Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation


1
Innere Medizin 2, Hämatologie und Onkologie, Universitätsklinikum Jena, 07747 Jena, Germany
2
Leibniz-Institute on Aging–Fritz Lipmann Institute (FLI), 07745 Jena, Germany
3
Dana-Farber Cancer Institute, Department of Pediatric Oncology, Harvard University, Boston, MA 02467, USA
4
Center for Immunology & Inflammatory Diseases, Massachusetts General Hospital, and Harvard Medical School, Boston, MA 02129, USA
*
Author to whom correspondence should be addressed.
Cells 2019, 8(8), 854; https://doi.org/10.3390/cells8080854
Submission received: 9 July 2019
/
Revised: 5 August 2019
/
Accepted: 6 August 2019
/
Published: 8 August 2019
(This article belongs to the Special Issue Aging and Regeneration)
{kind=link}
{kind=link}
Abstract
:Clonal alterations in hematopoietic cells occur during aging and are often associated with the establishment of a subclinical inflammatory environment. Several age-related conditions and diseases may be initiated or promoted by these alterations. JAK2 mutations are among the most frequently mutated genes in blood cells during aging. The most common mutation within the JAK2 gene is JAK2-V617F that leads to constitutive activation of the kinase and thereby aberrant engagement of downstream signaling pathways. JAK2 mutations can act as central drivers of myeloproliferative neoplasia, a pre-leukemic and age-related malignancy. Likewise, hyperactive JAK-signaling is a hallmark of immune diseases and critically influences inflammation, coagulation and thrombosis. In this review we aim to summarize the current knowledge on JAK2 in clonal hematopoiesis during aging, the role of JAK-signaling in inflammation and lymphocyte biology and JAK2 function in age-related diseases and malignant transformation.
1. Development of Clonal Hematopoiesis During Aging
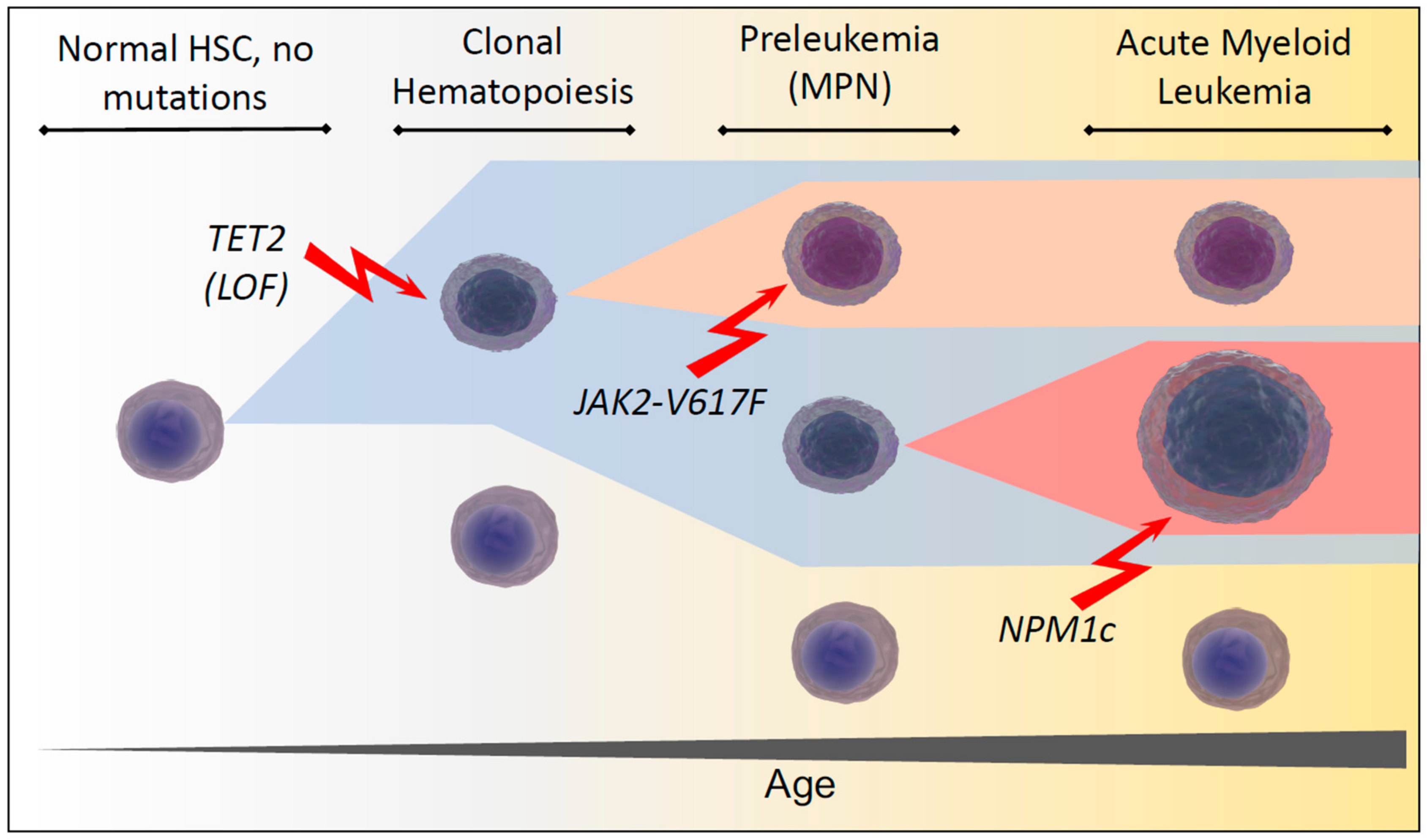
Normal hematopoietic stem cells (HSCs) show intact self-renewal and regeneration with a balanced differentiation potential towards myeloid and lymphoid progenitor cells. As the hematopoietic system ages, somatic mutations lead to a decline in the HSC function and skewing towards the myeloid compartment [1,2]. The first evidence supporting the idea that the hematopoietic system is subject to substantial clonal alterations during aging was provided by studies detecting an age-associated skewing of X-chromosome inactivation patterns in the blood cells of healthy females, particularly in the myeloid compartment [3,4]. The subsequent detection of age-related TET2 mutations in healthy individuals with X-inactivation skewing and hence clonal hematopoiesis provided one of the first proofs that somatic genetic aberrations occur in the healthy aging population and are associated with an age-related myeloid lineage bias. Furthermore, the detected mutations had been previously observed in patients with myeloid malignancies, suggesting that these clones may represent a premalignant state [5].
More recently, three large exome-analysis studies have shown age-related clonal hematopoiesis in healthy individuals, driven by mutations of genes recurrently mutated in myeloid neoplasms and associated with an increased risk of hematologic cancer and cardiovascular disease. Whilst healthy individuals younger than 50 years of age were rarely affected (<1%), about 10% of individuals over 65 years of age showed clonal hematopoiesis. All studies identified similar genes, with the majority of mutations affecting epigenetic modifiers such as DNMT3A, TET2 and ASXL1. Among other frequently mutated genes were those involved in RNA-splicing such as SF3B1 and SRSF2, and genes involved in signal transduction such as CBL and JAK2 [6,7,8]. Based on these findings, Steensma et al. [9] proposed the term clonal hematopoiesis of indeterminate potential (CHIP) for individuals who do not show conclusive morphologic and clinical evidence of malignant or pre-malignant disease, but harbor somatic mutations frequently detected in hematologic malignancies.
Interestingly, Young et al. have since used ultra-sensitive DNA-sequencing to demonstrate persisting leukemia-associated mutations at very low frequencies in 95% of healthy individuals aged 50 to 60 [10], suggesting that age-related clonal hematopoiesis is extremely common, if not inevitable. A prime example of the apparent paradox of healthy individuals carrying aberrations associated with myeloid malignancies, is the JAK2-V617F mutation, which has been considered to be the major culprit in causing polycythemia vera (PV) and other myeloproliferative neoplasms (MPN) for over a decade [11,12,13,14]. Yet recently it was shown that JAK2-V617F neoplasms develop from clonal hematopoiesis over many years with highly variable levels of clonal expansion [15]. Taken together these findings raise questions about the cascades of events necessary to transform clonal hematopoiesis from a seemingly common and benign to a relatively rare malignant state (Figure 1).
