ETF-QO Mutants Uncoupled Fatty Acid β-Oxidation and Mitochondrial Bioenergetics Leading to Lipid Pathology

 , , , and
, , , and
Abstract
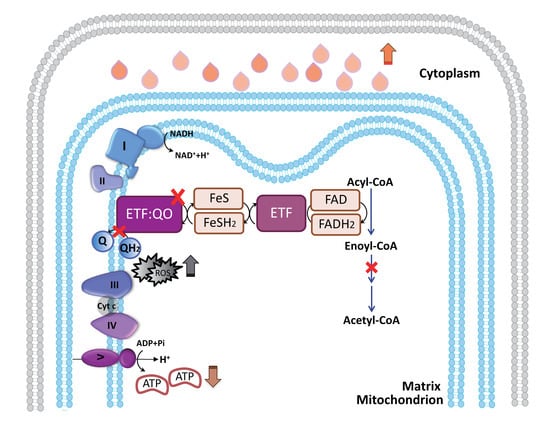
:
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Mutation Screening
2.3. Analysis of Blood Acyl-Carnitine Profiles
2.4. Histological and Histochemical Examination
2.5. Transmission Electron Microscope Examination
2.6. Cell Culture
2.7. Reverse-Transcription and Real-Time Quantitative PCR for ETFDH Expression
2.8. Western Blot Analysis for ETFDH Expression
2.9. ATP Assay
2.10. Flow Cytometry Analysis of Mitochondrial Membrane Potential (ΔΨm) and Lipid Droplet and Lipid Peroxide Levels
2.11. Seahorse XF24 Metabolic Flux Analysis for Mitochondrial Respiration
2.12. Oil Red O and Nile Red Staining for Lipid Droplets
2.12.1. Oil Red O Staining
2.12.2. Nile Red Staining
2.13. Cloning and Transfection of ETFDH Variants
2.14. Statistical Analysis
3. Results
3.1. Abnormal Profiles of Plasma Fatty Acid Composition and Metabolic Parameters in MADD Patients
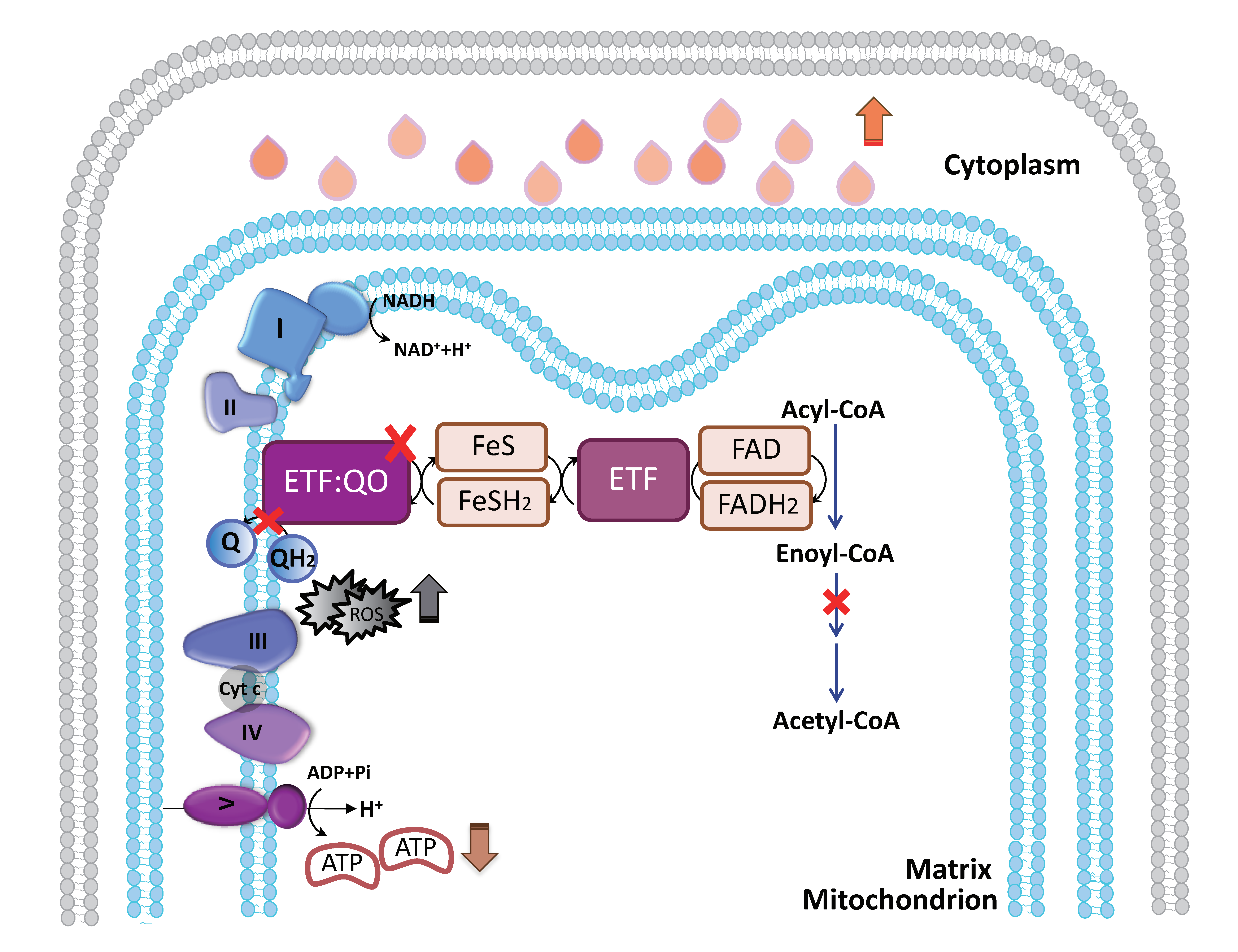
3.2. Increased Lipid Droplet Accumulation in the Sarcolemma in MADD Patient 1
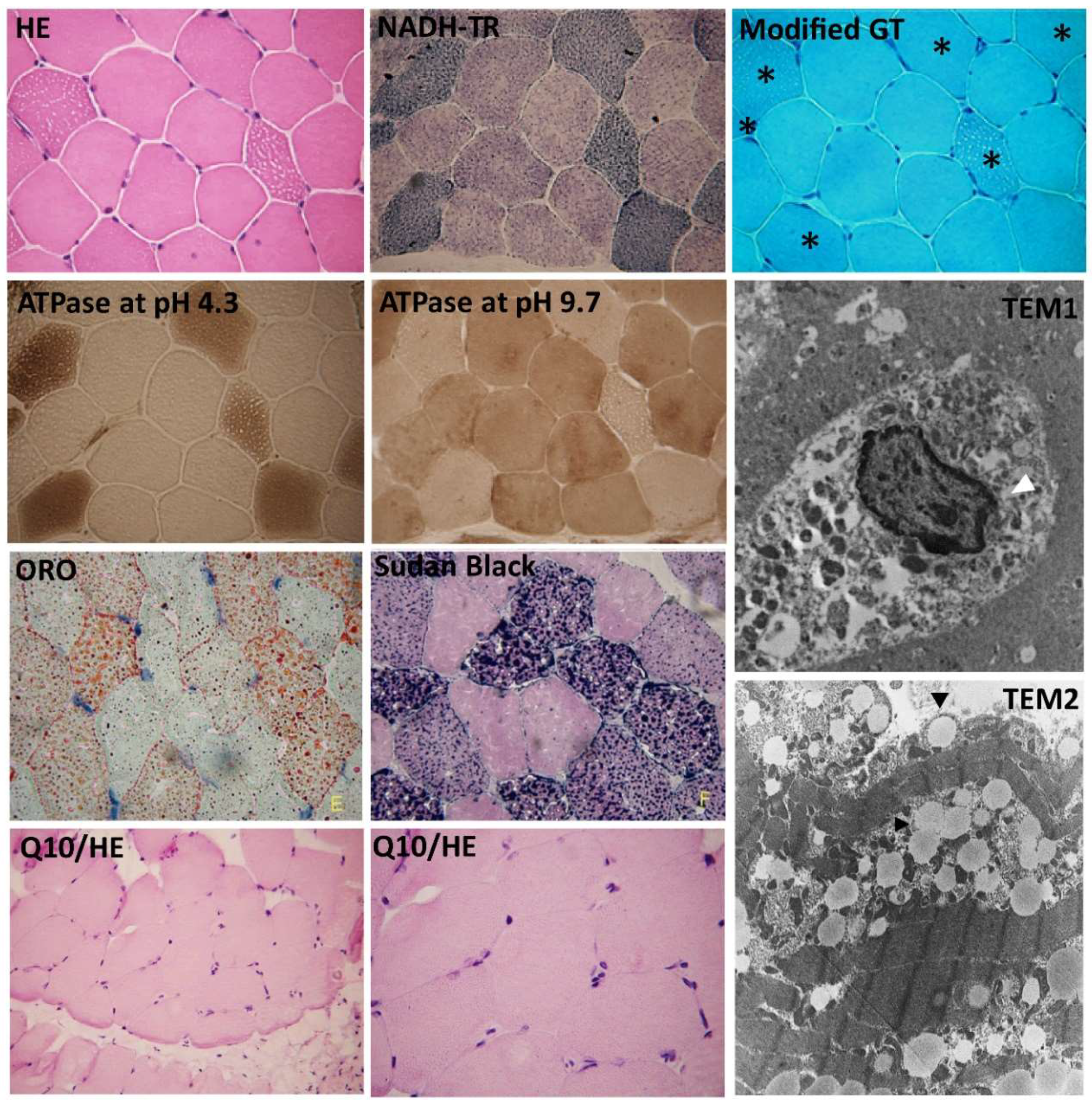
3.3. The Identification of the c.92C>T and c.250G>A Mutations in the Affected MADD Patients
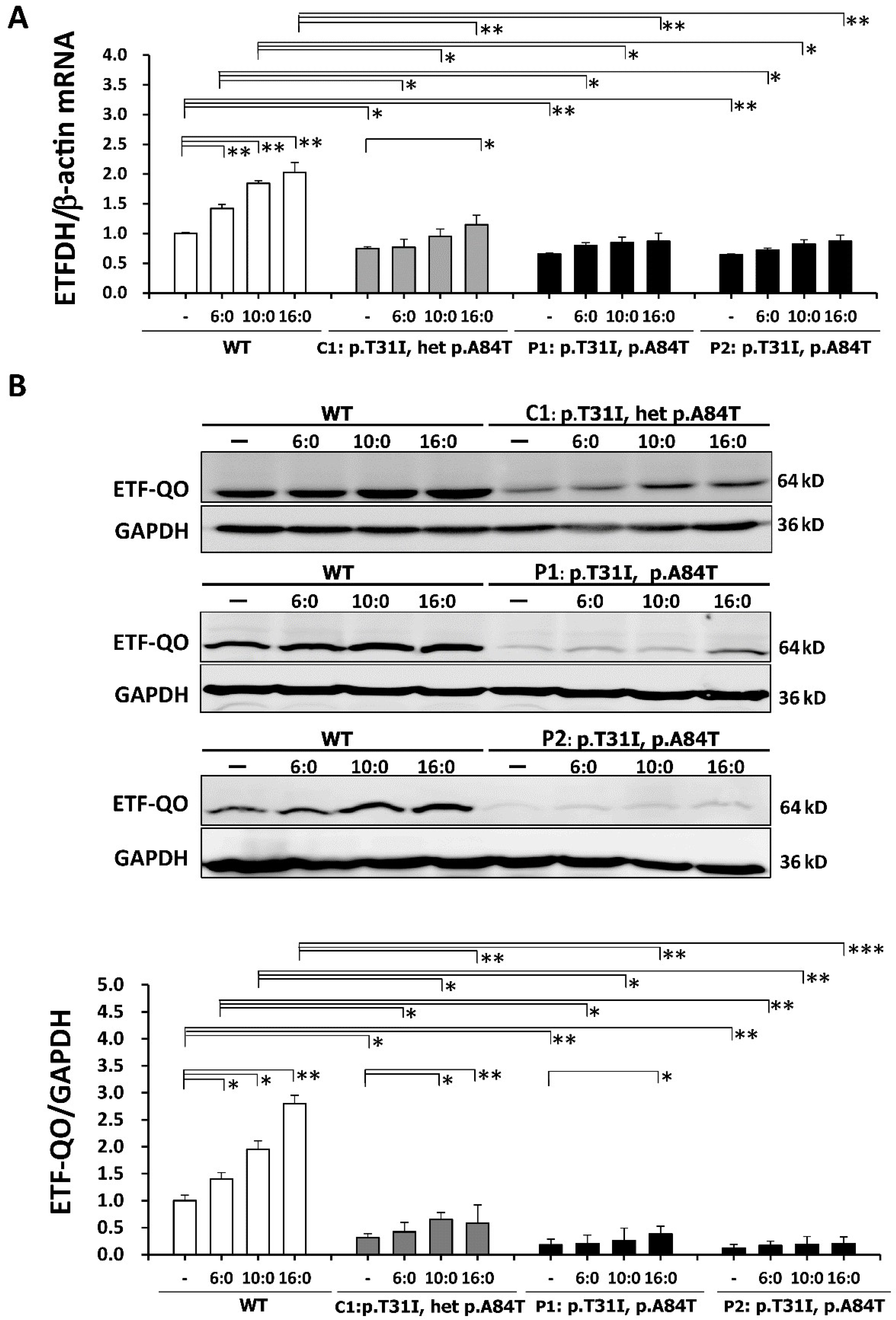
3.4. The Reduced Expression Levels of ETFDH mRNA and ETF: QO in the ETFDH-Mutated Lymphoblastoid Cells
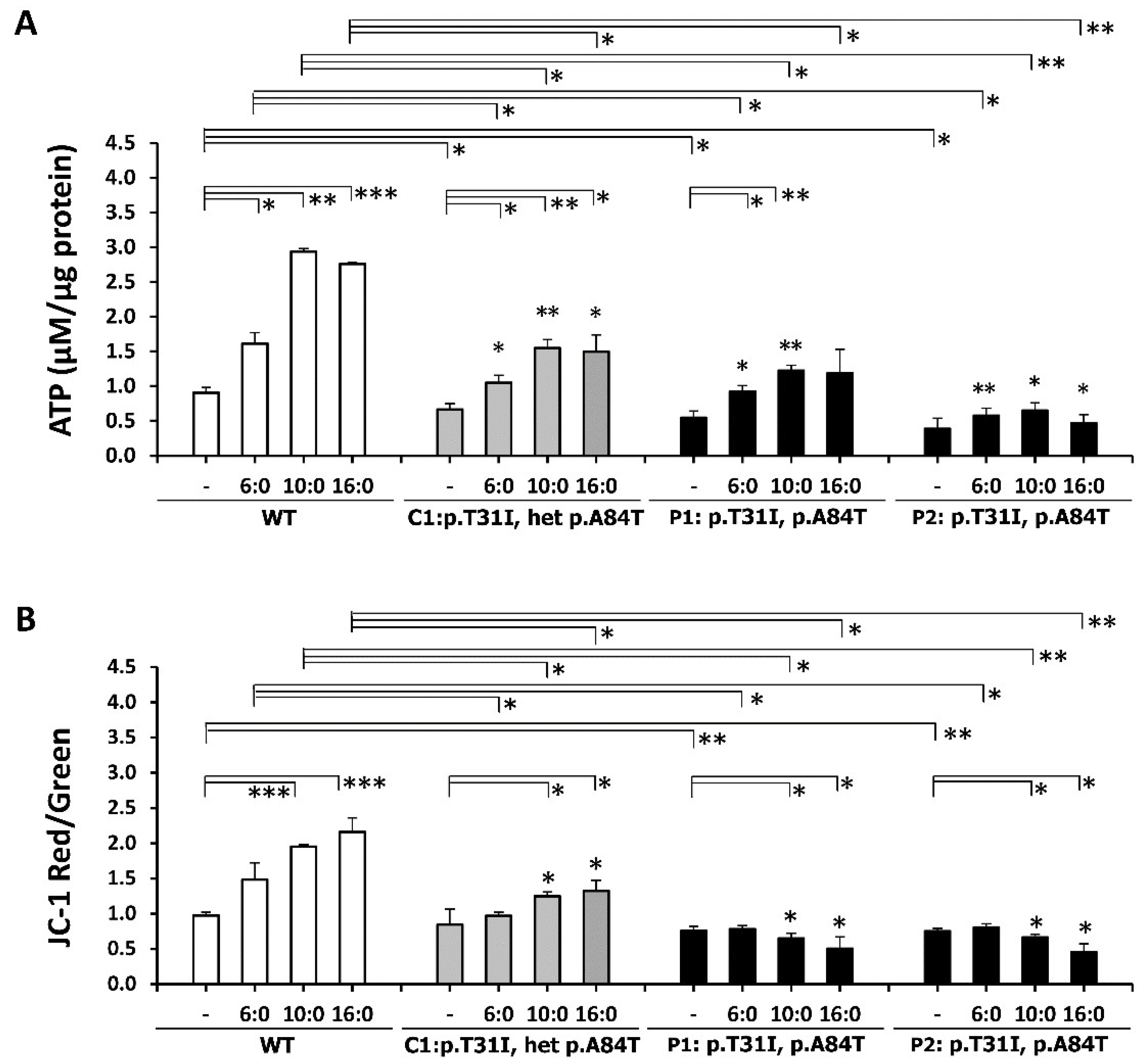
3.5. ETFDH Mutations Caused Mitochondrial Dysfunction
3.6. ETF-QO Is Involved in the Maintenance of Mitochondrial Bioenergetics and Respiratory Coupling
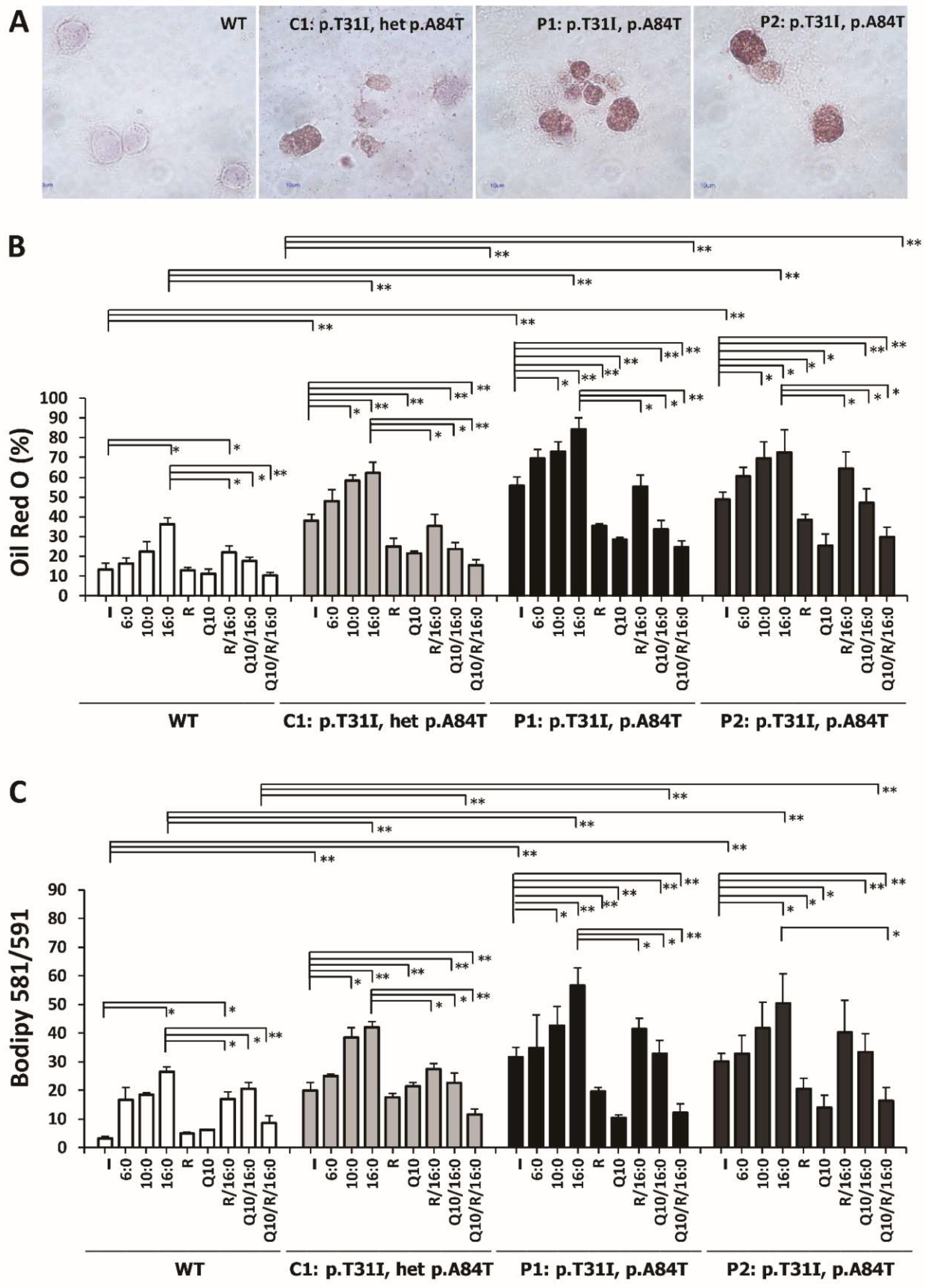
3.7. Riboflavin and Coenzyme Q10 Reduced the Intracellular Accumulation of Neutral Lipid Droplets and Lipid Peroxides in ETFDH-Mutated Cells
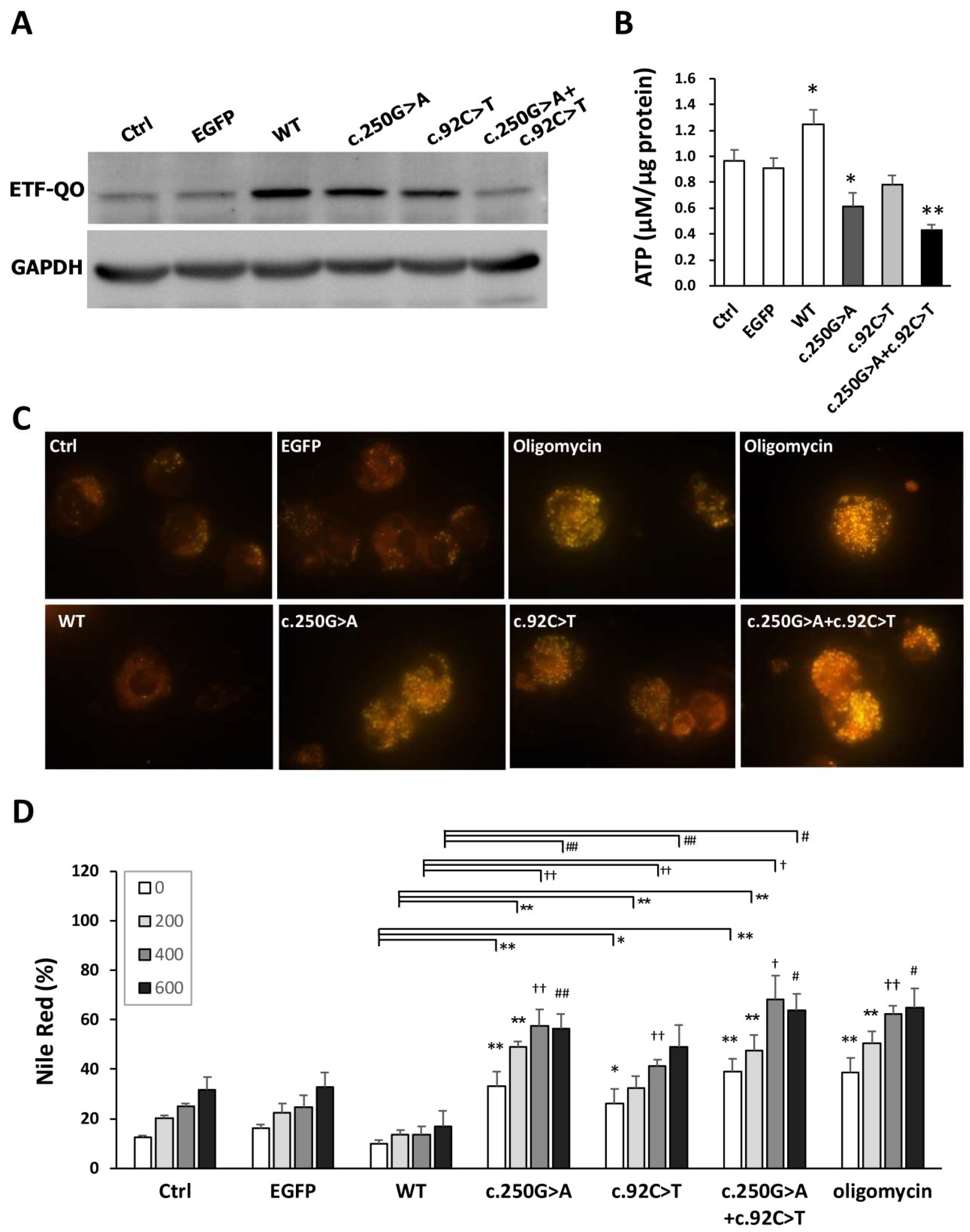
