelley, J.A.; Munroe, D.J.; Farrar, W.L. Interleukin 6 supports the maintenance of p53 tumor suppressor gene promoter methylation. Cancer Res. 2005, 65, 4673–4682. [Google Scholar] [CrossRef] [PubMed]
- Chiou, G.Y.; Chien, C.S.; Wang, M.L.; Chen, M.T.; Yang, Y.P.; Yu, Y.L.; Chien, Y.; Chang, Y.C.; Shen, C.C.; Chio, C.C.; et al. Epigenetic regulation of the mir142-3p/interleukin-6 circuit in glioblastoma. Mol. Cell 2013, 52, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Rokavec, M.; Wu, W.; Luo, J.L. Il6-mediated suppression of mir-200c directs constitutive activation of inflammatory signaling circuit driving transformation and tumorigenesis. Mol. Cell 2012, 45, 777–789. [Google Scholar] [CrossRef]
- Zhao, Y.; Xu, Y.; Li, Y.; Xu, W.; Luo, F.; Wang, B.; Pang, Y.; Xiang, Q.; Zhou, J.; Wang, X.; et al. Nf-kappab-mediated inflammation leading to emt via mir-200c is involved in cell transformation induced by cigarette smoke extract. Toxicol. Sci. 2013, 135, 265–276. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Cytokine | Master TFs | Epigenetic Mechanism | Effect |
|---|---|---|---|
| TGFβ | Smads | Induction of Snail, Slug, and Zeb TFs up-regulates mesenchymal genes through reprogramming of specific chromatin domains (reduction of histone methylation by LSD1 and KDM6B/JMJD3 demethylases) | EMT and cancer progression |
| TGFβ | Smads | Snail- and Zeb-dependent down-regulation of miR-34 and miR-200 (double negative feedback loops) SIRT1-dependent miR-200 down-regulation by histone deacetylation (negative feedback loop) | Induction of EMT and inhibition of MET |
| TGFβ | Various TFs | Several Smad-independent alterations, through MAPKs, PI-3K, and ΝF-κB pathways | EMT and cancer progression |
| TGFβ | Smad2/3–Smad4–Foxo | Reduced proliferation through induction of CDKN2B/p15ink4b, CDK4 inhibitor (removal of DNA methylation on CDKN2B promoter) | Tumor suppression |
| TNFα | NF-κB | Induction of ΝF-κB-regulated proteins (IL-6, IL-8, IL-18, iNOS, COX-2, 5-LOX, etc.) | Inflammation-dependent cancer development |
| TNFα | NF-κB | Induction of Twist1, Snail, Snail2, and Zebs leading to E-cadherin down-regulation and mesenchymal genes induction | EMT, migration and invasion |
| TNFα | NF-κB | DNMT1-dependent down-regulation of metastasis suppressor BRSM1 | Invasion and metastasis |
| TGFβ + TNFα | Smads + NF-κB | Induction of EMT TFs leading to epigenetic alterations that induce mesenchymal genes and several oncomiRs (miR-21, miR-31, miR-23a, etc.) | EMT and cancer progression |
| IL-1 | NF-κB | Expression of several inflammatory mediators (including IL-1, leading to an autocrine loop) | Inflammation-dependent cancer development |
| IL-1α | NF-κB + STAT3 | Induction of stem-cell associated genes bmi-1 and Nestin | EMT and CSC development |
| IL-1β | β-catenin | Expression of c-MYC, CCDN1, Snail1 and MMP2, and BIRC3. Twist1-dependent methylation of ESR1 gene promoter | EMT and cancer progression Resistance to chemotherapy |
| IL-1β | NF-κB | Induction of miR-181 or down-regulation of miR-506 | Cancer progression |
| IL-1β + TNFα + TGFβ | Several | Induction of inflammatory genes and several MMPs | EMT, migration and invasion |
| IL-6 | STAT3 | Induction of cell cycle regulators (E2Fs, JunB, c-Fos, etc.) and metabolic regulators (mTORC1) | Cancer progression |
| IL-6 | - | DNMT1 dependent DNA methylation of tumor suppressor gene promoters (CHFR, GATA5, and PAX6), including TP53 (negative feedback loop) | Cancer progression and CSC formation |
| IL-6 | ΝF-κB + STAT3 | Induction of EMT-TFs Snail, Slug, Twist, and Zeb (down-regulation of E-cadherin and up-regulation of mesenchymal genes and MMPs) | EMT, migration and invasion |
| IL-6 | ΝF-κB + STAT3 | Feedback loops involving IL-6, ΝF-κB, Lin28, and let-7 miRNA or IL-6, ΝF-κB, STAT3, miR-21 and miR-181b-1, and PTEN and CYLD | Cancer progression |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markopoulos, G.S.; Roupakia, E.; Marcu, K.B.; Kolettas, E. Epigenetic Regulation of Inflammatory Cytokine-Induced Epithelial-To-Mesenchymal Cell Transition and Cancer Stem Cell Generation. Cells 2019, 8, 1143. https://doi.org/10.3390/cells8101143
Markopoulos GS, Roupakia E, Marcu KB, Kolettas E. Epigenetic Regulation of Inflammatory Cytokine-Induced Epithelial-To-Mesenchymal Cell Transition and Cancer Stem Cell Generation. Cells. 2019; 8(10):1143. https://doi.org/10.3390/cells8101143
Chicago/Turabian StyleMarkopoulos, Georgios S., Eugenia Roupakia, Kenneth B. Marcu, and Evangelos Kolettas. 2019. "Epigenetic Regulation of Inflammatory Cytokine-Induced Epithelial-To-Mesenchymal Cell Transition and Cancer Stem Cell Generation" Cells 8, no. 10: 1143. https://doi.org/10.3390/cells8101143