
Cognitive Decline and BPSD Are Concomitant with Autophagic and Synaptic Deficits Associated with G9a Alterations in Aged SAMP8 Mice

 , , ,
, , ,  ,
,  , and
, and
Abstract
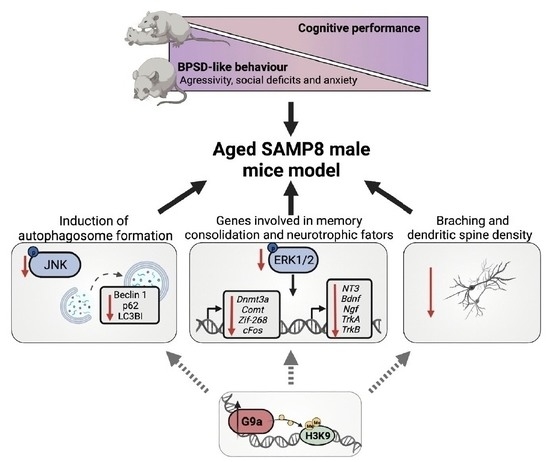
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Behavioural Experiments
2.2.1. Elevated Plus Maze (EPM)
2.2.2. Open Field Test (OF)
2.2.3. Novel Object-Recognition Test (NORT)
2.2.4. Three-Chamber Test (TCT)
2.2.5. Resident-Intruder Test (RI)
2.3. RNA Extraction and Gene Expression Determination
2.4. Western Blot (WB)
2.5. Dendritic Length, Spine Density and Golgi Stain Protocol
2.6. H3 Modification Patterns Quantification
2.7. Statistical Analysis
3. Results
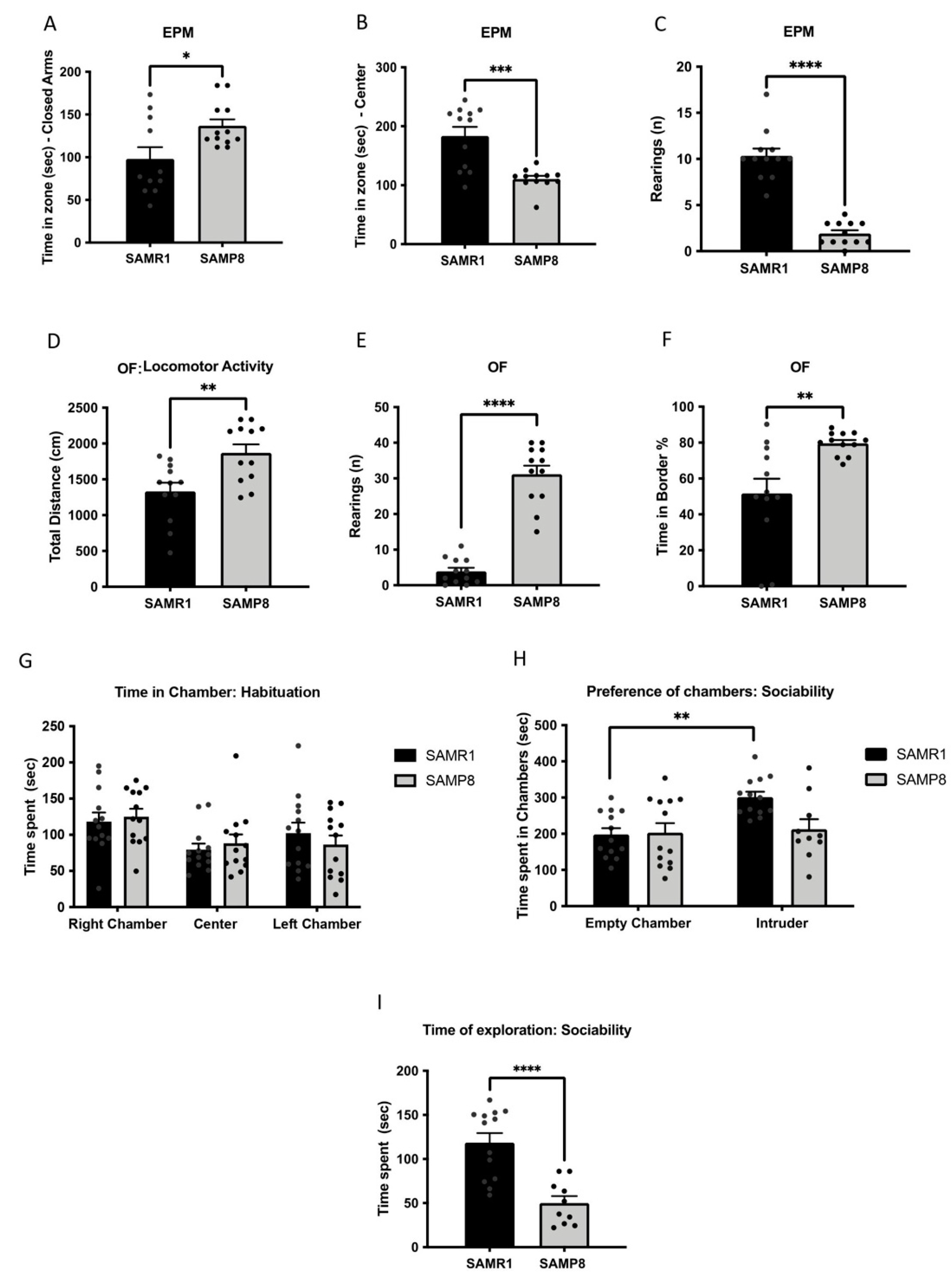
3.1. Aged SAMP8 Male Mice Exhibited Increased Aggressive and Anxiety-like Behaviours and Social Deficits
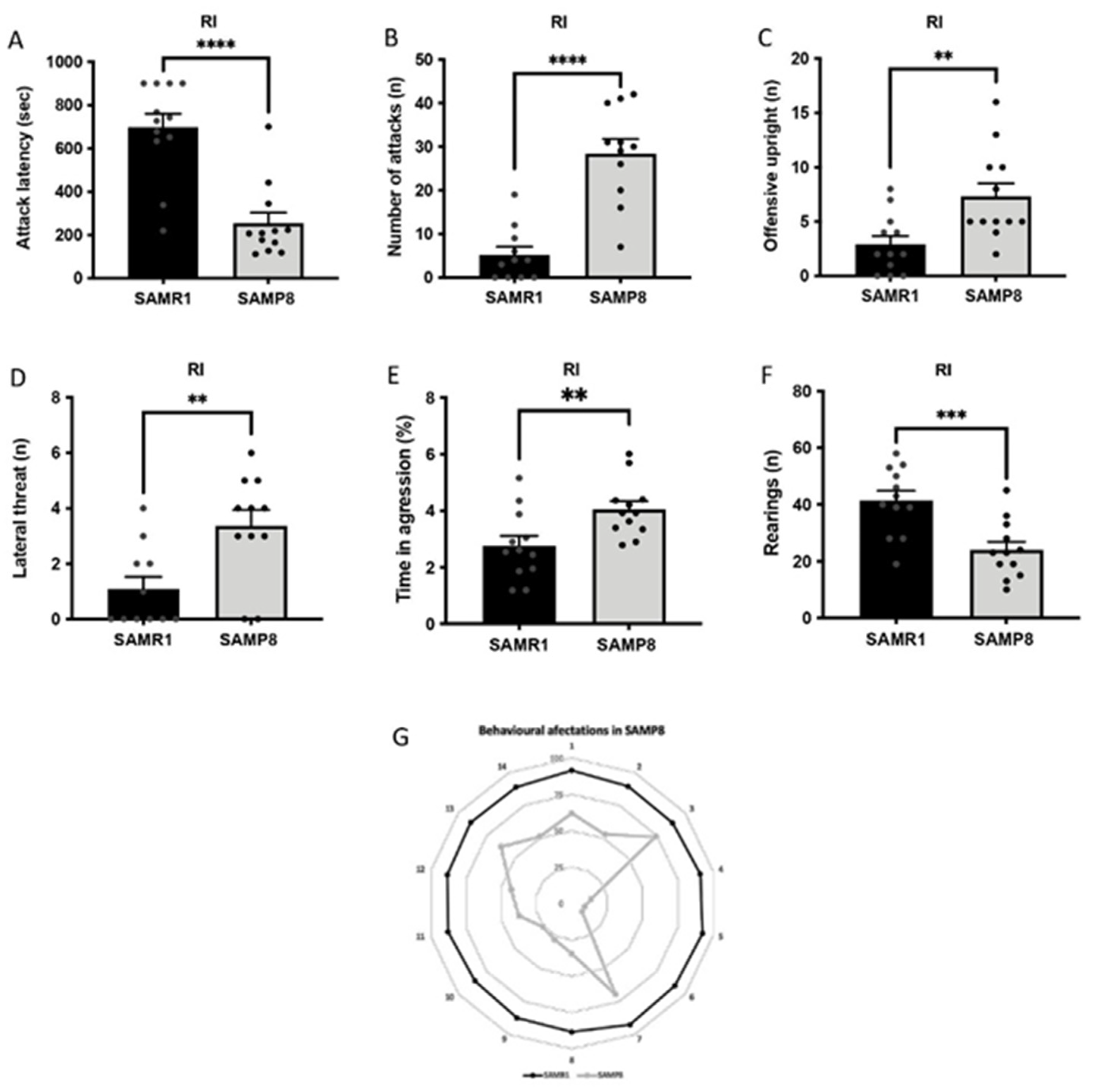
3.2. Aged SAMP8 Male Mice Present Increased Aggressive Behaviour
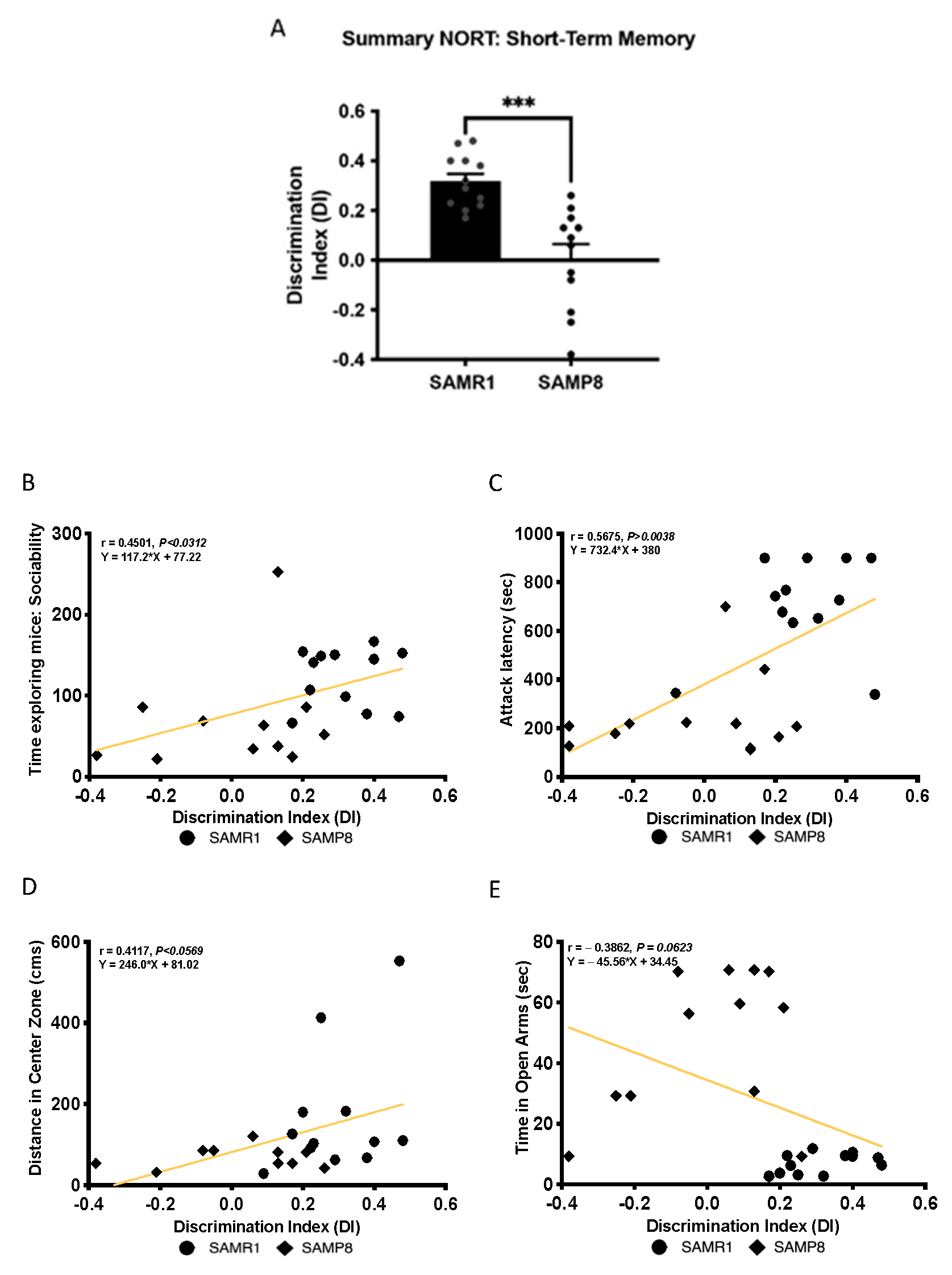
3.3. Cognitive Deficits Correlated with Behavioural Parameters in Aged SAMP8 Male Mice
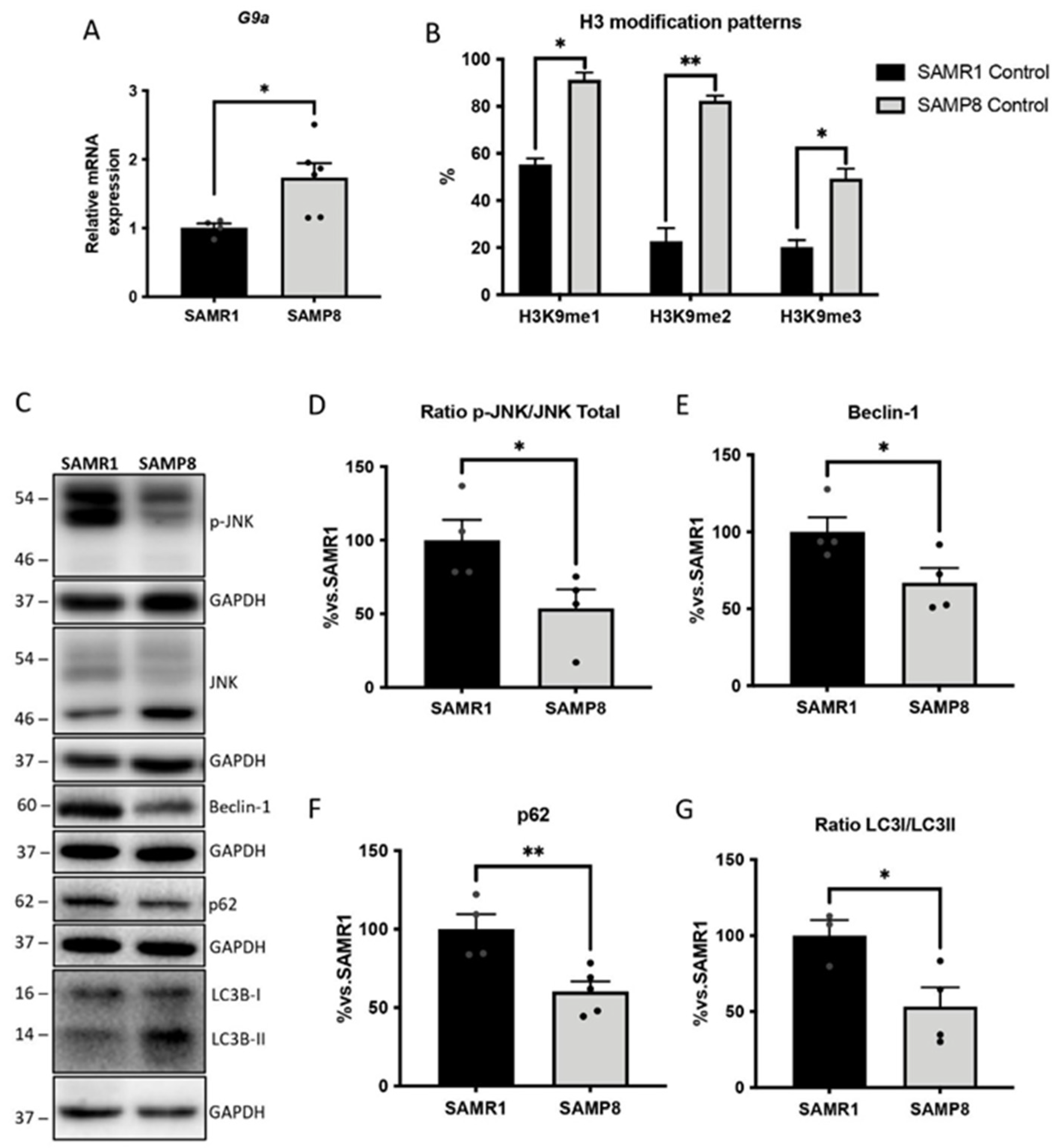
3.4. Upregulation of G9a Correlated with the Suppression of the JNK-Mediated Autophagy Process in Aged SAMP8 Mice
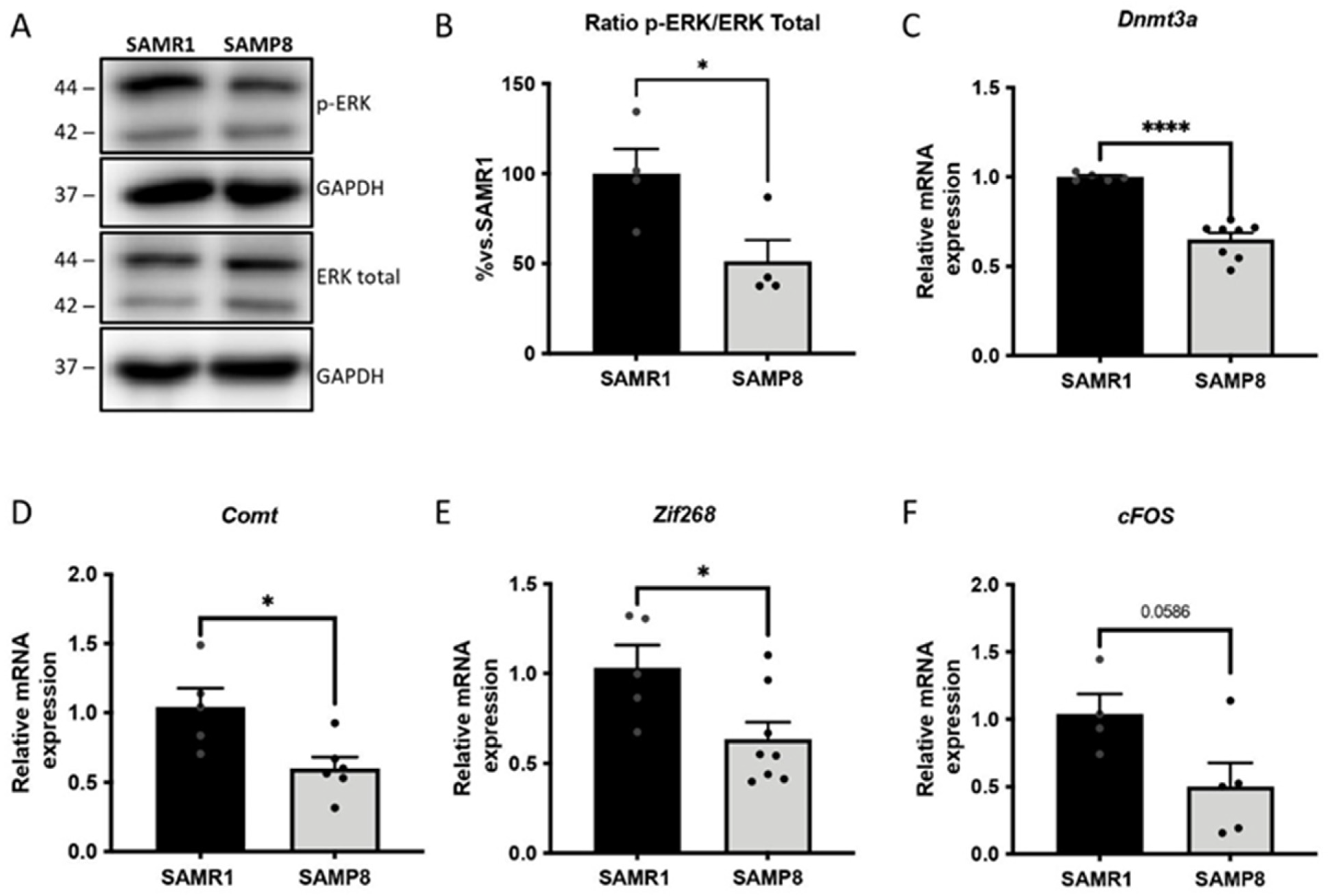
3.5. ERK Activity and Its Downstream Gene Targets in Memory Consolidation Are Downregulated in Aged SAMP8 Mice
3.6. Dendritic Morphological Abnormalities Accompanied Reduced Expression of Synaptic Plasticity-Related Genes in Aged SAMP8 Male Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Shinosaki, K.; Nishikawa, T.; Takeda, M. Neurobiological basis of behavioral and psychological symptoms in dementia of the Alzheimer type. Psychiatry Clin. Neurosci. 2000, 54, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.; Gobartt, A.L.; Balañá, M. Behavioural symptoms in patients with Alzheimer’s disease and their association with cognitive impairment. BMC Neurol. 2010, 10, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidzan, L.; Bidzan, M.; Pąchalska, M. Aggressive and impulsive behavior in Alzheimer’s disease and progression of dementia. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2012, 18, CR182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, J.-S.; Xie, H.; Ding, X. The role of epigenetic regulation in learning and memory. Exp. Neurol. 2015, 268, 30–36. [Google Scholar] [CrossRef]
- Jarome, T.J.; Lubin, F.D. Epigenetic mechanisms of memory formation and reconsolidation. Neurobiol. Learn. Mem. 2014, 115, 116–127. [Google Scholar] [CrossRef] [Green Version]
- Day, J.J.; Sweatt, J.D. Epigenetic mechanisms in cognition. Neuron 2011, 70, 813–829. [Google Scholar] [CrossRef] [Green Version]
