
Upregulation of Phospholipase C Gene Expression Due to Norepinephrine-Induced Hypertrophic Response


1
Asper Clinical Research Institute, St. Boniface Hospital, Winnipeg, MB R2H 2A6, Canada
2
Institute of Cardiovascular Sciences & Department of Physiology & Pathophysiology, Max Rady College of Medicine, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, MB R3T 2N2, Canada
*
Author to whom correspondence should be addressed.
Cells 2022, 11(16), 2488; https://doi.org/10.3390/cells11162488
Submission received: 11 July 2022
/
Revised: 4 August 2022
/
Accepted: 9 August 2022
/
Published: 11 August 2022
(This article belongs to the Special Issue Cardiac Growth Control and Heart Cell Death)
Abstract
:The activation of phospholipase C (PLC) is thought to have a key role in the cardiomyocyte response to several different hypertrophic agents such as norepinephrine, angiotensin II and endothelin-1. PLC activity results in the generation of diacylglycerol and inositol trisphosphate, which are downstream signal transducers for the expression of fetal genes, increased protein synthesis, and subsequent cardiomyocyte growth. In this article, we describe the signal transduction elements that regulate PLC gene expression. The discussion is focused on the norepinephrine- α1-adrenoceptor signaling pathway and downstream signaling processes that mediate an upregulation of PLC isozyme gene expression. Evidence is also indicated to demonstrate that PLC activities self-regulate the expression of PLC isozymes with the suggestion that PLC activities may be part of a coordinated signaling process for the perpetuation of cardiac hypertrophy. Accordingly, from the information provided, it is plausible that specific PLC isozymes could be targeted for the mitigation of cardiac hypertrophy.
1. Introduction
It is well established that cardiac hypertrophy is associated with concomitant alterations in the expression levels of many genes. In addition, cardiomyocyte growth is due to an increase in protein content in the cardiomyocyte as opposed to an increase in cell numbers because adult cardiomyocytes do not have the capacity to proliferate as they are terminally differentiated [1]. Accordingly, the cardiomyocyte hypertrophic response is characterized by increases in cardiomyocyte size, RNA concentration, and protein synthesis as well as expression of fetal genes including atrial natriuretic factor (ANF) [2,3,4,5,6,7,8,9,10,11,12,13] due to the activation of several transcription factors [14,15]. Furthermore, a transient activation of the c-Fos and c-Jun complex (AP-1) also occurs in the early response to hypertrophic stimuli [14,16]. In fact, cumulative evidence has determined that both AP-1 complex and ANF induce the activation of several hypertrophic genes [14,17,18,19].
The increases in c-Fos mRNA and protein synthesis in response to norepinephrine are attributed to the activation of the α1-adrenoceptor (AR) [14,18,20,21]. A transient increase of c-Jun expression has been reported in response to mechanical overload [22]. Cardiomyocyte transfection with a dominant negative c-Jun has been shown to prevent the increases in protein synthesis and atrial natriuretic peptide mRNA in response to the AR agonist, phenylephrine [23]. In contrast, a marked activation of the ANF gene promoter has been observed in cardiomyocytes overexpressing c-Jun [20]. On the other hand, pretreatment of neonatal rat cardiomyocytes, in vitro, with PLC antisense oligonucleotides was demonstrated to prevent the upregulation of c-Fos and c-Jun due to insulin-like growth factor-1 [24]. These authors suggested that signal transduction by specific phospholipase C (PLC) isozymes could have an important role in the regulation of these transcription factors.
The activation of PLC is considered a primary signaling event in the regulation of diverse cellular functions [25,26]. Several different agents, such as neurohormones and growth factors, induce phenotypic changes in cardiomyocytes that are characteristic of the hypertrophic response and activate PLC [27,28]. Mechanical stretching of isolated cardiomyocytes or in vivo due to pressure/volume overload also results in the activation of the PLC signal transduction pathway [4,29,30,31] as well as induces characteristic features of cardiac hypertrophy. Figure 1 depicts the norepinephrine-mediated signal transduction events that result in cardiomyocyte cell growth, for which PLC is an important and integral component. Indeed, the phosphorylation of target proteins [32], the activation of transcription factors, and subsequent gene expression [9,33] in the cardiomyocyte hypertrophic response to norepinephrine may all be initiated by PLC activation.