2. Consequences of Deregulated JAK2 Signaling in Hematopoiesis and Aging, CHIP-Driven Inflammation and Consequences of JAK2-Mutations
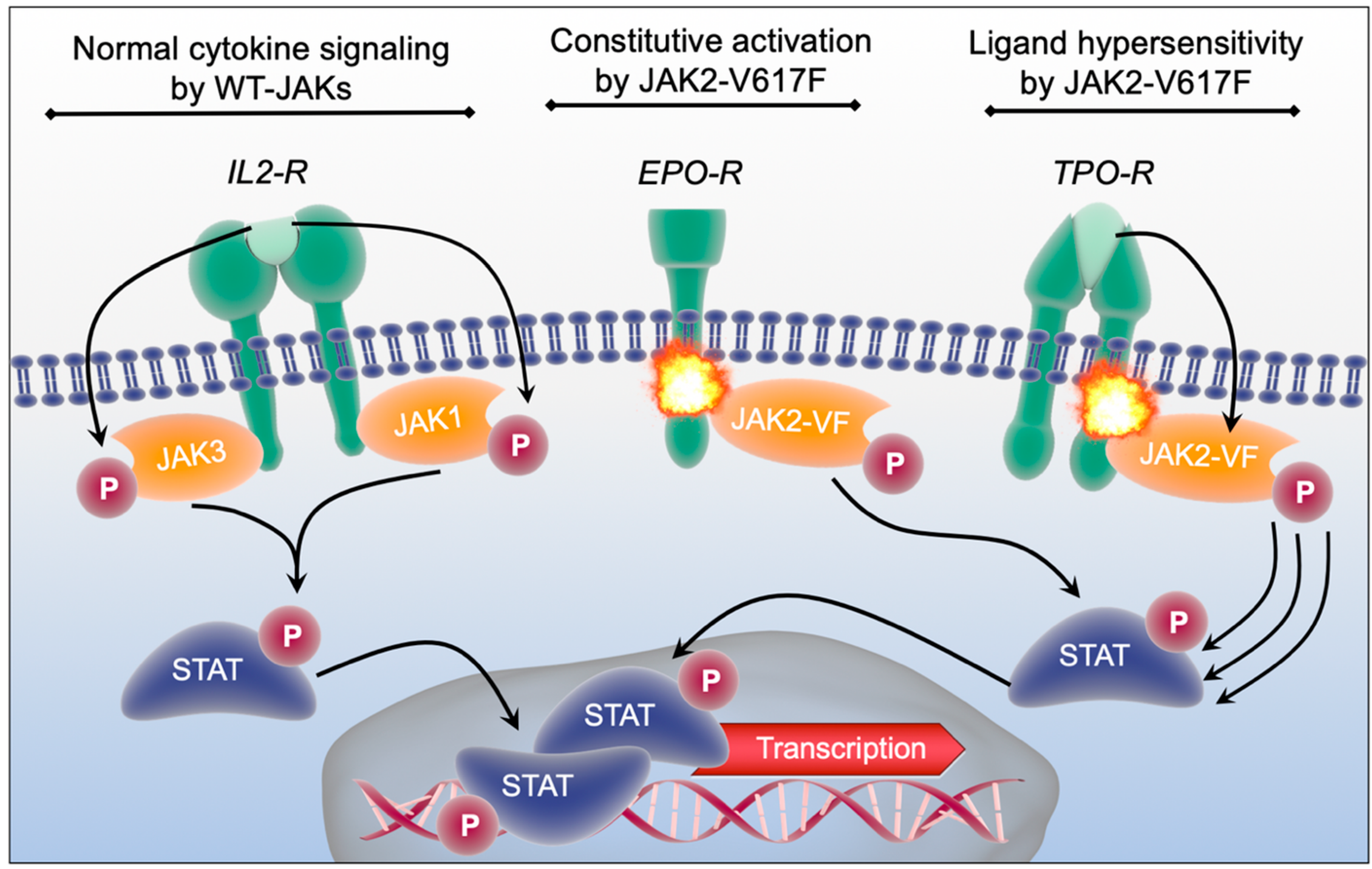
Even though the complex functions of age-related clonal alterations are just about to be explored by the scientific community one common feature of several described CHIP-mutations is the establishment of a sub-clinical inflammatory state. In addition to JAK2-mutations, particularly loss-of-function of TET2 has been described to be associated with chronic proinflammatory changes in vivo [16,17,18,19]. Of note, these indolent inflammatory alterations may become clinically apparent upon additional stress factors like infections. In 2018, Meisel et al. described the interplay between the TET2 loss-of-function and microbial stimuli to drive pre-leukemic myeloproliferation using a Tet2−/− knockout mouse model [20]. DNMT3A, the most frequently mutated gene in clonal hematopoiesis, has been identified as a critical mediator of mast cell responses [21]. Furthermore, also other genes that are frequently mutated in CHIP, like SF3B1 [22], CBL [23,24], GNB1 [25] and GNAS [26] have been recently explored as crucial mediators of inflammatory signals. Among the genes that are common subjects to age-related mutations JAK2 has been linked most clearly to inflammatory processes since it serves as a signal transmitter downstream of major cytokine receptors [27,28,29]. Therefore, multiple inflammatory conditions, including those driven by other genes mutated in CHIP, converge on the JAK signal transduction and are dependent on the JAK-activity. Type I and II cytokine receptors are a conserved family of transmembrane proteins including the receptors for interleukins, interferons, erythropoietin, thrombopoietin, growth hormone, leptin and colony stimulating factors (CSFs) [30,31]. These cytokine receptors lack an intrinsic kinase activity but are associated with “Janus kinases” (JAKs). The JAK-family consists of four members: JAK1, JAK2, JAK3 and TYK2 that have different association patterns to receptors [32,33,34,35,36,37]. JAK1 and JAK2 have non-redundant functions and are essential during development. Therefore, respective conventional knockout mouse models exhibit postnatal or embryonic lethality, respectively [38]. In the JAK2 knockout mice a complete absence of definitive erythropoiesis and interferon-gamma insensitivity was observed causing lethality at day 12.5 postcoitum [38]. The conventional JAK1 knockout allowed normal tissue development but caused postnatal lethality due to nursing insufficiency. In these animals the different interleukin and interferon responses were dramatically impaired [39]. Accordingly, inactivating germline mutations in JAK1 or JAK2 cannot be found in humans. Loss of the JAK3 function on the other hand is vital and causes a severe combined immunodeficiency (SCID) syndrome in patients and germline mutations of TYK2 can be found in patients and are a cause of an autosomal dominant form of the hyper-IgE syndrome [40].
Following activation of the cytokine receptors through binding of the respective ligand JAKs become phosphorylated and promote phosphorylation and activation of “Signal Transducers and Activators of Transcription” (STATs) (Figure 2, left scheme) [41,42,43,44,45]. STATs are DNA-binding proteins that contain seven members (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, STAT6). After dimerization of STATs through activation, they translocate from the membrane to the nucleus to regulate the transcription of target genes (Figure 2, bottom) [42,44]. In addition to the STATs, JAK downstream signaling is believed to be mainly executed by the “Mitogen-activated protein kinases” (MAPK) as well as the “Protein kinase B pathway” (PI3K/ AKT). Accordingly, JAKs can promote proliferation, differentiation and cytokine production in immune- and hematopoietic stem and progenitor cells [46,47]. These pathways can be pathologically activated by either excessive ligand binding (e.g., under chronic inflammatory conditions) or by activating mutations within JAK-genes. The JAK2-V617F point mutation is the most frequently detected somatic mutation in the JAK family that leads to constitutive activation of JAK2 and its downstream effectors independent from ligand availability as well as to a hypersensitivity of cytokine receptors upon ligand binding (Figure 2, middle and right schemes) [12,48]. Thereby, JAK2-V617F is a frequent driver of myeloproliferative neoplasms (MPN), a group of myeloid malignancies caused by an increased proliferation and cytokine production of different myeloid cell types [11,12,13,14]. Due to their involvement in multiple inflammatory pathways, JAKs are attractive targets for therapeutic intervention in inflammatory diseases [49,50,51,52]. Here, JAK-inhibition can lead to a reduction of symptom burden in MPN [53,54] as well as in different rheumatological diseases [49,55,56,57].
2.1. Dysfunctional JAK-Signaling and JAK-Inhibition in the Adaptive and Innate Immune System
As described in more detail above, JAKs have a crucial function in cytokine-driven signal transduction and are therefore central molecules in the immune system. Given the fact that cytokine signals induce differentiation and polarization of leukocyte subpopulations one can anticipate that alterations in the JAK activity and function may have profound consequences for white blood cell homeostasis. The activating JAK2-V617F mutation that drives MPN and is also detected in individuals with CHIP can indeed influence lymphocyte biology. In 2017, Nishanth et al. reported on a gain-of-function phenotype induced by mutations of JAK2 in T-cells [58]. In MPN the JAK2-V617F mutation arises from mutated HSC clones and is therefore not strictly committed to myeloid cells. The authors reported on 6/13 MPN patients harboring a JAK2-V617F mutation in a significant percentage of their CD4+ and CD8+ T-cells. Using a knock-in model of JAK2-V617F [59] with an inducible T-cell specific CD4-Cre-recombinase animals showed improved pathogen clearance during Listeria monocytogenes infection with increased cellular and serological responses. Otherwise, the thymic T-cell development was not significantly impaired. In contrast, increased numbers of neutrophils and erythroblasts could be detected in the spleens indicating a cell extrinsic effect of the JAK2-V617F+ T-cells on myeloid bystanders through cytokine secretion [58]. This experimental model provided first insights into how activating JAK2 mutations influence the lymphocyte function and confirmed the complexity of the interplay between the cell intrinsic and humoral effects of JAK2-V617F. In other reports Stijnis et al. assessed for the JAK2-V617F mutation in different lymphocyte subsets of MPN patients with co-existing chronic lymphocytic leukemia (B-CLL). The authors described JAK2-mutations in T-cells, NK-cells and polyclonal B-cells, but not within the monoclonal B-cell population (CD19+ CD5+ B-CLL-cells) [60]. Most notably, recent reports provided first evidence on how the JAK2-V617F mutated myeloid cells may influence T-cell responses. JAK2-V617F promoted the synthesis of PD-L1 in MPN cells leading to limited anti-neoplastic T-cell responses, metabolic changes in T-cells and eventually JAK2-V617F-driven immune-escape of MPN cells [61]. These findings may facilitate the use of immunotherapeutic approaches for JAK-mutated clones. The question how far the presence of JAK2-mutations in distinct lymphocyte subsets predict differential responses or outcomes in MPN patients or individuals with CHIP has not been addressed yet.
The consequences of JAK-inhibition on human immune cell function have been studied in more detail. In early clinical trials, increased numbers of viral infections have been described in MPN patients on treatment with the JAK1/2 inhibitor ruxolitinib [53,62]. JAK1/2 inhibition in MPN patients leads to a reduction in CD3+ T-cells and decreased cytokine production. Within the T-cell compartment regulatory T-cells (Treg) and Th1 cells seem to be most prominently affected [63,64,65]. Additionally, effector functions of CD8+ T-cells are impaired upon JAK1/2 inhibitor treatment [66]. Furthermore, JAK-inhibition compromises B-cell differentiation and antibody production [67,68] as well as dendritic cell function [69]. Even though the spectrum of the JAK-inhibition is broad, the suppression of cellular functions is cell type specific and incomplete. Therefore JAK-inhibition is relatively well tolerated by patients in randomized clinical trials [53,70,71,72] with a relatively low rate of infectious complications. Accordingly, JAK-inhibitor should be considered an immunomodulatory treatment rather than a classical immunosuppressive drug. In addition to the already mentioned attempts to utilize JAK-inhibitors in rheumatological diseases they are successfully used in the treatment of acute and chronic graft-versus-host disease (GvHD) after allogenic stem cell transplantation. In a mouse model of GvHD ruxolitinib improved survival and limited proinflammatory cytokine production as well as Th1 and Th17 polarization. A pilot trial of ruxolitinib in steroid refractory GvHD patients successfully limited cytokine production and clinical symptoms in this heavily pre-treated cohort [73]. Furthermore, advanced clinical trials investigating ruxolitinib for GvHD in a randomized fashion are currently under way. Interestingly, the graft-versus-leukemia effect which is required for the therapeutic efficacy remained unaffected [74], confirming the model of selective immunomodulatory functions of JAK. In how far different JAKs have redundant and non-redundant functions in immune cells is yet not fully understood. Since ruxolitinib is currently the only approved JAK inhibitor for the treatment of MPN but novel and more specific compounds are being developed, the question whether specificity of inhibitors for the JAK1 or JAK2 function (or both) are required for the T-cell function [75] is of utmost importance. We utilized two JAK1/2 inhibitors that are in clinical use (ruxolitinib and momelotinib) as well as a novel specific JAK2-inhibitor (BSK805). Interestingly, in vitro treatment of healthy donor T-cells with either JAK1/2 inhibitor resulted in the inhibition of proliferation, global activation (CD69), and STAT1 phosphorylation of CD4+ and CD8+ T-cells while selective JAK2-inhibition had no such effect. To confirm these findings we genetically inactivated either JAK1 or JAK2 in T-cells using RNAi. Only JAK1 depletion was sufficient to inhibit the global T-cell function in vitro. Consistently, JAK2 was dispensable for global T-cell effector functions in vivo in a mouse model of GvHD [75]. These findings highlight the importance of JAK-selectivity depending on the underlying condition or context. Selective JAK2 inhibitors are currently in clinical trials [76,77,78,79,80] and seem to be equally effective in symptom control and inhibition of systemic inflammation. The interplay between the different JAKs may limit the applicability of highly selective JAK-inhibitors [81]. Of note also JAK1, JAK3 and TYK2 selective inhibitors are currently under investigation in clinical trials for different inflammatory diseases [82,83,84,85,86,87,88,89,90,91,92,93].