- Morley, J.E.; Farr, S.A.; Kumar, V.B.; Armbrecht, H.J. The SAMP8 mouse: A model to develop therapeutic interventions for Alzheimer’s disease. Curr. Pharm. Des. 2012, 18, 1123–1130. [Google Scholar] [CrossRef]
- Griñán-Ferré, C.; Corpas, R.; Puigoriol-Illamola, D.; Palomera-Ávalos, V.; Sanfeliu, C.; Pallàs, M. Understanding epigenetics in the neurodegeneration of Alzheimer’s disease: SAMP8 mouse model. J. Alzheimer’s Dis. 2018, 62, 943–963. [Google Scholar] [CrossRef] [Green Version]
- Akiguchi, I.; Pallàs, M.; Budka, H.; Akiyama, H.; Ueno, M.; Han, J.; Yagi, H.; Nishikawa, T.; Chiba, Y.; Sugiyama, H. SAMP8 mice as a neuropathological model of accelerated brain aging and dementia: Toshio Takeda’s legacy and future directions. Neuropathology 2017, 37, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Pallàs, M. Senescence-Accelerated Mice P8: A Tool to Study Brain Aging and Alzheimer’s Disease in a Mouse Model. ISRN Cell Biol. 2012, 2012, 917167. [Google Scholar] [CrossRef] [Green Version]
- Puigoriol-Illamola, D.; Martínez-Damas, M.; Griñán-Ferré, C.; Pallàs, M. Chronic Mild Stress Modified Epigenetic Mechanisms Leading to Accelerated Senescence and Impaired Cognitive Performance in Mice. Int. J. Mol. Sci. 2020, 21, 1154. [Google Scholar] [CrossRef] [Green Version]
- Griñán-Ferré, C.; Sarroca, S.; Ivanova, A.; Puigoriol-Illamola, D.; Aguado, F.; Camins, A.; Sanfeliu, C.; Pallàs, M. Epigenetic mechanisms underlying cognitive impairment and Alzheimer disease hallmarks in 5XFAD mice. Aging 2016, 8, 664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griñan-Ferré, C.; Puigoriol-Illamola, D.; Palomera-Ávalos, V.; Pérez-Cáceres, D.; Companys-Alemany, J.; Camins, A.; Ortuño-Sahagún, D.; Rodrigo, M.T.; Pallàs, M. Environmental enrichment modified epigenetic mechanisms in SAMP8 mouse hippocampus by reducing oxidative stress and inflammaging and achieving neuroprotection. Front. Aging Neurosci. 2016, 8, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosín-Tomás, M.; Alvarez-López, M.J.; Sanchez-Roige, S.; Lalanza, J.F.; Bayod, S.; Sanfeliu, C.; Pallàs, M.; Escorihuela, R.M.; Kaliman, P. Epigenetic alterations in hippocampus of SAMP8 senescent mice and modulation by voluntary physical exercise. Front. Aging Neurosci. 2014, 6, 51. [Google Scholar] [PubMed]
- Maze, I.; Noh, K.-M.; Allis, C.D. Histone regulation in the CNS: Basic principles of epigenetic plasticity. Neuropsychopharmacology 2013, 38, 3–22. [Google Scholar] [CrossRef]
- Griñán-Ferré, C.; Marsal-García, L.; Bellver-Sanchis, A.; Kondengaden, S.M.; Turga, R.C.; Vázquez, S.; Pallàs, M. Pharmacological inhibition of G9a/GLP restores cognition and reduces oxidative stress, neuroinflammation and ß-Amyloid plaques in an early-onset Alzheimer’s disease mouse model. Aging 2019, 11, 11591–11608. [Google Scholar] [CrossRef]
