Epigenetic Changes as a Target in Aging Haematopoietic Stem Cells and Age-Related Malignancies


European Research Institute for the Biology of Ageing, University Medical Center Groningen, University of Groningen, 9700 Groningen, The Netherlands
*
Author to whom correspondence should be addressed.
Cells 2019, 8(8), 868; https://doi.org/10.3390/cells8080868
Submission received: 24 July 2019
/
Revised: 7 August 2019
/
Accepted: 9 August 2019
/
Published: 10 August 2019
(This article belongs to the Special Issue Aging and Regeneration)
Abstract
:Aging is associated with multiple molecular and functional changes in haematopoietic cells. Most notably, the self-renewal and differentiation potential of hematopoietic stem cells (HSCs) are compromised, resulting in myeloid skewing, reduced output of red blood cells and decreased generation of immune cells. These changes result in anaemia, increased susceptibility for infections and higher prevalence of haematopoietic malignancies. In HSCs, age-associated global epigenetic changes have been identified. These epigenetic alterations in aged HSCs can occur randomly (epigenetic drift) or are the result of somatic mutations in genes encoding for epigenetic proteins. Mutations in loci that encode epigenetic modifiers occur frequently in patients with haematological malignancies, but also in healthy elderly individuals at risk to develop these. It may be possible to pharmacologically intervene in the aberrant epigenetic program of derailed HSCs to enforce normal haematopoiesis or treat age-related haematopoietic diseases. Over the past decade our molecular understanding of epigenetic regulation has rapidly increased and drugs targeting epigenetic modifications are increasingly part of treatment protocols. The reversibility of epigenetic modifications renders these targets for novel therapeutics. In this review we provide an overview of epigenetic changes that occur in aging HSCs and age-related malignancies and discuss related epigenetic drugs.
1. Epigenetic Regulation of HSCs
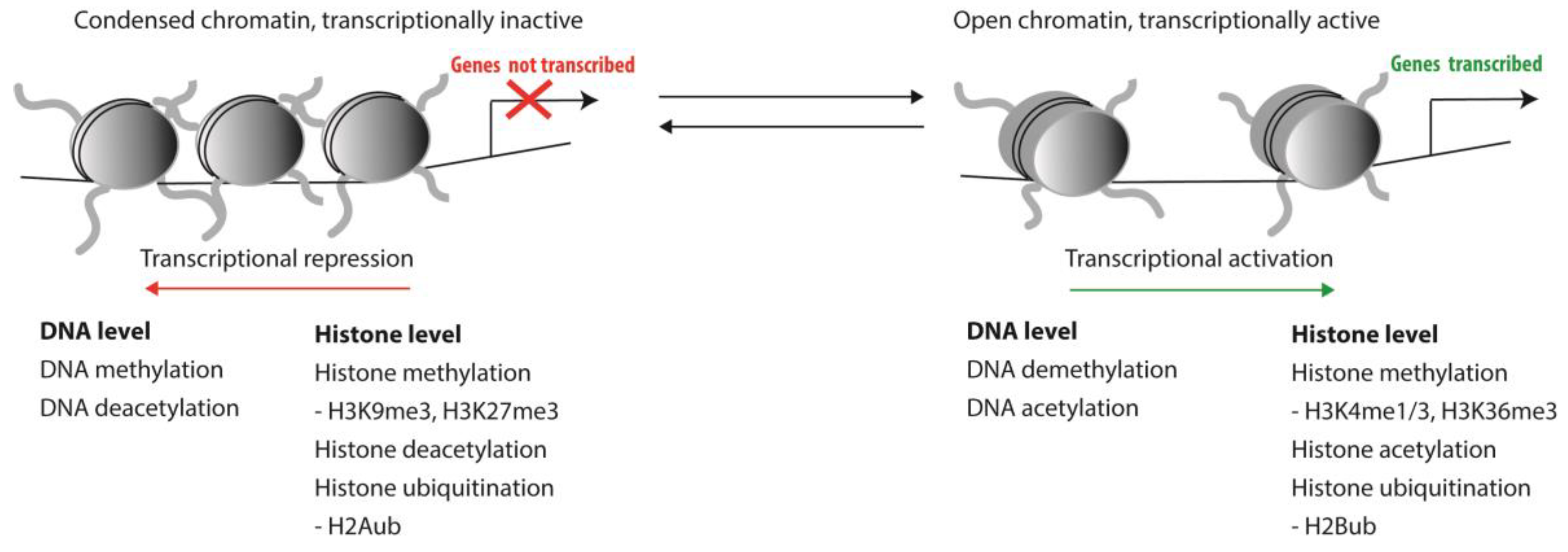
Haematopoiesis is a tightly regulated process and involves the control of transcription of genes that guard the balance between self-renewal of HSCs and their proper differentiation into all mature blood cell lineages. As primitive stem cells differentiate towards functionally mature peripheral blood cells, massive genome-wide changes in gene expression patterns occur [1]. In contrast, when HSCs self-renew at least one of the two daughter cells is expected to inherit a gene expression pattern that is highly similar, if not identical, to the mother stem cell. Genes involved in differentiation pathways must be repressed in HSCs, while self-renewal genes are repressed upon differentiation. Complex epigenetic control mechanisms play a crucial role in coordinating gene expression patterns that guard this balance between self-renewal and differentiation. Such epigenetic modifications involve both DNA methylation and chromatin conformation and, thus, impact the accessibility of DNA to transcription factors and the transcriptional machinery. Posttranslational modifications of histones that affect chromatin conformation mostly include methylation, acetylation and ubiquitination of histone tails (Figure 1).
Such epigenetic modifications can be actively deposited or removed by epigenetic enzymes. The role of these enzymes in haematopoiesis and leukemogenesis will be the main focus of this review. Passive (non-enzyme mediated) changes of epigenetic modifications also take place and are part of the replication process of cells. Every cell division changes in epigenetic landscape occur, most of which are temporary. For example, during DNA replication the newly formed strand is generated without the methylation marks present on the template strand, referred to as passive DNA methylation. In contrast to active DNA demethylation, DNA replication associated passive demethylation is not complete, since methylation marks on the parental strand remain. The parental methylation pattern on the newly formed strand is deposited by DNA methylases, such as DNMT1 [2]. Beyond DNA methylation, the parental epigenetic histone marks also have to be established on the newly synthesized histones. Replication stress can disturb posttranscriptional deposition of the parental chromatin marks and DNA methylation state and, thus, lead to epigenetic changes [3].
On the other hand, new DNA methylation patterns and histone modifications are actively deposited or removed by dedicated enzymes, or recognized by specific readers. Importantly, a large body of evidence has demonstrated an important, if not essential, role for many of these epigenetic writers, readers, and erasers in normal haematopoiesis. Multiple epigenetic proteins have been associated to control self-renewal capacity of HSCs. Table 1 provides an overview of the most prominent of such epigenetic haematopoietic stem cell genes.
2. Epigenetic Changes during Normal Aging of Murine HSCs
Genome-wide changes of epigenetic modifications are not only observed upon differentiation, but have also been identified during normal HSC aging. Studies in mice have shown that aged HSCs have low self-renewal potential [69,70,71,72,73], which appears to be inversely related to the number of cell divisions a HSC has undergone [74]. Importantly, the functional capacity of individual murine HSCs is highly variable and this variation increases during aging. Whereas the large majority of aged HSCs performs worse than their young counterparts, there are also aged HSC that function just fine [72,75,76]. This massive cell-to-cell variability is unlikely to result from the accumulation of random genetic mutations, as HSCs divide only very rarely during the lifespan of an organism [77]. Instead, functional heterogeneity is easier explained by random alterations of epigenetic modifications that may occur with each cell division.
Indeed, the functional heterogeneity and limited overlap of differentially expressed genes upon aging of murine HSCs supports the idea that deterioration of HSC functioning during aging may be caused by cumulative and random epigenetically inherited gene expression changes, rather than by mutations in specific loci [69,70,78,79,80,81,82,83,84,85,86,87,88,89],[Lazare et al. 2016(unpublished)]. Although a comprehensive overview of alterations of epigenetic histone and DNA modifications that are present in aged murine HSC is lacking, several age-associated epigenetic alterations have been identified. For example, in old HSCs trimethylation of lysine 4 of histone 3 (H3K4me3) is more widespread, while a gradual increase of global DNA methylation levels is observed. This is accompanied by a reduction of 5-hmC in old HSCs compared to young [79,90]. Concordantly, old HSCs have been reported to down-regulate expression of genes involved in chromatin regulation, such as chromatin remodelling genes (Smarca4, Smarcb1), histone deacetylases (Hdac 1, 5,6) and DNA methyltransferases (Dnmt3b) [69]. Intriguingly, chimeric mice produced from induced pluripotent stem (iPS) cells that were derived from aged HSCs presented with a rejuvenated haematopoietic system, suggesting that it is possible to reprogram aged HSCs and reverse the aging process [91]. These findings strongly suggest that it is indeed—potentially reversible—epigenetic changes that causally contribute to HSC aging.
3. Epigenetic Changes during Normal Aging of Human HSCs
Also in the haematopoietic system of humans there are indications that HSC function deteriorates with age. For example, inferior engraftment is seen when HSCs from older donors are transplanted compared to young donors [92]. Beyond differences in functionality, changes in the epigenetic landscape have been observed in aging human HSCs. Several factors are described to be related to these changes, such as loss of fidelity in copying the epigenetic marks during cell divisions, and genetic and environmental factors. Collectively, these lead to epigenetic changes by interfering with regulatory mechanisms [93,94]. The molecular mechanisms and the extent by which these factors contribute to epigenetic aging is still not completely clear. An example of epigenetic changes observed in both aged murine and human HSCs is the reduction in 5hmC levels [79,95].
A most striking observation has been the identification of strongly age-dependent differentially methylated genomic loci in peripheral blood leukocytes of healthy human donors. Indeed, it is now possible to accurately predict the chronological age of an individual based on the differentially methylated status of a very small number of loci in a peripheral blood cell sample [96,97]. Interestingly, this epigenetic age, based on DNA methylation patterns in leukocytes, appears to be cell-intrinsic and independent of the environment, as it does not change upon allogenic haematopoietic stem cell transplantation (HSCT) [98,99]. However, another study that investigated DNA methylation patterns after allogenic HSCT observed that in fact a short period of ‘rejuvenation’ occurs, followed by accelerated aging as measured by DNA methylation levels [100]. Patient groups, conditions, time points and follow-up differed between these studies. This epigenetic clock currently includes only DNA methylation patterns, but may become even more accurate when other age-related epigenetic changes can be included. It remains unclear to what extent this epigenetic clock reflects a history of cell proliferation, and whether it is first established in more primitive haematopoietic cell compartments, including stem cells.