2.2. Chronic Inflammation and JAK Signaling in Aging and Age-Related Degenerative Diseases
Aging and age-related diseases are accompanied by sterile, low grade and chronic inflammatory processes in different tissues [94,95,96,97,98,99,100,101]. Even though unbalanced inflammation during aging may lead to tissue degeneration and age related diseases, temporal and spatial orchestration of inflammatory stimuli is crucial for tissue homeostasis and regeneration [102,103,104,105,106,107]. In particular the importance of the JAK/STAT pathway for regenerative processes in multiple tissues has been highlighted [108,109,110,111,112,113]. In 2016, Shen et al. reported on their findings from a longitudinal study with 91 young and older individuals profiling immunological signatures and cytokine responsiveness over the course of three years. The authors found elevated baseline levels of activated STATs, most prominently in T-cells, leading to a reduced responsiveness to cytokine stimulation in the cohort of elderly individuals. Of note these changes where associated with cardiovascular diseases [114]. Based on these observations it is tempting to speculate in how far the chronic inflammatory state established by age related clonal mutations may participate to these changes in systemic homeostasis during aging.
Several studies investigated JAK-function and -inhibition in different degenerative aging related conditions. With regards to the issue of age-related cachexia one study using a respective mouse model pointed out that excessive activation of the JAK-STAT pathway in aged adipose tissue is inducing a senescence-associated secretory phenotype (SASP) with increased production of proinflammatory cytokines. Moreover, administration of a JAK1/2 inhibitor to aged mice for 10 weeks reduced inflammation and alleviated frailty [115]. Decreased or diminished regenerative capacity of skeletal muscle is another age-related symptom that can be challenged through pharmacological inhibition of JAK2 or STAT3 by stimulating symmetric expansion of satellite cells in vitro and their engraftment in vivo. Intramuscular injection of JAK-inhibitors resulted in a remarkable enhancement of muscle repair and regeneration [116]. In Parkinson´s and Alzheimer´s disease alpha-synuclein and Amyloid-beta accumulation is leading to activation and pre-sensitizing of microglia through the JAK/STAT pathway, resulting in an IFN-γ driven proinflammatory microglial phenotype promoting neurodegeneration. Treatment with JAK inhibitors prevented the degeneration of dopaminergic neurons in an in vivo model of Parkinson´s disease and attenuated the IFN-γ-induced changes in cultured microglia and in isolated microglia prepared from APP/PS1 Alzheimer´s mice [117,118]. In how far kinase inhibition could be utilized as an intervention for age related functional decline or chronic degenerative diseases is currently a subject to controversial discussions within the community [119,120]. For manifesting clinical conditions like neurodegenerative diseases some of those treatment approaches may enter clinical trials in the future due to the lack of alternative therapeutics and tolerability of potential side effects. For subclinical conditions like muscle or adipose tissue loss dietary and lifestyle interventions should primarily be considered to impact on health outcomes.
2.3. Pathophysiology of JAK2 in Malignant Transformation and Myeloproliferative Neoplasia
While JAK2-mutations are among the most common genetic aberrations detected in aging-associated clonal hematopoiesis they do not necessarily lead to development of myeloproliferative neoplasms or hematologic cancers [6,7,121,122]. Nevertheless, activating JAK2-V617F mutations are the most frequent driver mutations of myeloproliferative neoplasms (MPN) [123,124]. The vast majority of these activating JAK2 mutations are JAK2-V617F point mutations as mentioned above [14]. Furthermore, there is a small proportion of JAK2-V617F-negative MPN patients showing alterations in Exon 12 of the JAK2 gene leading to a similar disease phenotype [125]. Studies on primary human blood specimens had reported on the presence of other gene mutations before the acquisition of JAK2 mutations and raised the question whether the JAK2-mutation alone is sufficient for malignant transformation [126,127,128,129]. Most recently, however, elegant studies performed in murine models provided first evidence that in principle a single hematopoietic stem cell can be transformed by an activating JAK2-V617F-mutation and lead to erythrocytosis or thrombocytosis in vivo [130]. The presence of the JAK2-V617F-mutation in HSCs accelerated cell division and resulted in increased DNA damage, however, the penetrance of disease initiation in mouse models was low. Longitudinal studies focusing on the evolution of mutations in serial human samples found a low mutation rate of one mutation per 66 patient years [131] arguing against a strong hypermutable state in MPN. Of note, the strongest predictor of disease progression and outcome in these analyses was the number of somatic mutations that occurred in addition to driver mutations as JAK2. Therefore, individuals that exclusively harbor JAK2-mutations in the hematopoietic system, may experience long-term stability of the mutated clone or even clonal regression. In contrast, individuals with more than one mutation have a significantly higher risk of clonal expansion and progression [132,133,134]. The presence of clonal aberrations prior to the acquisition of the JAK2-V617F-mutation may therefore provide a ‘fertile ground’ for malignant transformation [131,135]. Once the clone has expanded and results in myeloproliferation, the presence, type and order of clonal aberrations will influence the disease phenotype and prognosis [136,137,138,139]. Although clinical markers are established to assess for prognosis of phenotypically defined subtypes of MPN, genomic classification can identify patients with common biologic factors that accurately define patients at risk for disease progression and further expansion of the malignant clone [140]. Consistent with these findings, mutations in genes such as TP53, TET2 or IDH1 have been frequently observed in primary samples of patients who had experienced transformation of MPN into blast phase or secondary acute myeloid leukemia [141,142,143] and suggest a role of these secondary events in leukemic transformation.
Although the dynamics of clonal expansion and mutational landscape have been defined in very detail, several aspects of disease development, maintenance and progression remain so far elusive. One unique aspect of JAK-V617F-mutations is the low dependency of mutated clones on the constitutive activity of the kinase. While in other cancers, inactivation of oncogenic kinases or receptors results in rapid and durable regression of the malignant clone [144,145], pharmacologic inhibition of JAK2 does not affect the overall disease burden or evolution of persistent clones to a major extent [146,147]. Combination with inhibitors of cell signaling or epigenetic compounds may facilitate targeting of the malignant clone. Likewise, the effects of extrinsic factors, such as inflammatory stimuli caused by co-morbidities or repeated infections dynamics on the JAK-mutated clones are incompletely understood.
The inflammatory phenotype that is observed in MPN patients is a major aspect of JAK2-induced pathophysiology and contributes to morbidity and mortality of the disease [28]. As outlined above in more detail, Janus-kinases are critical mediators of cytokine, chemokine and growth-hormone signaling. Constitutive activation of Janus-kinase due to mutations in the JAK-gene locus therefore results in inflammation that can promote myeloproliferation, thrombosis formation, and splenomegaly and is the major cause of constitutional symptoms [148,149] in patients with MPN. Even similarities of inflammatory symptoms and laboratory parameters with other reactive causes of inflammation (e.g., systemic inflammatory response syndrome–SIRS) have been described [28]. In murine models of MPN as well as in primary patient samples, high levels of pro-inflammatory cytokines could be detected [150,151,152,153]. Moreover, high levels of cytokine expression have been described to be prognostically relevant [152]. As one prominent example, tumor necrosis factor alpha (TNFa) was found to be highly expressed in JAK2-V617F-mutated MPN cells and to correlate with disease burden in primary patient cells [154]. Inactivation of TNFa in murine models of MPN or primary patient cells resulted in reduced disease development in vivo and abrogation of clonal growth, respectively. Recent publications provided first evidence that besides the JAK2-mutated clone also non-malignant ‘bystander’ cells can contribute to the secretion of pro-inflammatory cytokines [155]. Cytokine profiles of the JAK-mutated clone are depending on STAT-signaling and can partially overlap with those of non-malignant ‘bystanders’ or those produced by other driver mutations (e.g., thrombopoietin-receptor mutations). Secretion of pro-inflammatory cytokines and chemokines also results in tissue re-modeling in the bone marrow, specifically in the development of fibrosis and osteosclerosis. Moreover, the inflammatory milieu may by itself promote myeloproliferation and clonal evolution [27]. One model system that supports this hypothesis is overexpression of the transcription factor NF-E2, which has shown to be highly expressed in primary MPN samples [156,157,158]. The resulting inflammatory milieu promoted cellular proliferation and disease progression.
Anti-inflammatory compounds have been used to treat JAK2-V617F-driven MPN since decades. The early use of corticosteroids in patients with myelofibrosis was followed by the use of interferons, IMIDs and most recently JAK-inhibitors [123]. JAK-inhibitors have shown significant clinical activity on the reduction of inflammation [54,71]. Clinical use of JAK-inhibitors has shown to improve elevated levels of pro-inflammatory cytokines and associated symptoms such as weight loss, fever or splenomegaly.