Although animal models have facilitated in identifying alterations in gene expression during cardiac hypertrophy, the use of in vitro systems has permitted further delineation of the signal transduction processes and molecular events implicated in increasing cardiomyocyte growth during the response to different hypertrophic stimuli. Thus, in this review, it is intended to describe changes in PLC isozyme gene and protein expression as well as isozyme activities in cardiac hypertrophy due to different etiologies and in isolated cardiomyocytes in response to norepinephrine. In addition, we also discuss the in vivo as well as in vitro signal transduction elements and the identity of the transcription factors that regulate PLC gene expression. Furthermore, the available evidence is presented to show that PLC regulates its own gene expression. Overall, the contention of this article is that PLC might constitute a mechanism for the perpetuation of the hypertrophic process and ultimately its transition to heart failure and may therefore be an important molecular and pharmacological target.
2. Regulation of Cardiac PLC Isozymes
There are now 13 families of PLC isozymes that are categorized into 6 classes; PLC β, δ, γ, ε, ζ, and η isozymes [34,35], and are known to be differentially expressed in mammalian cells [36]. Phosphoinositide-specific PLC activity results in the hydrolysis of membrane phosphatidylinositol-4,5-bisphosphate (PIP2) to generate inositol-1,4,5-trisphosphate (IP3) and 1,2-diacylglycerol (DAG) [37]. Both these lipid molecules activate downstream signal transduction events for cardiac hypertrophy. In this regard, the interaction of IP3 with its receptors stimulates the release of Ca2+ from intracellular stores, whereas DAG activates protein kinase C (PKC) isoforms. Although there is now a large body of information that has established the involvement of specific PKC isoforms in cardiomyocyte growth [38,39], IP3/Ca2+ are also considered to be important signaling elements for cardiac hypertrophy [6,9,40,41].
PLC β, δ, γ, and ε [42,43,44,45,46,47] are the major myocardial PLC isozymes and are known to be activated by different mediators including G proteins, tyrosine kinases, and calcium [48]. The PLC β family has four types of isozymes (β1, β2, β3, and β4) [36]. PLC β4 has been reported to exhibit higher expression level in the heart relative to PLC β1, β2, and β3 [49]. The mechanisms involved in the activation of PLC β1 and PLC β3 isozymes are the most well studied among PLC isozymes in the heart. In this regard, norepinephrine and other α1-AR agonists are known to stimulate PLC β isozyme activities through Gqα [50]. In addition, unlike PLC β4, PLC β1-3 is also stimulated via the Gβγ dimer [51]. Furthermore, the Gβγ subunit can also directly activate PLC ε [52].
PLC γ1 is localized in the cell cytosol and is regarded as the predominant cardiac PLC isozyme [53]. This isozyme is activated by GPCR and growth factor receptor tyrosine kinases, although a non-tyrosine kinase-mediated stimulation of PLC γ1 has also been observed [54]. Indeed, some of our work has suggested the existence of a tyrosine kinase and Gqα subunit cross-talk in adult rat cardiomyocytes [55]. In contrast, PLC δ1 is viewed to be the major PLC isozyme located at the sarcolemmal (SL) membrane. This has been attributed to the presence of basic amino acids in the N-terminal region of the pleckstrin homology domain of PLC δ1, which have a strong affinity for membrane PIP2 [56,57]. While intracellular Ca2+ levels have been shown to modulate PLC δ1 activity, a direct α1-AR induced activation of PLC δ isozyme by the dimeric Gh protein has also been reported [58,59].
3. Upregulation of PLC Isozymes in Pathological Cardiomyocyte Growth
Transgenic and In Vivo Models of Cardiac Hypertrophy
The upregulation of PLC has been established in several diverse transgenic mice and other animal models of cardiac hypertrophy. In this regard, the activation of PLC-mediated signal transduction in cardiac hypertrophy in stroke-prone spontaneously hypertensive rats has been reported [60,61]. In volume overload-induced cardiac hypertrophy subsequent to an arteriovenous shunt, specific increases in PLC β1 and PLC γ1 isozyme gene, protein contents, and activities [62,63] have been observed. A similar activation of PLC β3 has been observed in pressure overload-induced cardiac hypertrophy due to aortic constriction in the rat [64]. In contrast, the activation of PKC isozymes has been observed in pressure overload cardiac hypertrophy in the guinea pig, subsequent to ligation of the descending thoracic artery, without any changes in PLC β1 and Gαq protein contents [65]. However, these investigators indicated that the stimulation of PKC isozymes could be a result of an increase in Gαq and PLC β1 activities instead of an upregulation of their expression [66]; it should be mentioned that PLC β1 activity was not measured in this study. Since mechanical stretch is an initiating feature for hypertrophy in response to hemodynamic overload, and that an increase in both Gqα and PLC β1 activities has been reported in stretched cardiomyocytes [29,30] as well as increased sympathetic nervous activity [67] in pressure-overload hypertrophy, it is therefore highly conceivable that stimulation of the α1-ARs increases PLC β isozyme activity in mechanical stress.
The stimulation of receptor-coupled Gqα in transgenic mouse models overexpressing Gqα results in cardiac hypertrophy [68,69,70,71], which potentially may be attributed to increases in PLC activity. However, the activation of PLC was found not to be associated with cardiac hypertrophy in two other different transgenic mouse lines expressing activated Gqα [72,73]. Although signal transduction mediated by specific PLC isozymes is an important component of the cardiomyocyte hypertrophic response, the abolition of signal transduction in PLC ε (−/−) mice results in cardiac hypertrophy that has been attributed to an increase in sensitivity to isoproterenol [66,74]. While the individual role of the different PLC isozymes in pathological cardiomyocyte growth still remains to be fully understood, from the information provided, it is apparent that PLC is an important contributor to the signaling processes leading to cardiac hypertrophy.
4. Isolated Cardiomyocytes for Assessing the Hypertrophic Response
The generation of IP3 in neonatal cardiomyocytes has been observed in response to norepinephrine and is considered to be attributed to the α1-AR-mediated activation of PLC β1 [75]. There are two splice variants of PLC β1 in the heart, PLC β1a and PLC β1b, which differ in their C-terminal amino acid sequences [76]. While PLC β1a is located in the cytosol, PLC β1b is localized in the SL membrane, particularly in regions rich in caveolae (membrane lipid rafts), and where the α1-ARs are also localized [76]. Therefore, α1-AR-mediated signal transduction in response to norepinephrine can be considered to be due to PLC β1b. As a consequence, targeting of PLC β1b is considered to have the potential to attenuate the development and progression of cardiac hypertrophy [76]. Indeed, an increase in cardiomyocyte growth with a concomitant increase in the ratio of protein to DNA as well as an increase in ANF levels have been reported in neonatal cardiomyocytes overexpressing PLC β1b [77]. These observations demonstrated the involvement of α1-AR-PLC β1b in the hypertrophic response and further strengthened the viability of PLC β1b as a target for prevention/restriction of cardiac hypertrophy [77]. As already mentioned, PLC β4 is highly expressed in human left ventricular tissue and in view of its reported increase in mouse HL-1 cardiomyocytes exposed to different hypertrophic agents, it is conceivable that PLC β4 may play a complementary role in the signal transduction processes for pathological cardiomyocyte growth [49].
Although there is a large body of evidence on the involvement of PLC β isozymes in cardiac hypertrophy, the contribution of other myocardial PLC isozymes to the cardiomyocyte hypertrophic response is less defined, but information is now emerging. In this regard, depletion of PLC ε with siRNA in neonatal cardiomyocytes has been observed to reduce the cardiomyocyte response to a variety of hypertrophic agents [74]. However, even though a reduction in PLC ε was shown not to attenuate IP3 production, it was proposed that regionalized PLC activity was essential for this response.
Our attention has focused on determining the contribution of the different PLC isozymes in cardiac hypertrophy. Although the involvement of the α1-AR- PLC signaling axis has been confirmed by antagonism of the α1-AR with prazosin, as well as inhibition of PLC activity with a compound, U73122, subsequent studies have been conducted to dissect the signal transduction events and transcriptional parameters that influence the expression levels of the PLC genes [78,79,80]. The data presented in Table 1 show specific increases in the transcription factors, c-Fos and c-Jun in adult rat cardiomyocytes treated with norepinephrine and with phenylephrine; no changes in the other transcription factors (NFAT3, NFκB, MEF2C, and MEF2D) were observed [78]. These findings were suggestive of a specific and early upregulation of both c-Fos and c-Jun due to stimulation of the α1-AR under our experimental conditions.
Cardiomyocytes were treated with 5 µM norepinephrine (NE) or 1 µM phenylephrine (PhE) for 2 h. Transcription factor mRNA levels were measured by semi-quantitative RT-PCR and the data are presented as a percentage of the control values. These values are means ± S.E. of 5 experiments conducted with 5 different cardiomyocyte preparations.
Subsequent studies demonstrated that both c-Fos and c-Jun have a regulatory role in the expression of PLC isozymes [79]. In this regard, by employing gene silencing techniques, it was found that transfection of cardiomyocytes with c-Fos siRNA prevented the norepinephrine-induced increases in PLC β1 and PLC β3 mRNA levels, but did not affect the norepinephrine-induced increases in PLC γ1 and PLC δ1 gene expression (Table 2A). In addition, silencing of c-Jun with siRNA not only inhibited the norepinephrine-induced increases in PLC β1 and PLC β3 mRNA levels but also prevented the increase in PLC δ1 mRNA levels in response to norepinephrine. Similarly, silencing of c-Jun did not attenuate PLC γ1 gene expression in response to norepinephrine (Table 2A). Furthermore, knockdown of both c-Fos and c-Jun inhibited the norepinephrine-induced activation of PLC isozymes as determined by the formation of inositol phosphates (Table 2B). These data demonstrated differential transcriptional regulation of PLC isozymes [79].
Since stimulation of the α1-AR was seen to result in a specific increase in the mRNA levels of both c-Fos and c-Jun, it is likely that α1-AR-PLC signal transduction is implicated in increases in c-Fos and c-Jun mRNA levels due to norepinephrine. It can be observed from Table 3 that both prazosin and U73122 blocked the increases in c-Fos and c-Jun mRNA levels in cardiomyocytes exposed to norepinephrine [78,79]. To further verify the participation of PLC in modulating transcription factor expression levels, cardiomyocytes were transfected with PLC isozyme-specific siRNA.
Cardiomyocytes were transfected with or without 5 nM siRNA and treated with NE (5 μM) for 2 h. Cardiomyocytes without any treatment served as control. PLC mRNA levels (A) were determined by semi-quantitative RT-PCR and data are presented as a percentage of the control value. PLC isozymes activities (B) were determined by measuring the hydrolysis of [3H]-PIP2 and are expressed as pmol/min/mg protein of inositol phosphates formed. These values are means ± S.E. of 5 experiments conducted with 5 different cardiomyocyte preparations.
Cardiomyocytes were exposed to NE (5 μM) without and with prazosin (2 μM), U73122 (1 nM), or after transfection with 5 nM PLC isozyme siRNA for 2 h. c-Fos and c-Jun mRNA levels were determined by semi-quantitative RT-PCR and data are presented as a percentage of the control value. These values are means ± S.E. of 5 experiments conducted with 5 different cardiomyocyte preparations.
With the exception of silencing of PLC γ1, knockdown of PLC β1, β3, and δ1 genes was observed to prevent the increases in the mRNA levels of c-Fos and c-Jun (Table 3). These data indicated that the norepinephrine- α1-AR-mediated increases in c-Fos and c-Jun mRNA may involve signal transduction via some specific PLC isozymes.
The signaling events that regulate the expression levels of PLC isozyme genes were delineated through pharmacological and gene silencing interventions [79,80]. While prazosin and U73122 attenuated the increases in PLC isozyme gene expression in response to norepinephrine, silencing of the PLC gene with siRNA also prevented the norepinephrine-induced increases in PLC gene expression. In view of these observations, it was hypothesized that PLC activity can increase its own gene expression in response to stimulation with norepinephrine in adult cardiomyocytes (Table 4). Furthermore, it was demonstrated that PLC isozyme self-regulation of gene expression, may involve downstream PKC- and ERK1/2- signaling processes (Table 5 [80]).
Cardiomyocytes were exposed to NE (5 μM) without and with prazosin (2 μM), U73122 (1 nM), or after transfection with 5 nM PLC isozyme siRNA for 2 h. PLC isozyme mRNA levels were determined by semi-quantitative RT-PCR and data are presented as a percentage of the control value. These values are means ± S.E. of 5–10 experiments conducted with 5–10 different cardiomyocyte preparations.
Cardiomyocytes were exposed to different concentrations of PMA (0.1, 1.0, and 10.0 μM) and to NE (5 μM) without and with varying concentrations of Bis-1 (50, 100, and 200 nM) and PD98059 (2, 10, and 25 nM) for 2 h. PLC mRNA levels were determined by semi-quantitative RT-PCR and data are presented as a percentage of the control value. These values are means ± S.E. of 5 experiments conducted with 5 different cardiomyocyte preparations.
Indeed, while the PKC activator (phorbol myristate acetate, PMA) increased PLC gene expression, the PKC activity inhibitor (bisindolylmaleimide, Bis-1) markedly attenuated the norepinephrine-induced increases in PLC isozyme mRNA level. Similarly, blockade of ERK1/2 with PD98059 abolished PLC gene expression in response to norepinephrine. From the aforementioned, it is proposed that PLC activation is an early response to α1-AR activation by norepinephrine and that subsequent signal transduction events that augment PLC gene expression and activities may constitute a sequence of cyclical events designed to perpetuate cardiac hypertrophy and facilitate its ultimate transition into heart failure (Figure 2).
5. Evidence for Regression of Abnormal Cardiomyocyte Growth by α1-AR Blockade
From the above-mentioned discussion, it is evident that the α1-AR-Gqα-PLC signal transduction pathway has an important contribution to the hypertrophic response to norepinephrine. Accordingly, there are some experimental studies that have demonstrated that blockade of the α1-AR with prazosin mitigates the transition of cardiac hypertrophy to heart failure [81,82,83,84,85].
In addition, we have observed prazosin and metoprolol (a β1-AR receptor blocker) to reverse cardiac remodeling in the failing rat heart [86,87]. Furthermore, labetalol, a non-selective β-AR blocker, has been reported to reverse cardiac hypertrophy [88,89].
Importantly, several clinical investigations have revealed the advantageous effects of α1-AR antagonists including prazosin in failing hearts due to different etiologies [90,91,92,93]. It should be mentioned that while co-administration of prazosin and metoprolol in heart failure was observed not to exert any additive effects [94], the results of the COMET trial (Carvedilol or Metoprolol European Trial), revealed a greater benefit of carvedilol in heart failure than with metoprolol alone [95]. Taken together, these lines of evidence suggest that agents that exhibit a capability to block both α- and β-ARs can provide an improved outcome by attenuation of cardiac hypertrophy. However, it should be noted that the use of α1-AR blockers in patients with heart failure has been reported to produce no improvement in the condition [96,97]. Nonetheless, signal transduction mediated by the α1-AR-PLC pathway can be considered to play an essential contributory role in pathological cardiomyocyte growth. Furthermore, it can be proposed that the activation of this signaling pathway perpetuates cardiac hypertrophy that eventually progresses to heart failure. Although the literature has focused largely on antagonism the role of the It should be mentioned that in cardiomyocytes isolated from spontaneously hypertensive rats, α2-AR signaling is markedly attenuated [98]. Moreover, it has been suggested that α2-AR-mediated signal transduction counterbalances PLC-mediated signaling [99] indicating that cardiac SL α2-ARs may also be an important target [100] and potentially for the mitigation of cardiac hypertrophy.
6. Conclusions
We have extensively reviewed the literature regarding the involvement of phospholipid-mediated signal transduction mechanisms in different myocardial diseases [101,102,103,104,105,106], and recently the involvement of PLC in the cardiomyocyte hypertrophic response to norepinephrine [107] as well as the role of PLC in the catecholamine-induced increase in cardiomyocyte protein synthesis [108] has also been reviewed. In the present article, the activation of PLC and the regulation of its gene expression in cardiomyocyte hypertrophic response have been addressed. From the evidence provided, it can be suggested that the activation of specific PLC isozymes by norepinephrine is an important aspect of the signal transduction cascade that stimulates abnormal cardiomyocyte growth and that this pathway may constitute a sequence of cyclical actions that allow for the continuation of cardiac hypertrophy (Figure 2). There are several protein kinases that are activated by the PLC pathway, these include PKC, ERK1/2, and Ca2+/calmodulin-dependent kinase. These kinases have an important role as they, in turn, phosphorylate and activate some transcription factors including c-Fos and c-Jun that leads to PLC gene expression.
The reciprocal relationship between PLC activities and PLC gene expression is a characteristic feature that augments cardiac hypertrophy. It should be noted that it is not the intention of this review to exclude the role of the β1-AR Gs-protein-adenylyl cyclase system, which is largely responsible for initiating the development of cardiac hypertrophy. However, it is our contention that signal transduction through the α1-AR-Gqα-PLC axis plays a critical and complementary role in the initial phase of abnormal cardiomyocyte growth, and that signal transduction through this pathway is more significant in the late stage of cardiac hypertrophy as the β1-AR is downregulated at this phase. It should also be mentioned that while the focus of this review has been on the α1-AR-Gqα-PLC, there are other GPCRs that mediate the hypertrophic response. In this regard, the angiotensin II receptor blocker losartan has been observed to diminish PLC gene expression with a concomitant regression of cardiac hypertrophy [62]. Accordingly, it can be suggested that PLC has the potential to be viewed as an additional target for limiting pathological cardiomyocyte growth.
Author Contributions
Conceptualization, P.S.T.; writing—original draft preparation, P.S.T.; responsibility for the final draft and the scientific background of the manuscript, N.S.D. and P.S.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Infrastructural support for this project was provided by the St. Boniface Hospital Albrechtsen Research Centre.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fiedler, B.; Wollert, K.C. Targeting calcineurin and associated pathways in cardiac hypertrophy and failure. Expert Opin. Ther. Targets 2005, 9, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Balakumar, P.; Jagadeesh, G. Multifarious molecular signaling cascades of cardiac hypertrophy: Can the muddy waters be cleared? Pharmacol. