
Etiopathogenesis, Diagnosis, and Treatment Strategies for Lymphomatoid Papulosis with Particular Emphasis on the Role of the Immune System

 , , , , , ,
, , , , , ,  , and
, and
Abstract
:1. Introduction
2. Epidemiology of Lyp in Terms of Histological Types
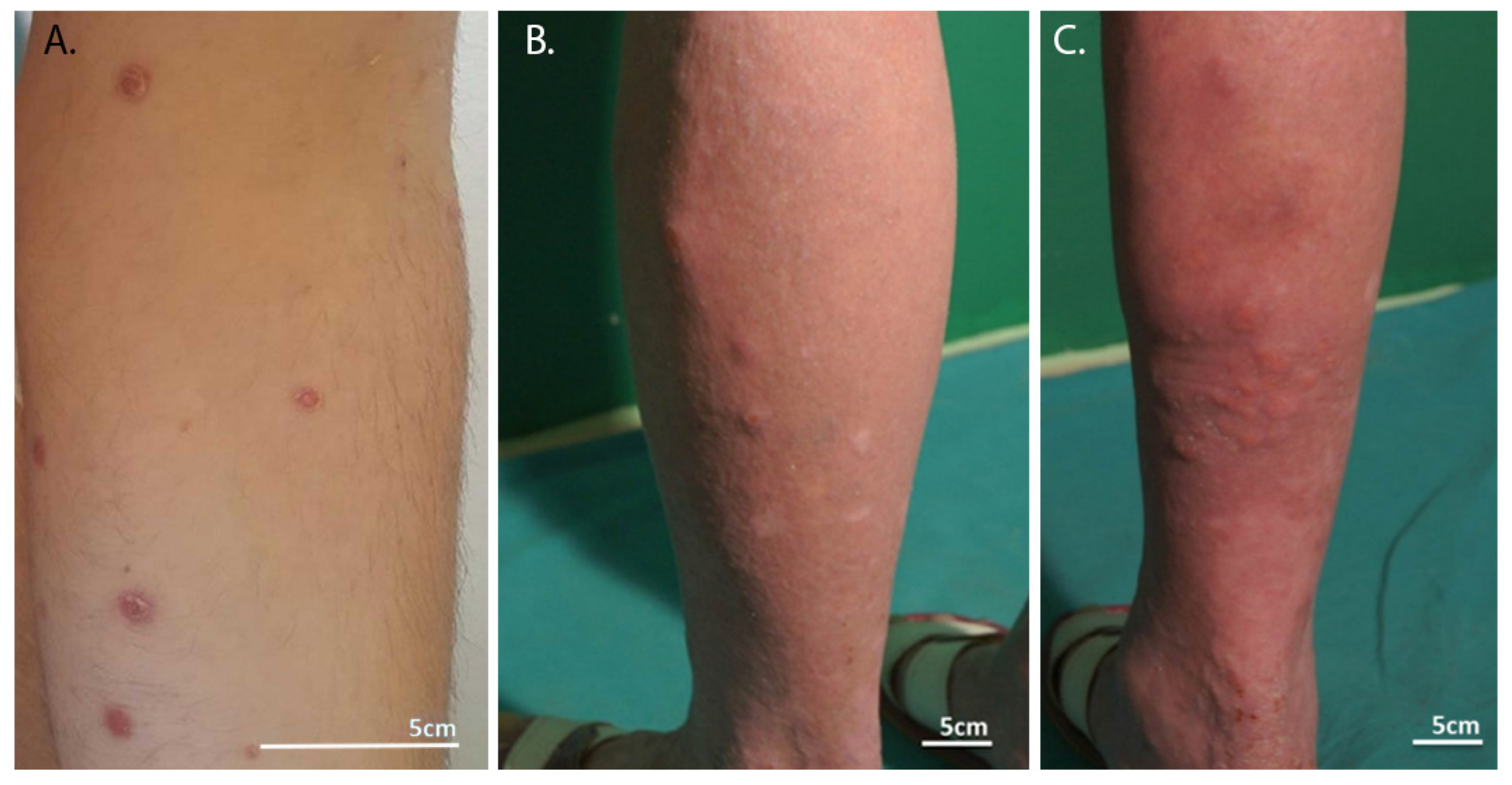
3. Clinical Manifestation in LyP
4. Etiopathogenesis and Role of the Immune System in the Development of LyP
5. Importance of Histopathological Immunophenotyping and Immunohistochemistry in the Diagnosis of LyP
5.1. Type A LyP
5.2. Type B LyP
5.3. Type C LyP
5.4. Type D LyP
5.5. Type E LyP
5.6. Type with Rearrangement 6p25.3
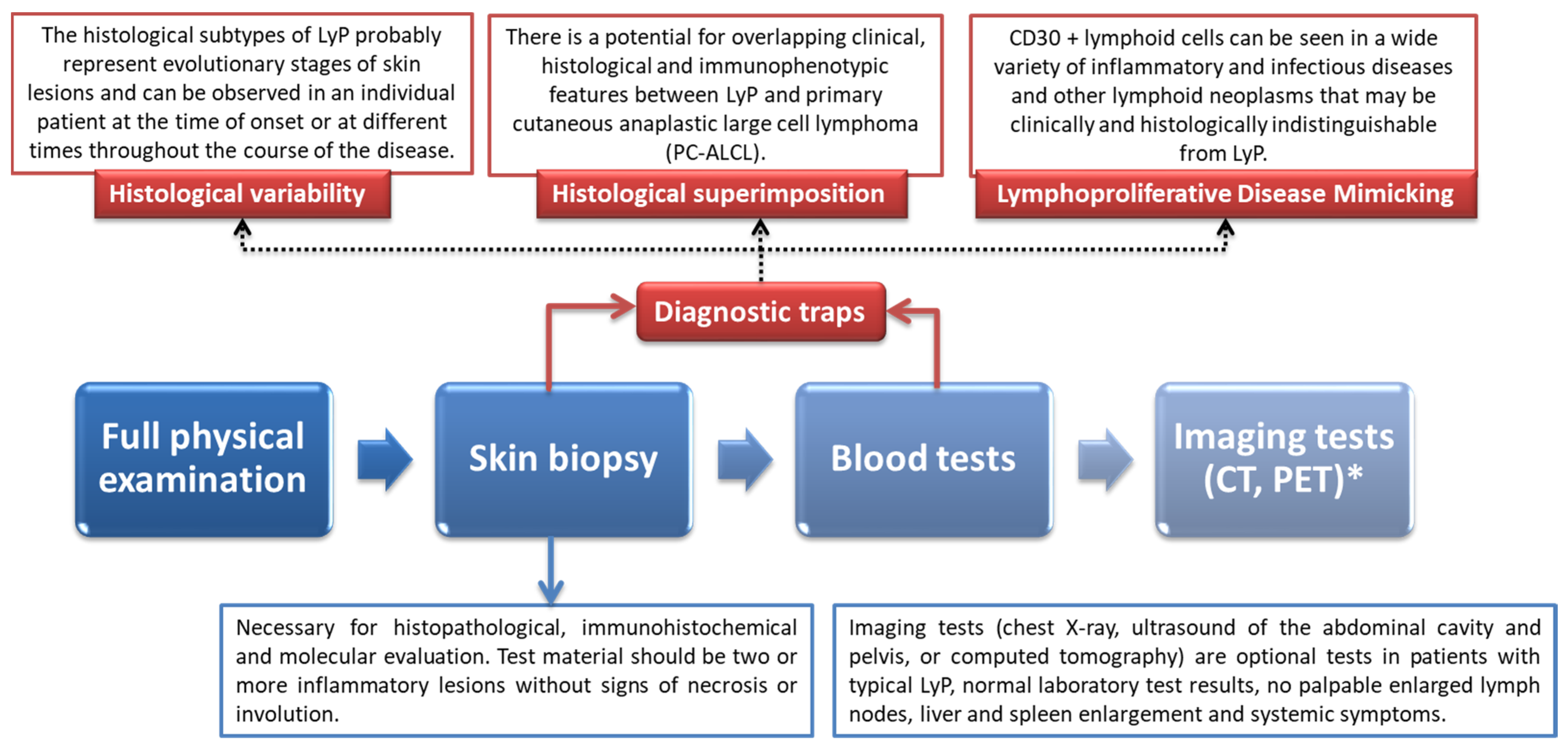
6. Diagnostic Procedure of LyP with Particular Emphasis on Differential Diagnosis and Underlying Histological Features
- (a)
- Overexpression of CD30 + T cells found during immunohistochemistry;
- (b)
- Infiltration of large atypical T cells (along with numerous other inflammatory cells including small lymphocytes, neutrophils, histiocytes, and eosinophils;
- (c)
- Clonal rearrangement of TCR genes (found in approximately 40–100% of cases).