3.8. Both the c.250G>A and c.92C>T ETFDH Variants Induced the Intracellular Accumulation of Neutral Lipid Droplets
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grunert, S.C. Clinical and genetical heterogeneity of late-onset multiple acyl-coenzyme A dehydrogenase deficiency. Orphanet. J. Rare Dis. 2014, 9, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennisi, E.M.; Garibaldi, M.; Antonini, G. Lipid Myopathies. J. Clin. Med. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, N.; Andresen, B.S.; Pedersen, C.B.; Olsen, R.K.; Corydon, T.J.; Bross, P. Mitochondrial fatty acid oxidation defects--remaining challenges. J. Inherit. Metab. Dis. 2008, 31, 643–657. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.K.; Olpin, S.E.; Andresen, B.S.; Miedzybrodzka, Z.H.; Pourfarzam, M.; Merinero, B.; Frerman, F.E.; Beresford, M.W.; Dean, J.C.; Cornelius, N.; et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain 2007, 130, 2045–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, M.; Rutledge, S.L.; Kelly, D.R.; Palmer, C.A.; Murdoch, G.; Majumder, N.; Nicholls, R.D.; Pei, Z.; Watkins, P.A.; Vockley, J. A new genetic disorder in mitochondrial fatty acid beta-oxidation: ACAD9 deficiency. Am. J. Hum. Genet. 2007, 81, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Zhu, X.; Qi, X.; Weijiang, D.; Yu, Y.; Wan, H.; Hong, D. Riboflavin-responsive multiple Acyl-CoA dehydrogenation deficiency in 13 cases, and a literature review in mainland Chinese patients. J. Hum. Genet. 2014, 59, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Wen, B.; Lin, J.; Zhu, W.; Luo, S.; Zhao, C.; Li, D.; Lin, P.; Lu, J.; Yan, C. Clinical features and ETFDH mutation spectrum in a cohort of 90 Chinese patients with late-onset multiple acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2014, 37, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.; Qiao, L.W.; Duan, C.Y.; Zhao, N.N.; Zhang, W.; Zhang, Q. A novel mutation in the ETFDH gene of an infant with multiple acyl-CoA dehydrogenase deficiency. Chin. J. Contemp. Pediatr. 2018, 20, 529–533. [Google Scholar] [CrossRef]
