1. Introduction
HCC is one of the deadliest tumors [
1]. China has a high incidence of HCC, and the overall survival rate is unsatisfactory [
2,
3]. Many clinical studies have shown that migration and invasion are the main reasons for this [
4,
5]. Migration and invasion are two major characteristics of HCC progression and recurrence. The cellular biological process is generally as follows: the primary tumor locally invades the extracellular matrix (ECM), then penetrates the basement membrane and the vascular endothelium, enters the blood, and, through circulation, reaches distant organs where it forms metastatic tumors [
6,
7]. In general, migration and invasion largely determine the success or failure of HCC treatments [
8]. Therefore, it is crucial to explore the mechanisms of the migration and invasion of HCC cells and identify effective targets.
The human
p62 gene is located on chromosome 5 [
9] and encodes a p62 protein with 440 amino acid residues, also known as
SQSTM1 [
10]. SQSTM1/p62 has multiple functional domains and is closely related to protein ubiquitination, selective autophagy, oxidative stress, and other reactions [
11,
12]. In recent years, several studies have confirmed that SQSTM1/p62 is overexpressed in various cancers [
13]. Increasing evidence shows that SQSTM1/p62 can affect the migration and invasion of tumors in a variety of ways [
13,
14,
15,
16]. However, the molecular mechanisms of SQSTM1/p62 involvement in migration and invasion are still unclear. Previous studies have shown that SQSTM1/p62 can release Nrf2 by combining with Keap1 [
17,
18]. In HCC, abnormal activation of Nrf2 promotes the expression of MMP2 and MMP9 [
19], which are matrix metalloproteinases that can degrade the basement membrane and promote tumor migration and invasion [
20,
21].
Cancer is also closely associated with inflammation. The inflammatory microenvironment favors tumor progression by accelerating tumor migration and invasion [
22,
23,
24,
25]. Several studies have reported that SQSTM1/p62 is significantly upregulated in the inflammatory microenvironment [
26,
27,
28,
29]. Therefore, it is important to further explore the effects of SQSTM1/p62 on the migration and invasion of HCC cells in the inflammatory microenvironment. Cisplatin, also known as diamminedichloroplatinum (DDP), is an effective drug for controlling tumor cell proliferation and inhibiting tumor cell migration and invasion [
30,
31]. In addition, DDP treatment significantly reduces the levels of SQSTM1/p62. Taken together, these results indicate that SQSTM1/p62 is regulated by many factors. However, it is not clear whether these environmental or pharmacological regulations of SQSTM1/p62 levels affect the migration and invasion of HCC.
The CRISPR/Cas9 system is a new and rapidly developing gene-editing technology. Gene editing systems are typically used to examine gene function and identify drug targets. In previous studies,
SQSTM1/p62 knockout was performed using CRISPR/Cas9 [
32,
33]. However, most of these studies have focused on autophagy. There is no record of
SQSTM1/p62 knockout and functional investigation into migration and invasion using the CRISPR/Cas9 system. In this study, we generated
SQSTM1/p62 wild-type HepG2 cells (
SQSTM1 WT HepG2 cells) and
SQSTM1/p62 knockout HepG2 cells (
SQSTM1 KO HepG2 cells) using the CRISPR/Cas9 system. The newly developed
SQSTM1/p62 knockout HepG2 cells may provide an important source for future research. We then studied the effects of
SQSTM1/p62 knockout on the migration and invasion of HCC cells in vivo and in vitro and investigated how SQSTM1/p62 regulates the migration and invasion of HCC through the Keap1/Nrf2/MMP2 signaling pathway. We also investigated the influence of the induced inflammatory microenvironment and antitumor drug DDP on the migration and invasion of HCC in the presence of SQSTM1/p62.
2. Materials and Methods
2.1. Bioinformatics Analysis
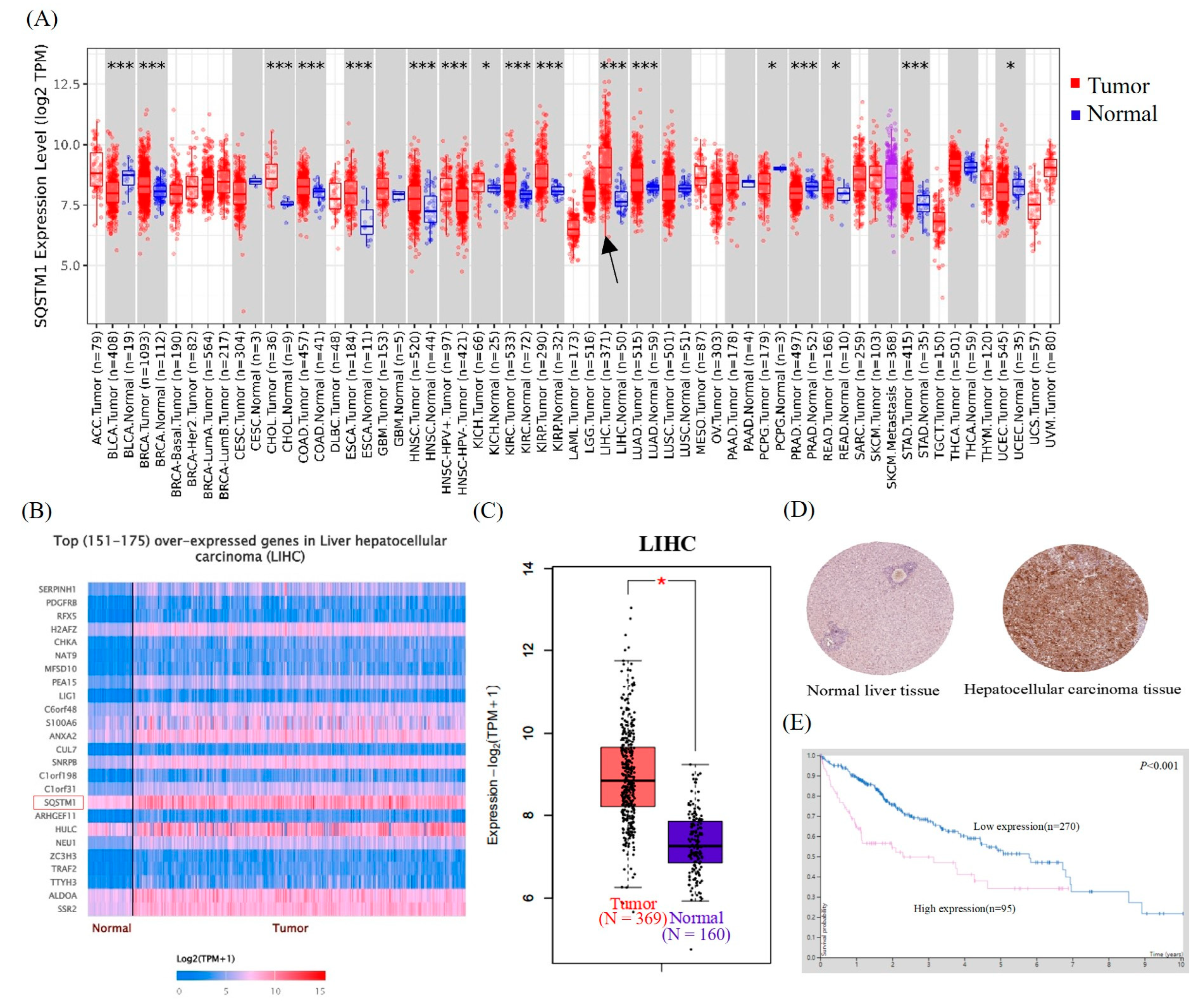
The Cancer Genome Atlas (TCGA) is a database that can provide important information about the mechanisms of cancer occurrence and development. TIMER2.0 (
http://timer.cistrome.org/, accessed on 25 July 2021) provides visualization functions [
34]. Using TIMER2.0, we mined the data of the expression of
SQSTM1/p62 in all forms of cancer in the TCGA database and evaluated the potential of this gene as a therapeutic target (
p < 0.001). The online analysis database Ualcan (
http://ualcan.path.uab.edu/index.html, accessed on 5 September 2021) and the powerful tool GEPIA2 (
http://gepia2.cancer-pku.cn/#index, accessed on 18 September 2021) [
35] were used to analyze the SQSTM1/p62 expression profile data in human liver cancer tissues and normal human liver tissues in TCGA and GTEx databases. Finally, SQSTM1/p62 immunohistochemical data and prognosis analyses were obtained from The Human Protein Atlas (HPA) (
https://www.proteinatlas.org, accessed on 27 September 2021).
2.2. Cell Culture and Treatment
HEK293T cells were supplied by the American Type Culture Collection (Manassas, VA, USA), HepG2 cells were provided by the Cell Bank of the Chinese Academy of Sciences (Shanghai, China), and THP-1 cells were purchased from the Procell Life Science & Technology Co., Ltd. (Wuhan, China). SQSTM1 WT HepG2 cells, SQSTM1 KO HepG2 cells, and SQSTM1 KO2 HepG2 cells were generated using the CRISPR/Cas9 system, as described in the following section. All cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA), which contained 1% penicillin-streptomycin antibiotics (Thermo Fisher Scientific, Waltham, MA, USA), 10% fetal bovine serum (FBS; Pan Biotech, Adenbach, German), and was incubated in a humidified atmosphere of 95% air/5% CO2 (vol/vol) at a stable temperature (37 °C) and constant pH (7.2–7.4). According to different experimental requirements, the cells were counted after digestion and inoculated in tissue culture plates (Jet Biofiltration Co., Ltd., Guangzhou, China). DDP (MedChemExpress, MCE, Monmouth Junction, NJ, USA) was added for 24 h after the cells had grown to 60–70% confluence. Lipopolysaccharide (LPS; Sigma-Aldrich, St. Louis, MO, USA) was added for 0–24 h after the cells had grown to 50–60% confluence to simulate the inflammatory microenvironment.