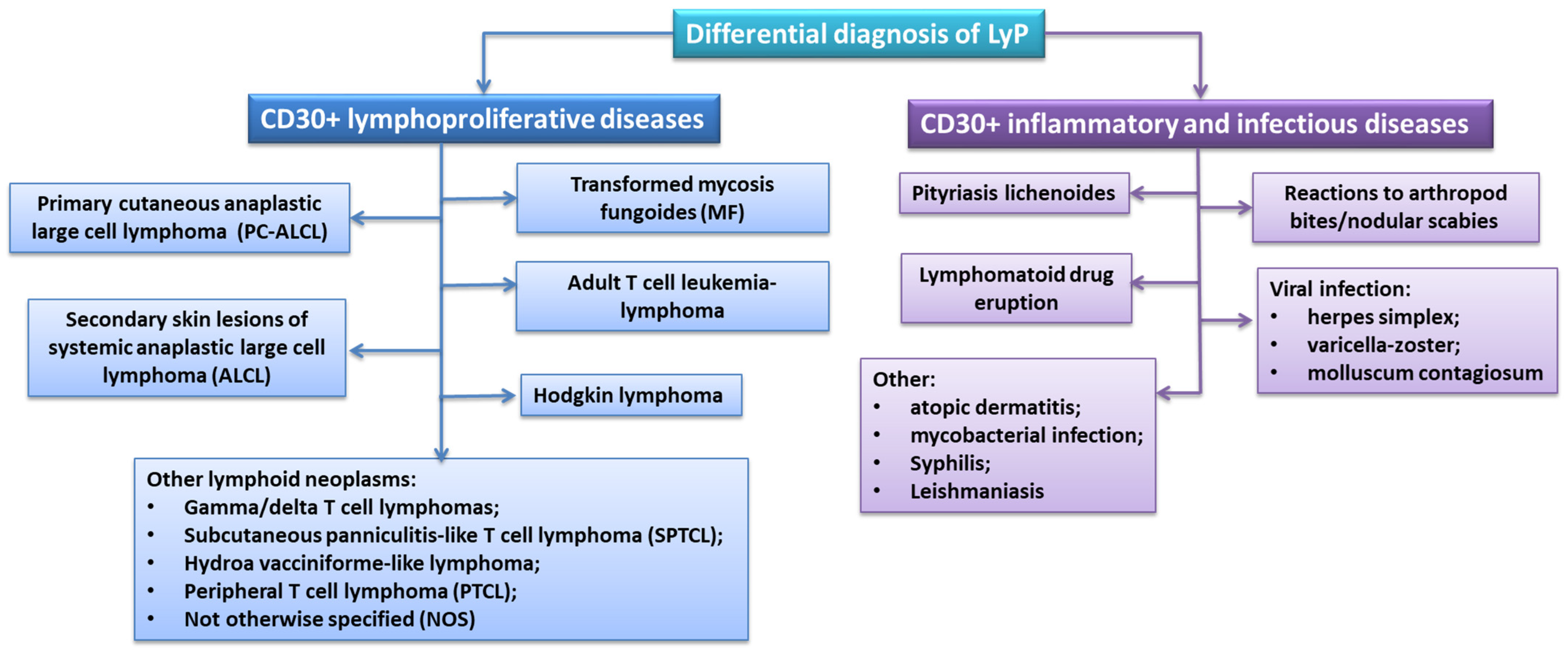
Differential Diagnosis of LyP
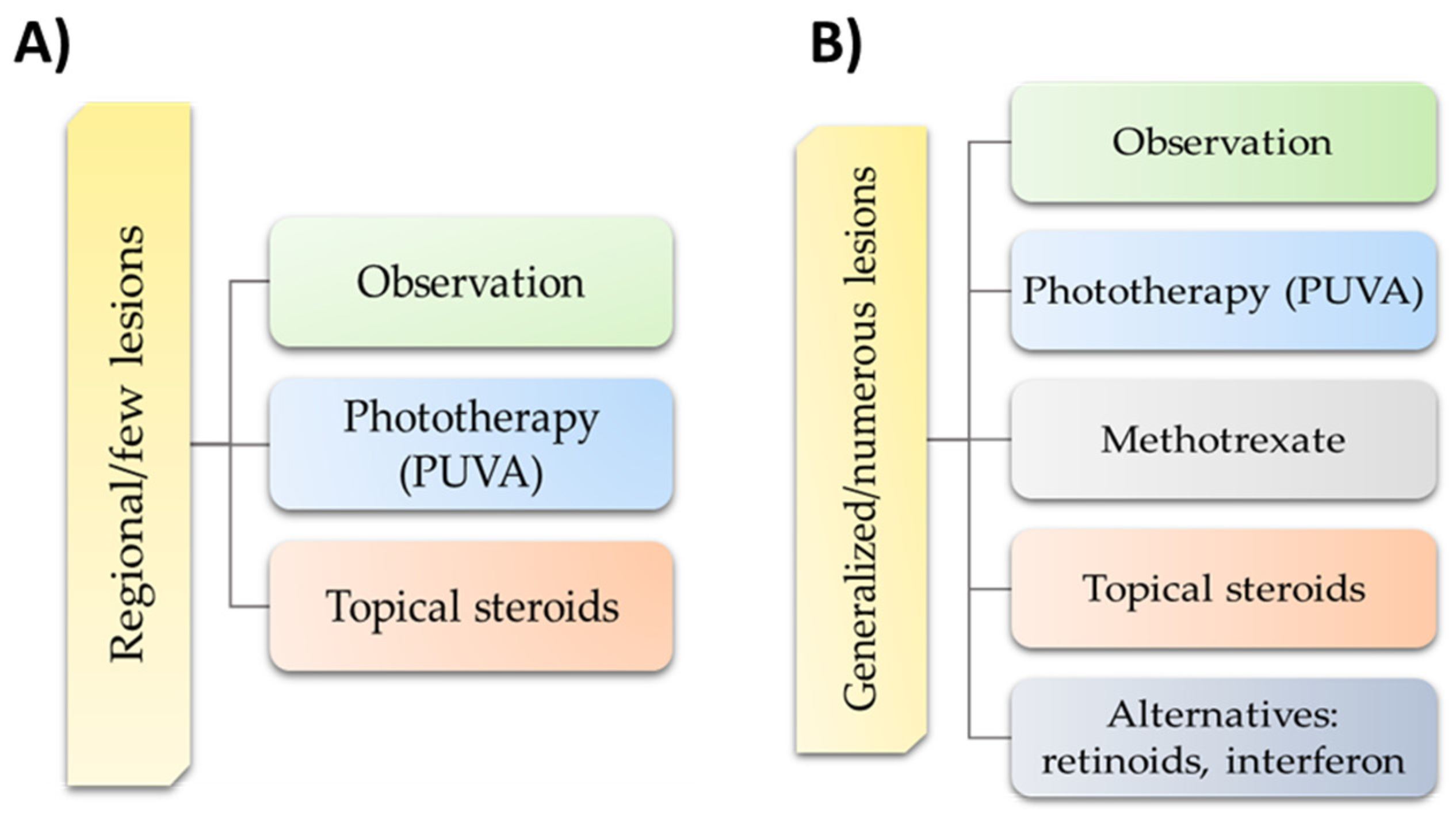
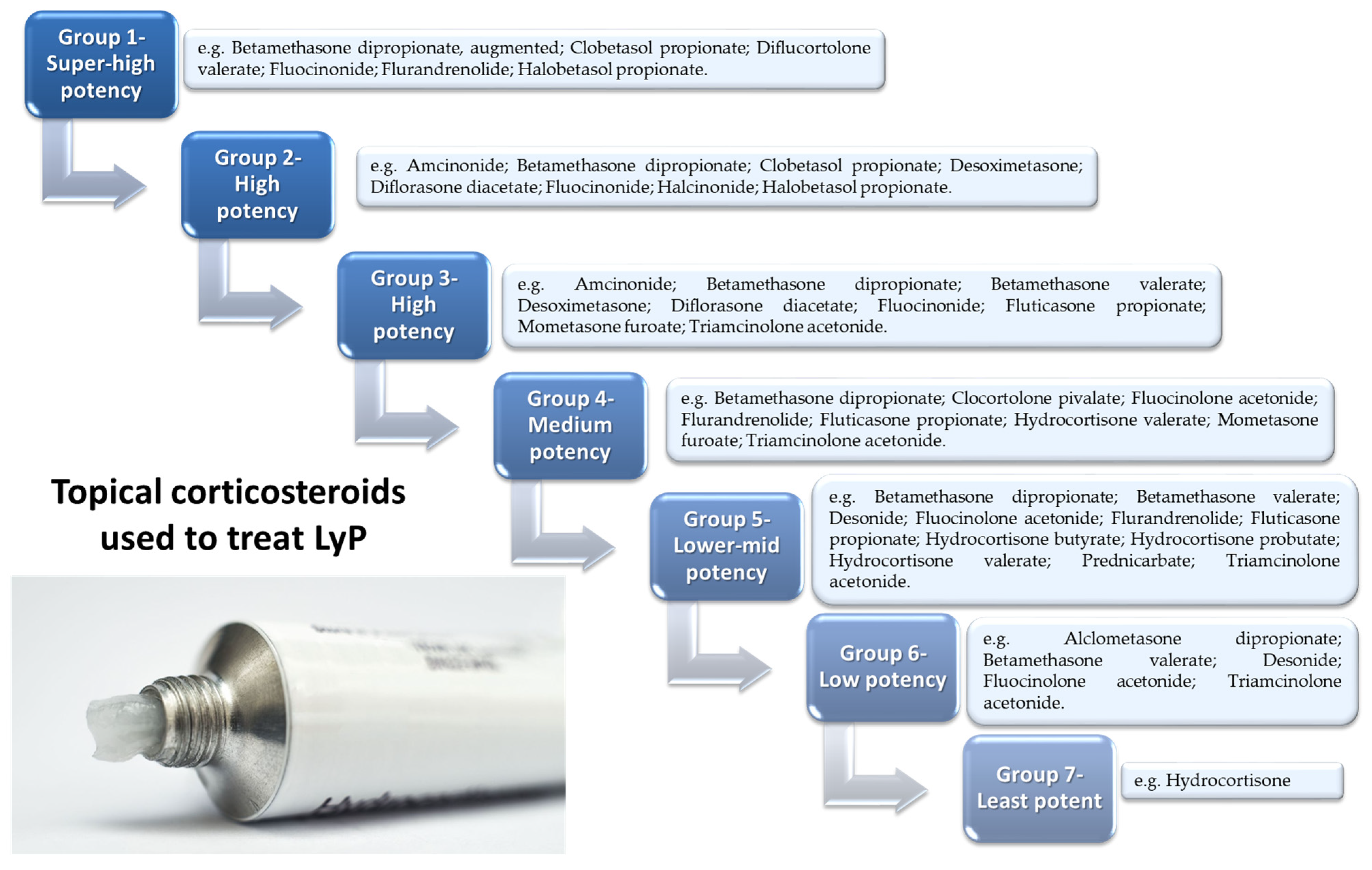
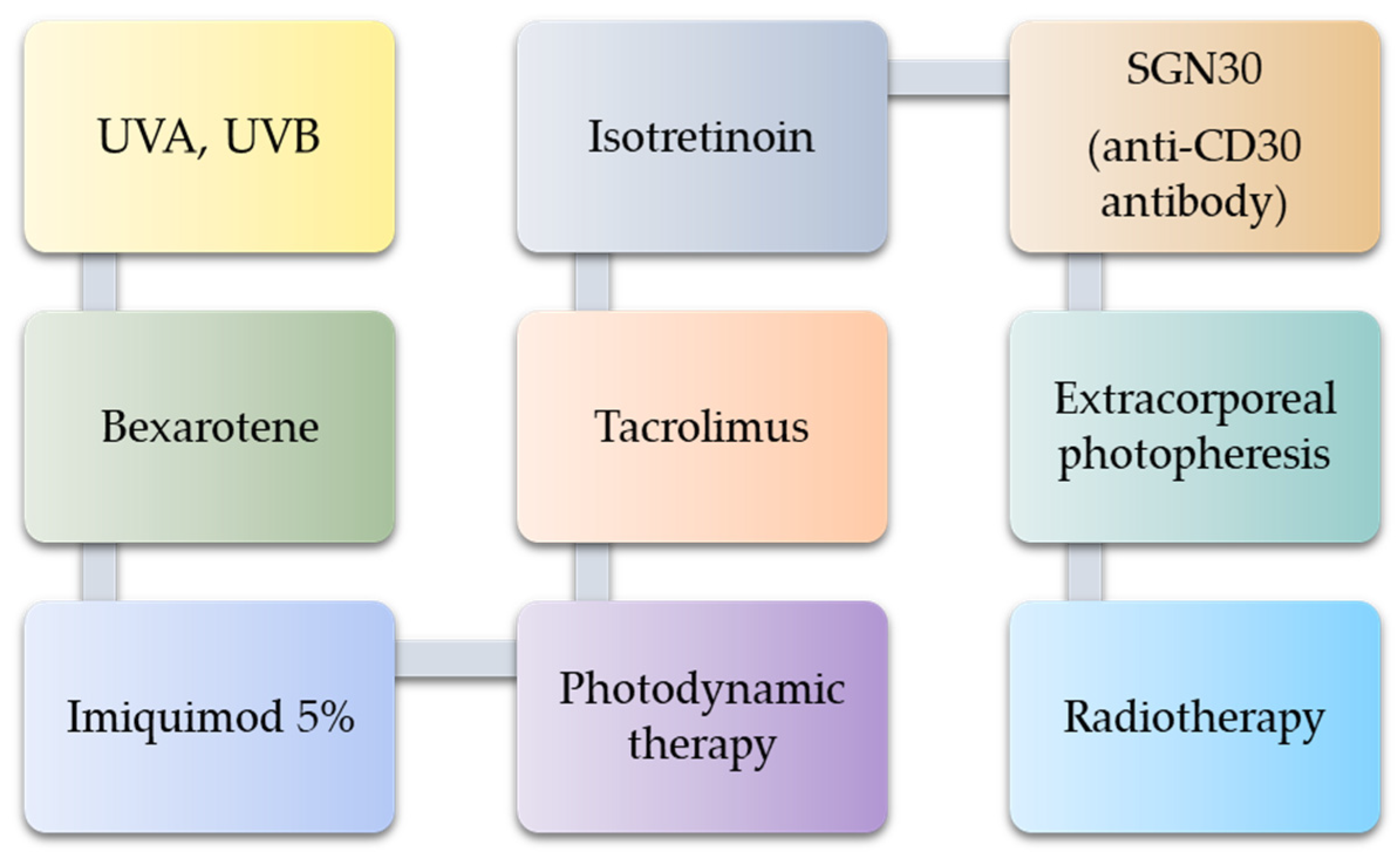
7. Possible Treatment Strategies
8. Materials and Methods
Search Strategy, Study Selection, and Data Extraction
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Assaf, C.; Gellrich, S.; Steinhoff, M.; Nashan, D.; Weiße, F.; Dippel, E.; Coors, E.; Stein, A.; Gollin, P.; Henke, U.; et al. Cutaneous lymphomas in Germany: An analysis of the Central Cutaneous Lymphoma Registry of the German Society of Dermatology (DDG). JDDG J. Der Dtsch. Dermatol. Ges. 2007, 5, 662–668. [Google Scholar] [CrossRef]
- Nijsten, T.; Curiel-Lewandrowski, C.; Kadin, M.E. Lymphomatoid Papulosis in Children: A Retrospective Cohort Study of 35 Cases. Arch. Dermatol. 2004, 140, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Wagner, G.; Rose, C.; Klapper, W.; Sachse, M.M. Lymphomatoid papulosis. J. Dtsch. Dermatol. Ges. 2020, 18, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Lymphomatoid Papulosis. Cutaneous Lymphoma Foundation. Available online: https://www.clfoundation.org/lymphomatoid-papulosis (accessed on 7 March 2022).