- Er, T.K.; Liang, W.C.; Chang, J.G.; Jong, Y.J. High resolution melting analysis facilitates mutation screening of ETFDH gene: Applications in riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Clin. Chim. Acta 2010, 411, 690–699. [Google Scholar] [CrossRef]
- Huang, H.P.; Chu, K.L.; Chien, Y.H.; Wei, M.L.; Wu, S.T.; Wang, S.F.; Hwu, W.L. Tandem mass neonatal screening in Taiwan--report from one center. J. Formos. Med. Assoc. 2006, 105, 882–886. [Google Scholar] [CrossRef]
- Lima, T.M.; Kanunfre, C.C.; Pompeia, C.; Verlengia, R.; Curi, R. Ranking the toxicity of fatty acids on Jurkat and Raji cells by flow cytometric analysis. Toxicol. In Vitro 2002, 16, 741–747. [Google Scholar] [CrossRef]
- Lan, M.Y.; Fu, M.H.; Liu, Y.F.; Huang, C.C.; Chang, Y.Y.; Liu, J.S.; Peng, C.H.; Chen, S.S. High frequency of ETFDH c.250G>A mutation in Taiwanese patients with late-onset lipid storage myopathy. Clin. Genet. 2010, 78, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Z.Q.; Zhao, D.Y.; Zheng, D.M.; Feng, J.; Song, L.C.; Luo, Y. AIF-mediated mitochondrial pathway is critical for the protective effect of diazoxide against SH-SY5Y cell apoptosis. Brain Res. 2011, 1370, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Er, T.K.; Chen, C.C.; Liu, Y.Y.; Chang, H.C.; Chien, Y.H.; Chang, J.G.; Hwang, J.K.; Jong, Y.J. Computational analysis of a novel mutation in ETFDH gene highlights its long-range effects on the FAD-binding motif. BMC Struct. Biol. 2011, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.C.; Ohkuma, A.; Hayashi, Y.K.; Lopez, L.C.; Hirano, M.; Nonaka, I.; Noguchi, S.; Chen, L.H.; Jong, Y.J.; Nishino, I. ETFDH mutations, CoQ10 levels, and respiratory chain activities in patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Neuromuscul. Disord. 