- Sharma, M.; Dierkes, T.; Sajikumar, S. Epigenetic regulation by G9a/GLP complex ameliorates amyloid-beta 1-42 induced deficits in long-term plasticity and synaptic tagging/capture in hippocampal pyramidal neurons. Aging Cell 2017, 16, 1062–1072. [Google Scholar] [CrossRef] [Green Version]
- Artal-Martinez de Narvajas, A.; Gomez, T.S.; Zhang, J.-S.; Mann, A.O.; Taoda, Y.; Gorman, J.A.; Herreros-Villanueva, M.; Gress, T.M.; Ellenrieder, V.; Bujanda, L. Epigenetic regulation of autophagy by the methyltransferase G9a. Mol. Cell. Biol. 2013, 33, 3983–3993. [Google Scholar] [CrossRef] [Green Version]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef] [Green Version]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive Involvement of Autophagy in Alzheimer Disease: An Immuno-Electron Microscopy Study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Glatigny, M.; Moriceau, S.; Rivagorda, M.; Ramos-Brossier, M.; Nascimbeni, A.C.; Lante, F.; Shanley, M.R.; Boudarene, N.; Rousseaud, A.; Friedman, A.K. Autophagy is required for memory formation and reverses age-related memory decline. Curr. Biol. 2019, 29, 435–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.-Y.; Yan, J.; Zukin, R.S. Autophagy and synaptic plasticity: Epigenetic regulation. Curr. Opin. Neurobiol. 2019, 59, 207–212. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Sidiropoulou, K.; Kallergi, E.; Dalezios, Y.; Tavernarakis, N. Modulation of autophagy by BDNF underlies synaptic plasticity. Cell Metab. 2017, 26, 230–242. [Google Scholar] [CrossRef] [Green Version]
- Kashyap, G.; Bapat, D.; Das, D.; Gowaikar, R.; Amritkar, R.E.; Rangarajan, G.; Ravindranath, V.; Ambika, G. Synapse loss and progress of Alzheimer’s disease-A network model. Sci. Rep. 2019, 9, 6555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karege, F.; Perret, G.; Bondolfi, G.; Schwald, M.; Bertschy, G.; Aubry, J.-M. Decreased serum brain-derived neurotrophic factor levels in major depressed patients. Psychiatry Res. 2002, 109, 143–148. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Prim. 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Vasilopoulou, F.; Bagan, A.; Rodriguez-Arevalo, S.; Escolano, C.; Griñán-Ferré, C.; Pallàs, M. Amelioration of BPSD-like Phenotype and Cognitive Decline in SAMP8 Mice Model Accompanied by Molecular Changes after Treatment with I2-Imidazoline Receptor Ligand MCR5. Pharmaceutics 2020, 12, 475. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cáceres, D.; Rodrigo, M.T.; Pubill, D.; Camins, A.; Camarasa, J.; Escubedo, E.; Pallàs, M. Depression-like behavior is dependent on age in male SAMP8 mice. Biogerontology 2013, 14, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, M. Characteristics of age-related behavioral changes in senescence-accelerated mouse SAMP8 and SAMP10. Exp. Gerontol. 1997, 32, 139–148. [Google Scholar] [CrossRef]