- Mational Institute of Health. Lymphomatoid Papulosis|Genetic and Rare Diseases Information Center (GARD)—An NCATS Program. Available online: https://rarediseases.info.nih.gov/diseases/6944/lymphomatoid-papulosis#ref_14862 (accessed on 7 March 2022).
- Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; Swerdlow, S.H., Ed.; International Agency for Research on Cancer: Lyon, France, 2008; Volume 2. [Google Scholar]
- Justiz Vaillant, A.A.; Stang, C.M. Lymphoproliferative Disorders. StatPearls Publishing. Available online: https://www.ncbi.nlm.nih.gov/books/NBK537162/ (accessed on 7 March 2022).
- Van Der Weyden, C.A.; Pileri, S.A.; Feldman, A.L.; Whisstock, J.; Prince, H.M. Understanding CD30 biology and therapeutic targeting: A historical perspective providing insight into future directions. Blood Cancer J. 2017, 7, e603. [Google Scholar] [CrossRef] [Green Version]
- Hansen, H.P.; Leme, A.F.P.; Hallek, M. Role of ADAM10 as a CD30 Sheddase in Classical Hodgkin Lymphoma. Front. Immunol. 2020, 11, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutaneous T-Cell Lymphoma. Mayo Clinic. Available online: https://www.mayoclinic.org/diseases-conditions/cutaneous-t-cell-lymphoma/symptoms-causes/syc-20351056 (accessed on 7 March 2022).
- Pulitzer, M. Cutaneous T-cell Lymphoma. Clin. Lab. Med. 2017, 37, 527–546. [Google Scholar] [CrossRef]
- Kempf, W.; Mitteldorf, C.; Karai, L.J.; Robson, A. Lymphomatoid papulosis-making sense of the alphabet soup: A proposal to simplify terminology. JDDG J. Der Dtsch. Dermatol. Ges. 2017, 15, 390–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willemze, R.; Cerroni, L.; Kempf, W.; Berti, E.; Facchetti, F.; Swerdlow, S.H.; Jaffe, E.S. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood 2019, 133, 1703–1714. [Google Scholar] [CrossRef]
- Sica, A.; Vitiello, P.; Sorriento, A.; Ronchi, A.; Calogero, A.; Sagnelli, C.; Troiani, T.; Fasano, M.; Dodaro, C.A.; Franco, R.; et al. Lymphomatoid papulosis. Minerva Med. 2020, 111, 166–172. [Google Scholar] [CrossRef]
- Bekkenk, M.W.; Geelen, F.A.; Vader, P.C.V.V.; Heule, F.; Geerts, M.L.; Van Vloten, W.A.; Meijer, C.J.; Willemze, R. Primary and secondary cutaneous CD30(+) lymphoproliferative disorders: A report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood 2000, 95, 3653–3661. [Google Scholar] [CrossRef]
- Sim, J.H.; Kim, Y.C. CD8+ Lymphomatoid Papulosis. Ann. Dermatol. 2011, 23, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Wieser, I.; Wohlmuth, C.; Nunez, C.A.; Duvic, M. Lymphomatoid Papulosis in Children and Adolescents: A Systematic Review. Am. J. Clin. Dermatol. 2016, 17, 319–327. [Google Scholar] [CrossRef]
- Martinez-Cabriales, S.A.; Walsh, S.; Sade, S.; Shear, N.H. Lymphomatoid papulosis: An update and review. J. Eur. Acad. Dermatol. Venereol. 2019, 34, 59–73. [Google Scholar] [CrossRef]
- Yip, L.; Darling, S.; Orchard, D. Lymphomatoid papulosis in children: Experience of five cases and the treatment efficacy of methotrexate. Australas. J. Dermatol. 2011, 52, 279–283. [Google Scholar] [CrossRef]
- Toumi, A.; Fazal, S.; Litaiem, N. Lymphomatoid Papulosis. StatPearls Publishing. Available online: https://www.ncbi.nlm.nih.gov/books/NBK532295/ (accessed on 7 March 2022).
- Wieser, I.; Oh, C.W.; Talpur, R.; Duvic, M. Lymphomatoid papulosis: Treatment response and associated lymphomas in a study of 180 patients. J. Am. Acad. Dermatol. 2015, 74, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Moy, A.; Sun, J.; Ma, S.; Seminario-Vidal, L. Lymphomatoid Papulosis and Other Lymphoma-Like Diseases. Dermatol. Clin. 2019, 37, 471–482. [Google Scholar] [CrossRef]
- Gruber, R.; Sepp, N.T.; Fritsch, P.O.; Schmuth, M. Prognosis of Lymphomatoid Papulosis. Oncologist 2006, 11, 955–957. [Google Scholar] [CrossRef] [PubMed]
- Beljaards, R.; Willemze, R. The prognosis of patients with lymphomatoid papulosis associated with malignant lymphomas. Br. J. Dermatol. 2008, 126, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Kunishige, J.H.; McDonald, H.; Alvarez, G.; Johnson, M.; Prieto, V.; Duvic, M. Lymphomatoid papulosis and associated lymphomas: A retrospective case series of 84 patients. Clin. Exp. Dermatol. 2009, 34, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Cordel, N.; Tressières, B.; D’Incan, M.; Machet, L.; Grange, F.; Estève, É.; Dalac, S.; Ingen-Housz-Oro, S.; Bagot, M.; Beylot-Barry, M.; et al. Frequency and Risk Factors for Associated Lymphomas in Patients With Lymphomatoid Papulosis. Oncologist 2015, 21, 76–83. [Google Scholar] [CrossRef]
- de Souza, A.; El-Azhary, R.; Camilleri, M.J.; Wada, D.A.; Appert, D.L.; Gibson, L.E. In search of prognostic indicators for lymphomatoid papulosis: A retrospective study of 123 patients. J. Am. Acad. Dermatol. 2012, 66, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Miquel, J.; Fraitag, S.; Hamel-Teillac, D.; Molina, T.; Brousse, N.; De Prost, Y.; Bodemer, C. Lymphomatoid papulosis in children: A series of 25 cases. Br. J. Dermatol. 2014, 171, 1138–1146. [Google Scholar] [CrossRef]
- Hallermann, C.; Niermann, C.; Fischer, R.; Schulze, H. Survival Data for 299 Patients with Primary Cutaneous Lymphomas: A Monocentre Study. Acta Derm. Venereol. 2011, 91, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Gheucă Solovăstru, L.; Vâţă, D.; Ciobanu, D. Stătescu, L.; Rotaru, M. The importance of histopathology findings in lymphomatoid papulosis. Rom. J. Morphol. Embryol. 2014, 55, 1527–1530. [Google Scholar] [PubMed]
- Lymphomatoid Papulosis. DermNet NZ. Available online: https://dermnetnz.org/topics/lymphomatoid-papulosis (accessed on 7 March 2022).