Beyond these age-related epigenetic changes, it has recently become apparent that in otherwise healthy elderly people a significant number of blood cells is derived from one, or only a few, dominant stem cell clones. This is referred to as clonal haematopoiesis of indeterminate potential (CHIP, [101]), or age-related clonal haematopoiesis (ARCH, [102]). Clonal haematopoiesis is characterized by the dominant presence of HSCs that carry somatic mutations in genes that are likely to drive the expansion of a genetically identical clone of haematopoietic cells. Large-scale analysis of blood-derived genetic data from individuals within the Cancer Genomes Atlas who did not display haematological malignancies showed age-related expansion of a haematopoietic stem- or progenitor cell clone and the concurrent presence of leukaemia/lymphoma-associated mutations in about 2% of individuals studied. This frequency increased to 5–6% for individuals who are at least 70 years of age [103]. These intriguing findings have been confirmed in multiple independent cohorts ([103,104,105] and many more). Healthy individuals with clonal haematopoiesis have a significantly higher change to develop haematological malignancies, or indeed other disease [106,107]. Although our molecular understanding of the mechanisms that lead to age-related clonal haematopoiesis at present is far from complete, it is interesting to note that mutations were found in genes encoding for proteins in involved in DNA (de-)methylation (DNMT3A and TET2) and chromatin compaction (ASXL1).
4. Epigenetic Changes in Age-Related Haematopoietic Malignancies
Mutations in these epigenetic modifiers in individuals who display CHIP or ARCH, are not only found in normal aging, but are also considered to be critical in the development of acute myeloid leukaemia (AML) and acute lymphoid leukaemia (ALL) [30]. Presumably, mutations in the aforementioned genes result in aberrant epigenetic regulation in HSCs, which derail proper control of HSC self-renewal programs, and thus constitute the initiating events in the pathogenesis of haematological malignancies. Aberrant epigenetic marks, epi-mutations, are prevalent in many myeloid leukaemias [18]. In leukaemia, (epigenetic) dysregulation may lead to the accumulation of immature, non-functioning pre-malignant cells. The malignant transformation subsequently results from the activation of oncogenes and self-renewal pathways or from silencing of tumour suppressor genes, repression of differentiation or apoptosis pathways. These molecular events can be induced by hypermethylation of promotor regions, increased Polycomb repression or aberrant chromatin conformation [108]. It has been shown in a cohort of 200 cases of de novo AML that 74% of patients had at least one mutation in an epigenetic modifier [109]. The three most recurrently mutated genes in AML affecting these aberrant DNA methylation patterns (DNMT3A and TET2) and histone modifications (ASXL1) will be discussed below.
4.1. Aberrant DNA Methylation
DNA methylation patterns have been used to stratify clinically relevant subgroups among AML patient samples [18]. Although clustering appeared possible, patterns were complex and not straightforward to interpret. Whereas ~25% of the differentially methylation clusters corresponded to known AML subtypes, four epigenetic subtypes were defined among patients who shared NPM1 mutations, some clusters were enriched for specific genetic abnormalities, and novel clusters appeared. Overall, while in most AML cases DNA hypomethylation was prevalent, there were also subgroups that showed hypermethylation [18]. Thus, aberrant hyper- as well as hypomethylation states are found in haematological malignancies.
DNMT3A, encoding for a de novo DNA methyltransferase, is one of the most mutated genes in AML. For de novo AML almost 20% of the patients carry mutations in this gene [110], while for de novo MDS this is ~13% [111]. Heterozygous mutations of DNMT3A have been observed to lead to the development of myeloid leukaemias, implying that DNMT3A qualifies as a haploinsufficient tumour suppressor gene [110,112,113,114]. DNMT3A mutations have been associated with poor prognosis (reviewed in [114]), however not in all studies [112]. Functional studies show that loss of the de novo methyltransferase DNMT3A increases HSC self-renewal in mice [8,9,10]. Correspondingly, most DNMT3A mutations are loss of function mutations and the most common DNMT3A mutation is associated with hypomethylation, although hypermethylated regions have also been found [115,116,117].
TET2 is another frequently mutated gene in haematopoietic malignancies. TET2 encodes a protein that catalyzes the conversion of methylcytosine to 5-hydroxymethylcytosine and constitutes the first step towards DNA demethylation. Loss-of-function mutations and deletions of Tet2 have been associated with DNA hypermethylation [118]. Loss of TET2 increased HSC self-renewal in functional studies in murine HSCs [13,18]. Restoration of TET2 function blocked aberrant self-renewal and leukaemia progression in chimeric mouse models [119]. TET2 mutations are detected in 7–23% of AML cases, 10–20% of MDS cases [120] and 50% of chronic myelomonocytic leukaemia (CMML) cases [121]. In de novo AML with TET2 mutations hypermethylated promotor regions were found, overlapping with IDH1/2 mutated leukaemias [14]. TET2 mutations are associated with an increased risk of MDS progression and confer a poor prognosis in AML [13,119]. Low TET2 mRNA levels are related to poor prognosis and TET2 mRNA levels might be a potential prognostic biomarker for AML since levels increase upon remission and decrease upon relapse [122].
4.2. Aberrant Histone Modifications
In addition to changes at the DNA level described earlier, multiple modifications of post-translational histone modifications have been found in haematological malignancies [123]. These aberrant modifications in leukaemia are diverse and often include altered methylation states of histone H3, mainly at lysine residues K4, K9, K27, K36 and K79 [124,125]. Perturbed histone modifications are presumably caused by altered expression of histone modifying enzymes. Indeed, high expression levels of histone deacetylases (HDAC) are found in several types of cancer [126]. In myeloid leukaemias the expression levels of HDACs are heterogenous, where HDAC levels were generally increased in CLL [127]. Also, overexpression of histone demethylases, such as LSD1 has been observed in leukaemia [128]. In addition, genes involved in histone methylation and demethylation, for example ASXL1, EZH2, and DOT1L are frequently mutated in myeloid malignancies.
ASXL1 is associated with the repressive H3K27me3 mark and is one of the three most commonly mutated epigenetic modifiers genes in AML. ASXL1 mutations were found to confer a global reduction of H3K27 trimethylation by disrupting normal recruitment of PRC2 [40]. Loss of Asxl1 was reported to lead to MDS-like disease in mice [41], whereas enforced overexpression of truncated forms of ASXL1 show gain-of-function and promote the pathogenesis of myeloid malignancies [129,130]. Acquired somatic mutations in ASXL1 occur in approximately 10–20% of patients with MDS and 15–25% in myeloproliferative neoplasms (MPN) and AML [131,132,133,134]. Mutations in ASXL1 are associated with poor prognosis in malignant myeloid diseases [131,135].
Beyond mutations in epigenetic histone modifiers, mutations affecting lysine residues in histone H3 genes have also been described. These have an inhibitory effect on EZH1/2, thereby causing reductions in H2K27me2/3. These mutations have a high prevalence in paediatric glioblastoma, but were recently also characterized in adult AML representing sporadic mutations and may co-occur with RUNX1 aberrations [136].
Although these studies clearly suggest a role for aberrant histone modifications in the aetiology of leukaemia, it is difficult to draw definitive conclusions as to how exactly each modification contributes to disease progression. Histone methylation can repress or activate gene expression, depending on which lysine is methylated. For definitive molecular insight it would be required to obtain single cell, single locus and single molecule resolution of histone modifications, and current technology does not permit such approaches. Additionally, discerning between levels of active and repressive marks is complicated by the fact that leukaemic mutations in methyltransferases that deposit active marks, such as the H3K4 and H3K36 methyltransferases, are both gain and loss of function, respectively [125]. Most mutations in genes encoding PRC2 proteins, involved in deposition of the repressive H3K27me3 mark, such as SUZ12, EED and EZH2, , are loss of function mutations. In contrast, mutations in genes encoding for proteins involved in the deposition of other repressive marks, such as H3K9me1 methyltransferases, turn out to be gain of function mutations [125]. Similar as the DNA hypo- and hypermethylated states that are found in AML (as described above), both gain and loss of H3K27me3 levels seems to promote malignant transformation. Similarly, both overexpression and loss of EZH2 are associated with malignant transformation [23,137]. Loss-of-function mutations have been described in CMML and in AML [137,138,139], whereas gain-of-function mutations are frequently encountered in lymphoma patients [140], but also in chronic myeloid leukaemia (CML) and lymphoid leukaemias [137]. In case of MDS and T-ALL, both overexpression and inactivating mutations have been described [137], indicating aberrant expression in either way may drive oncogenesis. It is conceivable therefore that any perturbation of the stochiometry of epigenetic writers, readers and erasers in large protein complexes may cause primitive haematopoietic cells to proliferate uncontrollably and, thus, results in malignancy.
5. Epigenome-Targeted Therapies
The presence of epigenetic modifications in leukaemia and the potential reversible nature of many epigenetic modifications suggest that compounds that affect the epigenetic machinery may be used for therapeutic purposes. Indeed, multiple pharmacological intervention strategies to reverse potentially pathological epigenetic modifications are in development (Table 2). DNA methyltransferase inhibitors (DNMTi) were the first FDA approved drugs targeting the epigenome, aimed to treat haematological diseases, such as MDS, AML and CMML [141]. In addition to drugs that affect DNA methylation, the clinical potential of compounds that target proteins involved in histone modifications is also actively explored. The first histone deacetylase inhibitors (HDACi), approved for multiple myeloma and cutaneous T cell lymphoma, are in clinical trials [142]. Histone methyltransferase inhibitors (HMTi) and histone demethylase inhibitors (HDMi) are emerging as strategies in epigenetic-based pharmacology. Small-molecule inhibitors of histone acetyltransferase (HATi) are investigated in pre-clinical studies. These approved DNMTi and HDACi, initially used for the treatment of haematological malignancies, are currently also being tested in clinical trials for other diseases, such as neurological diseases, latent viral infections and immune diseases [143].
5.1. DNA Methylation Inhibitors (DNMTi)
DNMTi are nucleoside analogues that form a covalent complex with DNA methyltransferases (DNMT) and thereby inhibit ongoing methylation. After their incorporation into DNA, these inhibitors sequester DNMT enzymes, leading to gradual and global demethylation upon cell division. The two most commonly used DNA methylation inhibitors, azacitidine and decitabine, have been evaluated in several clinical trials and are incorporated in clinical guidelines [141]. Azacitidine is approved for MDS and AML and provides an increase in overall survival in older AML patients with poor prognostic karyotypes and high risk MDS (AZA001 trial) [144]. However, it is important to note that only half of the treated patients show a response to azacitidine treatment. Criteria to stratify patients who will respond to these drugs have not yet been identified [145]. Overall survival of MDS patients in the AZA001 trial was superior to survival in cohorts of patients treated in normal clinical practice outside of clinical trials, discouraging routine treatment with azacitidine above enrolment in clinical trials [146]. Survival rates of patients with AML and MDS with cytogenetic abnormalities associated with unfavourable risk, with TP53 mutations, or with both, were similar to patients with an intermediate-risk cytogenetic profile after decitabine treatment [147]. This suggests a place for decitabine in treating these patients. In addition, these DNA hypomethylating agents were reported to have higher overall response rates in MDS and AML patients with TET2 mutations, associated with DNA hypermethylation [118]. Counterintuitively, hypomethylating agents also appear effective in DNMT3A mutated AML, which is associated with hypomethylation, and a small study suggests patients might respond even better [148]. These hypomethylating agents may target specific hypermethylated regions or may result in even further hypomethylation of the genome [114].
The disadvantage of DNMTi is that they induce genome-wide hypomethylation and lack locus specificity, which could lead to activation of oncogenes and/or increased genomic instability. Further research into the long term effects of treatment with DNMTi might learn us more about the therapy related effects and whether the association with therapy related malignancies is comparable to that of commonly used chemotherapeutics. Considering the heterogeneity of AML as a disease, the effectiveness of DNMTi will likely depend on the leukaemia subtype and is likely to vary from patient to patient. Insight into the molecular mechanism of aberrant DNA methylation patterns and the following development of criteria for responders can lead the way to use of these drugs for the right subgroups of AML patients. It is relevant to note that upon azanucleoside withdrawal DNA methylation levels return to pre-treatment levels [149]: demonstrating a continuous need for DNA methyltransferase inhibition.