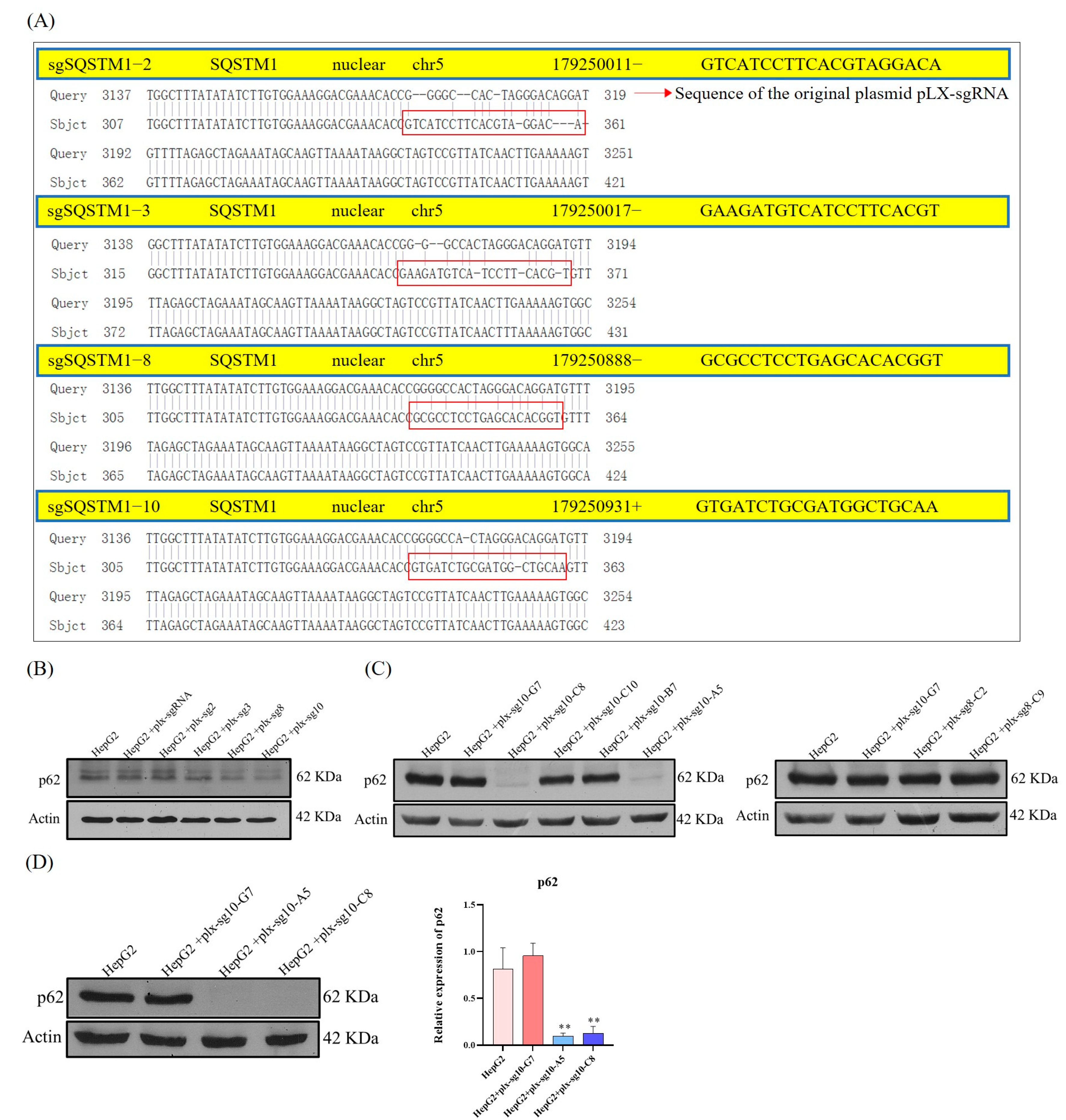
2.3. Generation of SQSTM1/p62 KO HepG2 Cells by CRISPR/Cas9 System
SQSTM1/p62 KO in HepG2 Cells was generated using the CRISPR/Cas9 system, as previously described, with slight modifications [
36]. First, the plasmid pLX-sgRNA (Addgene, Watertown, MA, USA) was expressed in
Escherichia coli (
E. coli) Stbl3 glycerol strain (Beyotime, Shanghai, China) and extracted using the TIANprep mini plasmid kit (Tiangen, Beijing, China) for amplification. Then, it was immediately used as the template to conduct multiple extension PCR with sgRNA-p62-2, sgRNA-p62-3, sgRNA-p62-8, and sgRNA-p62-10 (
Table A1), and the target PCR products were obtained. Subsequently, pLX-sgRNA and target PCR products were digested with the restriction endonuclease
Xho I and
Nhe l (TaKaRa, Mountain View, CA, USA) and retrieved using an agarose recovery kit (Tiangen, Beijing, China) after 0.8–1% agarose gel electrophoresis. Then, pLX-sgRNA was linked to the target fragments (460 bp) using T4 ligase, and recombinant plasmids were obtained.
Second, the recombinant plasmids (plx-sg2, plx-sg3, plx-sg8, plx-sg10) and pLX-sgRNA were transformed into E. coli DH5α (TaKaRa, Mountain View, CA, USA) and screened using 50 μg/mL ampicillin (AMP). Single colonies were selected from the plx-sg2, plx-sg3, plx-sg8, and plx-sg10 groups for amplification and gene sequencing. Single positive colonies with the correct sequencing results were selected for plasmid amplification.
Third, the encapsulated pCV-VSVg, packaged pMDLg/pRRE, and pRSV-Rev and pCW-Cas9 (Addgene, Watertown, MA, USA) were mixed and transfected into 293T cells to complete virus packaging, and the virus supernatant was collected. When HepG2 cells had grown to 50–70% confluence, a mixture of virus supernatant from 293T cells and DMEM (1:1) was added to HepG2 cells, which were infected with the virus for 3 h, and then screened continuously using 1–2 μg/mL puromycin (MCE, USA). Finally, HepG2-cas9 cells that produce Cas9 were generated.
Lastly, we delivered pLX-sgRNA empty and recombinant plasmids to HepG2-cas9 cells and constructed a HepG2-pLX-Cas9 cell line. Briefly, the encapsulated pCW-VSVg, packaged pMDLg/pRRE, and pRSV-Rev were mixed with empty pLX-sgRNA and recombinant plasmids (plx-sg2, plx-sg3, plx-sg8, plx-sg10) and transfected into 293T cells. After virus packaging, HepG2-Cas9 cells were infected with the virus solution as described above and screened using blasticidin (Genomeditech, Shanghai, China). Finally, the HepG2-pLX-Cas9 cell line, which expressed both sgRNA and Cas9, was obtained. Cas9 was induced using 2 μg/mL doxycycline (MCE, USA) for SQSTM1/p62 gene editing. The expression of SQSTM1/p62 in different cell types was determined using Western blotting (WB). Soon afterward, the cells with a significant decrease in SQSTM1/p62 expression levels were selected and underwent multiple cycles of enrichment using the single-cell culture method, and clones of SQSTM1/p62 KO HepG2 cells were obtained.
2.4. MTT Assay
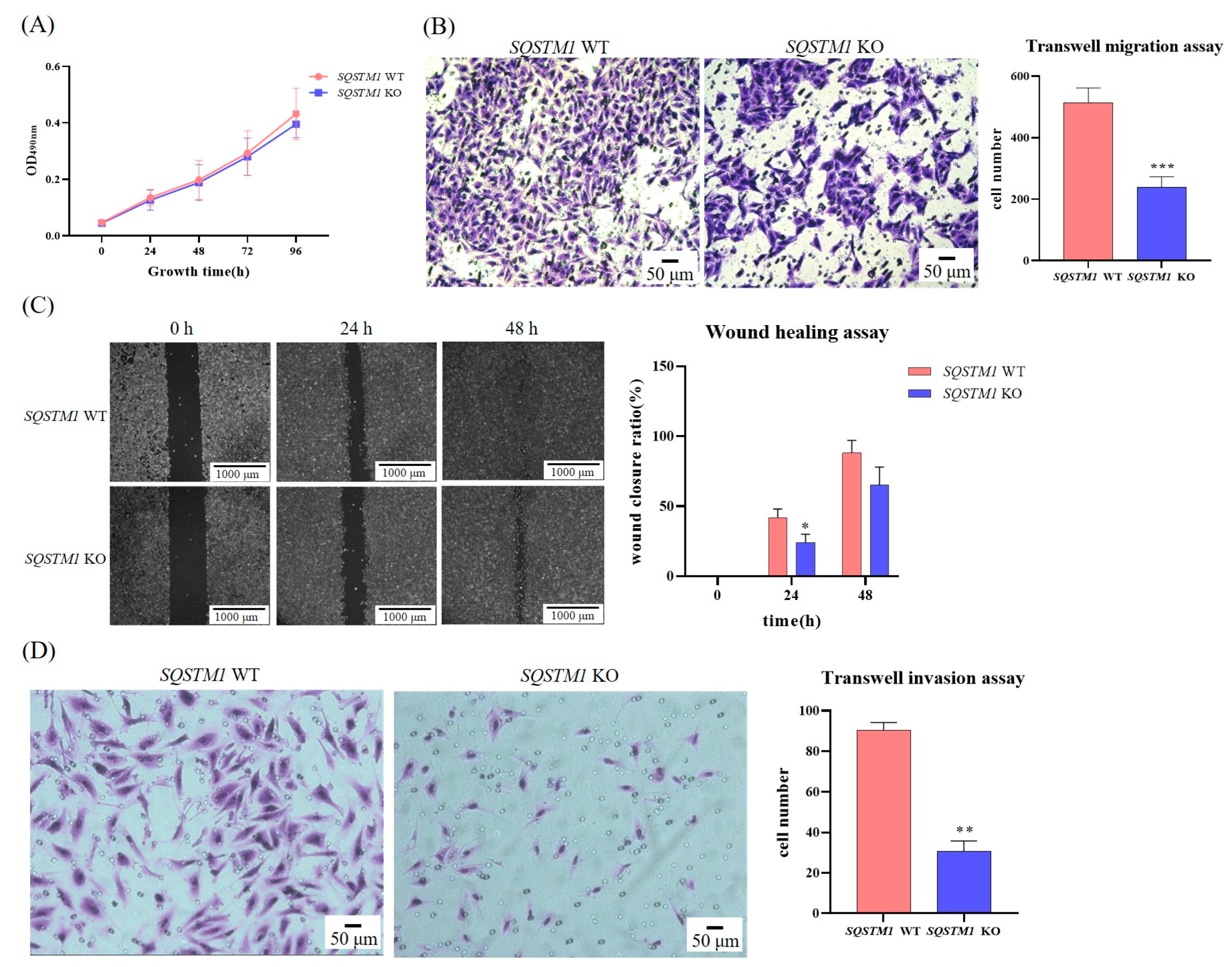
A thiazolyl blue tetrazolium bromide (MTT; Sangon Biotech, Shanghai, China) assay was used to determine cell growth. Briefly, SQSTM1 WT HepG2 cells and SQSTM1 KO HepG2 cells were inoculated into 96-well plates in equal numbers with DMEM containing 1% FBS, and five culture plates were inoculated in total. After 6 h, cells were incubated with DMEM containing 10% FBS for 0, 24, 48, 72, or 96 h. Subsequently, 20 μL sterilized MTT (5 mg/mL) were added to each well and incubated for 3 h at 37 °C. Finally, the medium was carefully removed, 100 µL dimethyl sulfoxide solution were added to fully dissolve the formazan, and OD490nm was measured using a spectrophotometer. The cytotoxicity of LPS or DDP was also analyzed using the MTT assay. Briefly, SQSTM1 WT HepG2 cells and SQSTM1 KO HepG2 cells were inoculated into 96-well plates in equal numbers with DMEM containing 10% FBS and LPS (1 μg/mL) or DDP (5 µM or 10 µM). After 24 h, cell growth was determined as described above.