Mutations of JAK2 have been linked with thromboembolic complications and alterations in hemostasis and cell adhesion in vitro and in vivo [159]. Several pathophysiologic aspects contribute to this thrombophilic state: (i) Hypercellularity induced increase in blood viscosity, (ii) JAK-induced alterations in plasmatic coagulation and vessel wall, (iii) JAK-induced changes of cell adhesion and function. Early observations reported increased rates of thromboembolic events in patients with myeloproliferative neoplasia [160,161]. Along these lines, control of hypercellularity (such as hematocrit control below 45% in polycythemia vera) results in reduction of thromboembolic complications. This therapeutic goal is achieved irrespective of the type of cytoreduction, through recurrent phlebotomies, pharmacologic cytoreduction using hydroxycarbamide or a combination of both [162]. Therefore, the cytoreductive treatment is a mainstay of the MPN therapy to prevent thromboembolic complications, especially in high risk situations defined by higher age or previous thromboembolic events [163]. However, although impairment of blood flow due to hypercellularity is an obvious risk factor for thrombus formation and development of arterial emboli, even patients in early phases of myeloproliferative neoplasia and with normal blood counts develop thrombotic events, e.g., of the splanchnic venous system [164]. Furthermore, the presence of cardiovascular risk factors increases the risk for thromboembolic complications in JAK2-V617F-mutated MPNs [165,166,167]. This indicates that other factors such as plasmatic coagulation or cell intrinsic alterations of JAK-mutated cells may contribute to thrombus formation. Most recently, murine models of MPN have given insight into JAK-induced pathophysiology regarding thrombotic complications. Expression of mutated JAK2 in endothelial cells (a finding that could be confirmed in the human system; [168,169,170]) resulted in abnormalities in blood flow and alteration of plasmatic coagulation [171]. Moreover, high expression of mutated JAK2 was associated with both, formation of unstable thrombi and bleeding events in vivo [171,172]. JAK-mutated platelets showed reduced activatability and moderate glycoprotein (GP) VI deficiency while a decreased proportion of high-molecular-weight von-Willebrand-factor multimers could reduce platelet adhesion [172]. Consistently, activation of JAK2-V617F-mutations in megakaryopoiesis resulted in hypersensitivity to thrombopoietin (Tpo) stimulation, higher mobility of megakaryocytes, elevated pro-platelet formation and increased thrombus formation due to enhanced platelet aggregation [173]. These findings are consistent with the clinical situation where JAK2-mutations indicate an increased risk for thromboembolic events per se compared to non-JAK-mutated MPN cases [174] and administration of salicylic acid is routinely used to prevent thrombotic complications [175]. When focusing on red blood cell (RBC) biology, other groups could show that abnormal adhesion of JAK2-V617F-mutated red blood cells is induced through upregulation adhesion molecules through Rap-Akt-signaling even in the absence of erythropoietin (Epo) [176]. In this model system, pharmacologic inhibition of Rap- or Akt-signaling was able to reduce abnormal adhesion of RBCs. In neutrophil biology, JAK2-V617F-mutations seem to affect various cellular functions. Primary patient neutrophils harboring activating JAK2-mutations show a significant increase in neutrophil extracellular trap (NET) formation [177]. Likewise, murine JAK2-V617F-mutated neutrophils contributed to increased thrombus formation following experimental venous ligation. Pharmacologic JAK-inhibitor treatment effectively reduced the phenotypes in both model systems. Of note, an extended clinical and molecular analysis on a large cohort of otherwise healthy individuals with clonal hematopoiesis (CHIP) revealed significantly higher rates of thromboembolic events in those harboring JAK2-mutations as clonal markers when compared to non-JAK-mutated CHIP [177]. Consistent with these findings, increased expression of beta1-integrins was described on granulocytes of MPN patients harboring JAK2-mutations as compared to age-matched healthy donor controls [178]. Functional studies using mouse models and primary human cells confirmed activation of integrins by JAK2 in a Rap1-GTPase dependent manner [179]. The activation of integrins may also contribute to the accumulation of myeloid cells in the spleen, as pharmacologic inhibition of the integrin function in mice resulted in improvement of splenomegaly and reduced cell numbers in the spleens. These results may facilitate a combination of Janus-kinase inhibitors with inhibitors of integrin activation or function to efficiently prevent thrombosis formation patients at risk.
3. Conclusions
Age-related somatic mutations in hematopoietic stem cells lead to the establishment of clonal sub-populations in the blood of elderly individuals and is frequently associated with chronic inflammatory changes. The presence of these clonal alterations is associated with cardiovascular risk and malignant transformation. Among the most frequently mutated genes during aging is the Janus kinase JAK2. The activating JAK2-V617F mutation is a driver of myeloproliferative neoplasia (MPN) a preleukemic blood disorder. Aberrant activation of JAK2 in MPN is associated with hyperproliferation of myeloid progenitor cells, abnormal inflammatory cytokine release as well as hyper-agglutination and thrombosis. The role of JAK mutations in clonal hematopoiesis of indeterminate potential (CHIP) on the other hand remains largely elusive. Of note even in the absence of a clinically manifest MPN JAK2-V617F appears to be associated with thromboembolic complications. Since most major cytokine and growth factor receptors utilize JAKs to transmit their signals this group of kinases form a common intersection in inflammatory signal transduction on where multiple cytokine driven responses converge. Therefore, pharmacological targeting of JAKs can serve as a therapeutic tool to modulate the activity of a broad range of immune functions. Importantly, inhibitors of JAKs are in clinical use for the treatment of MPN and graft compared to the host disease (GvHD) as well as for several rheumatological diseases, including rheumatoid arthritis, psoriasis and lupus erythematosus. Chronic inflammatory processes negatively influence on health outcomes during aging. One driver of this chronic sterile inflammation is the aberrant cytokine release and the respective responses of cells in different organs. Since JAKs transmit most of those signals they are centrally involved in mediating this alteration in systemic homeostasis. Recent evidence suggests that several age-associated functional alterations are driven or promoted by this chronic inflammation. Indeed, the decline in muscle, adipose tissue and neuronal function during aging can partially be challenged by the mediating JAK activity. Nevertheless, in how far drug treatment of these chronic age-related conditions may be beneficial in certain cases remains controversial.
Author Contributions
All authors contributed to the writing of the manuscript.
Funding
This work was funded by the Thuringian state program ProExzellenz (RegenerAging - FSU-I-03/14) of the Thuringian Ministry for Research (to F.H.H.) and in part by a grant of the German Research Council (DFG) (HE 6233/4-1 to F.H.H.).
Conflicts of Interest
F.H.H. has served as an advisory board member for Novartis, CTI and Celgene and has received research funding from Novartis.
References
- Beerman, I.; Bhattacharya, D.; Zandi, S.; Sigvardsson, M.; Weissman, I.L.; Bryder, D.; Rossi, D.J. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc. Natl. Acad. Sci. USA 2010, 107, 5465–5470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dykstra, B.; Olthof, S.; Schreuder, J.; Ritsema, M.; de Haan, G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J. Exp. Med. 2011, 208, 2691–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busque, L.; Mio, R.; Mattioli, J.; Brais, E.; Blais, N.; Lalonde, Y.; Maragh, M.; Gilliland, D.G. Nonrandom X-inactivation patterns in normal females: Lyonization ratios vary with age. Blood 1996, 88, 59–65. [Google Scholar] [PubMed]
- Gale, R.E.; Wheadon, H.; Boulos, P.; Linch, D.C. Tissue specificity of X-chromosome inactivation patterns. Blood 1994, 83, 2899–2905. [Google Scholar] [PubMed]
- Busque, L.; Patel, J.P.; Figueroa, M.E.; Vasanthakumar, A.; Provost, S.; Hamilou, Z.; Mollica, L.; Li, J.; Viale, A.; Heguy, A.; et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 2012, 44, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Kahler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Young, A.L.; Challen, G.A.; Birmann, B.M.; Druley, T.E. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 2016. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKerrell, T.; Park, N.; Chi, J.; Collord, G.; Moreno, T.; Ponstingl, H.; Dias, J.; Gerasimou, P.; Melanthiou, K.; Prokopiou, C.; et al. JAK2 V617F hematopoietic clones are present several years prior to MPN diagnosis and follow different expansion kinetics. Blood Adv. 2017, 1, 968–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389. [Google Scholar] [CrossRef] [PubMed]
- Cull, A.H.; Snetsinger, B.; Buckstein, R.; Wells, R.A.; Rauh, M.J. Tet2 restrains inflammatory gene expression in macrophages. Exp. Hematol. 2017, 55, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Kotzin, J.J.; Ramdas, B.; Chen, S.; Nelanuthala, S.; Palam, L.R.; Pandey, R.; Mali, R.S.; Liu, Y.; Kelley, M.R.; et al. Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell 2018. [Google Scholar] [CrossRef]
- Pan, W.; Zhu, S.; Qu, K.; Meeth, K.; Cheng, J.; He, K.; Ma, H.; Liao, Y.; Wen, X.; Roden, C.; et al. The DNA Methylcytosine Dioxygenase Tet2 Sustains Immunosuppressive Function of Tumor-Infiltrating Myeloid Cells to Promote Melanoma Progression. Immunity 2017, 47, 284–297. [Google Scholar] [CrossRef]
- Meisel, M.; Hinterleitner, R.; Pacis, A.; Chen, L.; Earley, Z.M.; Mayassi, T.; Pierre, J.F.; Ernest, J.D.; Galipeau, H.J.; Thuille, N.; et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 2018, 557, 580–584. [Google Scholar] [CrossRef]
- Leoni, C.; Montagner, S.; Rinaldi, A.; Bertoni, F.; Polletti, S.; Balestrieri, C.; Monticelli, S. Dnmt3a restrains mast cell inflammatory responses. Proc. Natl. Acad. Sci. USA 2017. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; North, K.; Kim, E.; Jang, E.; Obeng, E.; Lu, S.X.; Liu, B.; Inoue, D.; Yoshimi, A.; Ki, M.; et al. Synthetic Lethal and Convergent Biological Effects of Cancer-Associated Spliceosomal Gene Mutations. Cancer Cell 2018, 34, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Li, Y.Z.; An, H.T.; Wang, Y.L.; Chen, S.H.; Qian, Y.J.; Wang, K.; Zhen, J.L.; Fan, Z.; Gong, X.L.; et al. The E3 Ubiquitin Ligase c-Cbl Inhibits Microglia-Mediated CNS Inflammation by Regulating PI3K/Akt/NF-kappaB Pathway. CNS Neurosci. Ther. 2016, 22, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, C.; Flavell, R.A. c-Cbl deficiency leads to diminished lymphocyte development and functions in an age-dependent manner. Proc. Natl. Acad. Sci. USA 2010, 107, 8316–8321. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Ruengsinpinya, L.; Nakamura, E.; Takahata, Y.; Hata, K.; Okae, H.; Taniguchi, S.; Takahashi, M.; Nishimura, R. Cutting Edge: G Protein Subunit beta 1 Negatively Regulates NLRP3 Inflammasome Activation. J. Immunol. 2019, 202, 1942–1947. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Murray, F.; Koide, N.; Goldstone, J.; Dann, S.M.; Chen, J.; Bertin, S.; Fu, G.; Weinstein, L.S.; Chen, M.; et al. Divergent requirement for Gαs and cAMP in the differentiation and inflammatory profile of distinct mouse Th subsets. J. Clin. Investig. 2012, 122, 963–973. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: Is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 2012, 119, 3219–3225. [Google Scholar] [CrossRef]
- Koschmieder, S.; Mughal, T.I.; Hasselbalch, H.C.; Barosi, G.; Valent, P.; Kiladjian, J.J.; Jeryczynski, G.; Gisslinger, H.; Jutzi, J.S.; Pahl, H.L.; et al. Myeloproliferative neoplasms and inflammation: Whether to target the malignant clone or the inflammatory process or both. Leukemia 2016, 30, 1018–1024. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Kontzias, A.; Yamaoka, K.; Tanaka, Y.; Laurence, A. Janus kinase inhibitors in autoimmune diseases. Ann. Rheum. Dis. 2013. [Google Scholar] [CrossRef]
- Boulay, J.L.; O’Shea, J.J.; Paul, W.E. Molecular phylogeny within type I cytokines and their cognate receptors. Immunity 2003, 19, 159–163. [Google Scholar] [CrossRef]
- Drachman, J.G.; Millett, K.M.; Kaushansky, K. Thrombopoietin signal transduction requires functional JAK2, not TYK2. J. Biol. Chem. 1999, 274, 13480–13484. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Boulton, T.G.; Farruggella, T.; Ip, N.Y.; Davis, S.; Witthuhn, B.A.; Quelle, F.W.; Silvennoinen, O.; Barbieri, G.; Pellegrini, S.; et al. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science 1994, 263, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nature Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Marrero, M.B.; Schieffer, B.; Paxton, W.G.; Heerdt, L.; Berk, B.C.; Delafontaine, P.; Bernstein, K.E. Direct stimulation of Jak/STAT pathway by the angiotensin II AT1 receptor. Nature 1995, 375, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Witthuhn, B.A.; Silvennoinen, O.; Miura, O.; Lai, K.S.; Cwik, C.; Liu, E.T.; Ihle, J.N. Involvement of the Jak-3 Janus kinase in signalling by interleukins 2 and 4 in lymphoid and myeloid cells. Nature 1994, 370, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.M.; Johnston, J.A.; Noguchi, M.; Kawamura, M.; Bacon, C.M.; Friedmann, M.; Berg, M.; McVicar, D.W.; Witthuhn, B.A.; Silvennoinen, O.; et al. Interaction of IL-2R beta and gamma c chains with Jak1 and Jak3: Implications for XSCID and XCID. Science 1994, 266, 1042–1045. [Google Scholar] [CrossRef]
- Witthuhn, B.A.; Quelle, F.W.; Silvennoinen, O.; Yi, T.; Tang, B.; Miura, O.; Ihle, J.N. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell 1993, 74, 227–236. [Google Scholar] [CrossRef]
- Neubauer, H.; Cumano, A.; Muller, M.; Wu, H.; Huffstadt, U.; Pfeffer, K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell 1998, 93, 397–409. [Google Scholar] [CrossRef]
- Rodig, S.J.; Meraz, M.A.; White, J.M.; Lampe, P.A.; Riley, J.K.; Arthur, C.D.; King, K.L.; Sheehan, K.C.; Yin, L.; Pennica, D.; et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell 1998, 93, 373–383. [Google Scholar] [CrossRef]
- Casanova, J.L.; Holland, S.M.; Notarangelo, L.D. Inborn errors of human JAKs and STATs. Immunity 2012, 36, 515–528. [Google Scholar] [CrossRef]
- Larner, A.C.; David, M.; Feldman, G.M.; Igarashi, K.; Hackett, R.H.; Webb, D.S.; Sweitzer, S.M.; Petricoin, E.F., 3rd; Finbloom, D.S. Tyrosine phosphorylation of DNA binding proteins by multiple cytokines. Science 1993, 261, 1730–1733. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K.; Ziemiecki, A.; Wilks, A.F.; Harpur, A.G.; Sadowski, H.B.; Gilman, M.Z.; Darnell, J.E. Polypeptide signalling to the nucleus through tyrosine phosphorylation of Jak and Stat proteins. Nature 1993, 366, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.X.; Migone, T.S.; Tsang, M.; Friedmann, M.; Weatherbee, J.A.; Zhou, L.; Yamauchi, A.; Bloom, E.T.; Mietz, J.; John, S.; et al. The role of shared receptor motifs and common Stat proteins in the generation of cytokine pleiotropy and redundancy by IL-2, IL-4, IL-7, IL-13, and IL-15. Immunity 1995, 2, 331–339. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Gadina, M.; Schreiber, R.D. Cytokine signaling in 2002: New surprises in the Jak/Stat pathway. Cell 2002. [Google Scholar] [CrossRef]
- Jang, Y.N.; Baik, E.J. JAK-STAT pathway and myogenic differentiation. JAKSTAT 2013. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Murray, P.J. Cytokine signaling modules in inflammatory responses. Immunity 2008, 28, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Masse, A.; James, C.; Teyssandier, I.; Lecluse, Y.; Larbret, F.; Ugo, V.; Saulnier, P.; Koscielny, S.; Le Couedic, J.P.; et al. The JAK2 617V>F mutation triggers erythropoietin hypersensitivity and terminal erythroid amplification in primary cells from patients with polycythemia vera. Blood 2007, 110, 1013–1021. [Google Scholar] [CrossRef]
- Winthrop, K.L. The emerging safety profile of JAK inhibitors in rheumatic disease. Nature Rev. Rheumatol. 2017, 13, 234–243. [Google Scholar] [CrossRef]
- Damsky, W.; King, B.A. JAK inhibitors in dermatology: The promise of a new drug class. J. Am. Acad. Dermatol. 2017, 76, 736–744. [Google Scholar] [CrossRef]
- Pardanani, A.; Vannucchi, A.M.; Passamonti, F.; Cervantes, F.; Barbui, T.; Tefferi, A. JAK inhibitor therapy for myelofibrosis: Critical assessment of value and limitations. Leukemia 2011, 25, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.C.; Levine, R.L. Molecular pathways: Molecular basis for sensitivity and resistance to JAK kinase inhibitors. Clin. Cancer Res. 2014, 20, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Kiladjian, J.J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 2015, 372, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef]
- Schwartz, D.M.; Bonelli, M.; Gadina, M.; O’Shea, J.J. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nature Rev. Rheumatol. 2016, 12, 25–36. [Google Scholar] [CrossRef]
- Berekmeri, A.; Mahmood, F.; Wittmann, M.; Helliwell, P. Tofacitinib for the treatment of psoriasis and psoriatic arthritis. Expert Rev. Clin. Immunol 2018, 14, 719–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerrigan, S.A.; McInnes, I.B. JAK Inhibitors in Rheumatology: Implications for Paediatric Syndromes? Curr. Rheumatol. Rep. 2018. [Google Scholar] [CrossRef]
- Nishanth, G.; Wolleschak, D.; Fahldieck, C.; Fischer, T.; Mullally, A.; Perner, F.; Schnoder, T.M.; Just, S.; Heidel, F.H.; Schluter, D. Gain of function in Jak2(V617F)-positive T-cells. Leukemia 2017, 31, 1000–1003. [Google Scholar] [CrossRef]
- Mullally, A.; Lane, S.W.; Ball, B.; Megerdichian, C.; Okabe, R.; Al-Shahrour, F.; Paktinat, M.; Haydu, J.E.; Housman, E.; Lord, A.M.; et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell 2010, 17, 584–596. [Google Scholar] [CrossRef]
- Stijnis, C.; Kroes, W.G.; Balkassmi, S.; Marijt, E.W.; van Rossum, A.P.; Bakker, E.; Vlasveld, L.T. No evidence for JAK2(V617F) mutation in monoclonal B cells in 2 patients with polycythaemia vera and concurrent monoclonal B cell disorder. Acta Haematologica 2012, 128, 183–186. [Google Scholar] [CrossRef]
- Prestipino, A.; Emhardt, A.J.; Aumann, K.; O’Sullivan, D.; Gorantla, S.P.; Duquesne, S.; Melchinger, W.; Braun, L.; Vuckovic, S.; Boerries, M.; et al. Oncogenic JAK2(V617F) causes PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci. Transl. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lussana, F.; Cattaneo, M.; Rambaldi, A.; Squizzato, A. Ruxolitinib-associated infections: A systematic review and meta-analysis. Am.J. Haematol. 2018, 93, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Keohane, C.; Kordasti, S.; Seidl, T.; Perez Abellan, P.; Thomas, N.S.; Harrison, C.N.; McLornan, D.P.; Mufti, G.J. JAK inhibition induces silencing of T Helper cytokine secretion and a profound reduction in T regulatory cells. Br. J. Haematol. 2015, 171, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Parampalli Yajnanarayana, S.; Stubig, T.; Cornez, I.; Alchalby, H.; Schonberg, K.; Rudolph, J.; Triviai, I.; Wolschke, C.; Heine, A.; Brossart, P.; et al. JAK1/2 inhibition impairs T cell function in vitro and in patients with myeloproliferative neoplasms. Br. J. Haematol. 2015, 169, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Massa, M.; Rosti, V.; Campanelli, R.; Fois, G.; Barosi, G. Rapid and long-lasting decrease of T-regulatory cells in patients with myelofibrosis treated with ruxolitinib. Leukemia 2014, 28, 449–451. [Google Scholar] [CrossRef]