5.2. Histone Deacetylase Inhibitors (HDACi) and Histone Acetyltransferase (HATi)
Histone acetylation is an epigenetic mark that is typically associated with active gene expression. Concordantly, inhibition of histone deacetylases by HDACi has been shown to derepress expression of genes involved in cell death, growth arrest and differentiation in myeloid malignancies [142,150,151,152,153]. The mechanism of action of most HDACi is by binding to the catalytic site of the HDAC enzyme. HDACi can also affect acetylation of non-histone proteins potentially leading to more general effects [125], including the inhibition of DNA repair processes [154]. For example, DNA damage can be induced by the HDACi vorinostat, possibly by creating a more susceptible, open chromatin conformation. There are reports that suggest that HDACi-induced DNA damage can be repaired by normal cells but not by malignant cells [153]. Treatment of AML or MDS patients with HDACi only, showed low response rates in 10–20% [155].
The use of different combinations of drugs with HDACi is currently being explored. Cameron et al. have addressed the rationale for such combination, e.g., HDACi and DNMTi already in 1999 [156]. An approved HDACi combination is panobinostat combined with bortezomib and dexamethasone for treatment of multiple myeloma (FDA and EMA approved in 2015) [157]. Sequential administration of DNMTi and HDACi demonstrated clinical efficacy in patients with haematologic malignancies [158]. Additionally, pracinostat (SB939) combined with azacitidine is tested for efficacy and safety in a phase III AML trial (NCT03151408) [143]. Beyond inhibition of histone deacetylases, inhibition of histone acetyltransferases has also been evaluated [159]. This includes inhibition of p300 with small molecules to induce apoptosis in AML cells in pre-clinical setting [160].
5.3. Histone Methyltransferase Inhibitors (HMTi) and Histone Demethylase Inhibitors (HDMi)
Disruption of histone methylation patterns is another potential therapeutic strategy to (de)repress differentiation-inducing genes in cancer. The histone methyltransferase inhibitor tazemetostat, an EZH2 inhibitor, is tested for relapsed or refractory B cell NHL with EZH2 mutations [161,162]. Inhibitors targeting histone demethylases, such as the lysine specific demethylase (LSD1) inhibitor, INCB059872, are evaluated in phase I/II clinical MDS/AML trials [143].
5.4. Inhibiting Epigenetic Readers
Broadly acting epigenetic drugs will lead to genome wide alterations of epigenetic modifications. DNMTi, for example, have genome-wide effects on the DNA methylome, leading to acute genome-wide DNA demethylation of repetitive elements, as well as loss of methylation of specific tumour suppressor genes and normalization of cell growth [143]. To reduce these genome wide effects and associated side effects, more specific epigenetic targeting therapies are investigated in preclinical settings. Small molecules that specifically target the binding of epigenetic modifier proteins may be of use for this. The development of BET inhibitors, that preferentially prevent binding of bromodomain-containing epigenetic reader proteins to acetylated histones and transcription factors, served as a proof of principle that there are epigenetic drugs which are able to inhibit certain protein interactions more specifically [123,172]. Development of epigenetic drugs directed against specific protein-protein interactions and thereby influencing epigenetic regulation is ongoing, including compounds that prevent binding of PRC1/2 complexes (EZH2—[162], CBX7—[47,173,174,175]).
6. Future Prospects
It is important to remember that our knowledge of molecular mechanisms that contribute to HSC aging is essentially based on studies in mice, whereas the clinical implications (anaemia, increased susceptibility to infections and malignancies) are evident in human studies. We assume that the clinical observations made in human patients result from the molecular changes that have been well documented in mice, but at this point this remains to be proven. To cover this gap in knowledge, more insight in human HSC aging and robust mouse aging models are required. This also relates to the concept of age-related clonal haematopoiesis, which is very well described in humans, but much less in mice.
As less than one percent of elderly people with clonal haematopoiesis develop leukaemia, identification of those who will eventually develop haematological malignancies is of great interest. Once these individuals can be accurately identified it may be possible to prevent development of leukaemia by specifically targeting the emerging preleukaemic clones in elderly people. For reasons that we do not understand, elderly people with age-related clonal haematopoiesis also have increased risk to present with a wide variety of non-haematological conditions [106,107]. It is tempting to speculate that eradicating aberrant haematopoietic stem cell clones may also be beneficial to prevent these non-haematological diseases.
Once persistent and systematic epigenetic changes have been identified, the plasticity of such lesions renders them an interesting target for novel therapeutic strategies. Epigenetic therapy in AML is in development, many new targets and inhibitors are being explored. The first epigenetic compounds are already approved for clinical use in haematopoietic malignancies and more specific epigenetic therapies are in development and are studied in (pre)clinical studies. Finding ways to specifically target the aberrant epigenetic marks in abnormal HSCs while sparing healthy HSCs would potentially be of significant clinical interest. These newly developed epigenetic drugs may be beneficial as single agent, but also in combinations with commonly used chemotherapeutics, immune therapy or other epigenetic drugs [176]. Reactivation of epigenetically silenced apoptotic genes may increase efficacy of chemotherapy.
Unfortunately, development of new drugs for AML is impeded by the heterogeneity of this disease. The resulting variability in response rates warrants the need for disease stratification and biomarkers to discern which patients will benefit from new treatments. Epigenetic aberrations and mutations in epigenetic modifiers may be useful in this context [177,178,179]. Beyond patient stratification, epigenetic alterations may play a role in prognostic evaluations since mutations in several epigenetic modifiers are associated with clinical outcome in patients with malignant diseases (for example DNMT3A, TET2, IDH2, MLL, EZH2 and ASXL1; reviewed in [123]).
Upon aging many changes in the epigenetic landscape of HSCs occur, both in the murine and the human haematopoietic system. However, the functional consequences of these epigenetic perturbations, although likely to be relevant, remain unclear at this point. Elucidating the functional role of epigenetic modifications that occur during aging and during malignant transformation is partially provided by large-scale analysis of epigenetic modifications in patients with haematological disease (such as the BLUEPRINT project, part of The International Human Epigenome Consortium) [180]. Such important descriptive studies must be followed by functional studies unravelling how epigenetic changes are regulated and lead to aberrant gene expression and phenotypes.
Author Contributions
Writing—original draft preparation: S.B.; writing—review and editing: G.d.H.
Funding
This work was supported by the Dutch Cancer Society (RUG 2014-7178).
Acknowledgments
We apologize to the numerous people whose work we were unable to cite.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vedi, A.; Santoro, A.; Dunant, C.F.; Dick, J.E.; Laurenti, E. Molecular Landscapes of Human Hematopoietic Stem Cells in Health and Leukemia. Ann. N. Y. Acad. Sci. 2015, 1370, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Li, X.; Liang, D.; Li, T.; Zhu, P.; Guo, H.; Wu, X.; Wen, L.; Gu, T.-P.; Hu, B. Active and Passive Demethylation of Male and Female Pronuclear DNA in the Mammalian Zygote. Cell Stem Cell 2014, 15, 447–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khurana, S.; Oberdoerffer, P. Replication Stress: A Lifetime of Epigenetic Change. Genes 2015, 6, 858–877. [Google Scholar] [CrossRef] [PubMed]
- Trowbridge, J.J.; Snow, J.W.; Kim, J.; Orkin, S.H. DNA Methyltransferase 1 Is Essential for and Uniquely Regulates Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2009, 5, 442–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trowbridge, J.J.; Sinha, A.U.; Zhu, N.; Li, M.; Armstrong, S.A.; Orkin, S.H. Haploinsufficiency of Dnmt1 Impairs Leukemia Stem Cell Function through Derepression of Bivalent Chromatin Domains. Genes Dev. 2012, 26, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Bröske, A.-M.; Vockentanz, L.; Kharazi, S.; Huska, M.R.; Mancini, E.; Scheller, M.; Kuhl, C.; Enns, A.; Prinz, M.; Jaenisch, R.; et al. DNA Methylation Protects Hematopoietic Stem Cell Multipotency from Myeloerythroid Restriction. Nat. Genet. 2009, 41, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, Y.; Ema, H.; Okano, M.; Li, E.; Nakauchi, H. De Novo DNA Methyltransferase Is Essential for Self-Renewal, but Not for Differentiation, in Hematopoietic Stem Cells. J. Exp. Med. 2007, 204, 715–722. [Google Scholar] [CrossRef]
- Jeong, M.; Park, H.J.; Celik, H.; Ostrander, E.L.; Reyes, J.M.; Guzman, A.; Rodriguez, B.; Lei, Y.; Lee, Y.; Ding, L.; et al. Loss of Dnmt3a Immortalizes Hematopoietic Stem Cells In Vivo. Cell Rep. 2018, 23, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Challen, G.A.; Sun, D.; Mayle, A.; Jeong, M.; Luo, M.; Rodriguez, B.; Mallaney, C.; Celik, H.; Yang, L.; Xia, Z.; et al. Dnmt3a and Dnmt3b Have Overlapping and Distinct Functions in Hematopoietic Stem Cells. Cell Stem Cell 2014, 15, 350–364. [Google Scholar] [CrossRef] [Green Version]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a Is Essential for Hematopoietic Stem Cell Differentiation. Nat. Genet. 2011, 44, 23–31. [Google Scholar] [CrossRef]
- Trowbridge, J.J.; Orkin, S.H. Dnmt3a Silences Hematopoietic Stem Cell Self-Renewal. Nat. Genet. 2011, 44, 13–14. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, L.; Dawlaty, M.M.; Ndiaye-Lobry, D.; Yap, Y.S.; Bakogianni, S.; Yu, Y.; Bhattacharyya, S.; Shaknovich, R.; Geng, H.; Lobry, C.; et al. Tet1 Is a Tumor Suppressor of Hematopoietic Malignancy. Nat. Immunol. 2015, 16, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Guillamot, M.; Cimmino, L.; Aifantis, I. The Impact of DNA Methylation in Hematopoietic Malignancies. Trends Cancer 2016, 2, 70–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueroa, M.E.; Abdel-wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Thompson, E.C.; Hastie, R.; Tsangaratou, A.; Rajewsky, K.; Koralov, S.B.; Rao, A. Ten-Eleven-Translocation 2 (TET2) Negatively Regulates Homeostasis and Differentiation of Hematopoietic Stem Cells in Mice. PNAS 2011, 108, 14566–14571. [Google Scholar] [CrossRef] [PubMed]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Quivoron, C.; Couronné, L.; Della Valle, V.; Lopez, C.K.; Plo, I.; Wagner-Ballon, O.; Do Cruzeiro, M.; Delhommeau, F.; Arnulf, B.; Stern, M.-H.; et al. TET2 Inactivation Results in Pleiotropic Hematopoietic Abnormalities in Mouse and Is a Recurrent Event during Human Lymphomagenesis. Cancer Cell 2011, 20, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Lugthart, S.; Li, Y.; Erpelinck-verschueren, C.; Christos, P.J.; Schifano, E.; Booth, J.; Van Putten, W.; Campagne, F.; Mazumdar, M.; et al. DNA Methylation Signatures Identify Biologically Distinct Subtypes in Acute Myeloid Leukemia. Cancer Cell 2010, 17, 13–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, A.; Araujo Cruz, M.M.; Jyotsana, N.; Sharma, A.; Yun, H.; Görlich, K.; Wichmann, M.; Schwarzer, A.; Preller, M.; Thol, F.; et al. Mutant IDH1 Promotes Leukemogenesis in Vivo and Can Be Specifically Targeted in Human AML. Blood 2013, 122, 2877–2887. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Knobbe, C.B.; Munger, J.C.; Lind, E.F.; Brenner, D.; Brüstle, A.; Harris, I.S.; Holmes, R.; Wakeham, A.; Haight, J.; et al. IDH1(R132H) Mutation Increases Murine Haematopoietic Progenitors and Alters Epigenetics. Nature 2012, 488, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, I.; Herrera-Merchan, A.; Ligos, J.M.; Carramolino, L.; Nuñez, J.; Martinez, F.; Dominguez, O.; Torres, M.; Gonzalez, S. Ezh1 Is Required for Hematopoietic Stem Cell Maintenance and Prevents Senescence-like Cell Cycle Arrest. Cell Stem Cell 2012, 11, 649–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamminga, L.M.; Bystrykh, L.V.; de Boer, A.; Houwer, S.; Douma, J.; Weersing, E.; Dontje, B.; de Haan, G. The Polycomb Group Gene Ezh2 Prevents Hematopoietic Stem Cell Exhaustion. Blood 2006, 107, 2170–2179. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Merchan, A.; Arranz, L.; Ligos, J.M.; De Molina, A.; Dominguez, O.; Gonzalez, S. Ectopic Expression of the Histone Methyltransferase Ezh2 in Haematopoietic Stem Cells Causes Myeloproliferative Disease. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki-Kashio, M.; Wendt, G.R.; Iwama, A. Tumor Suppressor Function of the Polycomb Group Genes. Cell Cycle 2012, 11, 2043–2044. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; He, J.; Taranova, O.; Zhang, Y. Essential Role of DOT1L in Maintaining Normal Adult Hematopoiesis. Cell Res. 2011, 21, 1370–1373. [Google Scholar] [CrossRef]
- Feng, Y.; Yang, Y.; Ortega, M.M.; Copeland, J.N.; Zhang, M.; Jacob, J.B.; Fields, T.A.; Vivian, J.L.; Fields, P.E. Early Mammalian Erythropoiesis Requires the Dot1L Methyltransferase. Blood 2010, 116, 4483–4491. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.Y.; Granowicz, E.M.; Maillard, I.; Thomas, D.; Hess, J.L. Requirement for Dot1l in Murine Postnatal Hematopoiesis and Leukemogenesis by MLL Translocation. Blood 2011, 117, 4759–4768. [Google Scholar] [CrossRef]
- Gallipoli, P.; Giotopoulos, G.; Huntly, B.J.P. Epigenetic Regulators as Promising Therapeutic Targets in Acute Myeloid Leukemia. Ther. Adv. Hematol. 2015, 6, 103–119. [Google Scholar] [CrossRef]
- Guenther, M.G.; Lawton, L.N.; Rozovskaia, T.; Frampton, G.M.; Levine, S.S.; Volkert, T.L.; Croce, C.M.; Nakamura, T.; Canaani, E.; Young, R.A. Aberrant Chromatin at Genes Encoding Stem Cell Regulators in Human Mixed-Lineage Leukemia. Genes Dev. 2008, 22, 3403–3408. [Google Scholar] [CrossRef]
- Greenblatt, S.M.; Nimer, S.D. Chromatin Modifiers and the Promise of Epigenetic Therapy in Acute Leukemia. Leukemia 2014, 28, 1396–1406. [Google Scholar] [CrossRef]
- Smith, L.L.; Yeung, J.; Zeisig, B.B.; Popov, N.; Huijbers, I.; Barnes, J.; Wilson, A.J.; Taskesen, E.; Delwel, R.; Gil, J.; et al. Functional Crosstalk between Bmi1 and MLL/Hoxa9 Axis in Establishment of Normal Hematopoietic and Leukemic Stem Cells. Cell Stem Cell 2011, 8, 649–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, N.A.; Malouf, C.; Kapeni, C.; Bacon, W.A.; Giotopoulos, G.; Jacobsen, S.E.W.; Huntly, B.J.; Ottersbach, K. Mll-AF4 Confers Enhanced Self-Renewal and Lymphoid Potential during a Restricted Window in Development. Cell Rep. 2016, 16, 1039–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koide, S.; Oshima, M.; Takubo, K.; Yamazaki, S.; Nitta, E.; Saraya, A.; Aoyama, K.; Kato, Y.; Miyagi, S.; Nakajima-Takagi, Y.; et al. Setdb1 Maintains Hematopoietic Stem and Progenitor Cells by Restricting the Ectopic Activation of Non-Hematopoietic Genes. Blood 2016, 128, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Chittka, A.; Nitarska, J.; Grazini, U.; Richardson, W.D. Transcription Factor Positive Regulatory Domain 4 (PRDM4) Recruits Protein Arginine Methyltransferase 5 (PRMT5) to Mediate Histone Arginine Methylation and Control Neural Stem Cell Proliferation and Differentiation. J. Biol. Chem. 2012, 287, 42995–43006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stopa, N.; Krebs, J.E.; Shechter, D. The PRMT5 Arginine Methyltransferase: Many Roles in Development, Cancer and Beyond. Cell. Mol. Life Sci. 2015, 72, 2041–2059. [Google Scholar] [CrossRef] [PubMed]
- Tee, W.-W.; Pardo, M.; Theunissen, T.W.; Yu, L.; Choudhary, J.S.; Hajkova, P.; Surani, M.A. Prmt5 Is Essential for Early Mouse Development and Acts in the Cytoplasm to Maintain ES Cell Pluripotency. Genes Dev. 2010, 24, 2772–2777. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Cheng, G.; Hamard, P.-J.; Greenblatt, S.; Wang, L.; Man, N.; Perna, F.; Xu, H.; Tadi, M.; Luciani, L.; et al. Arginine Methyltransferase PRMT5 Is Essential for Sustaining Normal Adult Hematopoiesis. J. Clin. Investig. 2015, 125, 3532–3544. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.-I.; Hannah, R.L.; Dawson, M.A.; Pridans, C.; Foster, D.; Joshi, A.; Gottgens, B.; Van Deursen, J.M.; Huntly, B.J.P. The Transcriptional Coactivator Cbp Regulates Self-Renewal and Differentiation in Adult Hematopoietic Stem Cells. Mol. Cell. Biol. 2011, 31, 5046–5060. [Google Scholar] [CrossRef] [Green Version]
- Katsumoto, T.; Yoshida, N.; Kitabayashi, I. Roles of the Histone Acetyltransferase Monocytic Leukemia Zinc Finger Protein in Normal and Malignant Hematopoiesis. Cancer Sci. 2008, 99, 1523–1527. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 Mutations Promote Myeloid Transformation through Loss of PRC2-Mediated Gene Repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, Z.; He, Y.; Pan, F.; Chen, S.; Rhodes, S.; Nguyen, L.; Yuan, J.; Jiang, L.; Yang, X.; et al. Loss of Asxl1 Leads to Myelodysplastic Syndrome–like Disease in Mice. Blood 2014, 123, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Uni, M.; Masamoto, Y.; Sato, T.; Kamikubo, Y.; Arai, S.; Kurokawa, M. Modeling ASXL1 Mutation Revealed Impaired Hematopoiesis Caused by Derepression of P16Ink4a through Aberrant PRC1-Mediated Histone Modi Fi Cation. Leukemia 2018, 33, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Van Den Boom, V.; Rozenveld-geugien, M.; Bonardi, F.; Malanga, D.; Gosliga, V.; Heijink, A.M.; Viglietto, G.; Morrone, G.; Fusetti, F.; Schuringa, J.J.; et al. Nonredundant and Locus-Specific Gene Repression Functions of PRC1 Paralog Family Members in Human Hematopoietic Stem/Progenitor Cells. Blood 2013, 121, 2452–2461. [Google Scholar] [CrossRef] [PubMed]
- Core, N.; Bel, S.; Gaunt, S.J.; Aurrand-Lions, M.; Pearce, J.; Fisher, A.; Djabali, M. Altered Cellular Proliferation and Mesoderm Patterning in Polycomb-M33-Deficient Mice. Development 1997, 124, 721–729. [Google Scholar] [PubMed]
- Klauke, K.; Radulović, V.; Broekhuis, M.; Weersing, E.; Zwart, E.; Olthof, S.; Ritsema, M.; Bruggeman, S.; Wu, X.; Helin, K.; et al. Polycomb Cbx Family Members Mediate the Balance between Haematopoietic Stem Cell Self-Renewal and Differentiation. Nat. Cell Biol. 2013, 15, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.L.; Gil, J.; Hernando, E.; Teruya-Feldstein, J.; Narita, M.; Martínez, D.; Visakorpi, T.; Mu, D.; Cordon-Cardo, C.; Peters, G.; et al. Role of the Chromobox Protein CBX7 in Lymphomagenesis. PNAS 2007, 104, 5389–5394. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Buisman, S.C.; Weersing, E.; Dethmers-Ausema, A.; Zwart, E.; Schepers, H.; Dekker, M.R.; Lazare, S.S.; Hammerl, F.; Skokova, Y.; et al. CBX7 Induces Self-Renewal of Human Normal and Malignant Hematopoietic Stem and Progenitor Cells by Canonical and Non-Canonical Interactions. Cell Rep. 2019, 26, 1906–1918. [Google Scholar] [CrossRef]
- Calés, C.; Román-Trufero, M.; Pavón, L.; Serrano, I.; Melgar, T.; Endoh, M.; Pérez, C.; Koseki, H.; Vidal, M. Inactivation of the Polycomb Group Protein Ring1B Unveils an Antiproliferative Role in Hematopoietic Cell Expansion and Cooperation with Tumorigenesis Associated with Ink4a Deletion. Mol. Cell. Biol. 2008, 28, 1018–1028. [Google Scholar] [CrossRef]
- Park, I.; Qian, D.; Kiel, M.; Becker, M.W.; Pihalja, M.; Weissman, I.L.; Morrison, S.J.; Clarke, M.F. Bmi-1 Is Required for Maintenance of Adult Self-Renewing Haematopoietic Stem Cells. Nature 2003, 423, 302–305. [Google Scholar] [CrossRef]
- Lessard, J.; Sauvageau, G. Bmi-1 Determines the Proliferative Capacity of Normal and Leukaemic Stem Cells. Nature 2003, 423, 255–260. [Google Scholar] [CrossRef]
- Rizo, A.; Olthof, S.; Han, L.; Vellenga, E.; De Haan, G.; Schuringa, J.J. Repression of BMI1 in Normal and Leukemic Human CD34+ Cells Impairs Self-Renewal and Induces Apoptosis Repression of BMI1 in Normal and Leukemic Human CD34+ Cells Impairs Self-Renewal and Induces Apoptosis. Hematology 2009, 114, 1498–1504. [Google Scholar]
- Schuringa, J.J.; Vellenga, E. Role of the Polycomb Group Gene BMI1 in Normal and Leukemic Hematopoietic Stem and Progenitor Cells. Curr. Opin. Hematol. 2010, 17, 294–299. [Google Scholar] [CrossRef]
- Wada, T.; Koyama, D.; Kikuchi, J.; Honda, H.; Furukawa, Y. Overexpression of the Shortest Isoform of Histone Demethylase LSD1 Primes Hematopoietic Stem Cells for Malignant Transformation. Blood 2015, 125, 3731–3747. [Google Scholar] [CrossRef]
- Kerenyi, M.A.; Shao, Z.; Hsu, Y.-J.; Guo, G.; Luc, S.; O’Brien, K.; Fujiwara, Y.; Peng, C.; Nguyen, M.; Orkin, S.H. Histone Demethylase Lsd1 Represses Hematopoietic Stem and Progenitor Cell Signatures during Blood Cell Maturation. Elife 2013, 2. [Google Scholar] [CrossRef]
- Sprüssel, A.; Schulte, J.H.; Weber, S.; Necke, M.; Händschke, K.; Thor, T.; Pajtler, K.W.; Schramm, A.; König, K.; Diehl, L.; et al. Lysine-Specific Demethylase 1 Restricts Hematopoietic Progenitor Proliferation and Is Essential for Terminal Differentiation. Leukemia 2012, 26, 2039–2051. [Google Scholar] [CrossRef]
- Stewart, M.H.; Albert, M.; Sroczynska, P.; Cruickshank, V.A.; Guo, Y.; Rossi, D.J.; Helin, K.; Enver, T. The Histone Demethylase Jarid1b Is Required for Hematopoietic Stem Cell Self-Renewal in Mice. Blood 2015, 125, 2075–2078. [Google Scholar] [CrossRef]
- Cellot, S.; Hope, K.J.; Chagraoui, J.; Sauvageau, M.; Deneault, É.; MacRae, T.; Mayotte, N.; Wilhelm, B.T.; Landry, J.R.; Ting, S.B. RNAi Screen Identifies Jarid1b as a Major Regulator of Mouse HSC Activity. Blood 2013, 122, 1545–1555. [Google Scholar] [CrossRef]
- Van der Meulen, J.; Speleman, F.; Van Vlierberghe, P. The H3K27me3 Demethylase UTX in Normal Development and Disease. Epigenetics 2014, 9, 658–668. [Google Scholar] [CrossRef]
- Thieme, S.; Gyárfás, T.; Richter, C.; Özhan, G.; Fu, J.; Alexopoulou, D.; Muders, M.H.; Michalk, I.; Jakob, C.; Dahl, A.; et al. The Histone Demethylase UTX Regulates Stem Cell Migration and Hematopoiesis. Blood 2013, 121, 2462–2473. [Google Scholar] [CrossRef]
- Nakata, Y.; Yamasaki, N.; Ueda, T.; Ikeda, K.; Nagamachi, A.; Inaba, T.; Honda, H. JMJD3 Plays Essential Roles in the Maintenance of Hematopoietic Stem Cells and Leukemic Stem Cells through the Regulation of P16INK4a. Blood 2016, 128, 2653. [Google Scholar]
- Nijnik, A.; Clare, S.; Hale, C.; Raisen, C.; McIntyre, R.E.; Yusa, K.; Everitt, A.R.; Mottram, L.; Podrini, C.; Lucas, M.; et al. The Critical Role of Histone H2A-Deubiquitinase Mysm1 in Hematopoiesis and Lymphocyte Differentiation. Blood 2012, 119, 1370–1379. [Google Scholar] [CrossRef]
- Huo, Y.; Li, B.-Y.; Lin, Z.-F.; Wang, W.; Jiang, X.-X.; Chen, X.; Xi, W.-J.; Yang, A.-G.; Chen, S.-Y.; Wang, T. MYSM1 Is Essential for Maintaining Hematopoietic Stem Cell (HSC) Quiescence and Survival. Med. Sci. Monit. 2018, 24, 2541–2549. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Nandakumar, V.; Jiang, X.-X.; Jones, L.; Yang, A.-G.; Huang, X.F.; Chen, S.-Y. The Control of Hematopoietic Stem Cell Maintenance, Self-Renewal, and Differentiation by Mysm1-Mediated Epigenetic Regulation. Blood 2013, 122, 2812–2822. [Google Scholar] [CrossRef]
- Wang, H.; Diao, D.; Shi, Z.; Zhu, X.; Gao, Y.; Gao, S.; Liu, X.; Wu, Y.; Rudolph, K.L.; Liu, G.; et al. SIRT6 Controls Hematopoietic Stem Cell Homeostasis through Epigenetic Regulation of Wnt Signaling. Cell Stem Cell 2016, 18, 495–507. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Williams, C.A.; Klarmann, K.; Burkett, S.S.; Keller, J.R.; Oberdoerffer, P. Sirt1 Ablation Promotes Stress-Induced Loss of Epigenetic and Genomic Hematopoietic Stem and Progenitor Cell Maintenance. J. Exp. Med. 2013, 210, 987–1001. [Google Scholar] [CrossRef]
- Brown, K.; Xie, S.; Qiu, X.; Mohrin, M.; Shin, J.; Liu, Y.; Zhang, D.; Scadden, D.T.; Chen, D. SIRT3 Reverses Aging-Associated Degeneration. Cell Rep. 2013, 3, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Mohrin, M.; Shin, J.; Liu, Y.; Brown, K.; Luo, H.; Xi, Y.; Haynes, C.M.; Chen, D. A Mitochondrial UPR-Mediated Metabolic Checkpoint Regulates Hematopoietic Stem Cell Aging. Science 2015, 347, 1374–1377. [Google Scholar] [CrossRef]
- Luo, H.; Mu, W.-C.; Karki, R.; Chiang, H.-H.; Mohrin, M.; Shin, J.J.; Ohkubo, R.; Ito, K.; Kanneganti, T.-D.; Chen, D. Mitochondrial Stress-Initiated Aberrant Activation of the NLRP3 Inflammasome Regulates the Functional Deterioration of Hematopoietic Stem Cell Aging. Cell Rep. 2019, 26, 945–954. [Google Scholar] [CrossRef]
- Chambers, S.M.; Shaw, C.A.; Gatza, C.; Fisk, C.J.; Donehower, L.A.; Goodell, M.A. Aging Hematopoietic Stem Cells Decline in Function and Exhibit Epigenetic Dysregulation. PLoS Biol. 2007, 5, 1750–1762. [Google Scholar] [CrossRef]
- Rossi, D.J.; Bryder, D.; Zahn, J.M.; Ahlenius, H.; Sonu, R.; Wagers, A.J.; Weissman, I.L. Cell Intrinsic Alterations Underlie Hematopoietic Stem Cell Aging. PNAS 2005, 102, 9194–9199. [Google Scholar] [CrossRef]
- Beerman, I.; Bhattacharya, D.; Zandi, S.; Sigvardsson, M.; Weissman, I.L.; Bryder, D. Functionally Distinct Hematopoietic Stem Cells Modulate Hematopoietic Lineage Potential during Aging by a Mechanism of Clonal Expansion. PNAS 2010, 107. [Google Scholar] [CrossRef]
- Dykstra, B.; Olthof, S.; Schreuder, J.; Ritsema, M.; De Haan, G. Clonal Analysis Reveals Multiple Functional Defects of Aged Murine Hematopoietic Stem Cells. J. Exp. Med. 2011, 208, 2691–2703. [Google Scholar] [CrossRef]
- Sudo, B.K.; Ema, H.; Morita, Y.; Nakauchi, H. Age-Associated Characteristics of Murine Hematopoietic Stem Cells. J. Exp. Med. 2000, 192, 1273–1280. [Google Scholar] [CrossRef] [Green Version]
- Säwén, P.; Lang, S.; Mandal, P.; Rossi, D.J.; Soneji, S.; Bryder, D. Mitotic History Reveals Distinct Stem Cell Populations and Their Contributions to Hematopoiesis. Cell Rep. 2016, 14, 2809–2818. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, R.; Wilkinson, A.C.; Ooehara, J.; Nakauchi, Y.; Pritchard, J.K.; Yamamoto, R.; Wilkinson, A.C.; Ooehara, J.; Lan, X.; Lai, C.; et al. Large-Scale Clonal Analysis Resolves Aging of the Mouse Hematopoietic Stem Cell Compartment Resource Large-Scale Clonal Analysis Resolves Aging of the Mouse Hematopoietic Stem Cell Compartment. Cell Stem Cell 2018, 22, 600–607. [Google Scholar] [CrossRef]
- Cho, R.H.; Sieburg, H.B.; Muller-Sieburg, C.E. A New Mechanism for the Aging of Hematopoietic Stem Cells: Aging Changes the Clonal Composition of the Stem Cell Compartment but Not Individual Stem Cells. Blood 2008, 111, 5553–5562. [Google Scholar] [CrossRef]
- Bernitz, J.M.; Kim, H.S.; MacArthur, B.; Sieburg, H.; Moore, K. Hematopoietic Stem Cells Count and Remember Self-Renewal Divisions. Cell 2016, 167, 1296–1309. [Google Scholar] [CrossRef]
- Quéré, R.; Saint-paul, L.; Carmignac, V.; Martin, R.Z.; Chrétien, M. Tif1 γ Regulates the TGF- β 1 Receptor and Promotes Physiological Aging of Hematopoietic Stem Cells. PNAS 2014, 111, 10592–10597. [Google Scholar]
- Sun, D.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic Profiling of Young and Aged HSCs Reveals Concerted Changes during Aging That Reinforce Self-Renewal. Cell Stem Cell 2014, 14, 673–688. [Google Scholar] [CrossRef]
- Wahlestedt, M.; Norddahl, G.L.; Sten, G.; Ugale, A.; Frisk, M.A.M.; Mattsson, R.; Deierborg, T.; Sigvardsson, M.; Bryder, D. An Epigenetic Component of Hematopoietic Stem Cell Aging Amenable to Reprogramming into a Young State. Blood 2013, 121, 4257–4264. [Google Scholar] [CrossRef]
- Maryanovich, M.; Zahalka, A.H.; Pierce, H.; Pinho, S.; Nakahara, F.; Asada, N.; Wei, Q.; Wang, X.; Ciero, P.; Xu, J.; et al. Adrenergic Nerve Degeneration in Bone Marrow Drives Aging of the Hematopoietic Stem Cell Niche. Nat. Med. 2018, 24, 782–791. [Google Scholar] [CrossRef]
- Beerman, I.; Rossi, D.J. Epigenetic Regulation of Hematopoietic Stem Cell Aging. Exp. Cell Res. 2014, 329, 192–199. [Google Scholar] [CrossRef]
- Beerman, I.; Bock, C.; Garrison, B.S.; Smith, Z.D.; Gu, H.; Meissner, A.; Rossi, D.J. Article Proliferation-Dependent Alterations of the DNA Methylation Landscape Underlie Hematopoietic Stem Cell Aging. Stem Cell 2013, 12, 413–425. [Google Scholar]
- Bersenev, A.; Rozenova, K.; Balcerek, J.; Jiang, J.; Wu, C.; Tong, W. Lnk Deficiency Partially Mitigates Hematopoietic Stem Cell Aging. Aging Cell 2012, 11, 949–959. [Google Scholar] [CrossRef]
- Flach, J.; Bakker, S.T.; Mohrin, M.; Conroy, P.C.; Pietras, E.M.; Reynaud, D.; Alvarez, S.; Diolaiti, M.E.; Ugarte, F.; Camilla, E.; et al. Replication Stress Is a Potent Driver of Functional Decline in Ageing Haematopoietic Stem Cells. Nature 2014, 512, 198–202. [Google Scholar] [CrossRef]
- Kirschner, K.; Chandra, T.; Kiselev, V.; Hemberg, M.; Reik, W.; Green, A.R.; Kirschner, K.; Chandra, T.; Kiselev, V.; Cruz, D.F.; et al. Proliferation Drives Aging-Related Functional Decline in a Subpopulation of the Hematopoietic Stem Cell Compartment Report Proliferation Drives Aging-Related Functional Decline in a Subpopulation of the Hematopoietic Stem Cell Compartment. Cell Rep. 2017, 19, 1503–1511. [Google Scholar] [CrossRef]
- Kowalczyk, M.S.; Tirosh, I.; Heckl, D.; Rao, T.N.; Dixit, A.; Haas, B.J.; Schneider, R.K.; Wagers, A.J.; Ebert, B.L.; Regev, A. Single-Cell RNA-Seq Reveals Changes in Cell Cycle and Differentiation Programs upon Aging of Hematopoietic Stem Cells. Genome Res. 2015, 25, 1860–1872. [Google Scholar] [CrossRef]
- Noda, S.; Ichikawa, H.; Miyoshi, H. Hematopoietic Stem Cell Aging Is Associated with Functional Decline and Delayed Cell Cycle Progression. Biochem. Biophys. Res. Commun. 2009, 383, 210–215. [Google Scholar] [CrossRef]
- Norddahl, G.L.; Pronk, C.J.; Wahlestedt, M.; Sten, G.; Nygren, J.M.; Ugale, A.; Sigvardsson, M.; Bryder, D. Accumulating Mitochondrial DNA Mutations Drive Premature Hematopoietic Aging Phenotypes Distinct from Physiological Stem Cell Aging. Stem Cell 2011, 8, 499–510. [Google Scholar] [CrossRef] [Green Version]
- Kramer, A.; Challen, G.A. The Epigenetic Basis of Hematopoietic Stem Cell Aging Functional and Molecular Manifestations of HSC Aging. Semin. Hematol. 2017, 54, 19–24. [Google Scholar] [CrossRef]
- Wahlestedt, M.; Bryder, D. The Slippery Slope of Hematopoietic Stem Cell Aging. Exp. Hematol. 2017, 56, 1–6. [Google Scholar] [CrossRef]
- Pang, W.W.; Schrier, S.L.; Weissman, I.L. Age-Associated Changes in Human Hematopoietic Stem Cells. Semin. Hematol. 2017, 54, 39–42. [Google Scholar] [CrossRef]
- Huidobro, C.; Fernandez, A.F.; Fraga, M.F. Aging Epigenetics: Causes and Consequences. Mol. Aspects Med. 2013, 34, 765–781. [Google Scholar] [CrossRef]
- Zane, L.; Sharma, V.; Misteli, T. Common Features of Chromatin in Aging and Cancer: Cause or Coincidence? Trends Cell Biol. 2014, 24, 686–694. [Google Scholar] [CrossRef]
- Fuke, C.; Shimabukuro, M.; Petronis, A.; Sugimoto, J.; Oda, T.; Miura, K.; Miyazaki, T.; Ogura, C.; Okazaki, Y.; Jinno, Y. Age Related Changes in 5-Methylcytosine Content in Human Peripheral Leukocytes and Placentas: An HPLC-Based Study. Ann. Hum. Genet. 2004, 68, 196–204. [Google Scholar] [CrossRef]
- Wagner, W. Epigenetic Aging Clocks in Mice and Men. Genome Biol. 2017, 18, 17–19. [Google Scholar] [CrossRef]
- Horvath, S. Erratum to DNA Methylation Age of Human Tissues and Cell Types. Genome Biol. 2015, 16. [Google Scholar] [CrossRef]
- Søraas, A.; Matsuyama, M.; de Lima, M.; Wald, D.; Buechner, J.; Gedde-Dahl, T.; Søraas, C.L.; Chen, B.; Ferrucci, L.; Dahl, J.A.; et al. Epigenetic Age Is a Cell-Intrinsic Property in Transplanted Human Hematopoietic Cells. Aging Cell 2019. [Google Scholar] [CrossRef]
- Weidner, C.I.; Ziegler, P.; Hahn, M.; Brümmendorf, T.H.; Ho, A.D.; Dreger, P.; Wagner, W. Epigenetic Aging upon Allogeneic Transplantation: The Hematopoietic Niche Does Not Affect Age-Associated DNA Methylation. Leukemia 2015, 29, 985–988. [Google Scholar] [CrossRef]
- Stölzel, F.; Brosch, M.; Horvath, S.; Kramer, M.; Thiede, C.; von Bonin, M.; Ammerpohl, O.; Middeke, M.; Schetelig, J.; Ehninger, G.; et al. Dynamics of Epigenetic Age Following Hematopoietic Stem Cell Transplantation. Haematologica 2017, 102, e321–e323. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal Hematopoiesis of Indeterminate Potential and Its Distinction from Myelodysplastic Syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef]
- Shlush, L.I. Age-Related Clonal Hematopoiesis. Blood 2018, 131, 496–504. [Google Scholar] [CrossRef]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-Related Mutations Associated with Clonal Hematopoietic Expansion and Malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Steensma, D.P. Clinical Consequences of Clonal Hematopoiesis of Indeterminate Potential. Blood 2018, 2, 264–269. [Google Scholar]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic Plasticity and the Hallmarks of Cancer. Science 2017, 357. [Google Scholar] [CrossRef]
- Cancer Genome Atlas. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A Mutations in Acute Myeloid Leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of Genetic Lesions in 944 Patients with Myelodysplastic Syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef]
- Gaidzik, V.I.; Schlenk, R.F.; Paschka, P.; Stölzle, A.; Späth, D.; Kuendgen, A.; von Lilienfeld-Toal, M.; Brugger, W.; Derigs, H.G.; Kremers, S.; et al. Clinical Impact of DNMT3A Mutations in Younger Adult Patients with Acute Myeloid Leukemia: Results of the AML Study Group (AMLSG). Blood 2013, 121, 4769–4777. [Google Scholar] [CrossRef]
- Gale, R.E.; Lamb, K.; Allen, C.; El-Sharkawi, D.; Stowe, C.; Jenkinson, S.; Tinsley, S.; Dickson, G.; Burnett, A.K.; Hills, R.K.; et al. Simpson’s Paradox and the Impact of Different DNMT3A Mutations on Outcome in Younger Adults With Acute Myeloid Leukemia. J. Clin. Oncol. 2015, 33, 2072–2083. [Google Scholar] [CrossRef]
- Brunetti, L.; Gundry, M.C.; Goodell, M.A. DNMT3A in Leukemia. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef]
- Spencer, D.H.; Al-Khalil, B.; Russler-Germain, D.; Lamprecht, T.; Havey, N.; Fulton, R.S.; O’Laughlin, M.; Fronick, C.; Wilson, R.K.; Ley, T.J. Whole-Genome Bisulfite Sequencing of Primary AML Cells with the DNMT3A R882H Mutation Identifies Regions of Focal Hypomethylation That Are Associated with Open Chromatin. Blood 2014, 124, 608. [Google Scholar]
- Spencer, D.H.; Russler-Germain, D.A.; Ketkar, S.; Helton, N.M.; Lamprecht, T.L.; Fulton, R.S.; Fronick, C.C.; O’Laughlin, M.; Heath, S.E.; Shinawi, M.; et al. CpG Island Hypermethylation Mediated by DNMT3A Is a Consequence of AML Progression. Cell 2017, 168, 801–816. [Google Scholar] [CrossRef]
- Yang, L.; Rodriguez, B.; Mayle, A.; Park, H.J.; Lin, X.; Luo, M.; Jeong, M.; Curry, C.V.; Kim, S.-B.; Ruau, D.; et al. DNMT3A Loss Drives Enhancer Hypomethylation in FLT3-ITD-Associated Leukemias. Cancer Cell 2016, 29, 922–934. [Google Scholar] [CrossRef] [Green Version]
- Bejar, R.; Lord, A.; Stevenson, K.; Bar-Natan, M.; Pérez-Ladaga, A.; Zaneveld, J.; Wang, H.; Caughey, B.; Stojanov, P.; Getz, G.; et al. TET2 Mutations Predict Response to Hypomethylating Agents in Myelodysplastic Syndrome Patients. Blood 2014, 124, 2705–2712. [Google Scholar] [CrossRef]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Valle, V. Della; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in Myeloid Cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Kosmider, O.; Gelsi-Boyer, V.; Ciudad, M.; Racoeur, C.; Jooste, V.; Vey, N.; Quesnel, B.; Fenaux, P.; Bastie, J.-N.; Beyne-Rauzy, O.; et al. TET2 Gene Mutation Is a Frequent and Adverse Event in Chronic Myelomonocytic Leukemia. Haematologica 2009, 94, 1676–1681. [Google Scholar] [CrossRef]
- Zhang, T.; Zhou, J.-D.; Yang, D.; Wang, Y.; Wen, X.; Guo, H.; Yang, L.; Lian, X.; Lin, J. TET2 Expression Is a Potential Prognostic and Predictive Biomarker in Cytogenetically Normal Acute Myeloid Leukemia. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef]
- Fong, C.Y.; Morison, J.; Dawson, M.A. Epigenetics in the Hematologic Malignancies. Hematologica 2014, 99, 1079–1095. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, Erasing and Reading Histone Lysine Methylations. Exp. Mol. Med. 2017, 49. [Google Scholar] [CrossRef]
- Ungerstedt, J.S. Epigenetic Modifiers in Myeloid Malignancies: The Role of Histone Deacetylase Inhibitors. Int. J. Mol. Sci. 2018, 19, 3091. [Google Scholar] [CrossRef]
- Nakagawa, M.; Oda, Y.; Eguchi, T.; Aishima, S.I.; Yao, T.; Hosoi, F.; Basaki, Y.; Ono, M.; Kuwano, M.; Tanaka, M.; et al. Expression Profile of Class I Histone Deacetylases in Human Cancer Tissues. Oncol. Rep. 2007, 18, 769–774. [Google Scholar] [CrossRef]
- Yang, H.; Maddipoti, S.; Quesada, A.; Bohannan, Z.; Cabrero Calvo, M.; Colla, S.; Wei, Y.; Estecio, M.; Wierda, W.; Bueso-Ramos, C.; et al. Analysis of Class I and II Histone Deacetylase Gene Expression in Human Leukemia. Leuk. Lymphoma 2015, 56, 3426–3433. [Google Scholar] [CrossRef]
- Castelli, G.; Pelosi, E.; Testa, U. Targeting Histone Methyltransferase and Demethylase in Acute Myeloid Leukemia Therapy. Onco. Targets. Ther. 2018, 11, 131–155. [Google Scholar] [CrossRef]
- Nagase, R.; Inoue, D.; Pastore, A.; Fujino, T.; Hou, H.A.; Yamasaki, N.; Goyama, S.; Saika, M. Expression of Mutant Asxl1 Perturbs Hematopoiesis and Promotes Susceptibility to Leukemic Transformation. J. Exp. Med. 2018, 215, 1729–1747. [Google Scholar] [CrossRef]
- Yang, H.; Kurtenbach, S.; Guo, Y.; Lohse, I.; Durante, M.A.; Li, J.; Li, Z.; Al-Ali, H.; Li, L.; Chen, Z.; et al. Gain of Function of ASXL1 Truncating Protein in the Pathogenesis of Myeloid Malignancies. Blood 2018, 131, 328–341. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [Green Version]
- Boultwood, J.; Perry, J.; Pellagatti, A.; Fernandez-Mercado, M.; Fernandez-Santamaria, C.; Calasanz, M.J.; Larrayoz, M.J.; Garcia-Delgado, M.; Giagounidis, A.; Malcovati, L.; et al. Frequent Mutation of the Polycomb-Associated Gene ASXL1 in the Myelodysplastic Syndromes and in Acute Myeloid Leukemia. Leukemia 2010, 24, 1062–1065. [Google Scholar] [CrossRef]
- Gelsi-Boyer, V.; Trouplin, V.; Adélaïde, J.; Bonansea, J.; Cervera, N.; Carbuccia, N.; Lagarde, A.; Prebet, T.; Nezri, M.; Sainty, D.; et al. Mutations of Polycomb-associated Gene ASXL1 in Myelodysplastic Syndromes and Chronic Myelomonocytic Leukaemia. Br. J. Haematol. 2009, 145, 788–800. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Pardanani, A.; Patel, J.; Wadleigh, M.; Lasho, T.; Heguy, A.; Beran, M.; Gilliland, D.G.; Levine, R.L.; Tefferi, A. Concomitant Analysis of EZH2 and ASXL1 Mutations in Myelofibrosis, Chronic Myelomonocytic Leukaemia and Blast-Phase Myeloproliferative Neoplasms. Leukemia 2011, 25, 1200–1202. [Google Scholar] [CrossRef]
- Gelsi-Boyer, V.; Brecqueville, M.; Devillier, R.; Murati, A.; Mozziconacci, M.J.; Birnbaum, D. Mutations in ASXL1 Are Associated with Poor Prognosis across the Spectrum of Malignant Myeloid Diseases. J. Hematol. Oncol. 2012, 5. [Google Scholar] [CrossRef]
- Lehnertz, B.; Zhang, Y.W.; Boivin, I.; Mayotte, N.; Tomellini, E.; Chagraoui, J.; Lavallée, V.P.; Hébert, J.; Sauvageau, G. H3K27M/I Mutations Promote Context-Dependent Transformation in Acute Myeloid Leukemia with RUNX1 Alterations. Blood 2017, 130, 2204–2214. [Google Scholar] [CrossRef]
- Safaei, S.; Baradaran, B.; Hagh, M.F.; Alivand, M.R.; Talebi, M.; Gharibi, T.; Solali, S. Double Sword Role of EZH2 in Leukemia. Biomed. Pharmacother. 2018, 98, 626–635. [Google Scholar] [CrossRef]
- Xu, F.; Liu, L.; Chang, C.; He, Q.; Wu, L.; Zhang, Z. Genomic Loss of EZH2 Leads to Epigenetic Modifications and Overexpression of the HOX Gene Clusters in Myelodysplastic Syndrome. Oncotarget 2016, 7, 8119–8130. [Google Scholar] [CrossRef]
- Sashida, G.; Harada, H.; Matsui, H.; Oshima, M.; Yui, M.; Harada, Y.; Tanaka, S.; Mochizuki-Kashio, M.; Wang, C.; Saraya, A.; et al. Ezh2 Loss Promotes Development of Myelodysplastic Syndrome but Attenuates Its Predisposition to Leukaemic Transformation. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Huet, S.; Xerri, L.; Tesson, B.; Mareschal, S.; Taix, S.; Mescam-Mancini, L.; Sohier, E.; Carrère, M.; Lazarovici, J.; Casasnovas, O. EZH2 Alterations in Follicular Lymphoma: Biological and Clinical Correlations. Blood Cancer J. 2017, 7. [Google Scholar] [CrossRef]
- Navada, S.C.; Steinmann, J.; Lübbert, M.; Silverman, L.R. Clinical Development of Demethylating Agents in Hematology. J. Clin. Investig. 2014, 124, 40–46. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Berdasco, M.; Esteller, M. Clinical Epigenetics: Seizing Opportunities for Translation. Nat. Rev. Genet. 2018, 20, 109–127. [Google Scholar] [CrossRef]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. International Phase 3 Study of Azacitidine vs. Conventional Care Regimens in Older Patients with Newly Diagnosed AML with > 30% Blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef]
- Stahl, M.; Zeidan, A.M.; Komrokji, R.S.; Sekeres, M.A.; Gore, S.D.; Steensma, D.P. A Call for Action: Increasing Enrollment of Untreated Patients with Higher-Risk Myelodysplastic Syndromes in First-Line Clinical Trials. Cancer 2017, 123, 3662–3672. [Google Scholar]
- Stahl, M.; Davidoff, A.J.; Zeidan, A.M.; Huntington, S.; DeVeaux, M.; Ma, X.; Podoltsev, N.; Wang, R.; Gore, S.D.; Giri, S. Counseling Patients with Higher-Risk MDS Regarding Survival with Azacitidine Therapy: Are We Using Realistic Estimates? Blood Cancer J. 2018, 8, 4–7. [Google Scholar]
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef]
- Traina, F.; Visconte, V.; Elson, P.; Tabarroki, A.; Jankowska, A.M.; Hasrouni, E.; Sugimoto, Y.; Szpurka, H.; Makishima, H.; O’Keefe, C.L.; et al. Impact of Molecular Mutations on Treatment Response to DNMT Inhibitors in Myelodysplasia and Related Neoplasms. Leukemia 2014, 28, 78–87. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in Human Disease and Prospects for Epigenetic Therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef]
- Insinga, A.; Monestiroli, S.; Ronzoni, S.; Gelmetti, V.; Marchesi, F.; Viale, A.; Altucci, L.; Nervi, C.; Minucci, S.; Pelicci, P.G. Inhibitors of Histone Deacetylases Induce Tumor-Selective Apoptosis through Activation of the Death Receptor Pathway. Nat. Med. 2005, 11, 71–76. [Google Scholar] [CrossRef]
- Nebbioso, A.; Clarke, N.; Voltz, E.; Germain, E.; Ambrosino, C.; Bontempo, P.; Alvarez, R.; Schiavone, E.M.; Ferrara, F.; Bresciani, F.; et al. Tumor-Selective Action of HDAC Inhibitors Involves TRAIL Induction in Acute Myeloid Leukemia Cells. Nat. Med. 2005, 11, 77–84. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and Emerging HDAC Inhibitors for Cancer Treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef]
- Lee, J.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone Deacetylase Inhibitor Induces DNA Damage, Which Normal but Not Transformed Cells Can Repair. PNAS 2010, 107, 14639–14644. [Google Scholar] [CrossRef]
- Robert, C.; Rassool, F.V. Roles of DNA Damage and Repair. In Histone Deacetylase Inhibitors as Cancer Therapeutics; Academic Press: San Diego, CA, USA, 2012; Volume 116, pp. 87–129. [Google Scholar]
- Quintas-Cardama, A.; Santos, F.P.S.; Garcia-Manero, G. Histone Deacetylase Inhibitors for the Treatment of Myelodysplastic Syndrome and Acute Myeloid Leukemia. Leukemia 2011, 25, 226–235. [Google Scholar] [CrossRef]
- Cameron, E.E.; Bachman, K.E.; Myöhänen, S.; Herman, J.G.; Baylin, S.B. Synergy of Demethylation and Histone Deacetylase Inhibition in the Re-Expression of Genes Silenced in Cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef]
- Novartis Pharmaceuticals Corporation. Farydak (panobinostat) [prescribing information]. Available online: https://us.farydak.com/ accessed on 22-07-2019 (accessed on 22 July 2019).