2.5. Wound Healing Assay
The wound healing assay, also known as the scratch experiment, is a basic way to study cell migration and directly observe this dynamic process. Briefly, when the cells grew to a density of more than 90%, straight and regular wounds were made in the cells using a sterilized toothpick. The cells were rinsed with phosphate-buffered solution (PBS) and then cultured in DMEM containing 1% FBS for 48 h. Images at 50×/100× magnification were captured under a microscope at 0, 24, and 48 h, and the scratch width and cell migration rate were calculated.
2.6. Transwell Migration Assay
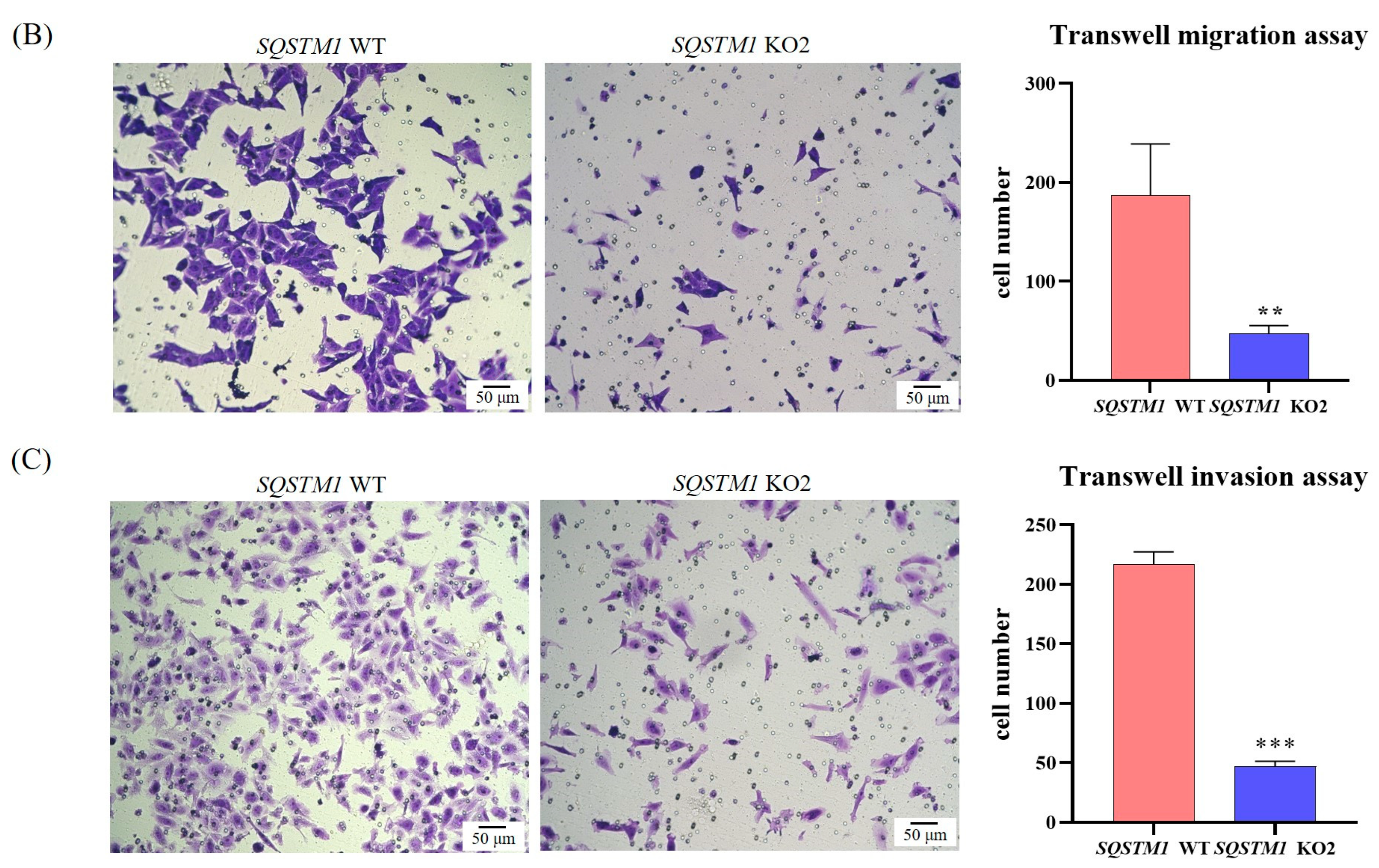
When SQSTM1 WT HepG2 cells and SQSTM1 KO HepG2 cells were in the logarithmic growth period, they were inoculated into Transwell inserts with a pore size of 8 μm (Jet Biofiltration Co., Ltd., Guangzhou, China) at the same density with 200 μL serum-free medium (SFM). The Transwell inserts were then placed in 24-well plates filled with 600 μL DMEM and 20% FBS and cultured in a humidified tissue culture incubator at 37 °C in a 5% CO2 atmosphere for 24 h. The cells were then fixed in 4% paraformaldehyde for 20 min in the dark and stained with 0.1% crystal violet for 30 min in the dark. Finally, the unmigrated cells were washed under slow water flow and wiped off with a cotton swab. After air-drying, images were captured at 200× magnification under a microscope. The number of migrating cells was measured using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
2.7. Transwell Invasion Assay
The invasive ability and metastatic potential of SQSTM1 WT HepG2 cells and SQSTM1 KO HepG2 cells were assessed using Corning® BioCoat™ Matrigel® Invasion Chambers with a pore size of 8 μm (Corning® BioCoat™, Bedford, MA, USA). Briefly, warm SFM (37 °C) was added to the interior of the 24-well Matrigel® Invasion Chambers and the bottom of the wells for 2 h to rehydrate the matrigel. After rehydration, the 24-well Matrigel® Invasion Chambers were transferred to the wells containing DMEM with 20% FBS, SQSTM1 WT HepG2 cells, and SQSTM1 KO HepG2 cells and were inoculated into the chambers at the same density in SFM. Immediately, the Matrigel Invasion Chambers were incubated for 24 h in a humidified atmosphere of 5% CO2 at 37 °C, followed by the Transwell migration assay.
2.8. Quantitative Real-Time Polymerase Chain Reaction (RT-PCR) Assay
Total cellular RNA was extracted using RNAiso Plus (TaKaRa Biotechnology, Dalian, China), according to previous studies [
37,
38]. Total RNA (500 ng) was immediately used to synthesize cDNA using the
Evo M-MLV RT Mix Kit (Accurate Biology, Changsha, China) according to the manufacturer’s instructions, and then the expression levels of mRNAs were quantitatively analyzed with the SYBR
® Green Premix Pro Taq HS qPCR Kit (Accurate Biology, Changsha, China). Finally, relative gene expression was normalized to β-actin and calculated using the 2
−ΔΔCt method. The target primers were synthesized by GENEWIZ (Cambridge, MA, USA) and are as follows: h-β-actin-F/R: CATGTACGTTGCTATCCAGGC/CTCCTTAATGTCACGCACGAT; h-NFE2L2-F/R: TCCAGTCAGAAACCAGTGGAT/GAATGTCTGCGCCAAAAGC-TG; h-MMP2-F/R: CCCATGCGGTTTTCTCGAAT/CAAAGGGGTATCCATCGCCAT; h-MMP9-F/R: AGACCTGGGCAGATTCCAAAC/CGGCAAGTCTTCCGAGTAGT; h-CD11b-F/R: CAGCCTTTGACCTTATGTCATGG/CCTGTGCTGTAGTCGCACT; h-CD68-F/R: CGAGCATCATTCTTTCACCAGCT/ATGAGAGGCAGCAAGATGGACC.