- Griñan-Ferré, C.; Palomera-Ávalos, V.; Puigoriol-Illamola, D.; Camins, A.; Porquet, D.; Plá, V.; Aguado, F.; Pallàs, M. Behaviour and cognitive changes correlated with hippocampal neuroinflammaging and neuronal markers in female SAMP8, a model of accelerated senescence. Exp. Gerontol. 2016, 80, 57–69. [Google Scholar] [CrossRef] [Green Version]
- Griñán-Ferré, C.; Codony, S.; Pujol, E.; Yang, J.; Leiva, R.; Escolano, C.; Puigoriol-Illamola, D.; Companys-Alemany, J.; Corpas, R.; Sanfeliu, C. Pharmacological inhibition of soluble epoxide hydrolase as a new therapy for Alzheimer’s disease. Neurotherapeutics 2020, 17, 1825–1835. [Google Scholar] [CrossRef]
- Companys-Alemany, J.; Turcu, A.L.; Bellver-Sanchis, A.; Loza, M.I.; Brea, J.M.; Canudas, A.M.; Leiva, R.; Vázquez, S.; Pallàs, M.; Griñán-Ferré, C. A novel NMDA receptor antagonist protects against cognitive decline presented by senescent mice. Pharmaceutics 2020, 12, 284. [Google Scholar] [CrossRef] [Green Version]
- Maes, T.; Mascaró, C.; Rotllant, D.; Lufino, M.M.P.; Estiarte, A.; Guibourt, N.; Cavalcanti, F.; Griñan-Ferré, C.; Pallàs, M.; Nadal, R. Modulation of KDM1A with vafidemstat rescues memory deficit and behavioral alterations. PLoS ONE 2020, 15, e0233468. [Google Scholar] [CrossRef]
- Pentkowski, N.S.; Rogge-Obando, K.K.; Donaldson, T.N.; Bouquin, S.J.; Clark, B.J. Anxiety and Alzheimer’s disease: Behavioral analysis and neural basis in rodent models of Alzheimer’s-related neuropathology. Neurosci. Biobehav. Rev. 2021, 127, 647–658. [Google Scholar] [CrossRef]
- Kosel, F.; Pelley, J.M.S.; Franklin, T.B. Behavioural and psychological symptoms of dementia in mouse models of Alzheimer’s disease-related pathology. Neurosci. Biobehav. Rev. 2020, 112, 634–647. [Google Scholar] [CrossRef]
- Boyer, F.; Jaouen, F.; Ibrahim, E.C.; Gascon, E. Deficits in social behavior precede cognitive decline in middle-aged mice. Front. Behav. Neurosci. 2019, 13, 55. [Google Scholar] [CrossRef] [Green Version]
- An, P.N.T.; Shimaji, K.; Tanaka, R.; Yoshida, H.; Kimura, H.; Fukusaki, E.; Yamaguchi, M. Epigenetic regulation of starvation-induced autophagy in Drosophila by histone methyltransferase G9a. Sci. Rep. 2017, 7, 7343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Zeng, J.; Gao, Y.; Guan, Z.; Ma, Z.; Shi, Q.; Du, C.; Jia, J.; Xu, S.; Wang, X. G9a inhibition induces autophagic cell death via AMPK/mTOR pathway in bladder transitional cell carcinoma. PLoS ONE 2015, 10, e0138390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, L.O.; MacKeigan, J.P.; Blenis, J. A network of immediate early gene products propagates subtle differences in mitogen-activated protein kinase signal amplitude and duration. Mol. Cell. Biol. 2004, 24, 144–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, S.-W.; Futter, M.; Rosenblum, K.; Webber, M.J.; Hunt, S.P.; Bliss, T.V.P.; Bramham, C.R. Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: Requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J. Neurosci. 2002, 22, 1532–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, S.J.; Watson, J.J.; Dawbarn, D. The neurotrophins and their role in Alzheimer’s disease. Curr. Neuropharmacol. 2011, 9, 559–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narisawa-Saito, M.; Wakabayashi, K.; Tsuji, S.; Takahashi, H.; Nawa, H. Regional specificity of alterations in NGF, BDNF and NT-3 levels in Alzheimer’s disease. Neuroreport 1996, 7, 2925–2928. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.C.; Lee, D.Y.; Jhoo, J.H.; Kim, K.W.; Choo, I.H.; Woo, J.I. Prevalence of neuropsychiatric syndromes in Alzheimer’s disease (AD). Arch. Gerontol. Geriatr. 2011, 52, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Cerejeira, J.; Lagarto, L.; Mukaetova-Ladinska, E.B. Behavioral and psychological symptoms of dementia. Front. Neurol. 2012, 3, 73. [Google Scholar] [CrossRef] [Green Version]
- Kosel, F.; Munoz, P.T.; Yang, J.R.; Wong, A.A.; Franklin, T.B. Age-related changes in social behaviours in the 5xFAD mouse model of Alzheimer’s disease. Behav. Brain Res. 2019, 362, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Filali, M.; Lalonde, R.; Rivest, S. Cognitive and non-cognitive behaviors in an APPswe/PS1 bigenic model of Alzheimer’s disease. Genes Brain Behav. 2009, 8, 143–148. [Google Scholar] [CrossRef]
- Chen, G.-H.; Wang, C.; Yangcheng, H.-Y.; Liu, R.-Y.; Zhou, J.-N. Age-related changes in anxiety are task-specific in the senescence-accelerated prone mouse 8. Physiol. Behav. 2007, 91, 644–651. [Google Scholar] [CrossRef]
- Flood, J.F.; Farr, S.A.; Uezu, K.; Morley, J.E. Age-related changes in septal serotonergic, GABAergic and glutamatergic facilitation of retention in SAMP8 mice. Mech. Ageing Dev. 1998, 105, 173–188. [Google Scholar] [CrossRef]
- Miyamoto, M.; Takahashi, H.; Ohta, H.; Sakamoto, J. Animal model of brain aging: Senescence-accelerated mouse (SAM). CNS Drug Rev. 1998, 4, 361–375. [Google Scholar] [CrossRef]
- Meeker, H.C.; Chadman, K.K.; Heaney, A.T.; Carp, R.I. Assessment of social interaction and anxiety-like behavior in senescence-accelerated-prone and-resistant mice. Physiol. Behav. 2013, 118, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, M. Emotional disorders and memory deficits in senescence-accelerated mice, SAMP8 and SAMP10. In International Congress Series; Elsevier: Amsterdam, The Netherlands, 2004; Volume 1260, pp. 99–106. [Google Scholar]
- Vasilopoulou, F.; Rodríguez-Arévalo, S.; Bagán, A.; Escolano, C.; Griñán-Ferré, C.; Pallàs, M. Disease-modifying treatment with I2 imidazoline receptor ligand LSL60101 in an Alzheimer’s disease mouse model: A comparative study with donepezil. Br. J. Pharmacol. 2021, 178, 3017–3033. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.L.; Mallory, R. Aggression and violence among elderly patients, a growing health problem. J. Gen. Intern. Med. 2009, 24, 1167–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, M.F.; Martín, M.G. Epigenetic mechanisms related to cognitive decline during aging. J. Neurosci. Res. 2020, 98, 234–246. [Google Scholar] [CrossRef]
- Lin, E.; Tsai, S.-J. Epigenetics and depression: An update. Psychiatry Investig. 2019, 16, 654. [Google Scholar] [CrossRef]
- Januar, V.; Ancelin, M.-L.; Ritchie, K.; Saffery, R.; Ryan, J. BDNF promoter methylation and genetic variation in late-life depression. Transl. Psychiatry 2015, 5, e619. [Google Scholar] [CrossRef]
- Simmons, J.G.; Schwartz, O.S.; Bray, K.; Deane, C.; Pozzi, E.; Richmond, S.; Smith, J.; Vijayakumar, N.; Byrne, M.L.; Seal, M.L. Study protocol: Families and childhood transitions study (FACTS)—A longitudinal investigation of the role of the family environment in brain development and risk for mental health disorders in community based children. BMC Pediatr. 2017, 17, 153. [Google Scholar] [CrossRef] [Green Version]
- Cittaro, D.; Lampis, V.; Luchetti, A.; Coccurello, R.; Guffanti, A.; Felsani, A.; Moles, A.; Stupka, E.; D’Amato, F.R.; Battaglia, M. Histone modifications in a mouse model of early adversities and panic disorder: Role for Asic1 and neurodevelopmental genes. Sci. Rep. 2016, 6, 25131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulle, F.; Van Den Hove, D.L.A.; Jakob, S.B.; Rutten, B.P.; Hamon, M.; Van Os, J.; Lesch, K.P.; Lanfumey, L.; Steinbusch, H.W.; Kenis, G. Epigenetic regulation of the BDNF gene: Implications for psychiatric disorders. Mol. Psychiatry 2012, 17, 584–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsankova, N.; Renthal, W.; Kumar, A.; Nestler, E.J. Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 2007, 8, 355–367. [Google Scholar] [CrossRef]
- Roopra, A.; Qazi, R.; Schoenike, B.; Daley, T.J.; Morrison, J.F. Localized domains of G9a-mediated histone methylation are required for silencing of neuronal genes. Mol. Cell 2004, 14, 727–738. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, A.; Wang, Z.J.; Cao, Q.; Wang, W.; Lin, L.; Ma, K.; Zhang, F.; Wei, J.; Matas, E.; et al. Inhibition of EHMT1/2 rescues synaptic and cognitive functions for Alzheimer’s disease. Brain 2019, 142, 787–807. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Ortega, K.; Garcia-Esparcia, P.; Gil, L.; Lucas, J.J.; Ferrer, I. Altered machinery of protein synthesis in Alzheimer’s: From the nucleolus to the ribosome. Brain Pathol. 2016, 26, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Gupta-Agarwal, S.; Franklin, A.V.; DeRamus, T.; Wheelock, M.; Davis, R.L.; McMahon, L.L.; Lubin, F.D. G9a/GLP histone lysine dimethyltransferase complex activity in the hippocampus and the entorhinal cortex is required for gene activation and silencing during memory consolidation. J. Neurosci. 2012, 32, 5440–5453. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Holsinger, R.M.D. Exercise-induced brain-derived neurotrophic factor expression: Therapeutic implications for Alzheimer’s dementia. Ageing Res. Rev. 2018, 48, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, T.; Gal-Ben-Ari, S.; Dieterich, D.C.; Kreutz, M.R.; Ziv, N.E.; Gundelfinger, E.D.; Rosenblum, K. The roles of protein expression in synaptic plasticity and memory consolidation. Front. Mol. Neurosci. 2014, 7, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hock, C.; Heese, K.; Müller-Spahn, F.; Hulette, C.; Rosenberg, C.; Otten, U. Decreased trkA neurotrophin receptor expression in the parietal cortex of patients with Alzheimer’s disease. Neurosci. Lett. 1998, 241, 151–154. [Google Scholar] [CrossRef]
- Buhusi, M.; Etheredge, C.; Granholm, A.-C.; Buhusi, C.V. Increased hippocampal ProBDNF contributes to memory impairments in aged mice. Front. Aging Neurosci. 2017, 9, 284. [Google Scholar] [CrossRef]
- Jiao, S.S.; Shen, L.L.; Zhu, C.; Bu, X.L.; Liu, Y.H.; Liu, C.H.; Yao, X.Q.; Zhang, L.L.; Zhou, H.D.; Walker, D.G. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl. Psychiatry 2016, 6, e907. [Google Scholar] [CrossRef]
- Lu, B.; Nagappan, G.; Guan, X.; Nathan, P.J.; Wren, P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 401–416. [Google Scholar] [CrossRef]
- Peng, S.; Garzon, D.J.; Marchese, M.; Klein, W.; Ginsberg, S.D.; Francis, B.M.; Mount, H.T.J.; Mufson, E.J.; Salehi, A.; Fahnestock, M. Decreased brain-derived neurotrophic factor depends on amyloid aggregation state in transgenic mouse models of Alzheimer’s disease. J. Neurosci. 2009, 29, 9321–9329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambigapathy, G.; Zheng, Z.; Keifer, J. Regulation of BDNF chromatin status and promoter accessibility in a neural correlate of associative learning. Epigenetics 2015, 10, 981–993. [Google Scholar] [CrossRef] [Green Version]
- Alonso, M.; Medina, J.H.; Pozzo-Miller, L. ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Learn. Mem. 2004, 11, 172–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hof, P.R.; Morrison, J.H. The aging brain: Morphomolecular senescence of cortical circuits. Trends Neurosci. 2004, 27, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Dorostkar, M.M.; Zou, C.; Blazquez-Llorca, L.; Herms, J. Analyzing dendritic spine pathology in Alzheimer’s disease: Problems and opportunities. Acta Neuropathol. 2015, 130, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masliah, E.; Crews, L.; Hansen, L. Synaptic remodeling during aging and in Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 91–99. [Google Scholar] [CrossRef]
- Patel, M.R.; Weaver, A.M. Astrocyte-derived small extracellular vesicles promote synapse formation via fibulin-2-mediated TGF-β signaling. Cell Rep. 2021, 34, 108829. [Google Scholar] [CrossRef]
- Medendorp, W.E.; Petersen, E.D.; Pal, A.; Wagner, L.-M.; Myers, A.R.; Hochgeschwender, U.; Jenrow, K.A. Altered behavior in mice socially isolated during adolescence corresponds with immature dendritic spine morphology and impaired plasticity in the prefrontal cortex. Front. Behav. Neurosci. 2018, 12, 87. [Google Scholar] [CrossRef]
- Radley, J.J.; Rocher, A.B.; Rodriguez, A.; Ehlenberger, D.B.; Dammann, M.; McEwen, B.S.; Morrison, J.H.; Wearne, S.L.; Hof, P.R. Repeated stress alters dendritic spine morphology in the rat medial prefrontal cortex. J. Comp. Neurol. 2008, 507, 1141–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norrholm, S.D.; Ouimet, C.C. Altered dendritic spine density in animal models of depression and in response to antidepressant treatment. Synapse 2001, 42, 151–163. [Google Scholar] [CrossRef]
- Maze, I.; Covington, H.E., III; Dietz, D.M.; Laplant, Q.; Renthal, W.; Russo, S.J.; Mechanic, M.; Mouzon, E.; Neve, R.L.; Stephen, J.; et al. Essential Role of the Histone Methyltransferase G9a in Cocaine- induced Plasticity. Science 2010, 327, 213–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasilopoulou, F.; Bellver-Sanchis, A.; Companys-Alemany, J.; Jarne-Ferrer, J.; Irisarri, A.; Palomera-Ávalos, V.; Gonzalez-Castillo, C.; Ortuño-Sahagún, D.; Sanfeliu, C.; Pallàs, M.; et al. Cognitive Decline and BPSD Are Concomitant with Autophagic and Synaptic Deficits Associated with G9a Alterations in Aged SAMP8 Mice. Cells 2022, 11, 2603. https://doi.org/10.3390/cells11162603
Vasilopoulou F, Bellver-Sanchis A, Companys-Alemany J, Jarne-Ferrer J, Irisarri A, Palomera-Ávalos V, Gonzalez-Castillo C, Ortuño-Sahagún D, Sanfeliu C, Pallàs M, et al. Cognitive Decline and BPSD Are Concomitant with Autophagic and Synaptic Deficits Associated with G9a Alterations in Aged SAMP8 Mice. Cells. 2022; 11(16):2603. https://doi.org/10.3390/cells11162603
Chicago/Turabian StyleVasilopoulou, Foteini, Aina Bellver-Sanchis, Júlia Companys-Alemany, Júlia Jarne-Ferrer, Alba Irisarri, Verónica Palomera-Ávalos, Celia Gonzalez-Castillo, Daniel Ortuño-Sahagún, Coral Sanfeliu, Mercè Pallàs, and et al. 2022. "Cognitive Decline and BPSD Are Concomitant with Autophagic and Synaptic Deficits Associated with G9a Alterations in Aged SAMP8 Mice" Cells 11, no. 16: 2603. https://doi.org/10.3390/cells11162603