- Kempf, W.; Kazakov, D.V.; Baumgartner, H.-P.; Kutzner, H. Follicular lymphomatoid papulosis revisited: A study of 11 cases, with new histopathological findings. J. Am. Acad. Dermatol. 2013, 68, 809–816. [Google Scholar] [CrossRef] [Green Version]
- Chimenti, S.; Fargnoli, M.C.; Pacifico, A.; Peris, K. Mucosal involvement in a patient with lymphomatoid papulosis. J. Am. Acad. Dermatol. 2001, 44, 339–341. [Google Scholar] [CrossRef]
- Pujol, R.M.; Muret, M.P.G.; Bergua, P.; Bordes, R.; Alomar, A. Oral involvement in lymphomatoid papulosis. Report of two cases and review of the literature. Dermatology 2005, 210, 53–57. [Google Scholar] [CrossRef]
- Marques-Piubelli, M.; Ferrufino-Schmidt, M.; Miranda, R. Lymphomatoid Papulosis. Available online: https://www.pathologyoutlines.com/topic/skintumornonmelanocyticlymphomatoidpapulosis.html (accessed on 7 March 2022).
- Fernández-Guarino, M.; Carrillo-Gijón, R.; Jaén-Olasolo, P. Lymphomatoid papulosis: Clinical and pathological findings in 18 patients. Actas Dermosifiliogr. 2012, 103, 388–393. [Google Scholar] [CrossRef]
- Killoran, E.; Mehta-Shah, N.; Musiek, A. Lymphomatoid Papulosis. JAMA Dermatol. 2020, 156, 360. [Google Scholar] [CrossRef]
- Schwartz, Z.; Coleman, M.; Toyohara, J.P.; Freedman, P.D.; Magro, C.M. Oral Lymphomatoid papulosis type C: A diagnostic pitfall, often confused with T-cell lymphoma. Ann. Diagn. Pathol. 2017, 31, 50–55. [Google Scholar] [CrossRef]
- Sciubba, J.; Said-Al-Naief, N.; Fantasia, J. Critical review of lymphomatoid papulosis of the oral cavity with case report. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2000, 90, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Korpusik, D.; Ruzicka, T. Klinische Verlaufsformen und Therapie der lymphomatoiden Papulose. Der Hautarzt 2007, 58, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.L.; Hoppe, R.T.; Kohler, S.; Harvell, J.D.; Reddy, S.; Kim, Y.H. Cd30+ cutaneous lymphoproliferative disorders: The stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J. Am. Acad. Dermatol. 2003, 49, 1049–1058. [Google Scholar] [CrossRef]
- Fujimura, T.; Lyu, C.; Tsuchiyama, K.; Aiba, S. CD30-Positive Angioinvasive Lymphomatoid Papulosis (Type E) Developing from Parapsoriasis en Plaque. Case Rep. Oncol. 2018, 11, 850–854. [Google Scholar] [CrossRef]
- Steinhoff, M.; Assaf, C.; Sterry, W. Persistent agmination of lymphomatoid papulosis: Not a new entity, but localized lymphomatoid papulosis. J. Am. Acad. Dermatol. 2008, 59, 164–165. [Google Scholar] [CrossRef]
- Sureda, N.; Thomas, L.; Bathelier, E.; Balme, B.; Depaepe, L.; Dalle, S. Bullous lymphomatoid papulosis. Clin. Exp. Dermatol. 2011, 36, 800–801. [Google Scholar] [CrossRef]
- Kavvalou, E.; Stefanidou, M.; Krueger-Krasagakis, S.E.; Evangelou, G.; Koumaki, D.; Marinos, L.; Tzardi, M.; Krasagakis, K. Lymphomatoid Papulosis Type A: A Case Report of the “Wait-and-See Strategy” in a 27-Year-Old Male Patient with Extensive Disease. Case Rep. Dermatol. Med. 2019, 2019, 1765210. [Google Scholar] [CrossRef]
- Willemze, R. Cutaneous T-cell lymphoma: Epidemiology, etiology, and classification. Leuk. Lymphoma 2003, 44, S49–S54. [Google Scholar] [CrossRef]
- Kempf, W.; Kadin, M.E.; Dvorak, A.M.; Lord, C.C.; Burg, G.; Letvin, N.L.; Koralnik, I.J. Endogenous retroviral elements, but not exogenous retroviruses, are detected in CD30-positive lymphoproliferative disorders of the skin. Carcinogenesis 2003, 24, 301–306. [Google Scholar] [CrossRef] [Green Version]
- Ghazawi, F.; AlGhazawi, N.; Le, M.; Netchiporouk, E.; Glassman, S.J.; Sasseville, D.; Litvinov, I.V. Environmental and Other Extrinsic Risk Factors Contributing to the Pathogenesis of Cutaneous T Cell Lymphoma (CTCL). Front. Oncol. 2019, 9, 300. [Google Scholar] [CrossRef]
- Namba, H.; Hamada, T.; Iwatsuki, K. Human T-cell leukemia virus type 1-positive lymphomatoid papulosis. Eur. J. Dermatol. 2016, 26, 194–195. [Google Scholar] [CrossRef] [PubMed]
- Sokołowska-Wojdyło, M.; Olek-Hrab, K.; Ruckemann-Dziurdzińska, K. Primary cutaneous lymphomas: Diagnosis and treatment. Adv. Dermatol. Allergol. 2015, 32, 368–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, N.J.; McQuaid, A.J.; Sobande, T.; Kissane, S.; Agius, E.; Jackson, S.E.; Salmon, M.; Falciani, F.; Yong, K.; Rustin, M.H.; et al. Different Proliferative Potential and Migratory Characteristics of Human CD4+ Regulatory T Cells That Express either CD45RA or CD45RO. J. Immunol. 2010, 184, 4317–4326. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Miyoshi, H.; Asano, N.; Yoshida, N.; Yamada, K.; Yanagida, E.; Moritsubo, M.; Nakata, M.; Umeno, T.; Suzuki, T.; et al. Human leukocyte antigen class II expression is a good prognostic factor in adult T-cell leukemia/lymphoma. Haematologica 2019, 104, 1626–1632. [Google Scholar] [CrossRef]
- Seliger, B.; Kloor, M.; Ferrone, S. HLA class II antigen-processing pathway in tumors: Molecular defects and clinical relevance. OncoImmunology 2017, 6, e1171447. [Google Scholar] [CrossRef]
- Bataller, A.; Montalban-Bravo, G.; Soltysiak, K.A.; Garcia-Manero, G. The role of TGFβ in hematopoiesis and myeloid disorders. Leukemia 2019, 33, 1076–1089. [Google Scholar] [CrossRef]
- Kadin, M.E.; Levi, E.; Kempf, W. Progression of Lymphomatoid Papulosis to Systemic Lymphoma Is Associated with Escape from Growth Inhibition by Transforming Growth Factor-β and CD30 Ligand. Ann. N. Y. Acad. Sci. 2006, 941, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Greenland, C.; Dastugue, N.; Touriol, C.; Lamant, L.; Delsol, G.; Brousset, P. Anaplastic large cell lymphoma with the t(2;5)(p23;q35) NPM/ALK chromosomal translocation and duplication of the short arm of the non-translocated chromosome 2 involving the full length of the ALK gene. J. Clin. Pathol. 2001, 54, 152–154. [Google Scholar] [CrossRef] [Green Version]
- Mathas, S.; Kreher, S.; Meaburn, K.J.; Jöhrens, K.; Lamprecht, B.; Assaf, C.; Sterry, W.; Kadin, M.E.; Daibata, M.; Joos, S.; et al. Gene deregulation and spatial genome reorganization near breakpoints prior to formation of translocations in anaplastic large cell lymphoma. Proc. Natl. Acad. Sci. USA 2009, 106, 5831–5836. [Google Scholar] [CrossRef] [Green Version]
- Kuravi, S.; Kunchala, P.; Ganguly, S.; Abhyankar, S.; Saunthararajah, Y.; Elenitoba-Johnson, K.S.J.; Roy, J.; McGuirk, J.; Balusu, R. NPM1-TYK2 Fusion Is an Oncogene and a Novel HSP90-Client Protein. Blood 2016, 128, 4127. [Google Scholar] [CrossRef]