2.9. Western Blotting Assay
Western blotting analysis was performed as previously described with slight modifications [
37,
38]. Briefly, the treated cells were lysed with lysis buffer, collected, and centrifuged, and the total protein concentration was measured using the Coomassie Brilliant Blue G250 (Beyotime, Shanghai, China) method. Subsequently, heat-denatured protein samples were separated using 10% (g/mL) sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) at 135 V for 2 h and then transferred (200 mA, 2 h) to nitrocellulose membranes (NC; Pall, NY, USA) in an ice bath. Immediately, the membranes were blocked using 5% (g/mL) defeat dried milk for 2 h at 25 °C and then probed with primary antibodies overnight at 4 °C. After washing four times (10 min/wash) with TBST buffer (TBS with 0.5% Tween-20), the membranes were incubated with the respective secondary antibody for 1.5 h at 25 °C. After washing three times (15 min each time) with TBST, the protein blots were visualized using a chemiluminescence solution (Thermo Fisher Scientific, Waltham, MA, USA). Finally, the relative grey density values of the protein blots were quantified using ImageJ software and normalized to the density of β-actin. The following antibodies were used in this study: β-actin (Sigma-Aldrich, A1978, Mouse, 1:5000), SQSTM1/p62 (ABclonal Technology, Wuhan, China, A19700, Rabbit, 1:1000), Keap1 (ABclonal Technology, Wuhan, China, A21724, Rabbit, 1:1000), Nrf2 (ABclonal Technology, Wuhan, China, A0674, Rabbit, 1:1000), MMP2 (GenXspan, AL, USA, GXP328105, Rabbit, 1:1000), MMP9 (Cell Signal Technology, Boston, MA, USA, #13667, Rabbit, 1:1000), mouse secondary antibody (Cell Signal Technology, Boston, MA, USA, #7076S, 1:5000), and rabbit secondary antibody (Cell Signal Technology, Boston, MA, USA, #7074P2, 1:5000).
2.10. Gelatin Zymography Assay
Concerning the previous method [
39,
40], after minor adjustments, the activity of MMP2 was determined by gelatin zymography. Generally, the cells were cultured in 12-well plates in equal numbers. When the cells were grown to a density of 60–70%, they were cultured in 800 μL serum-free DMEM for 24 h. Then, 500 μL of supernatant were collected in ultrafiltration centrifuge tubes (3 kDa, Merck, Darmstadt, Germany), and 30–50 μL concentrated solutions were acquired after centrifugation at 14,000×
g at 4 °C for 30 min and centrifugation at 2000×
g and 4 °C for 5 min. Next, 30 μL samples containing 22.5 μL concentrated solutions and 7.5 μL 5× loading buffer (
Table A2) were electrophoresed using 10% (g/mL) SDS-PAGE containing 0.1% gelatin (Sangon Biotech, Shanghai, China) at 135 V for 1–2 h. After electrophoresis, the gel was incubated in an eluent (
Table A3) and washed twice (40 min/wash) while shaking at a slow speed. Immediately, the gel was washed twice (20 min/time) with a rinsing solution (
Table A4) and incubated with fresh incubation buffer (
Table A5) at 37 °C for 48 h. Then, Coomassie brilliant blue staining solution (30% Methanol, 10% Acetic Acid, and 0.05% Coomassie Brilliant Blue) was added to the gel at 37 °C for 3 h, and the gel was destained with decolorizing solution (V
Methanol:V
Acetic acid:V
Water = 5:1:4) while shaking at a slow speed for 1–2 h. Finally, the gel was taken, and the active area of matrix metalloproteinase was displayed as a transparent band under the dark blue background.
2.11. siRNA Transfection
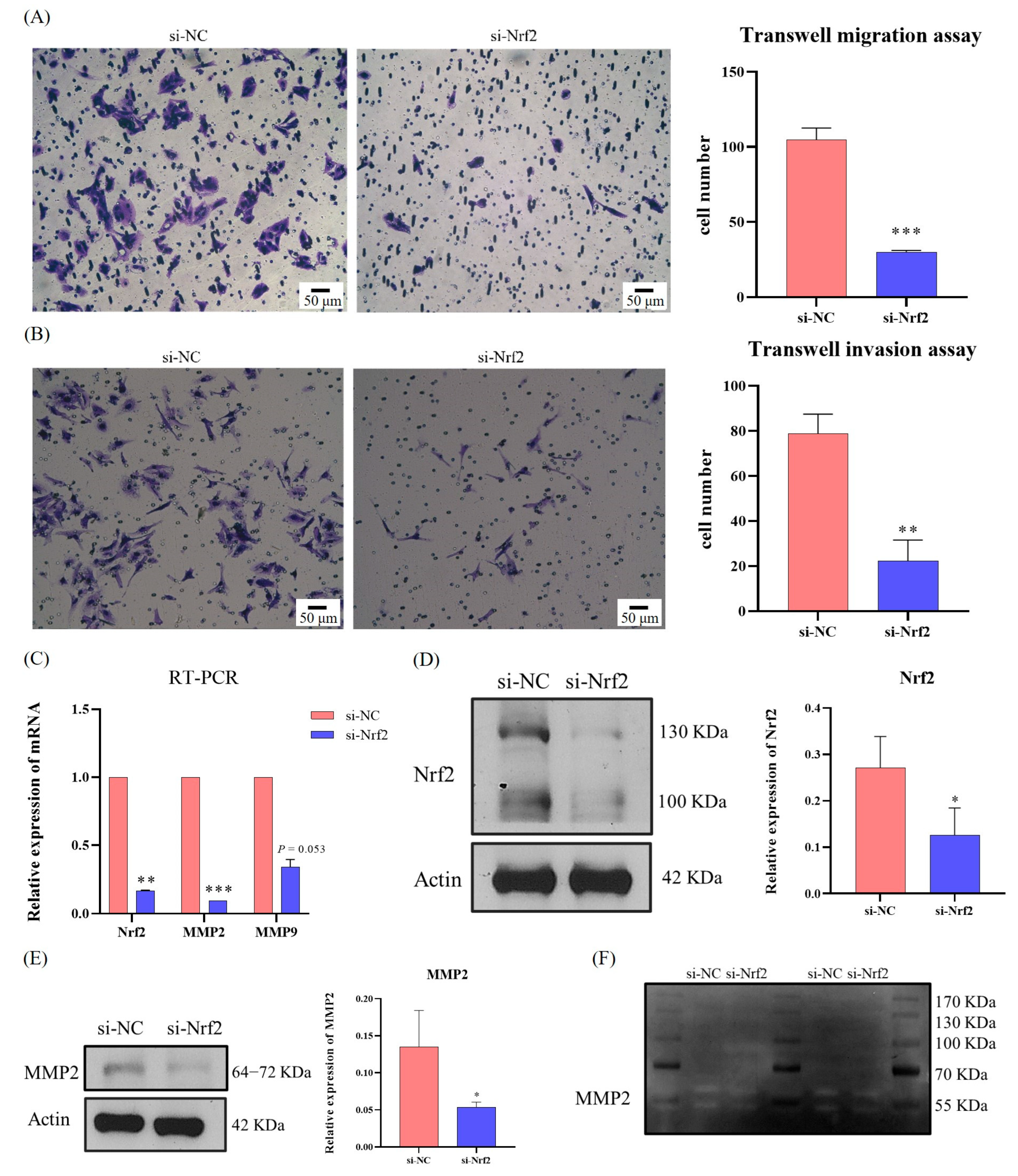
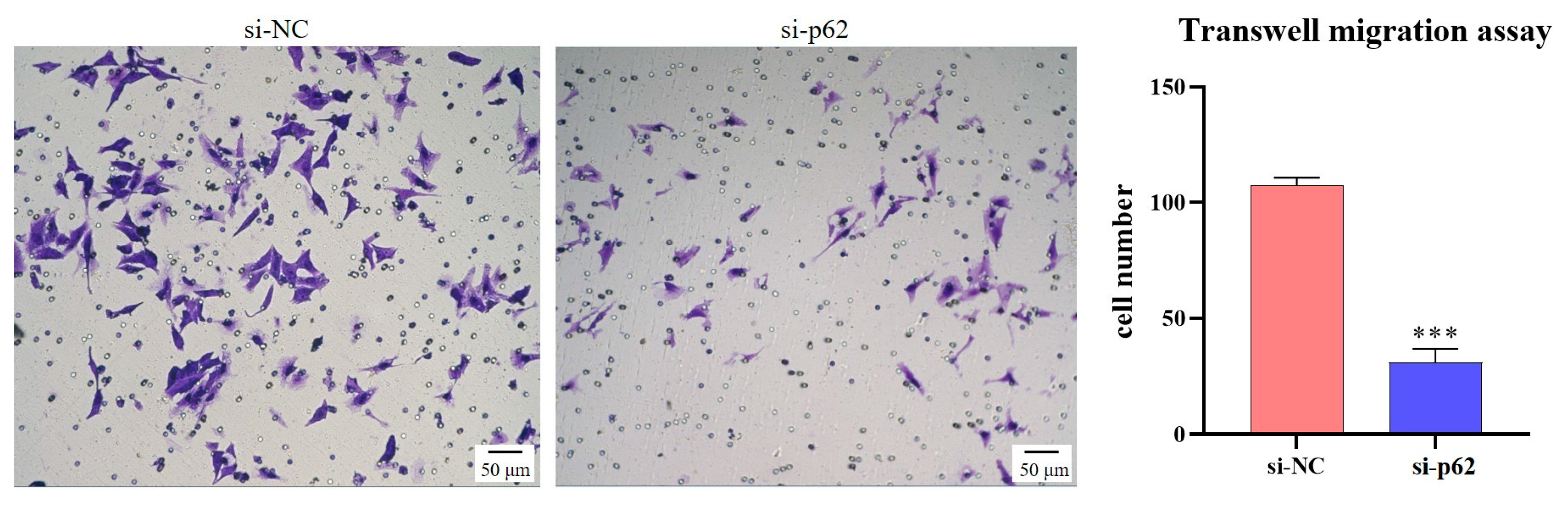
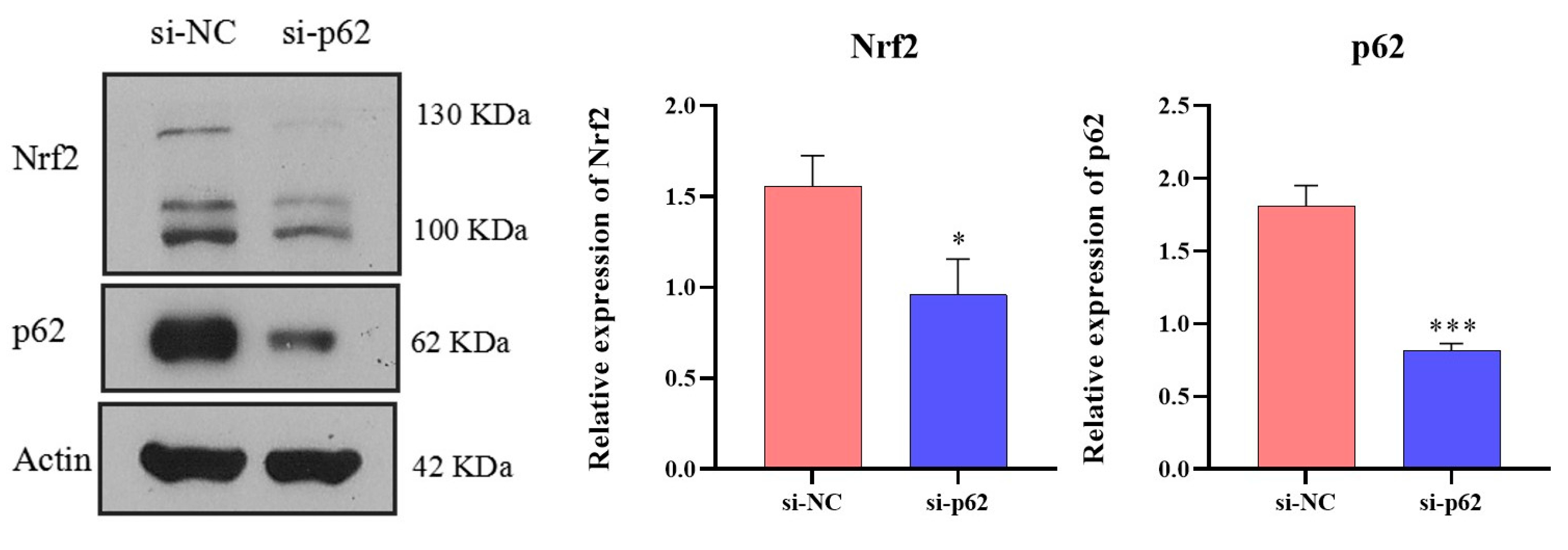
Transfection is an effective method for verifying target and signaling pathways. Taking 6-well plates as an example, 5 μL Lipofectamine® 3000 (ThermoFisher Scientific, Waltham, MA, USA) and 245 μL Opti-MEM® (ThermoFisher Scientific, Waltham, MA, USA) were mixed and left to stand for 5 min to obtain the transfection reagent. Then, si-NC (a common negative control without homology to the sequence of the target gene) or siRNA-Target gene (e.g., siRNA-p62 and siRNA-Nrf2; RiboBio, Guangzhou, China) were mixed with 245 μL Opti-MEM®, and the siRNA solution was obtained. The two solutions were then mixed and allowed to stand for 20 min. Then, 500 μL Opti-MEM® of the mixed solution were added to each well of cells at 60% confluence, and the final concentration of siRNA remained at 100 nM. After 12–48 h of transfection, the cells were used for Transwell migration, Transwell invasion, gelatin zymography, and RNA and protein assays.