- Kelly, T.K.; De Carvalho, D.D.; Jones, P.A. Epigenetics Modifications as Therapeutic Targets. Nat. Biotechnol. 2010, 28, 1069–1078. [Google Scholar] [CrossRef]
- Bowers, E.M.; Yan, G.; Mukherjee, C.; Orry, A.; Wang, L.; Holbert, M.A.; Crump, N.T.; Hazzalin, C.A.; Liszczak, G.; Yuan, H.; et al. Virtual Ligand Screening of the P300/CBP Histone Acetyltransferase: Identification of a Selective Small Molecule Inhibitor. Chem. Biol. 2010, 17, 471–482. [Google Scholar] [CrossRef]
- Gao, X.; Lin, J.; Ning, Q.; Gao, L.; Yao, Y.; Zhou, J.; Li, Y.; Wang, L.; Yu, L. A Histone Acetyltransferase P300 Inhibitor C646 Induces Cell Cycle Arrest and Apoptosis Selectively in AML1-ETO-Positive AML Cells. PLoS ONE 2013, 2. [Google Scholar] [CrossRef]
- Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.J.; Abascal, F.; Hall, M.W.J.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M.R.; et al. Somatic Mutant Clones Colonize the Human Esophagus with Age. Science 2018. [Google Scholar] [CrossRef]
- Italiano, A.; Soria, J.-C.; Toulmonde, M.; Michot, J.-M.; Lucchesi, C.; Varga, A.; Coindre, J.-M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 Inhibitor, in Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma and Advanced Solid Tumours: A First-in-Human, Open-Label, Phase 1 Study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Wouters, B.J.; Delwel, R. Epigenetics and Approaches to Targeted Epigenetic Therapy in Acute Myeloid Leukemia. Blood 2016, 127, 42–52. [Google Scholar] [CrossRef]
- Galkin, M.; Jonas, B.A. Enasidenib in the Treatment of Relapsed/Refractory Acute Myeloid Leukemia: An Evidence-Based Review of Its Place in Therapy. Core Evid. 2019, 14, 3–17. [Google Scholar] [CrossRef]
- Nassereddine, S.; Lap, C.J.; Haroun, F.; Tabbara, I. The Role of Mutant IDH1 and IDH2 Inhibitors in the Treatment of Acute Myeloid Leukemia. Ann. Hematol. 2017, 96, 1983–1991. [Google Scholar] [CrossRef]
- Goldman, S.L.; Hassan, C.; Khunte, M.; Soldatenko, A.; Jong, Y.; Afshinnekoo, E.; Mason, C.E. Epigenetic Modifications in Acute Myeloid Leukemia: Prognosis, Treatment, and Heterogeneity. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Picaud, S.; Fedorov, O.; Thanasopoulou, A.; Leonards, K.; Jones, K.; Meier, J.; Olzscha, H.; Monteiro, O.; Martin, S.; Philpott, M.; et al. Generation of a Selective Small Molecule Inhibitor of the CBP/P300 Bromodomain for Leukemia Therapy. Cancer Res. 2015, 75, 5106–5119. [Google Scholar] [CrossRef]
- Magliulo, D.; Bernardi, R.; Messina, S. Lysine-Specific Demethylase 1A as a Promising Target in Acute Myeloid Leukemia. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Nakagawa, M.; Fujita, S.; Katsumoto, T.; Yamagata, K.; Ogawara, Y.; Hattori, A.; Kagiyama, Y.; Honma, D.; Araki, K.; Inoue, T.; et al. Dual Inhibition of Enhancer of Zeste Homolog 1/2 Overactivates WNT Signaling to Deplete Cancer Stem Cells in Multiple Myeloma. Cancer Sci. 2018, 110, 194–208. [Google Scholar] [CrossRef]
- Fujita, S.; Honma, D.; Adachi, N.; Araki, K.; Takamatsu, E.; Katsumoto, T.; Yamagata, K.; Akashi, K.; Aoyama, K.; Iwama, A.; et al. Dual Inhibition of EZH1/2 Breaks the Quiescence of Leukemia Stem Cells in Acute Myeloid Leukemia. Leukemia 2018, 32, 855–864. [Google Scholar] [CrossRef]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L Inhibitor Pinometostat Reduces H3K79 Methylation and Has Modest Clinical Activity in Adult Acute Leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.-I.; Robson, S.C.; Chung, C.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET Recruitment to Chromatin as an Effective Treatment for MLL-Fusion Leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef]
- Ren, C.; Smith, S.G.; Yap, K.; Li, S.; Li, J.; Mezei, M.; Rodriguez, Y.; Vincek, A.; Aguilo, F.; Walsh, M.J.; et al. Structure-Guided Discovery of Selective Antagonists for the Chromodomain of Polycomb Repressive Protein CBX7. ACS Med. Chem. Lett. 2016, 7, 601–605. [Google Scholar] [CrossRef] [Green Version]
- Stuckey, J.I.; Dickson, B.M.; Cheng, N.; Liu, Y.; Norris, J.L.; Cholensky, S.H.; Tempel, W.; Qin, S.; Huber, K.G.; Sagum, C.; et al. A Cellular Chemical Probe Targeting the Chromodomains of Polycomb Repressive Complex 1. Nat. Chem. Biol. 2016, 12, 180–187. [Google Scholar] [CrossRef]
- Ren, C.; Morohashi, K.; Plotnikov, A.N.; Jakoncic, J.; Smith, S.G.; Li, J.; Zeng, L.; Rodriguez, Y.; Stojanoff, V.; Walsh, M.; et al. Small Molecule Modulators of Methyl-Lysine Binding for the CBX7 Chromodomain. Chem. Biol. 2015, 22, 161–168. [Google Scholar] [CrossRef]
- Bewersdorf, J.P.; Shallis, R.; Stahl, M.; Zeidan, A.M. Epigenetic Therapy Combinations in Acute Myeloid Leukemia: What Are the Options? Ther. Adv. Hematol. 2019, 10, 1–19. [Google Scholar] [CrossRef]
- Claes, B.; Buysschaert, I.; Lambrechts, D. Pharmaco-Epigenomics: Discovering Therapeutic Approaches and Biomarkers for Cancer Therapy. Heredity 2010, 105, 152–160. [Google Scholar] [CrossRef]
- Majchrzak-Celińska, A.; Baer-Dubowska, W. Pharmacoepigenetics: An Element of Personalized Therapy? Expert Opin. Drug Metab. Toxicol. 2017, 13, 387–398. [Google Scholar] [CrossRef]
- Kronfol, M.M.; Dozmorov, M.G.; Huang, R.; Slattum, P.W.; McClay, J.L. The Role of Epigenomics in Personalized Medicine. Expert Rev. Precis. Med. Drug Dev. 2017, 2, 33–45. [Google Scholar] [CrossRef]
- Stunnenberg, H.G.; Abrignani, S.; Adams, D.; de Almeida, M.; Altucci, L.; Amin, V.; Amit, I.; Antonarakis, S.E.; Aparicio, S.; Arima, T.; et al. The International Human Epigenome Consortium: A Blueprint for Scientific Collaboration and Discovery. Cell 2016, 167, 1145–1149. [Google Scholar] [CrossRef]
Figure 1.
Epigenetic modifications and their effect on DNA accessibility.


{kind=link}
Table 1.
Overview of major epigenetic writers, readers and erasers that play a role in HSC self-renewal.
Table 1.
Overview of major epigenetic writers, readers and erasers that play a role in HSC self-renewal.
| Gene | Function | Effect on HSC | Reference |
|---|---|---|---|
| DNA modifications | |||
| DNMT1 | DNA methyltransferase: maintenance parental cell methylation patterns. | Required for HSC self-renewal, niche retention and progression from multipotent to myeloid progenitors. Deletion leads to lineage skewing towards myelopoiesis and defective self-renewal. | [4,5,6] |
| DNMT3A/B | DNA methyltransferase: de novo DNA methylation. | Essential for HSC self-renewal and deletion increases HSC life span. DNMT3A and DNMT3B show complementary de novo methylation patterns responsible for silencing of self-renewal genes in HSCs. | [7,8,9,10,11] |
| TET1/2 | Catalyse the oxidation of 5-methylcytosine into 5-hydroxymethylcytosine, resulting in DNA demethylation. | TET1 deficiency increases HSC self-renewal potential. TET2 deletion resulted in enhanced HSC self-renewal and enhanced myelopoiesis. TET2 mutations are mutually exclusive with gain of function mutations IDH1/2 in AML. | [12,13,14,15,16,17] |
| IDH1/2 | Isocitrate dehydrogenase 1/2 enzymes (IDH1/2), required for conversion of isocitrate into a-ketoglutarate, a TET2 cofactor. | IDH1 mutations impair histone demethylation and result in hypermethylation. TET2 function becomes disrupted and differentiation is inhibited. Mutant IDH1/2 overexpression in primary bone marrow cells disturbs haematopoietic differentiation and leads to elevated numbers of HSPCs. | [18,19,20] |
| Histone modifications | |||
| EZH1/2 | Histone lysine methyltransferase, PRC2 member. | Important for HSC maintenance. Deletion decreases self-renewal potential. EZH2 deletion alone does not compromise self-renewal, possibly due to complementary EZH1 activity. | [21,22,23,24] |
| DOT1L | H3K79 methyltransferase | Crucial for embryonic erythropoiesis. Not essential for adult haematopoiesis. | [25,26,27,28] |
| MLL proteins | H3Kmethyltransferases | MLL-fusion enriched target genes play a role in HSC function, MLL mediated activation of HOX genes has been described, MLL fusions are potent inducers of leukaemia. | [29,30,31,32] |
| SETDB1 | H3K9 methyltransferase | Essential for HSC function. | [33] |
| PMRT4/5 | Protein arginine methyltransferases | PMRT4 blocks myeloid differentiation. PMRT5 inhibits expression of differentiation associated genes, PMRT5 deletion results in HSPC exhaustion. | [34,35,36,37] |
| CBP/p300 MOZ | Histone acetyltransferases | CBP/p300 regulates self-renewal and differentiation in adult HSCs. MOZ maintains the generation and development of HSCs. | [38,39] |
| ASXL1 | Polycomb chromatin-binding protein, associates with PRC1 and PRC2. | Loss results in impaired self-renewal. | [40,41,42] |
| CBX2 | Chromobox 2, PRC1 member. Reads H3K27me3. | Impairs HSC and progenitor proliferation. | [43,44,45] |
| CBX7 | Chromobox 7, PRC1 member. Reads H3K27me3 and other trimethylated proteins. | Overexpression increases HSC self-renewal. | [45,46,47] |
| Ring1B | E3 ubiquitin-protein ligase. | Antiproliferative role in progenitor expansion. | [48] |
| BMI1/MEL-18 | Polycomb ring finger protein, PRC1 member. | Repression impairs self-renewal. Frequently overexpressed in malignant haematopoiesis. Loss enhances HSC self-renewal. | [49,50,51,52] |
| LSD1 | H3K4/9 demethylase | Overexpression of the short isoform increases self-renewal potential. Loss causes defects in long-term HSC self-renewal and impaired differentiation. | [53,54,55] |
| JARID1b | H3K4 demethylase | Described as a positive regulator of HSC self-renewal capacity. | [56,57] |
| KDM6A/UTX | H3K27 demethylase | Knock out leads to myelodysplasia, suppressed megakaryocytopoiesis and extramedullary haematopoiesis, indicating a regulatory role in haematopoiesis. | [58,59] |
| KDM6B/JMJD3 | H3K27 demethylase | Loss impairs HSC self-renewal potential following proliferative stress. | [59,60] |
| MYSM1 | Histone H2A deubiquitinase | Maintenance of HSC function. Deletion results in impaired self-renewal. | [61,62,63] |
| SIRT1/2/3/7 | NAD-dependent deacetylases | Sirt1 loss increases HSC numbers. Sirt2 and 3 are not required for HSC maintenance at young age, but knockout does compromise HSC function at old age. Sirt6-deficient HSCs exhibited impaired self-renewal ability. Sirt7 overexpression increases constitutive capacity of HSCs. | [64,65,66,67,68] |
Table 2.
Examples of epigenetic drugs and their stage of development for treatment of haematological malignancies.
Table 2.
Examples of epigenetic drugs and their stage of development for treatment of haematological malignancies.
| Type of Drug | Target Enzyme | Compound | Stage of Development | References |
|---|---|---|---|---|
| DNMTi | DNA methyltransferase | Azacitidine, decitabine | EMA and FDA approval for MDS, AML and CMML | [163] |
| IDH1/2 inhibitors | Pre-clinical and clinical | [164,165,166] | ||
| Ivosidenib (IDH1) | EMA and FDA approval for IDH1 mutated and R/R AML | |||
| Enasidenib (IDH2) | EMA and FDA approval for R/R AML | |||
| HDACi | Histone deacetylases | Panobinostat | EMA and FDA approval for MM, CTCL | [125,142,152,154] |
| Romidepsin | EMA and FDA approval for CTCL | |||
| HATi | Histone acetyltransferases | P300 inhibitors | Pre-clinical | [159,160,167] |
| HDMi | Histone demethylases | LSD1 inhibitors | Pre-clinical and clinical | [128,168] |
| HMTi | Histone methyltransferases | EZH1/2 inhibitors Tazemetostat (EZH2) | Pre-clinical and clinical | [169,170] |
| DOT1L inhibitors | Pre-clinical and clinical | [128,171] | ||
| MENIN1-MLL interaction inhibitors | Pre-clinical | [128] | ||
| HATi | Histone acetyl readers | BET-inhibitors | Pre-clinical and clinical | [163,166] |
| (myelodysplastic syndrome (MDS), relapsed/refractory (R/R) acute myeloid leukaemia (AML), chronic myelomonocytic leukaemia (CMML), multiple myeloma (MM), cutaneous T cell lymphoma (CTCL)) | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Buisman, S.C.; de Haan, G. Epigenetic Changes as a Target in Aging Haematopoietic Stem Cells and Age-Related Malignancies. Cells 2019, 8, 868. https://doi.org/10.3390/cells8080868
AMA Style
Buisman SC, de Haan G. Epigenetic Changes as a Target in Aging Haematopoietic Stem Cells and Age-Related Malignancies. Cells. 2019; 8(8):868. https://doi.org/10.3390/cells8080868
Chicago/Turabian StyleBuisman, Sonja C., and Gerald de Haan. 2019. "Epigenetic Changes as a Target in Aging Haematopoietic Stem Cells and Age-Related Malignancies" Cells 8, no. 8: 868. https://doi.org/10.3390/cells8080868
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.