- Velusamy, T.; Kiel, M.J.; Sahasrabuddhe, A.A.; Rolland, D.C.; Dixon, C.A.; Bailey, N.G.; Betz, B.L.; Brown, N.A.; Hristov, A.C.; Wilcox, R.A.; et al. Novel Gene Translocations Involving TYK2 in Cutaneous CD30-Positive Lymphoproliferative Disorders. Blood 2014, 124, 3032. [Google Scholar] [CrossRef]
- Badje, E.D.; Tejasvi, T.; Hristov, A. γδ lymphomatoid papulosis type D: A histologic mimic of primary cutaneous γδ T-cell lymphoma. JAAD Case Rep. 2019, 5, 264–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greisser, J.; Palmedo, G.; Sander, C.; Kutzner, H.; Kazakov, D.V.; Roos, M.; Burg, G.; Kempf, W. Detection of clonal rearrangement of T-cell receptor genes in the diagnosis of primary cutaneous CD30+ lymphoproliferative disorders. J. Cutan. Pathol. 2006, 33, 711–715. [Google Scholar] [CrossRef]
- Kempf, W.; Pfaltz, K.; Vermeer, M.H.; Cozzio, A.; Ortiz-Romero, P.L.; Bagot, M.; Olsen, E.; Kim, Y.H.; Dummer, R.; Pimpinelli, N.; et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: Lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma*. Blood 2011, 118, 4024–4035. [Google Scholar] [CrossRef] [Green Version]
- Saggini, A.; Gulia, A.; Argenyi, Z.; Fink-Puches, R.; Lissia, A.; Magaña, M.; Requena, L.; Simonitsch, I.; Cerroni, L. A Variant of Lymphomatoid Papulosis Simulating Primary Cutaneous Aggressive Epidermotropic CD8+ Cytotoxic T-cell Lymphoma. Description of 9 Cases. Am. J. Surg. Pathol. 2010, 34, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Kempf, W. A new era for cutaneous CD30-positive T-cell lymphoproliferative disorders. Semin. Diagn. Pathol. 2017, 34, 22–35. [Google Scholar] [CrossRef]
- Kempf, W.; Kazakov, D.V.; Schärer, L.; Rütten, A.; Mentzel, T.; Paredes, B.E.; Palmedo, G.; Panizzon, R.G.; Kutzner, H. Angioinvasive lymphomatoid papulosis: A new variant simulating aggressive lymphomas. Am. J. Surg. Pathol. 2013, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kartan, S.; Johnson, W.T.; Sokol, K.; Alpdogan, O.; Gru, A.A.; Nikbakht, N.; Porcu, P. The spectrum of CD30+ T cell lymphoproliferative disorders in the skin. Chin. Clin. Oncol. 2019, 8, 3. [Google Scholar] [CrossRef]
- El Shabrawi-Caelen, L.; Kerl, H.; Cerroni, L. Lymphomatoid papulosis: Reappraisal of clinicopathologic presentation and classification into subtypes A, B, and C. Arch. Dermatol. 2004, 140, 441–447. [Google Scholar] [CrossRef]
- Guitart, J.; Querfeld, C. Cutaneous CD30 lymphoproliferative disorders and similar conditions: A clinical and pathologic prospective on a complex issue. Semin. Diagn. Pathol. 2009, 26, 131–140. [Google Scholar] [CrossRef]
- Goodlad, J.R. The many faces of lymphomatoid papulosis. Diagn. Histopathol. 2014, 20, 263–270. [Google Scholar] [CrossRef]
- El-Sayed, A.M.; El-Borai, M.H.; Bahnassy, A.A.; El-Gerzawi, S.M. Flow cytometric immunophenotyping (FCI) of lymphoma: Correlation with histopathology and immunohistochemistry. Diagn. Pathol. 2008, 3, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, T.; Dewan, K.; Mann, N. Comparing flow cytometry immunophenotypic and immunohistochemical analyses in diagnosis and prognosis of chronic lymphoproliferative disorders: Experience from a Tertiary Care Center. Clin. Cancer Investig. J. 2015, 4, 707. [Google Scholar] [CrossRef]
- Willemze, R.; Jaffe, E.S.; Burg, G.; Cerroni, L.; Berti, E.; Swerdlow, S.H.; Ralfkiaer, E.; Chimenti, S.; Diaz-Perez, J.L.; Duncan, L.M.; et al. WHO-EORTC Classification for Cutaneous Lymphomas. Blood 2005, 105, 3768–3785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ba, W.; Yin, G.; Yang, J.; Zhang, Z.; Wang, W.; Zhao, Z.; Chen, H.; Li, C. Lymphomatoid papulosis type E with a CD56+ immunophenotype presenting with purpura-like lesions. J. Cutan. Pathol. 2019, 46, 542–545. [Google Scholar] [CrossRef]
- Fletcher, C.D.M. Diagnostic Histopathology of Tumors, 5th ed.; Elsevier Health Sciences: Amsterdam, The Netherlands, 2019; Volume 1, xiii, 2310p. [Google Scholar]
- Stonestrom, A.J. Function of Bromodomain and Extra-Terminal Motif Proteins (Bets) in Gata1-Mediated Transcription; University of Pennsylvania: Philadelphia, PA, USA, 2015. [Google Scholar]
- Medeiros, L.J.; Miranda, R.N. (Eds.) Lymphomatoid Papulosis. In Diagnostic Pathology: Lymph Nodes and Extranodal Lymphomas, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 722–727. [Google Scholar]
- McQuitty, E.; Curry, J.L.; Tetzlaff, M.T.; Prieto, V.G.; Duvic, M.; Torres-Cabala, C. The differential diagnosis of CD8-positive (“type D”) lymphomatoid papulosis. J. Cutan. Pathol. 2013, 41, 88–100. [Google Scholar] [CrossRef]
- Magro, C.M.; Morrison, C.; Merati, K.; Porcu, P.; Wright, E.D.; Crowson, A.N. CD8+ Lymphomatoid Papulosis and Its Differential Diagnosis. Am. J. Clin. Pathol. 2006, 125, 490–501. [Google Scholar] [CrossRef]
- Bertolotti, A.; Pham-Ledard, A.-L.; Vergier, B.; Parrens, M.; Bedane, C.; Beylot-Barry, M. Lymphomatoid papulosis type D: An aggressive histology for an indolent disease. Br. J. Dermatol. 2013, 169, 1157–1159. [Google Scholar] [CrossRef]
- Temel, A.B.; Unal, B.; Şanlı, H.E.; Duygulu, Ş.; Uzun, S. A Severe Case of Lymphomatoid Papulosis Type E Successfully Treated with Interferon-Alfa 2a. Case Rep. Dermatol. Med. 2017, 2017, 3194738. [Google Scholar] [CrossRef]
- Lee, G.H.; Bae, G.H.; Rieger, K.E.; Kim, Y.H.; Chiou, A.S. Angiodestructive lymphomatoid papulosis lasting more than 45 years. JAAD Case Rep. 2019, 5, 767–769. [Google Scholar] [CrossRef]
- Wu, W.-M.; Tsai, H.-J. Lymphomatoid Papulosis Histopathologically Simulating Angiocentric and Cytotoxic T-cell Lymphoma. Am. J. Dermatopathol. 2004, 26, 133–135. [Google Scholar] [CrossRef]
- Massone, C.; Chott, A.; Metze, D.; Kerl, K.; Citarella, L.; Vale, E.; Kerl, H.; Cerroni, L. Subcutaneous, blastic natural killer (NK), NK/T-cell, and other cytotoxic lymphomas of the skin: A morphologic, immuno-phenotypic, and molecular study of 50 patients. Am. J. Surg. Pathol. 2004, 28, 719–735. [Google Scholar] [CrossRef]
- Tao, J.; Shelat, S.G.; Jaffe, E.S.; Bagg, A. Aggressive Epstein-Barr Virus-Associated, CD8+, CD30+, CD56+, Surface CD3−, Natural Killer (NK)-Like Cytotoxic T-Cell Lymphoma. Am. J. Surg. Pathol. 2002, 26, 111–118. [Google Scholar] [CrossRef]
- Agnarsson, B.A.; Vonderheid, E.C.; Kadin, M.E. Cutaneous T cell lymphoma with suppressor/cytotoxic (CD8) phenotype: Identification of rapidly progressive and chronic subtypes. J. Am. Acad. Dermatol. 1990, 22, 569–577. [Google Scholar] [CrossRef]
- Swamy, M.R.; Pollock, S.; Goldberg, L.J.; Shen, L. A case of lymphomatoid papulosis type E in a young adult: An uncommon entity. J. Cutan. Pathol. 2021, 48, 694–700. [Google Scholar] [CrossRef]
- Georgesen, C.; Magro, C. Lymphomatoid papulosis in children and adolescents: A clinical and histopathologic retrospective cohort. Ann. Diagn. Pathol. 2020, 46, 151486. [Google Scholar] [CrossRef]
- Karai, L.J.; Kadin, M.E.; Hsi, E.D.; Sluzevich, J.C.; Ketterling, R.P.; Knudson, R.A.; Feldman, A.L. Chromosomal Rearrangements of 6p25.3 Define a New Subtype of Lymphomatoid Papulosis. Am. J. Surg. Pathol. 2013, 37, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Onaindia, A.; Montes-Moreno, S.; Rodríguez-Pinilla, S.M.; Batlle, A.; De Villambrosía, S.G.; Rodríguez, A.M.; Alegre, V.; Bermúdez, G.M.; González-Vela, C.; Piris, M.A. Primary cutaneous anaplastic large cell lymphomas with 6p25.3 rearrangement exhibit particular histological features. Histopathology 2014, 66, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-H.; Mao, Y.-N.; Jhuang, J.-Y.; Chen, B.-J. Lymphomatoid papulosis with DUSP22-IRF4 rearrangement on 6p25-3: A case report. Dermatol. Sin. 2021, 39, 212. [Google Scholar] [CrossRef]
- Ba, W.; Yang, Y.; Zhang, Z.; Wei, X.; Wang, W.; Zhao, Z.; Zheng, L.; Li, C. Lymphomatoid papulosis with folliculotropism, eccrinotropism and neurotropism. J. Cutan. Pathol. 2018, 45, 530–534. [Google Scholar] [CrossRef]
- Kempf, W. CD30+ lymphoproliferative disorders: Histopathology, differential diagnosis, new variants, and simulators. J. Cutan. Pathol. 2006, 33, 58–70. [Google Scholar] [CrossRef]
- Xue, D.; Li, X.; Ren, Y.; Liu, Q.; Yen, Y.; Xue, L. Primary Cutaneous Anaplastic Large Cell Lymphoma With Positive ALK Expression and a Rapidly Progressive Cutaneous Nodule. Int. J. Surg. Pathol. 2015, 23, 333–335. [Google Scholar] [CrossRef]
- Sasaki, K.; Sugaya, M.; Fujita, H.; Takeuchi, K.; Torii, H.; Asahina, A.; Tamaki, K. A case of primary cutaneous anaplastic large cell lymphoma with variant anaplastic lymphoma kinase translocation. Br. J. Dermatol. 2004, 150, 1202–1207. [Google Scholar] [CrossRef]
- Pulitzer, M.; Ogunrinade, O.; Lin, O.; Steinherz, P. ALK -positive (2p23 rearranged) anaplastic large cell lymphoma with localization to the skin in a pediatric patient. J. Cutan. Pathol. 2014, 42, 182–187. [Google Scholar] [CrossRef] [Green Version]
- VisualDx-Lymphomatoid Papulosis. Available online: https://www.visualdx.com/visualdx/diagnosis/lymphomatoid+papulosis?diagnosisId=51912&moduleId=101 (accessed on 4 November 2022).
- Humme, D.; Lukowsky, A.; Steinhoff, M.; Beyer, M.; Walden, P.; Sterry, W.; Assaf, C. Dominance of Nonmalignant T-Cell Clones and Distortion of the TCR Repertoire in the Peripheral Blood of Patients with Cutaneous CD30+ Lymphoproliferative Disorders. J. Investig. Dermatol. 2009, 129, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo, M.M.D.L.G.; Patel, K.P.; Loghavi, S.; Curry, J.L.; Cabala, C.A.T.; Cason, R.C.; Gangar, P.; Prieto, V.G.; Medeiros, L.J.; Duvic, M.; et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin. Hum. Pathol. 2015, 46, 558–569. [Google Scholar] [CrossRef]
- Durer, C.; Babiker, H.M. Adult T Cell Leukemia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Molgó, M.; Espinoza-Benavides, L.; Rojas, P.; González, S. Mycosis Fungoides, Lymphomatoid Papulosis and Hodgkin’s Lymphoma in the Same Patient: Apropos of a Possible Monoclonal Origin. Indian J. Dermatol. 2020, 65, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Kempf, W.; Kazakov, D.V.; Palmedo, G.; Fraitag, S.; Schaerer, L.; Kutzner, H. Pityriasis Lichenoides et Varioliformis Acuta with Numerous CD30(+) Cells: A Variant Mimicking Lymphomatoid Papulo-sis and Other Cutaneous Lymphomas. A Clinicopathologic, Immunohistochemical, and Molecular Biological Study of 13 Cases. Am. J. Surg. Pathol. 2012, 36, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Walton, S.F.; Oprescu, F.I. Immunology of scabies and translational outcomes: Identifying the Missing Links. Curr. Opin. Infect. Dis. 2013, 26, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Werner, B.; Massone, C.; Kerl, H.; Cerroni, L. Large CD30-positive cells in benign, atypical lymphoid infiltrates of the skin. J. Cutan. Pathol. 2008, 35, 1100–1107. [Google Scholar] [CrossRef]
- Jung, J.; Levin, E.C.; Jarrett, R.; Lu, D.; Mann, C. Lymphomatoid Drug Reaction to Ustekinumab. Arch. Dermatol. 2011, 147, 992–993. [Google Scholar] [CrossRef]
- Saeed, S.A.M.; Bazza, M.; Zaman, M.; Ryatt, K.S. Cefuroxime induced lymphomatoid hypersensitivity reaction. Postgrad. Med. J. 2000, 76, 577–579. [Google Scholar] [CrossRef] [Green Version]
- Leinweber, B.; Kerl, H.; Cerroni, L. Histopathologic Features of Cutaneous Herpes Virus Infections (Herpes Simplex, Herpes Varicella/Zoster): A Broad Spectrum of Presentations with Common Pseudolymphomatous Aspects. Am. J. Surg. Pathol. 2006, 30, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Kadin, M.E. Current management of primary cutaneous CD30+ T-cell lymphoproliferative disorders. Oncology 2009, 23, 1158–1164. [Google Scholar] [PubMed]
- Fernández-De-Misa, R.; Hernández-Machín, B.; Servitje, O.; Valentí-Medina, F.; Maroñas-Jiménez, L.; Ortiz-Romero, P.L.; Schmidt, J.S.; Pujol, R.M.; Gallardo, F.; Pau-Charles, I.; et al. First-line treatment in lymphomatoid papulosis: A retrospective multicentre study. Clin. Exp. Dermatol. 2017, 43, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollina, U. Lymphomatoid papulosis treated with extracorporeal photochemotherapy. Oncol. Rep. 1998, 5, 57–66. [Google Scholar] [CrossRef]
- Hearn, R.; Kerr, A.; Rahim, K.; Ferguson, J.; Dawe, R. Incidence of skin cancers in 3867 patients treated with narrow-band ultraviolet B phototherapy. Br. J. Dermatol. 2008, 159, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Calzavara-Pinton, P.; Venturini, M.; Sala, R. Medium-dose UVA1 therapy of lymphomatoid papulosis. J. Am. Acad. Dermatol. 2005, 52, 530–532. [Google Scholar] [CrossRef]
- Sina, B. Lymphomatoid Papulosis: Case Reports and Literature Review. Arch. Dermatol. 1983, 119, 189–197. [Google Scholar] [CrossRef]
- Coelho, M.M.V.; Apetato, M. The dark side of the light: Phototherapy adverse effects. Clin. Dermatol. 2016, 34, 556–562. [Google Scholar] [CrossRef]
- Arimatsu, A.; Tomii, K.; Fujiwara, H.; Hasegawa, G.; Shigehara, Y.; Tachibana, T. Photodynamic therapy can prevent recurrence of lymphomatoid papulosis. Photodiagnosis Photodyn. Ther. 2019, 25, 334–335. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, J.S.; Jaworsky, C. Topical methotrexate for lymphomatoid papulosis. J. Am. Acad. Dermatol. 2003, 49, 937–939. [Google Scholar] [CrossRef]
- Romero-Maté, A.; Martín-Fragueiro, L.; Miñano-Medrano, R.; Martinez-Morán, C.; Arias-Palomo, D.; Borbujo, J. Persistent agmination of lymphomatoid papulosis evolving to classical lesions of lymphomatoid papulosis. J. Am. Acad. Dermatol. 2009, 61, 1087–1088. [Google Scholar] [CrossRef]
- Bruijn, M.S.; Horváth, B.; Vader, P.C.V.V.; Willemze, R.; Vermeer, M. Recommendations for treatment of lymphomatoid papulosis with methotrexate: A report from the Dutch Cutaneous Lymphoma Group. Br. J. Dermatol. 2015, 173, 1319–1322. [Google Scholar] [CrossRef]
- Vonderheid, E.C.; Sajjadian, A.; Kadin, M.E. Methotrexate is effective therapy for lymphomatoid papulosis and other primary cutaneous CD30-positive lymphoproliferative disorders. J. Am. Acad. Dermatol. 1996, 34, 470–481. [Google Scholar] [CrossRef]
- Newland, K.M.; McCormack, C.J.; Twigger, R.; Buelens, O.; Hughes, C.F.; Lade, S.; Dickinson, M.; Yap, L.M.; Ryan, G.; Prince, H.M. The efficacy of methotrexate for lymphomatoid papulosis. J. Am. Acad. Dermatol. 2015, 72, 1088–1090. [Google Scholar] [CrossRef]
- Shen, S.; O’Brien, T.; Yap, L.M.; Prince, H.M.; McCormack, C.J. The use of methotrexate in dermatology: A review. Australas. J. Dermatol. 2011, 53, 1–18. [Google Scholar] [CrossRef]
- Bangert, C.A.; Costner, M.I. Methotrexate in dermatology. Dermatol. Ther. 2007, 20, 216–228. [Google Scholar] [CrossRef]
- Fujita, H.; Nagatani, T.; Miyazawa, M.; Wada, H.; Koiwa, K.; Komatsu, H.; Ikezawa, Z. Primary cutaneous anaplastic large cell lymphoma successfully treated with low-dose oral methotrexate. Eur. J. Dermatol. 2008, 18, 360–361. [Google Scholar] [CrossRef]
- Assaf, C.; Hirsch, B.; Wagner, F.; Lucka, L.; Grünbaum, M.; Gellrich, S.; Lukowsky, A.; Sterry, W.; Stein, H.; Dürkop, H. Differential Expression of TRAF1 Aids in the Distinction of Cutaneous CD30-Positive Lymphoproliferations. J. Investig. Dermatol. 2007, 127, 1898–1904. [Google Scholar] [CrossRef]
- Krathen, R.A.; Ward, S.; Duvic, M. Bexarotene Is a New Treatment Option for Lymphomatoid Papulosis. Dermatology 2003, 206, 142–147. [Google Scholar] [CrossRef]
- Hughes, P.S. Treatment of lymphomatoid papulosis with imiquimod 5% cream. J. Am. Acad. Dermatol. 2006, 54, 546–547. [Google Scholar] [CrossRef] [PubMed]
- Wyss, M.; Dummer, R.; Dommann, S.; Joller-Jemelka, H.; Dours-Zimmermann, M.; Gilliet, F.; Burg, G. Lymphomatoid Papulosis–Treatment with Recombinant Interferon Alfa-2a and Etretinate. Dermatology 1995, 190, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; McCormack, C.; Yap, L.-M.; Prince, H.M.; Roberts, H.; Williams, R.; Foley, P. Successful treatment of lymphomatoid papulosis with photodynamic therapy. Australas. J. Dermatol. 2009, 50, 129–132. [Google Scholar] [CrossRef]
- Duvic, M.; Reddy, S.A.; Pinter-Brown, L.; Korman, N.J.; Zic, J.; Kennedy, D.A.; Lorenz, J.; Sievers, E.L.; Kim, Y.H. A Phase II Study of SGN-30 in Cutaneous Anaplastic Large Cell Lymphoma and Related Lymphoproliferative Disorders. Clin. Cancer Res. 2009, 15, 6217–6224. [Google Scholar] [CrossRef] [Green Version]
- Yagi, H.; Tokura, Y.; Furukawa, F.; Takigawa, M. Th2 Cytokine mRNA Expression in Primary Cutaneous CD30-Positive Lymphoproliferative Disorders: Successful Treatment With Recombinant Interferon-γ. J. Investig. Dermatol. 1996, 107, 827–832. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Cluster of Differentiation | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| CD30 | CD2 | CD3 | CD4 | CD5 | CD7 | CD8 | TIA1 | References | |
| Type A LyP | + | +/− | + | + | +/− | +/− | − | + | [11] |
| Type B LyP | +/− | +/− | + | + | +/− | − | − | + | [11] |
| Type C LyP | + | +/− | + | + | +/− | +/− | − | + | [11] |
| Type D LyP | + | + | + | − | − | − | + | + | [63] |
| Type E LyP | + | + | + | +/− | + | +/− | + | + | [63] |
| Type with rearrangement 6p25.3 | + | +/− | + | + | +/− | +/− | +/− | − | [63] |
| Differential Diagnosis | ||||
|---|---|---|---|---|
| Lymphomatoid Papulosis | Main Differential Diagnosis | Similarities | Differences | Reference |
| Each subtype | Primary cutaneous ALCL |
|
| [75,90] |
| Type A | Classic Hodgkin’s lymphoma, Primary cutaneous ALCL, tumor stage MF |
|
| [75,90,91] |
| Type B | Plaque stage MF |
| - | [75] |
| Type C | Primary cutaneous ALCL, transformed MF |
|
| [65,75] |
| Type D | PCAETL resembling pagetoid reticulosis |
|
| [61,76] |
| Type E | NK/T-cell lymphoma nasal type, extranodal CGD-TCL ALCL/borderline CD30+ cutaneous LPD |
|
| [70,92,93,94,95,96,97] |
| Type with 6p25.3 rearrangement | ALCL |
| - | [61,77] |
| The Authors of the Study | Description of the Research and Its Results | Reference |
|---|---|---|
| Vonderheid et al. |
| [118] |
| Bruijn et al. |
| [117] |
| Newland et al. |
| [119] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nowicka, D.; Mertowska, P.; Mertowski, S.; Hymos, A.; Forma, A.; Michalski, A.; Morawska, I.; Hrynkiewicz, R.; Niedźwiedzka-Rystwej, P.; Grywalska, E. Etiopathogenesis, Diagnosis, and Treatment Strategies for Lymphomatoid Papulosis with Particular Emphasis on the Role of the Immune System. Cells 2022, 11, 3697. https://doi.org/10.3390/cells11223697
Nowicka D, Mertowska P, Mertowski S, Hymos A, Forma A, Michalski A, Morawska I, Hrynkiewicz R, Niedźwiedzka-Rystwej P, Grywalska E. Etiopathogenesis, Diagnosis, and Treatment Strategies for Lymphomatoid Papulosis with Particular Emphasis on the Role of the Immune System. Cells. 2022; 11(22):3697. https://doi.org/10.3390/cells11223697
Chicago/Turabian StyleNowicka, Danuta, Paulina Mertowska, Sebastian Mertowski, Anna Hymos, Alicja Forma, Adam Michalski, Izabela Morawska, Rafał Hrynkiewicz, Paulina Niedźwiedzka-Rystwej, and Ewelina Grywalska. 2022. "Etiopathogenesis, Diagnosis, and Treatment Strategies for Lymphomatoid Papulosis with Particular Emphasis on the Role of the Immune System" Cells 11, no. 22: 3697. https://doi.org/10.3390/cells11223697