2.12. Animal Experiments
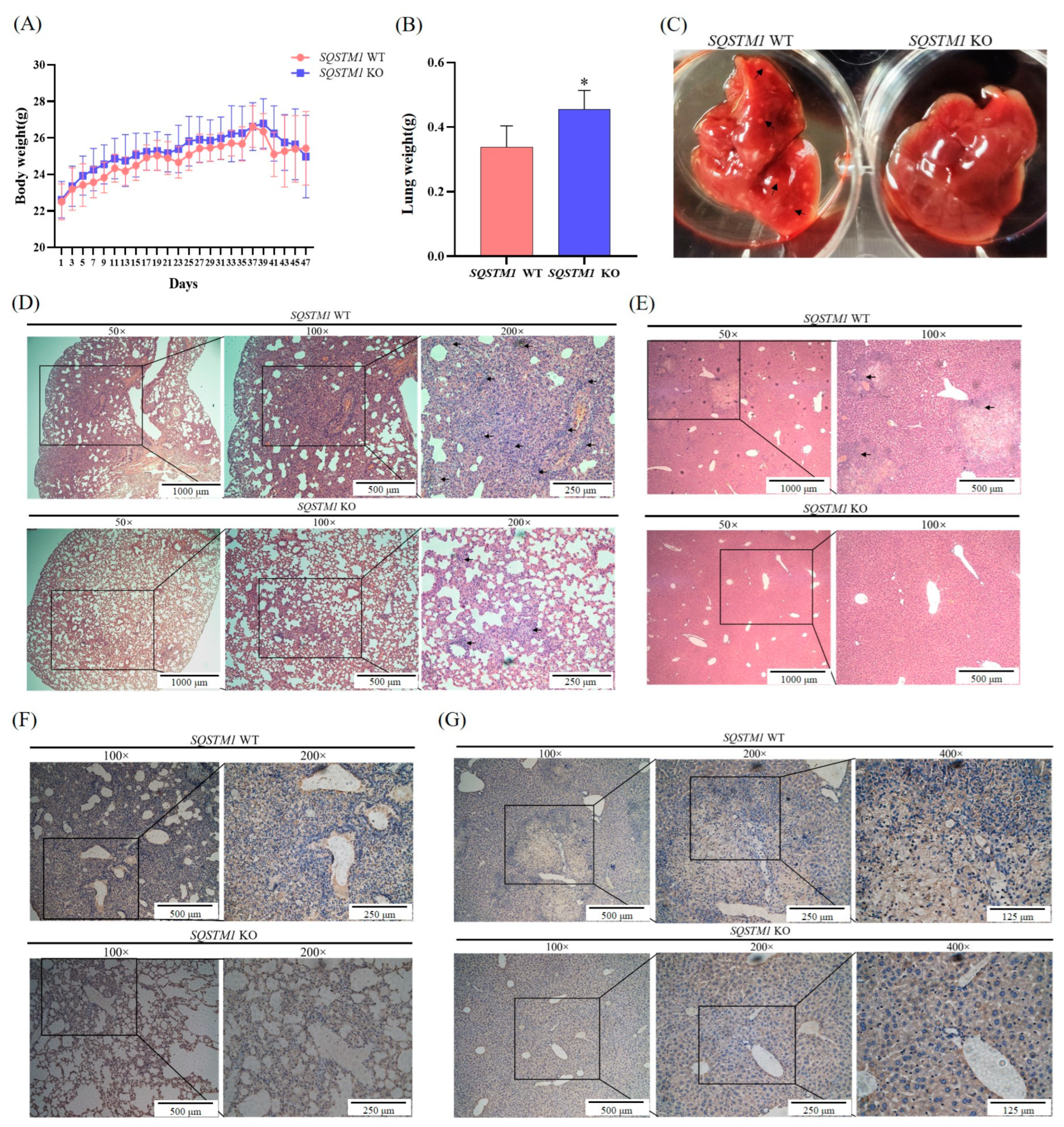
BALB/c-nu/nu nude mice [SPF grade, Male, six weeks old, Certified No. SCXK (Guang-dong) 2022-0002], chow diets, and corncob padding were supplied by the Guangdong Medical Laboratory Animal Center (Guangzhou, China). The animals were fed in a closed animal rearing cabinet (F2005001725) in a quarantine room (ADM-024) in a suitable environment (humidity: 40–70%, temperature: 20–26 °C, light illumination, 12 h/day). This study strictly followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals to ensure animal welfare and ethical standards. The protocol was approved by the Bioethics Committee of the Shenzhen International Graduate School of Tsinghua University (Ethics issue (2022) No. F109). In this study, a lung metastasis model of HCC in nude mice was established by tail vein injection. Briefly, nude mice were randomly divided into control and experimental groups based on their weight (n = 5 per group). SQSTM1 WT HepG2 cells (1.5 × 106) and SQSTM1 KO HepG2 cells (1.5 × 106) were injected into the tail vein of mice in the control and experimental group, respectively. The weight and general condition of the mice were recorded every other day. After 7 weeks of inoculation, all nude mice were killed humanely and dissected to observe metastasis in the lungs and liver. In addition, all lung and liver tissues were weighed, and portions were produced as 4% paraformaldehyde-fixed paraffin-embedded samples for conventional hematoxylin and eosin (H&E) staining and immunohistochemical analysis (MMP2).
2.13. Statistical Analysis
In this study, the experimental data were statistically analyzed using GraphPad Prism 8.4.3 (GraphPad Software Inc., Waltham, MA, USA) and are shown as mean ± standard deviation (SD). Differences with statistical significance between different groups were evaluated using one-way ANOVA with Tukey’s post hoc test. Statistical significance was set at p < 0.05.
4. Discussion
HCC is the sixth most common malignant tumor worldwide, with high mortality and invasiveness [
1,
43]. Although various treatment methods for HCC have been developed and improved [
44], the survival rate and recurrence of HCC have not been properly resolved owing to the inconspicuous clinical symptoms at the early stage of HCC. Extensive research has found that migration and invasion are signs of HCC entering the late stage and seriously affect the success rate of HCC treatment [
6,
7]. Therefore, inhibiting the migration and invasion of HCC cells is the main trend of current research in the field of HCC treatment.
This study first found that SQSTM1/p62 was highly expressed in tumor tissues with strong invasion and migration ability, especially in HCC tissues, through the analysis of large data of clinical samples. It was hypothesized that SQSTM1/p62 plays an important role in these processes. Recently, many studies have confirmed that SQSTM1/p62 regulates the occurrence and development of various tumors [
13,
14,
15,
16], but most studies have focused on autophagy. However, in HCC, whether SQSTM1/p62 regulates tumor migration and invasion and the mechanisms involved remain unclear. Therefore, in this study, we constructed
SQSTM1 WT HepG2 cells and
SQSTM1 KO HepG2 cells using the CRISPR/Cas9 system, studied the effects of SQSTM1/p62 on migration and invasion of HCC cells in vivo and in vitro, and determined whether SQSTM1/p62 can serve as an important target for cancer treatment.