2009, 19, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.I.; Binard, R.J.; Woontner, M.R.; Frerman, F.E. Glutaric acidemia type II: Gene structure and mutations of the electron transfer flavoprotein:ubiquinone oxidoreductase (ETF:QO) gene. Mol. Genet. Metab. 2002, 77, 86–90. [Google Scholar] [CrossRef]
- Law, L.K.; Tang, N.L.; Hui, J.; Fung, S.L.; Ruiter, J.; Wanders, R.J.; Fok, T.F.; Lam, C.W. Novel mutations in ETFDH gene in Chinese patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Clin. Chim. Acta 2009, 404, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Nascimbeni, A.C.; Cenacchi, G.; Tasca, E. Lipolysis and lipophagy in lipid storage myopathies. Biochim. Biophys. Acta 2016, 1862, 1367–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, W.C.; Nishino, I. State of the art in muscle lipid diseases. Acta Myol. 2010, 29, 351–356. [Google Scholar] [PubMed]
- Wanders, R.J.; Tager, J.M. Peroxisomal fatty acid beta-oxidation in relation to adrenoleukodystrophy. Dev. Neurosci. 1991, 13, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Dessein, A.F.; Fontaine, M.; Andresen, B.S.; Gregersen, N.; Brivet, M.; Rabier, D.; Napuri-Gouel, S.; Dobbelaere, D.; Mention-Mulliez, K.; Martin-Ponthieu, A.; et al. A novel mutation of the ACADM gene (c.145C>G) associated with the common c.985A>G mutation on the other ACADM allele causes mild MCAD deficiency: A case report. Orphanet. J. Rare Dis. 2010, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Brivet, M.; Slama, A.; Saudubray, J.M.; Legrand, A.; Lemonnier, A. Rapid diagnosis of long chain and medium chain fatty acid oxidation disorders using lymphocytes. Ann. Clin. Biochem. 1995, 32 Pt 2, 154–159. [Google Scholar] [CrossRef]
- Karsten, S.; Schafer, G.; Schauder, P. Cytokine production and DNA synthesis by human peripheral lymphocytes in response to palmitic, stearic, oleic, and linoleic acid. J. Cell Physiol. 1994, 161, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Spieker-Polet, H.; Polet, H. Requirement of a combination of a saturated and an unsaturated free fatty acid and a fatty acid carrier protein for in vitro growth of lymphocytes. J. Immunol. 1981, 126, 949–954. [Google Scholar] [PubMed]
- Anel, A.; Naval, J.; Gonzalez, B.; Torres, J.M.; Mishal, Z.; Uriel, J.; Pineiro, A. Fatty acid metabolism in human lymphocytes. I. Time-course changes in fatty acid composition and membrane fluidity during blastic transformation of peripheral blood lymphocytes. Biochim. Biophys. Acta 1990, 1044, 323–331. [Google Scholar] [CrossRef]
- Olsen, R.K.; Broner, S.; Sabaratnam, R.; Doktor, T.K.; Andersen, H.S.; Bruun, G.H.; Gahrn, B.; Stenbroen, V.; Olpin, S.E.; Dobbie, A.; et al. The ETFDH c.158A>G variation disrupts the balanced interplay of ESE- and ESS-binding proteins thereby causing missplicing and multiple Acyl-CoA dehydrogenation deficiency. Hum. Mutat. 2014, 35, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, N.; Frerman, F.E.; Corydon, T.J.; Palmfeldt, J.; Bross, P.; Gregersen, N.; Olsen, R.K. Molecular mechanisms of riboflavin responsiveness in patients with ETF-QO variations and multiple acyl-CoA dehydrogenation deficiency. Hum. Mol. Genet. 2012, 21, 3435–3448. [Google Scholar] [CrossRef] [Green Version]
- Yotsumoto, Y.; Hasegawa, Y.; Fukuda, S.; Kobayashi, H.; Endo, M.; Fukao, T.; Yamaguchi, S. Clinical and molecular investigations of Japanese cases of glutaric acidemia type 2. Mol. Genet. Metab. 2008, 94, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Selak, M.A.; Watson, C.T.; Coutts, C.; Scherer, P.C.; Panzer, J.A.; Gibbs, S.; Scott, M.O.; Willer, G.; Gregg, R.G.; et al. Mechanisms underlying metabolic and neural defects in zebrafish and human multiple acyl-CoA dehydrogenase deficiency (MADD). PLoS ONE 2009, 4, e8329. [Google Scholar] [CrossRef]
- Muralidhara, B.K.; Rathinakumar, R.; Wittung-Stafshede, P. Folding of Desulfovibrio desulfuricans flavodoxin is accelerated by cofactor fly-casting. Arch. Biochem. Biophys. 2006, 451, 51–58. [Google Scholar] [CrossRef]
- Saijo, T.; Tanaka, K. Isoalloxazine ring of FAD is required for the formation of the core in the Hsp60-assisted folding of medium chain acyl-CoA dehydrogenase subunit into the assembly competent conformation in mitochondria. J. Biol. Chem. 1995, 270, 1899–1907. [Google Scholar] [CrossRef]
- Gempel, K.; Topaloglu, H.; Talim, B.; Schneiderat, P.; Schoser, B.G.; Hans, V.H.; Palmafy, B.; Kale, G.; Tokatli, A.; Quinzii, C.; et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007, 130, 2037–2044. [Google Scholar] [CrossRef] [Green Version]
- Wen, B.; Li, D.; Shan, J.; Liu, S.; Li, W.; Zhao, Y.; Lin, P.; Zheng, J.; Li, D.; Gong, Y.; et al. Increased muscle coenzyme Q10 in riboflavin responsive MADD with ETFDH gene mutations due to secondary mitochondrial proliferation. Mol. Genet. Metab. 2013, 109, 154–160. [Google Scholar] [CrossRef]
- Boveris, A.; Chance, B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 1973, 134, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Cornelius, N.; Corydon, T.J.; Gregersen, N.; Olsen, R.K. Cellular consequences of oxidative stress in riboflavin responsive multiple acyl-CoA dehydrogenation deficiency patient fibroblasts. Hum. Mol. Genet. 2014. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, N.; Byron, C.; Hargreaves, I.; Guerra, P.F.; Furdek, A.K.; Land, J.; Radford, W.W.; Frerman, F.; Corydon, T.J.; Gregersen, N.; et al. Secondary coenzyme Q10 deficiency and oxidative stress in cultured fibroblasts from patients with riboflavin responsive multiple Acyl-CoA dehydrogenation deficiency. Hum. Mol. Genet. 2013, 22, 3819–3827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welte, M.A. Expanding roles for lipid droplets. Curr Biol 2015, 25, R470–481. [Google Scholar] [CrossRef] [PubMed]
- Krahmer, N.; Farese, R.V., Jr.; Walther, T.C. Balancing the fat: Lipid droplets and human disease. EMBO Mol. Med. 2013, 5, 973–983. [Google Scholar] [CrossRef]
- Olsen, R.K.; Andresen, B.S.; Christensen, E.; Bross, P.; Skovby, F.; Gregersen, N. Clear relationship between ETF/ETFDH genotype and phenotype in patients with multiple acyl-CoA dehydrogenation deficiency. Hum. Mutat. 2003, 22, 12–23. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test | Patient 1 (µmol/L) | Patient 2 (µmol/L) | Reference Range (µmol/L) |
|---|---|---|---|
| Alanine | 165.93 | 182.88 | <409.20 |
| Arginine | 6.55 | 2.49 | <16.57 |
| Citrulline | 10.99 | 9.53 | 3.21–19.50 |
| Glycine | 71.07 | 62.83 | <431.08 |
| Leucine/Isoleucine | 144.03 | 115.21 | 36.59–185.76 |
| Methionine | 19.93 | 18.56 | 8.11–27.49 |
| Ornithine | 16.15 | 9.86 | <53.40 |
| Phenylalanine | 49.46 | 37.33 | 33.61–81.56 |
| Tyrosine | 41.59 | 24.97 | <84.40 |
| Valine | 162.68 | 163.22 | <283.37 |
| Free carnitine | 36.14 | 12.22 | 13.34–53.92 |
| C2-acylcarnitine | 12.11 | 5.75 | 8.03–31.02 |
| C3-acylcarnitine | 1.60 | 0.50 | <4.01 |
| C3DC-acylcarnitine | 0.09 | 0.09 | <0.27 |
| C4-acylcarnitine | 0.57 | 0.50 | <0.62 |
| C4DC-acylcarnitine | 0.33 | 0.24 | <0.37 |
| C5-acylcarnitine | 0.40 | 0.58 | <0.2 |
| C5DC-acylcarnitine | 0.02 | 0.03 | <0.05 |
| C5OH-acylcarnitine | 0.20 | 0.21 | <0.59 |
| C6-acylcarnitine | 0.46 | 0.21 | <0.32 |
| C8-acylcarnitine | 0.56 | 0.33 | <0.31 |
| C8:1-acylcarnitine | 0.18 | 0.05 | <0.41 |
| C10-acylcarnitine | 0.88 | 0.32 | <0.3 |
| C10:1-acylcarnitine | 0.23 | 0.14 | <0.45 |
| C12-acylcarnitine | 1.18 | 0.62 | <0.39 |
| C12:1-acylcarnitine | 0.14 | 0.22 | <0.15 |
| C14-acylcarnitine | 0.55 | 0.52 | <0.22 |
| C14:1-acylcarnitine | 0.54 | 0.86 | <0.32 |
| C14OH-acylcarnitine | 0.01 | 0.01 | <0.09 |
| C16-acylcarnitine | 1.42 | 1.66 | 0.27–1.87 |
| C16:1-acylcarnitine | 0.40 | 0.68 | <1.13 |
| C16OH-acylcarnitine | 0.01 | 0.04 | <0.1 |
| C18-acylcarnitine | 1.07 | 1.18 | 0.18–1.47 |
| C18:1-acylcarnitine | 1.13 | 1.45 | 0.18–2.15 |
| C18OH-acylcarnitine | 0.03 | 0.04 | <0.11 |
| C3/C2 | 0.13 | 0.09 | <0.27 |
| C5DC/C16 | 0.02 | 0.02 | <0.19 |
| C8/C10 | 0.63 | 1.06 | <0.53 |
| Phe/Tyr | 1.19 | 1.49 | <2.82 |
| C0/(C16+C18) | 14.51 | 4.30 | 7.65–48.65 |
| C4OH | 0.00 | 0.00 | <0.53 |
| C5:1 | 0.00 | 0.00 | <0.25 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chokchaiwong, S.; Kuo, Y.-T.; Hsu, S.-P.; Hsu, Y.-C.; Lin, S.-H.; Zhong, W.-B.; Lin, Y.-F.; Kao, S.-H. ETF-QO Mutants Uncoupled Fatty Acid β-Oxidation and Mitochondrial Bioenergetics Leading to Lipid Pathology. Cells 2019, 8, 106. https://doi.org/10.3390/cells8020106
Chokchaiwong S, Kuo Y-T, Hsu S-P, Hsu Y-C, Lin S-H, Zhong W-B, Lin Y-F, Kao S-H. ETF-QO Mutants Uncoupled Fatty Acid β-Oxidation and Mitochondrial Bioenergetics Leading to Lipid Pathology. Cells. 2019; 8(2):106. https://doi.org/10.3390/cells8020106
Chicago/Turabian StyleChokchaiwong, Suphannee, Yung-Ting Kuo, Sung-Po Hsu, Yi-Ching Hsu, Shih-Hsiang Lin, Wen-Bin Zhong, Yung-Feng Lin, and Shu-Huei Kao. 2019. "ETF-QO Mutants Uncoupled Fatty Acid β-Oxidation and Mitochondrial Bioenergetics Leading to Lipid Pathology" Cells 8, no. 2: 106. https://doi.org/10.3390/cells8020106