- Xing, L.; Dai, Z.; Jabbari, A.; Cerise, J.E.; Higgins, C.A.; Gong, W.; de Jong, A.; Harel, S.; DeStefano, G.M.; Rothman, L.; et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat. Med. 2014, 20, 1043–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.P.; Iwata, S.; Nakayamada, S.; Sakata, K.; Yamaoka, K.; Tanaka, Y. Tofacitinib, a JAK inhibitor, inhibits human B cell activation in vitro. Ann. Rheum. Dis. 2014, 73, 2213–2215. [Google Scholar] [CrossRef]
- Rizzi, M.; Lorenzetti, R.; Fischer, K.; Staniek, J.; Janowska, I.; Troilo, A.; Strohmeier, V.; Erlacher, M.; Kunze, M.; Bannert, B.; et al. Impact of tofacitinib treatment on human B-cells in vitro and in vivo. J. Autoimm. 2017, 77, 55–66. [Google Scholar] [CrossRef]
- Heine, A.; Held, S.A.; Daecke, S.N.; Wallner, S.; Yajnanarayana, S.P.; Kurts, C.; Wolf, D.; Brossart, P. The JAK-inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood 2013, 122, 1192–1202. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.W.; Miller, C.B.; Silver, R.T.; Talpaz, M.; et al. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J. Hematol. Oncol. 2017. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.W.; Miller, C.B.; Silver, R.T.; et al. Efficacy, safety and survival with ruxolitinib in patients with myelofibrosis: Results of a median 2-year follow-up of COMFORT-I. Haematologica 2013, 98, 1865–1871. [Google Scholar] [CrossRef] [PubMed]
- Spoerl, S.; Mathew, N.R.; Bscheider, M.; Schmitt-Graeff, A.; Chen, S.; Mueller, T.; Verbeek, M.; Fischer, J.; Otten, V.; Schmickl, M.; et al. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood 2014, 123, 3832–3842. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Cooper, M.L.; Alahmari, B.; Ritchey, J.; Collins, L.; Holt, M.; DiPersio, J.F. Pharmacologic blockade of JAK1/JAK2 reduces GvHD and preserves the graft-versus-leukemia effect. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Perner, F.; Schnoder, T.M.; Ranjan, S.; Wolleschak, D.; Ebert, C.; Pils, M.C.; Frey, S.; Polanetzki, A.; Fahldieck, C.; Schonborn, U.; et al. Specificity of JAK-kinase inhibition determines impact on human and murine T-cell function. Leukemia 2016, 30, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Gotlib, J.R.; Jamieson, C.; Cortes, J.E.; Talpaz, M.; Stone, R.M.; Silverman, M.H.; Gilliland, D.G.; Shorr, J.; Tefferi, A. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J. Clin. Oncol. 2011, 29, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Tefferi, A.; Jamieson, C.; Gabrail, N.Y.; Lebedinsky, C.; Gao, G.; Liu, F.; Xu, C.; Cao, H.; Talpaz, M. A phase 2 randomized dose-ranging study of the JAK2-selective inhibitor fedratinib (SAR302503) in patients with myelofibrosis. Blood Cancer J. 2015. [Google Scholar] [CrossRef]
- Verstovsek, S.; Talpaz, M.; Ritchie, E.; Wadleigh, M.; Odenike, O.; Jamieson, C.; Stein, B.; Uno, T.; Mesa, R.A. A phase I, open-label, dose-escalation, multicenter study of the JAK2 inhibitor NS-018 in patients with myelofibrosis. Leukemia 2017, 31, 393–402. [Google Scholar] [CrossRef]
- Verstovsek, S.; Tam, C.S.; Wadleigh, M.; Sokol, L.; Smith, C.C.; Bui, L.A.; Song, C.; Clary, D.O.; Olszynski, P.; Cortes, J.; et al. Phase I evaluation of XL019, an oral, potent, and selective JAK2 inhibitor. Leukemia Res. 2014, 38, 316–322. [Google Scholar] [CrossRef]
- Harrison, C.N.; Schaap, N.; Vannucchi, A.M.; Kiladjian, J.J.; Tiu, R.V.; Zachee, P.; Jourdan, E.; Winton, E.; Silver, R.T.; Schouten, H.C.; et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): A single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017. [Google Scholar] [CrossRef]
- Zhang, Y.; Liang, R.; Chen, C.W.; Mallano, T.; Dees, C.; Distler, A.; Reich, A.; Bergmann, C.; Ramming, A.; Gelse, K.; et al. JAK1-dependent transphosphorylation of JAK2 limits the antifibrotic effects of selective JAK2 inhibitors on long-term treatment. Ann. Rheum. Dis. 2017, 76, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.O.; Talpaz, M.; Gupta, V.; Foltz, L.M.; Savona, M.R.; Paquette, R.; Turner, A.R.; Coughlin, P.; Winton, E.; Burn, T.C.; et al. Primary analysis of a phase II open-label trial of INCB039110, a selective JAK1 inhibitor, in patients with myelofibrosis. Haematologica 2017, 102, 327–335. [Google Scholar] [CrossRef] [PubMed]
- De Vries, L.C.S.; Ludbrook, V.J.; Hicks, K.J.; D’Haens, G.R. GSK2586184, a JAK1 selective inhibitor, in two patients with ulcerative colitis. BMJ Case Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, A.; Kremer, J.; Ponce, L.; Cseuz, R.; Reshetko, O.V.; Stanislavchuk, M.; Greenwald, M.; Van der Aa, A.; Vanhoutte, F.; Tasset, C.; et al. Filgotinib (GLPG0634/GS-6034), an oral selective JAK1 inhibitor, is effective as monotherapy in patients with active rheumatoid arthritis: Results from a randomised, dose-finding study (DARWIN 2). Ann. Rheum. Dis. 2017, 76, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Smolen, J.S.; Weinblatt, M.E.; Burmester, G.R.; Meerwein, S.; Camp, H.S.; Wang, L.; Othman, A.A.; Khan, N.; Pangan, A.L.; et al. Efficacy and Safety of ABT-494, a Selective JAK-1 Inhibitor, in a Phase IIb Study in Patients With Rheumatoid Arthritis and an Inadequate Response to Methotrexate. Arthritis Rheumatol. 2016, 68, 2857–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahl, L.; Patel, J.; Layton, M.; Binks, M.; Hicks, K.; Leon, G.; Hachulla, E.; Machado, D.; Staumont-Salle, D.; Dickson, M.; et al. Safety, tolerability, efficacy and pharmacodynamics of the selective JAK1 inhibitor GSK2586184 in patients with systemic lupus erythematosus. Lupus 2016, 25, 1420–1430. [Google Scholar] [CrossRef] [PubMed]
- Ludbrook, V.J.; Hicks, K.J.; Hanrott, K.E.; Patel, J.S.; Binks, M.H.; Wyres, M.R.; Watson, J.; Wilson, P.; Simeoni, M.; Schifano, L.A.; et al. Investigation of selective JAK1 inhibitor GSK2586184 for the treatment of psoriasis in a randomized placebo-controlled phase IIa study. Br. J. Dermatol. 2016, 174, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.; Coates, L.C.; Helliwell, P.S.; Stanislavchuk, M.; Rychlewska-Hanczewska, A.; Dudek, A.; Abi-Saab, W.; Tasset, C.; Meuleners, L.; Harrison, P.; et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active psoriatic arthritis (EQUATOR): Results from a randomised, placebo-controlled, phase 2 trial. Lancet 2018, 392, 2367–2377. [Google Scholar] [CrossRef]
- van der Heijde, D.; Baraliakos, X.; Gensler, L.S.; Maksymowych, W.P.; Tseluyko, V.; Nadashkevich, O.; Abi-Saab, W.; Tasset, C.; Meuleners, L.; Besuyen, R.; et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active ankylosing spondylitis (TORTUGA): Results from a randomised, placebo-controlled, phase 2 trial. Lancet 2018, 392, 2378–2387. [Google Scholar] [CrossRef]
- Vermeire, S.; Schreiber, S.; Petryka, R.; Kuehbacher, T.; Hebuterne, X.; Roblin, X.; Klopocka, M.; Goldis, A.; Wisniewska-Jarosinska, M.; Baranovsky, A.; et al. Clinical remission in patients with moderate-to-severe Crohn’s disease treated with filgotinib (the FITZROY study): Results from a phase 2, double-blind, randomised, placebo-controlled trial. Lancet 2017, 389, 266–275. [Google Scholar] [CrossRef]
- Genovese, M.C.; van Vollenhoven, R.F.; Pacheco-Tena, C.; Zhang, Y.; Kinnman, N. VX-509 (Decernotinib), an Oral Selective JAK-3 Inhibitor, in Combination With Methotrexate in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.M.; Damjanov, N.S.; Kivitz, A.J.; Legedza, A.; Hoock, T.; Kinnman, N. A randomized, double-blind, placebo-controlled, twelve-week, dose-ranging study of decernotinib, an oral selective JAK-3 inhibitor, as monotherapy in patients with active rheumatoid arthritis. Arthritis Rheumatol. 2015, 67, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.; Gordon, K.; Thaci, D.; Morita, A.; Gooderham, M.; Foley, P.; Girgis, I.G.; Kundu, S.; Banerjee, S. Phase 2 Trial of Selective Tyrosine Kinase 2 Inhibition in Psoriasis. N. Engl. J. Med. 2018, 379, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Finch, C.E.; Crimmins, E.M. Inflammatory exposure and historical changes in human life-spans. Science 2004, 305, 1736–1739. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zheng, X.; Zheng, Y. Age-associated loss of lamin-B leads to systemic inflammation and gut hyperplasia. Cell 2014, 159, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Cesari, M.; Anton, S.; Marzetti, E.; Giovannini, S.; Seo, A.Y.; Carter, C.; Yu, B.P.; Leeuwenburgh, C. Molecular inflammation: Underpinnings of aging and age-related diseases. Ageing Res. Rev. 2009, 8, 18–30. [Google Scholar] [CrossRef] [Green Version]
- d’Avila, J.C.; Siqueira, L.D.; Mazeraud, A.; Azevedo, E.P.; Foguel, D.; Castro-Faria-Neto, H.C.; Sharshar, T.; Chretien, F.; Bozza, F.A. Age-related cognitive impairment is associated with long-term neuroinflammation and oxidative stress in a mouse model of episodic systemic inflammation. J. Neuroinflamm. 2018. [Google Scholar] [CrossRef]
- Josephson, A.M.; Bradaschia-Correa, V.; Lee, S.; Leclerc, K.; Patel, K.S.; Muinos Lopez, E.; Litwa, H.P.; Neibart, S.S.; Kadiyala, M.; Wong, M.Z.; et al. Age-related inflammation triggers skeletal stem/progenitor cell dysfunction. Proc. Natl. Acad. Sci. USA 2019, 116, 6995–7004. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Clevers, H. Reparative inflammation takes charge of tissue regeneration. Nature 2016, 529, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Wu, L.W.; Grivennikov, S.I.; de Jong, P.R.; Lian, I.; Yu, F.X.; Wang, K.; Ho, S.B.; Boland, B.S.; Chang, J.T.; et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 2015, 519, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Kyritsis, N.; Kizil, C.; Zocher, S.; Kroehne, V.; Kaslin, J.; Freudenreich, D.; Iltzsche, A.; Brand, M. Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science 2012, 338, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and metabolism in tissue repair and regeneration. Science 2017, 356, 1026–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.K.; O’Brien, M.; Mau, T.; Yung, R. Toll-like receptor 4 (TLR4) deficient mice are protected from adipose tissue inflammation in aging. Aging 2017, 9, 1971–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2017. [Google Scholar] [CrossRef]
- Herrera, S.C.; Bach, E.A. JAK/STAT signaling in stem cells and regeneration: From Drosophila to vertebrates. Development 2019. [Google Scholar] [CrossRef]
- Elsaeidi, F.; Bemben, M.A.; Zhao, X.F.; Goldman, D. Jak/Stat signaling stimulates zebrafish optic nerve regeneration and overcomes the inhibitory actions of Socs3 and Sfpq. J. Neurosci. 2014, 34, 2632–2644. [Google Scholar] [CrossRef]
- Richmond, C.A.; Rickner, H.; Shah, M.S.; Ediger, T.; Deary, L.; Zhou, F.; Tovaglieri, A.; Carlone, D.L.; Breault, D.T. JAK/STAT-1 Signaling Is Required for Reserve Intestinal Stem Cell Activation during Intestinal Regeneration Following Acute Inflammation. Stem Cell Rep. 2018, 10, 17–26. [Google Scholar] [CrossRef]
- Doles, J.D.; Olwin, B.B. The impact of JAK-STAT signaling on muscle regeneration. Nat. Med. 2014, 20, 1094–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Fortezza, M.; Schenk, M.; Cosolo, A.; Kolybaba, A.; Grass, I.; Classen, A.K. JAK/STAT signalling mediates cell survival in response to tissue stress. Development 2016, 143, 2907–2919. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Patel, P.H.; Kohlmaier, A.; Grenley, M.O.; McEwen, D.G.; Edgar, B.A. Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell 2009, 137, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Shen-Orr, S.S.; Furman, D.; Kidd, B.A.; Hadad, F.; Lovelace, P.; Huang, Y.W.; Rosenberg-Hasson, Y.; Mackey, S.; Grisar, F.A.; Pickman, Y.; et al. Defective Signaling in the JAK-STAT Pathway Tracks with Chronic Inflammation and Cardiovascular Risk in Aging Humans. Cell Syst. 2016, 3, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA 2015. [Google Scholar] [CrossRef] [PubMed]
- Price, F.D.; von Maltzahn, J.; Bentzinger, C.F.; Dumont, N.A.; Yin, H.; Chang, N.C.; Wilson, D.H.; Frenette, J.; Rudnicki, M.A. Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat. Med. 2014, 20, 1174–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, H.; Buckley, J.A.; Li, X.; Liu, Y.; Fox, T.H., 3rd; Meares, G.P.; Yu, H.; Yan, Z.; Harms, A.S.; Li, Y.; et al. Inhibition of the JAK/STAT Pathway Protects Against alpha-Synuclein-Induced Neuroinflammation and Dopaminergic Neurodegeneration. J. Neurosci. 2016, 36, 5144–5159. [Google Scholar] [CrossRef]
- Jones, R.S.; Minogue, A.M.; Fitzpatrick, O.; Lynch, M.A. Inhibition of JAK2 attenuates the increase in inflammatory markers in microglia from APP/PS1 mice. Neurobiol. Aging 2015, 36, 2716–2724. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Tchkonia, T.; Kirkland, J.L. Perspective: Targeting the JAK/STAT pathway to fight age-related dysfunction. Pharmacol. Res. 2016, 111, 152–154. [Google Scholar] [CrossRef] [Green Version]
- Cano, M.; Ayala, A.; Marotta, F.; Arguelles, S. Application of Kinase Inhibitors for Anti-aging Intervention. Curr. Pharm. Des. 2017, 23, 4351–4368. [Google Scholar] [CrossRef]
- Sidon, P.; El Housni, H.; Dessars, B.; Heimann, P. The JAK2V617F mutation is detectable at very low level in peripheral blood of healthy donors. Leukemia 2006. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, Q.; Luo, J.; Xing, S.; Li, Q.; Krantz, S.B.; Fu, X.; Zhao, Z.J. JAK2(V617F): Prevalence in a large Chinese hospital population. Blood 2007, 109, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Mead, A.J.; Mullally, A. Myeloproliferative neoplasm stem cells. Blood 2017, 129, 1607–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoi, K.; Cross, N.C. Genomics of Myeloproliferative Neoplasms. J. Clin. Oncol. 2017, 35, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Beer, P.A.; Delhommeau, F.; LeCouedic, J.P.; Dawson, M.A.; Chen, E.; Bareford, D.; Kusec, R.; McMullin, M.F.; Harrison, C.N.; Vannucchi, A.M.; et al. Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood 2010, 115, 2891–2900. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Masse, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Schaub, F.X.; Jager, R.; Looser, R.; Hao-Shen, H.; Hermouet, S.; Girodon, F.; Tichelli, A.; Gisslinger, H.; Kralovics, R.; Skoda, R.C. Clonal analysis of deletions on chromosome 20q and JAK2-V617F in MPD suggests that del20q acts independently and is not one of the predisposing mutations for JAK2-V617F. Blood 2009, 113, 2022–2027. [Google Scholar] [CrossRef]
- Schaub, F.X.; Looser, R.; Li, S.; Hao-Shen, H.; Lehmann, T.; Tichelli, A.; Skoda, R.C. Clonal analysis of TET2 and JAK2 mutations suggests that TET2 can be a late event in the progression of myeloproliferative neoplasms. Blood 2010, 115, 2003–2007. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, P.; Takizawa, H.; Kubovcakova, L.; Guo, G.; Hao-Shen, H.; Dirnhofer, S.; Orkin, S.H.; Manz, M.G.; Skoda, R.C. Myeloproliferative neoplasms can be initiated from a single hematopoietic stem cell expressing JAK2-V617F. J. Exp. Med. 2014, 211, 2213–2230. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquelin, S.; Straube, J.; Cooper, L.; Vu, T.; Song, A.; Bywater, M.; Baxter, E.; Heidecker, M.; Wackrow, B.; Porter, A.; et al. Jak2V617F and Dnmt3a loss cooperate to induce myelofibrosis through activated enhancer-driven inflammation. Blood 2018, 132, 2707–2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mora, B.; Giorgino, T.; Guglielmelli, P.; Rumi, E.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; Kiladjian, J.J.; et al. Value of cytogenetic abnormalities in post-polycythemia vera and post-essential thrombocythemia myelofibrosis: A study of the MYSEC project. Haematologica 2018, 103. [Google Scholar] [CrossRef] [PubMed]
- Bartels, S.; Faisal, M.; Busche, G.; Schlue, J.; Kreipe, H.; Lehmann, U. Fibrotic progression in Polycythemia vera is associated with early concomitant driver-mutations besides JAK2. Leukemia 2018, 32, 556–558. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.; Sajid, Z.; Pedersen, R.K.; Gudmand-Hoeyer, J.; Ellervik, C.; Skov, V.; Kjaer, L.; Pallisgaard, N.; Kruse, T.A.; Thomassen, M.; et al. Mathematical modelling as a proof of concept for MPNs as a human inflammation model for cancer development. PLoS ONE 2017. [Google Scholar] [CrossRef] [PubMed]
- Nangalia, J.; Nice, F.L.; Wedge, D.C.; Godfrey, A.L.; Grinfeld, J.; Thakker, C.; Massie, C.E.; Baxter, J.; Sewell, D.; Silber, Y.; et al. DNMT3A mutations occur early or late in patients with myeloproliferative neoplasms and mutation order influences phenotype. Haematologica 2015, 100, 438–442. [Google Scholar] [CrossRef]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Manshouri, T.; Patel, J.; Harris, K.; Yao, J.; Hedvat, C.; Heguy, A.; Bueso-Ramos, C.; Kantarjian, H.; Levine, R.L.; et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010, 70, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Harutyunyan, A.; Klampfl, T.; Cazzola, M.; Kralovics, R. p53 lesions in leukemic transformation. N. Engl. J. Med. 2011, 364, 488–490. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Lasho, T.; Finke, C.; Oh, S.T.; Gotlib, J.; Tefferi, A. LNK mutation studies in blast-phase myeloproliferative neoplasms, and in chronic-phase disease with TET2, IDH, JAK2 or MPL mutations. Leukemia 2010, 24, 1713–1718. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.P.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S.; et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017, 376, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, F.A.; Rodrigues Pereira, J.; Ciuleanu, T.; Tan, E.H.; Hirsh, V.; Thongprasert, S.; Campos, D.; Maoleekoonpiroj, S.; Smylie, M.; Martins, R.; et al. Erlotinib in previously treated non-small-cell lung cancer. N. Engl. J. Med. 2005, 353, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.; Radich, J.; Burn, T.C.; Huber, R.; Paranagama, D.; Verstovsek, S. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood 2015, 126, 1551–1554. [Google Scholar] [Green Version]
- Harrison, C.N.; Vannucchi, A.M.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Knoops, L.; Cervantes, F.; Jones, M.M.; Sun, K.; McQuitty, M.; et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia 2016, 30, 1701–1707. [Google Scholar] [CrossRef] [Green Version]
- Geyer, H.L.; Kosiorek, H.; Dueck, A.C.; Scherber, R.; Slot, S.; Zweegman, S.; Te Boekhorst, P.A.; Senyak, Z.; Schouten, H.C.; Sackmann, F.; et al. Associations between gender, disease features and symptom burden in patients with myeloproliferative neoplasms: An analysis by the MPN QOL International Working Group. Haematologica 2017, 102, 85–93. [Google Scholar] [CrossRef]
- Lubberich, R.K.; Walenda, T.; Goecke, T.W.; Strathmann, K.; Isfort, S.; Brummendorf, T.H.; Koschmieder, S.; Wagner, W. Serum of myeloproliferative neoplasms stimulates hematopoietic stem and progenitor cells. PLoS ONE 2018. [Google Scholar] [CrossRef]
- Rampal, R.; Al-Shahrour, F.; Abdel-Wahab, O.; Patel, J.P.; Brunel, J.P.; Mermel, C.H.; Bass, A.J.; Pretz, J.; Ahn, J.; Hricik, T.; et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 2014, 123, 123–133. [Google Scholar] [CrossRef]
- Schnoder, T.M.; Eberhardt, J.; Koehler, M.; Bierhoff, H.B.; Weinert, S.; Pandey, A.D.; Nimmagadda, S.C.; Wolleschak, D.; Johrens, K.; Fischer, T.; et al. Cell autonomous expression of CXCL-10 in JAK2V617F-mutated MPN. J. Cancer Res. Clin. Oncol. 2017, 143, 807–820. [Google Scholar] [PubMed]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: A comprehensive cytokine profiling study. J. Clin. Oncol. 2011, 29, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; LaTocha, D.H.; Yang, F.; Press, R.D.; et al. TNFalpha facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 2011, 118, 6392–6398. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Jutzi, J.S.; Bogeska, R.; Nikoloski, G.; Schmid, C.A.; Seeger, T.S.; Stegelmann, F.; Schwemmers, S.; Grunder, A.; Peeken, J.C.; Gothwal, M.; et al. MPN patients harbor recurrent truncating mutations in transcription factor NF-E2. J. Exp. Med. 2013, 210, 1003–1019. [Google Scholar] [CrossRef] [PubMed]
- Jutzi, J.S.; Pahl, H.L. The Hen or the Egg: Inflammatory Aspects of Murine MPN Models. Mediators Inflamm. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wehrle, J.; Seeger, T.S.; Schwemmers, S.; Pfeifer, D.; Bulashevska, A.; Pahl, H.L. Transcription factor nuclear factor erythroid-2 mediates expression of the cytokine interleukin 8, a known predictor of inferior outcome in patients with myeloproliferative neoplasms. Haematologica 2013, 98, 1073–1080. [Google Scholar] [CrossRef] [Green Version]
- Griesshammer, M.; Kiladjian, J.J.; Besses, C. Thromboembolic events in polycythemia vera. Ann. Hematol. 2019, 98, 1071–1082. [Google Scholar] [CrossRef] [Green Version]
- Chievitz, E.; Thiede, T. Complications and causes of death in polycythaemia vera. Acta Med. Scand. 1962, 172, 513–523. [Google Scholar] [CrossRef]
- Landolfi, R.; Marchioli, R.; Kutti, J.; Gisslinger, H.; Tognoni, G.; Patrono, C.; Barbui, T. European Collaboration on Low-Dose Aspirin in Polycythemia Vera, I. Efficacy and safety of low-dose aspirin in polycythemia vera. N. Engl. J. Med. 2004, 350, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Marchioli, R.; Finazzi, G.; Specchia, G.; Cacciola, R.; Cavazzina, R.; Cilloni, D.; De Stefano, V.; Elli, E.; Iurlo, A.; Latagliata, R.; et al. Cardiovascular events and intensity of treatment in polycythemia vera. N. Engl. J. Med. 2013, 368, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Barbui, T.; Cervantes, F.; Harrison, C.; Kiladjian, J.J.; Kroger, N.; Thiele, J.; Buske, C.; Committee, E.G. Philadelphia chromosome-negative chronic myeloproliferative neoplasms: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Boissinot, M.; Lippert, E.; Girodon, F.; Dobo, I.; Fouassier, M.; Masliah, C.; Praloran, V.; Hermouet, S. Latent myeloproliferative disorder revealed by the JAK2-V617F mutation and endogenous megakaryocytic colonies in patients with splanchnic vein thrombosis. Blood 2006, 108, 3223–3224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Stefano, V.; Carobbio, A.; Di Lazzaro, V.; Guglielmelli, P.; Iurlo, A.; Finazzi, M.C.; Rumi, E.; Cervantes, F.; Elli, E.M.; Randi, M.L.; et al. Benefit-risk profile of cytoreductive drugs along with antiplatelet and antithrombotic therapy after transient ischemic attack or ischemic stroke in myeloproliferative neoplasms. Blood Cancer J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gugliotta, L.; Iurlo, A.; Gugliotta, G.; Tieghi, A.; Specchia, G.; Gaidano, G.; Scalzulli, P.R.; Rumi, E.; Dragani, A.; Martinelli, V.; et al. Unbiased pro-thrombotic features at diagnosis in 977 thrombocythemic patients with Philadelphia-negative chronic myeloproliferative neoplasms. Leukemia Res. 2016, 46, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.; Pancholy, N.; Thomas, L.; Rai, A.; Kher, A.; Peters, C.; Amin, A.; Patel, T.M.; Pancholy, S. Effect of Chronic Hematologic Malignancies on In-Hospital Outcomes of Patients With ST-Segment Elevation Myocardial Infarction. Am. J. Cardiol. 2019, 124, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Rosti, V.; Villani, L.; Riboni, R.; Poletto, V.; Bonetti, E.; Tozzi, L.; Bergamaschi, G.; Catarsi, P.; Dallera, E.; Novara, F.; et al. Spleen endothelial cells from patients with myelofibrosis harbor the JAK2V617F mutation. Blood 2013, 121, 360–368. [Google Scholar] [CrossRef]
- Teofili, L.; Martini, M.; Iachininoto, M.G.; Capodimonti, S.; Nuzzolo, E.R.; Torti, L.; Cenci, T.; Larocca, L.M.; Leone, G. Endothelial progenitor cells are clonal and exhibit the JAK2(V617F) mutation in a subset of thrombotic patients with Ph-negative myeloproliferative neoplasms. Blood 2011, 117, 2700–2707. [Google Scholar] [CrossRef]
- Sozer, S.; Fiel, M.I.; Schiano, T.; Xu, M.; Mascarenhas, J.; Hoffman, R. The presence of JAK2V617F mutation in the liver endothelial cells of patients with Budd-Chiari syndrome. Blood 2009, 113, 5246–5249. [Google Scholar] [CrossRef]
- Etheridge, S.L.; Roh, M.E.; Cosgrove, M.E.; Sangkhae, V.; Fox, N.E.; Chen, J.; Lopez, J.A.; Kaushansky, K.; Hitchcock, I.S. JAK2V617F-positive endothelial cells contribute to clotting abnormalities in myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Lamrani, L.; Lacout, C.; Ollivier, V.; Denis, C.V.; Gardiner, E.; Ho Tin Noe, B.; Vainchenker, W.; Villeval, J.L.; Jandrot-Perrus, M. Hemostatic disorders in a JAK2V617F-driven mouse model of myeloproliferative neoplasm. Blood 2014, 124, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, C.M.; Manning, H.; Bennett, C.; Vasquez, L.; Severin, S.; Brain, L.; Mazharian, A.; Guerrero, J.A.; Li, J.; Soranzo, N.; et al. JAK2V617F leads to intrinsic changes in platelet formation and reactivity in a knock-in mouse model of essential thrombocythemia. Blood 2013, 122, 3787–3797. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Larran, A.; Perez-Encinas, M.; Ferrer-Marin, F.; Hernandez-Boluda, J.C.; Ramirez, M.J.; Martinez-Lopez, J.; Magro, E.; Cruz, Y.; Mata, M.I.; Aragues, P.; et al. Risk of thrombosis according to need of phlebotomies in patients with polycythemia vera treated with hydroxyurea. Haematologica 2017, 102, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Finazzi, G.; Caruso, V.; Marchioli, R.; Capnist, G.; Chisesi, T.; Finelli, C.; Gugliotta, L.; Landolfi, R.; Kutti, J.; Gisslinger, H.; et al. Acute leukemia in polycythemia vera: An analysis of 1638 patients enrolled in a prospective observational study. Blood 2005, 105, 2664–2670. [Google Scholar] [CrossRef] [PubMed]
- De Grandis, M.; Cambot, M.; Wautier, M.P.; Cassinat, B.; Chomienne, C.; Colin, Y.; Wautier, J.L.; Le Van Kim, C.; El Nemer, W. JAK2V617F activates Lu/BCAM-mediated red cell adhesion in polycythemia vera through an EpoR-independent Rap1/Akt pathway. Blood 2013, 121, 658–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolach, O.; Sellar, R.S.; Martinod, K.; Cherpokova, D.; McConkey, M.; Chappell, R.J.; Silver, A.J.; Adams, D.; Castellano, C.A.; Schneider, R.K.; et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Edelmann, B.; Schnoeder, T.M.; Saalfeld, F.C.; Wolleschak, D.; Kliche, S.; Schraven, B.; Heidel, F.H.; Fischer, T. JAK2-V617F activates beta1-integrin-mediated adhesion of granulocytes to vascular cell adhesion molecule 1. Leukemia 2017, 31, 1223–1226. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, B.; Gupta, N.; Schnoeder, T.M.; Oelschlegel, A.M.; Shahzad, K.; Goldschmidt, J.; Philipsen, L.; Weinert, S.; Ghosh, A.; Saalfeld, F.C.; et al. JAK2-V617F promotes venous thrombosis through beta1/beta2 integrin activation. J. Clin. Investig. 2018, 128, 4359–4371. [Google Scholar] [CrossRef]
Figure 1.
Schematic of clonal development in hematopoietic stem and progenitor cells during aging. Representative example of the sequence of TET2 loss of function (LOF), Janus Kinase 2 (JAK2-V617F) and Nucleophosmin (NPM1c) mutations leading to clonal hematopoiesis, a myeloproliferative neoplasia (MPN) and finally to the development of acute myeloid leukemia (AML) respectively.
Figure 1.
Schematic of clonal development in hematopoietic stem and progenitor cells during aging. Representative example of the sequence of TET2 loss of function (LOF), Janus Kinase 2 (JAK2-V617F) and Nucleophosmin (NPM1c) mutations leading to clonal hematopoiesis, a myeloproliferative neoplasia (MPN) and finally to the development of acute myeloid leukemia (AML) respectively.

Figure 2.
Schematic representation of normal and aberrant JAK-signal transduction. The IL2-receptor (IL2-R) associated with JAK1 and JAK3 serves in this cartoon as an example for physiological JAK-signaling dependent on cytokine binding. JAK2-V617F (JAK2-VF) is representatively depicted in association with the Erythropoetin-receptor (EPO-R) and the Thrombopoetin-receptor (TPO-R) where it induces constitutive activation and ligand hypersensitivity, respectively.
Figure 2.
Schematic representation of normal and aberrant JAK-signal transduction. The IL2-receptor (IL2-R) associated with JAK1 and JAK3 serves in this cartoon as an example for physiological JAK-signaling dependent on cytokine binding. JAK2-V617F (JAK2-VF) is representatively depicted in association with the Erythropoetin-receptor (EPO-R) and the Thrombopoetin-receptor (TPO-R) where it induces constitutive activation and ligand hypersensitivity, respectively.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Perner, F.; Perner, C.; Ernst, T.; Heidel, F.H. Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation. Cells 2019, 8, 854. https://doi.org/10.3390/cells8080854
AMA Style
Perner F, Perner C, Ernst T, Heidel FH. Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation. Cells. 2019; 8(8):854. https://doi.org/10.3390/cells8080854
Chicago/Turabian StylePerner, Florian, Caroline Perner, Thomas Ernst, and Florian H. Heidel. 2019. "Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation" Cells 8, no. 8: 854. https://doi.org/10.3390/cells8080854
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.