In this study, we first showed that
SQSTM1/p62 knockout could significantly inhibit the migration and invasion of HCC cells using wound healing and Transwell assays. SQSTM1/p62 can promote Keap1 degradation and cancel the inhibition activity on Nrf2. So, SQSTM1/p62 can promote Nrf2 activity. Recent studies have shown that Nrf2 overexpression can mediate the survival and development of cancer cells through multiple signaling pathways [
45,
46]. Nrf2 is highly expressed in HCC and plays an inhibitory role in the initial stage of hepatocarcinogenesis. However, with the gradual progression of liver cancer, Nrf2 will promote the expression of MMP9 [
19] and is a key factor in promoting the proliferation, metastasis, invasion, and other malignant biological behaviors of HCC [
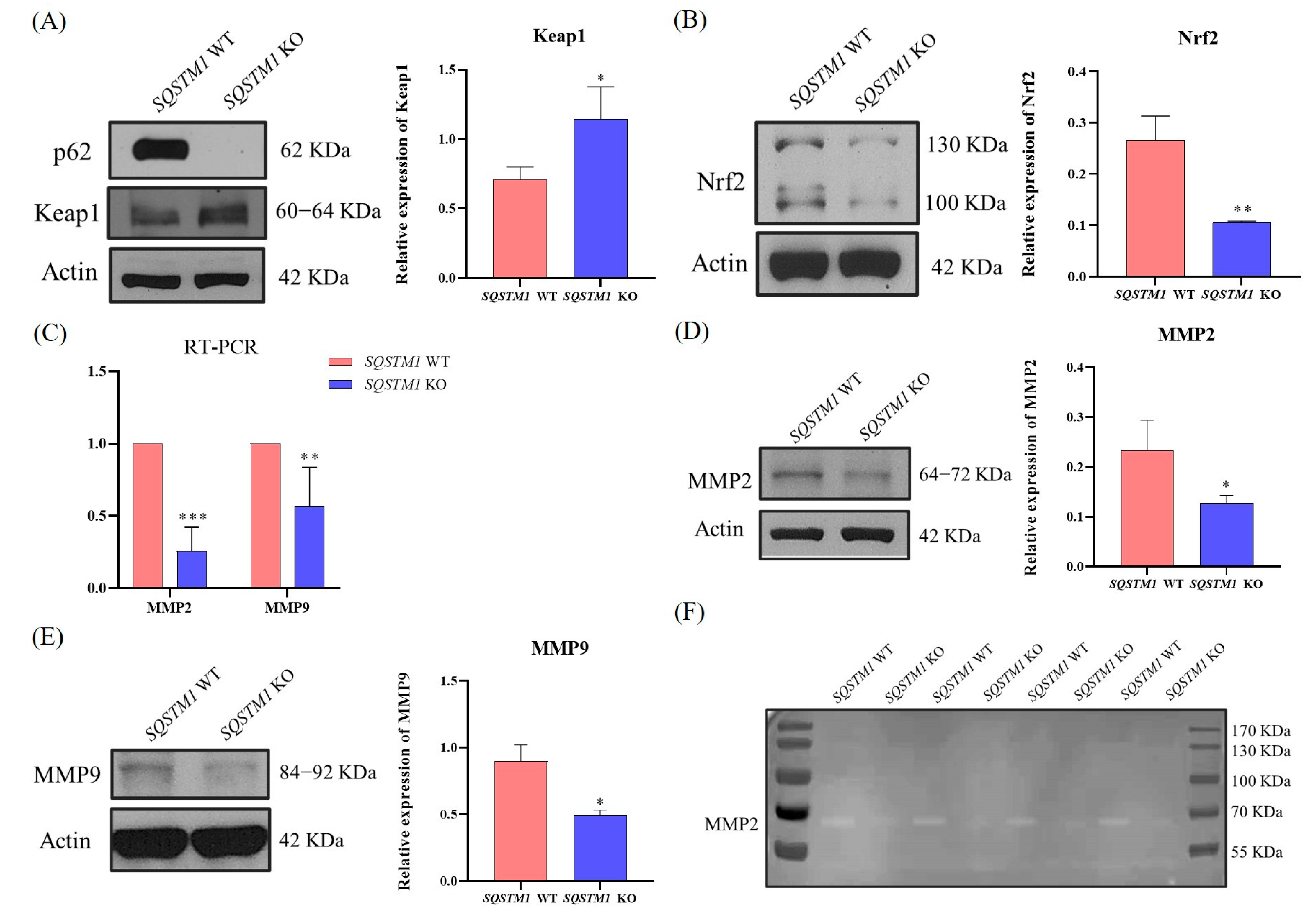
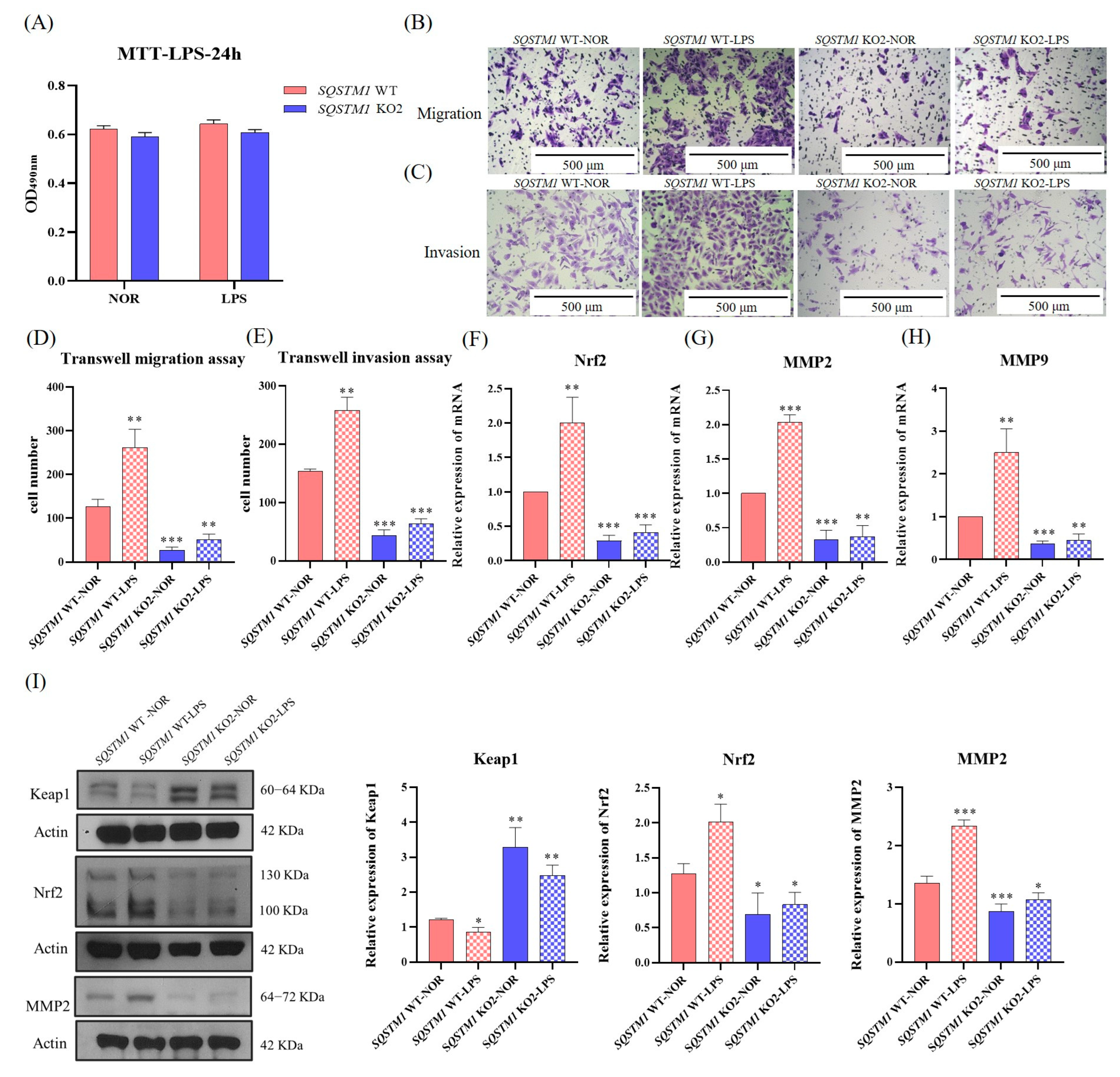
47]. We suspected that SQSTM1/p62 might act in the progression of HCC through the Keap1/Nrf2/MMP2 signaling pathway, but this requires further validation.
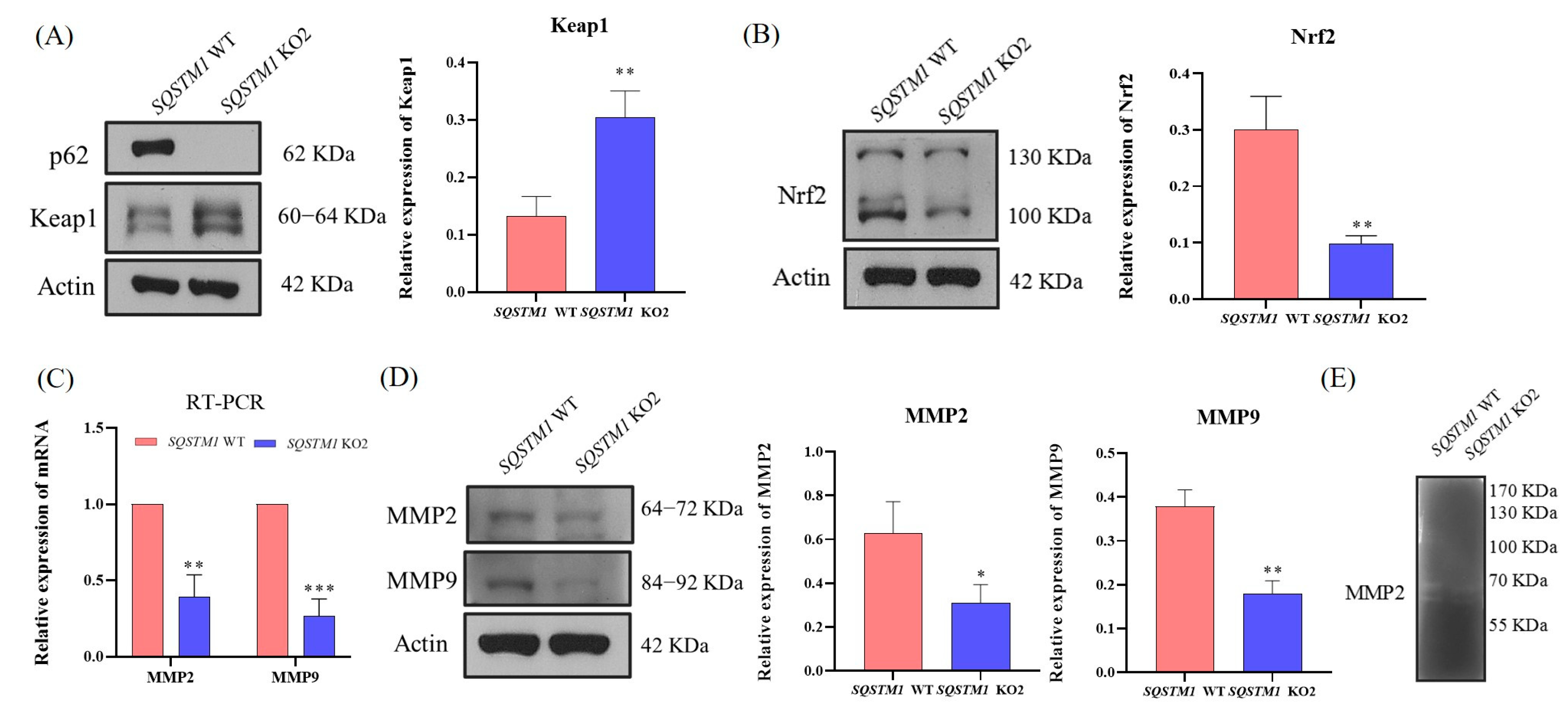
Further experiments showed that in HCC,
SQSTM1/p62 knockout resulted in a significant increase in the expression of Keap1 and a significant reduction in the expression of Nrf2. Furthermore, the mRNA and protein levels of MMP2 and MMP9 were significantly decreased. MMP2 is a Zn
2+-dependent proteolytic enzyme that hydrolyzes type IV collagen, activates MMP9 to degrade ECM, helps tumor cells break through the basement membrane, and plays a very important role in tumor migration and invasion [
48,
49,
50,
51]. Relevant experiments indicated that the enzymatic activity of MMP2 was significantly reduced, which further clarified the molecular mechanism of inhibiting the migration and invasion of HCC after
SQSTM1/p62 knockout. Furthermore, we verified that after Nrf2 silencing, the expression and activity of MMP2 declined significantly, indicating that Nrf2 affects MMP2 activity. Through an in vivo study, we also found that
SQSTM1/p62 knockout affected HCC migration, and the mechanisms might be associated with the Nrf2/MMP pathway. Therefore, this study demonstrated that SQSTM1/p62 could promote the migration and invasion of HCC cells through the Keap1/Nrf2/MMP2 signaling pathway.
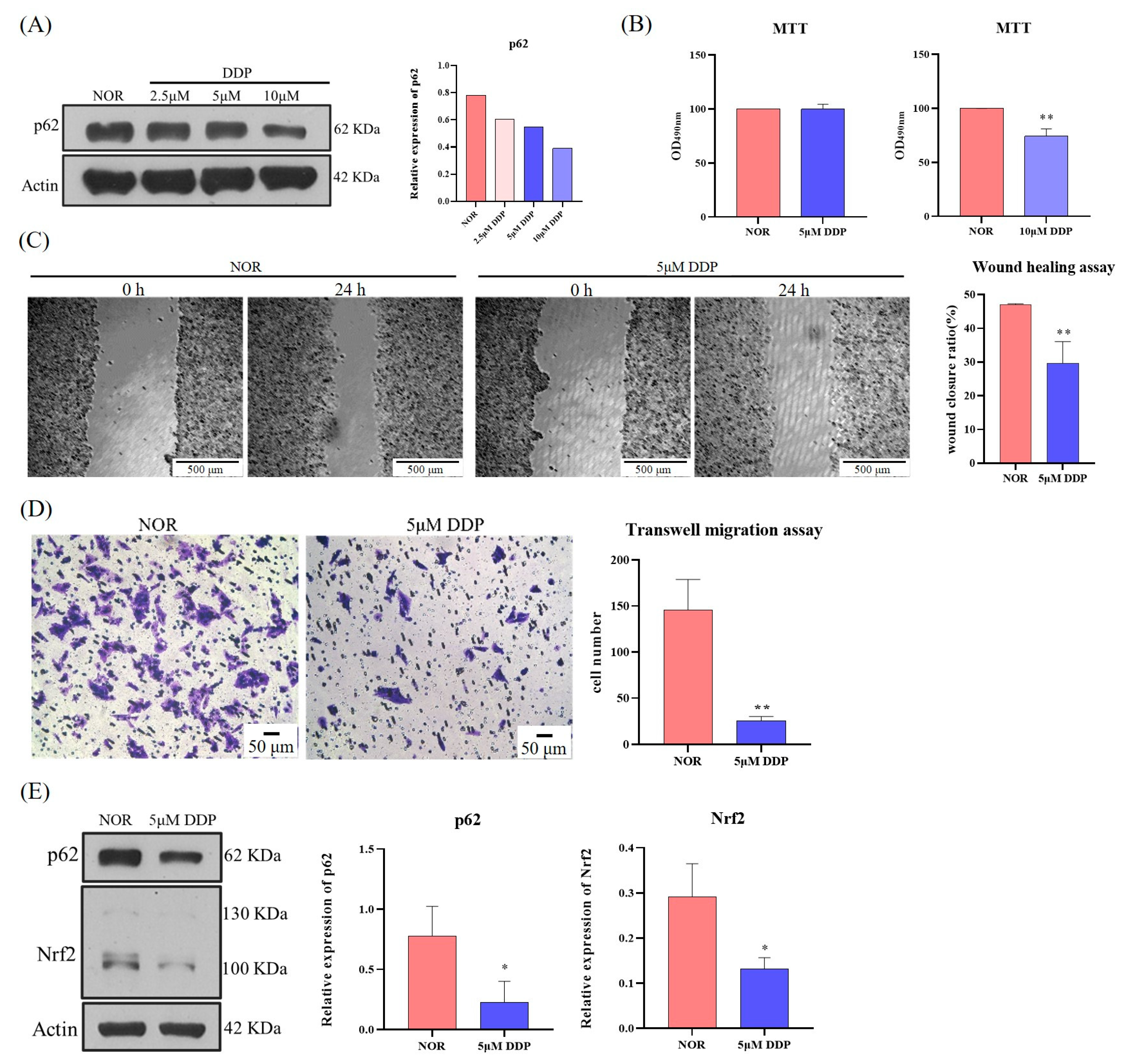
SQSTM1/p62 appears to mediate the migration and invasion of tumors and may serve as a promising target for drug treatment. However, drugs that regulate SQSTM1/p62 remain unclear. Therefore, we searched for relevant drugs targeting SQSTM1/p62. Interestingly, this study revealed that DDP, an antitumor drug, significantly inhibited the expression of SQSTM1/p62 and slowed the migration and invasion of HCC cells. According to the domestic literature, there have been many reports on the impact of cisplatin on tumor occurrence and development, but they mainly focus on drug resistance, cytotoxicity, inhibition of proliferation, and apoptosis [
52,
53,
54,
55]. However, there are no reports on the impact of DDP on the migration and invasion of HCC cells through SQSTM1/p62. Therefore, another novelty of this study is that it suggests a new treatment strategy to inhibit the migration and invasion of HCC.
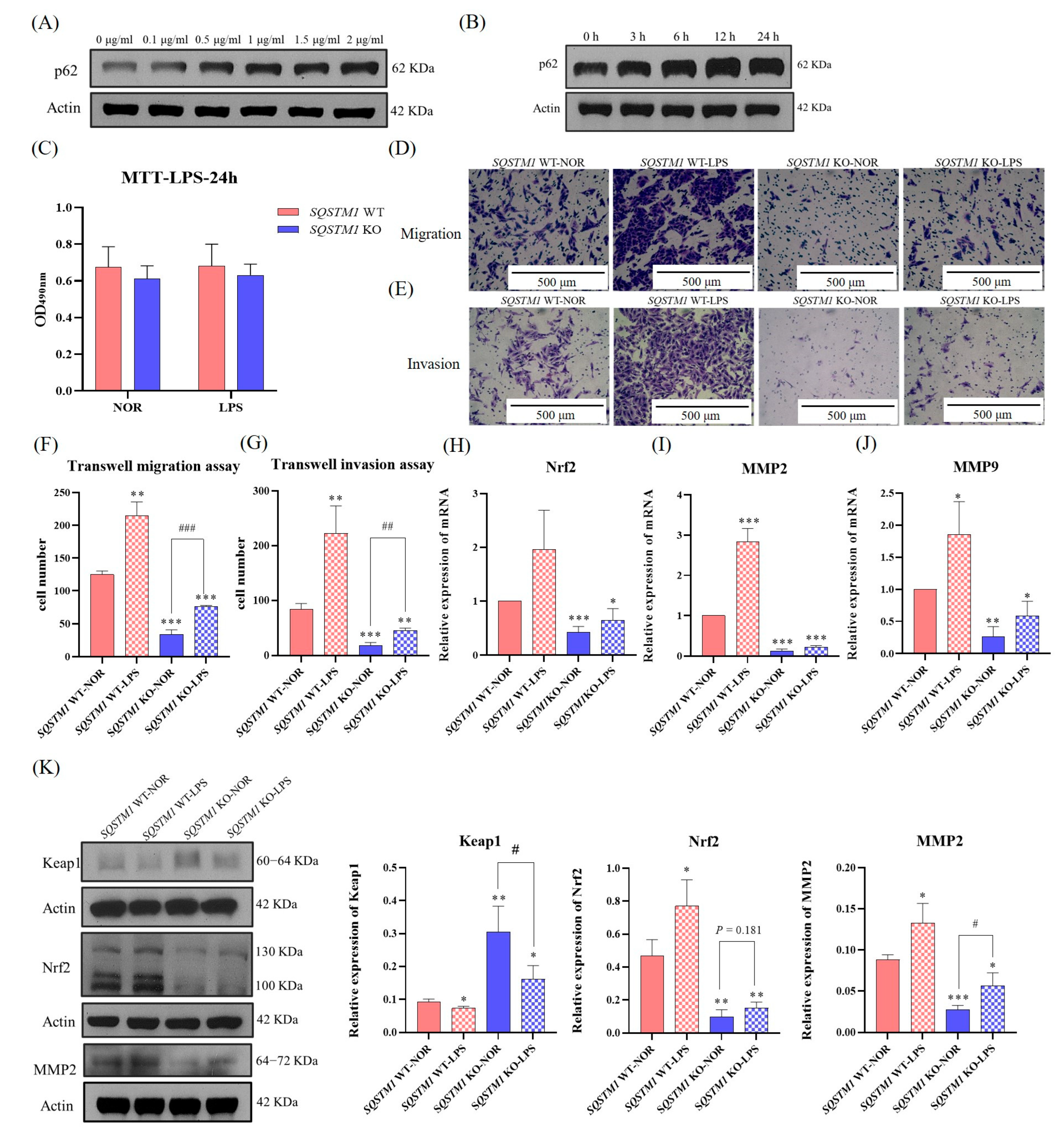
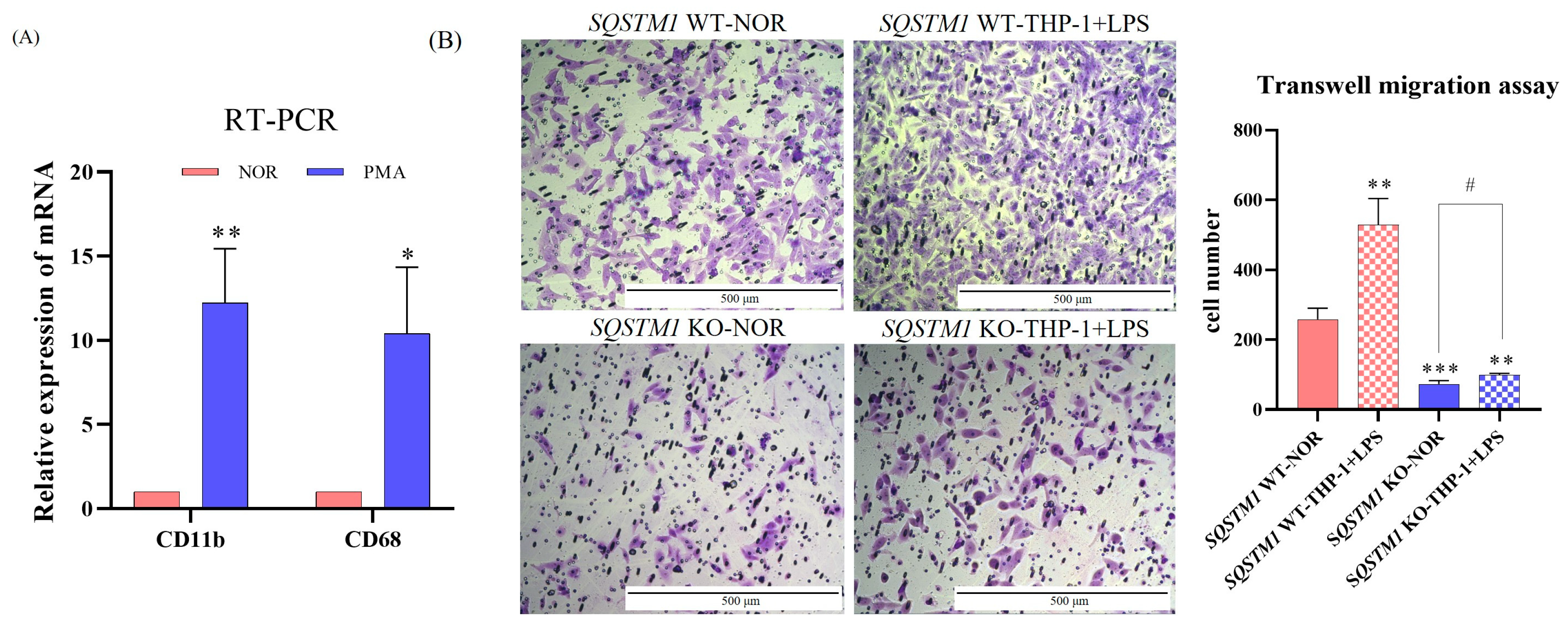
In addition, emerging evidence indicates that the inflammatory microenvironment plays an important role in tumor progression [
25]. Several studies have reported that SQSTM1/p62 is significantly upregulated in the inflammatory microenvironment [
26,
27,
28,
29]. Therefore, this study further explored the impact and mechanism of SQSTM1/p62 on the migration and invasion of HCC cells in an inflammatory microenvironment. It was found that the inflammatory microenvironment simulated by LPS treatment upregulated SQSTM1/p62 and significantly affected the migration and invasion of HCC. This effect of inflammation on the migration and invasion of HCC seems to depend on SQSTM1/p62, but further validation is required. Taken together, the pharmacological or pathological changes in SQSTM1/p62 levels can affect the migration and invasion of HCC cells.
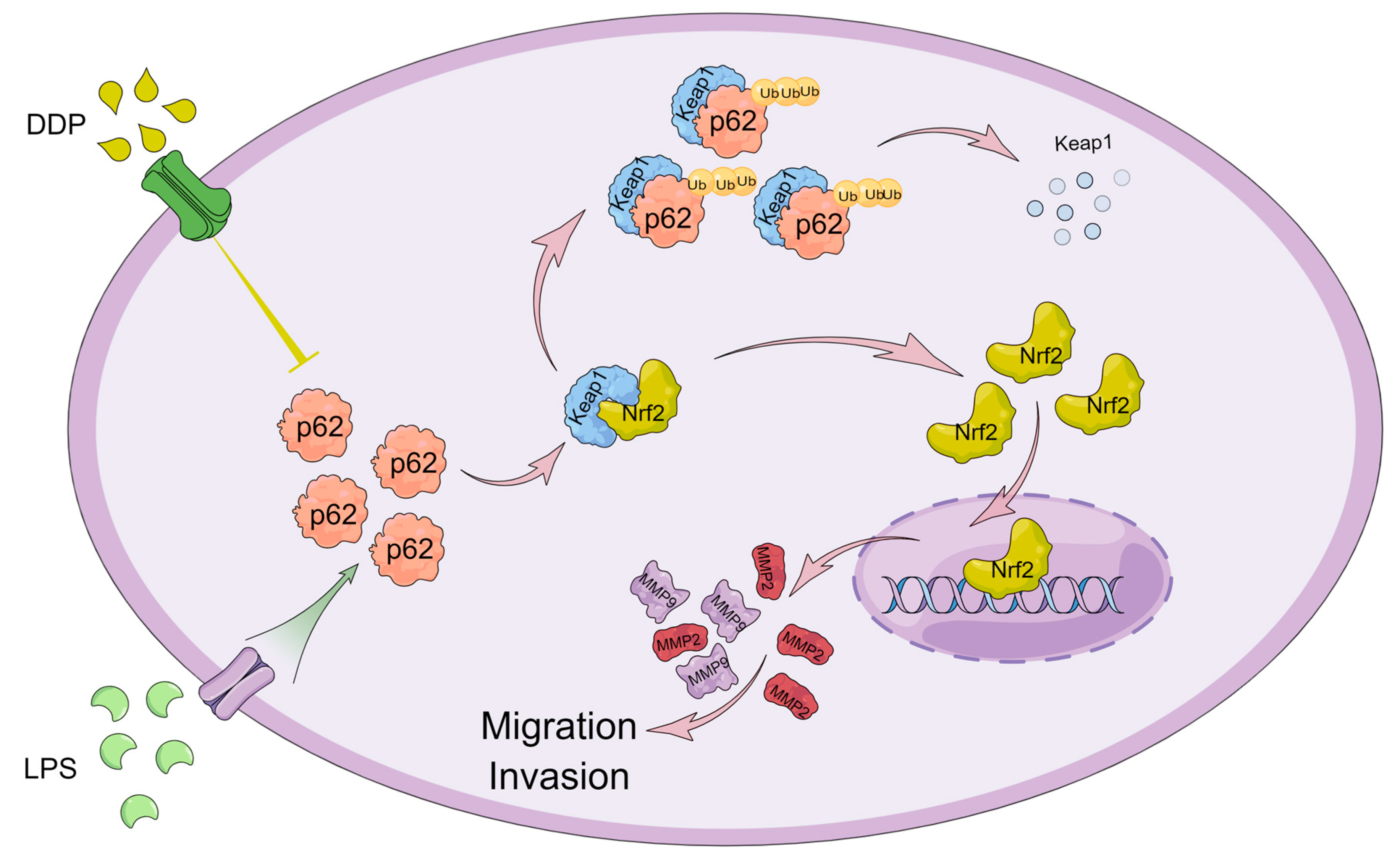
In conclusion, in this study, we first found that SQSTM1/p62 plays a key role in the migration and invasion of HCC in vitro and in vivo using the CRISPR/Cas9 system (
Figure 9).
SQSTM1/p62 knockout may inhibit the migration and invasion of HCC through the Keap1/Nrf2/MMP2 signaling pathway. The pharmacological or pathological regulation of SQSTM1/p62 can affect the migration and invasion of HCC cells (
Figure 9). This study indicated that SQSTM1/p62 is a promising target for the treatment of HCC migration and invasion. However, considering that only one cell line (HepG2) is used in the functional experiments in this study, which has certain limitations, further validation studies should be conducted in the future.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}















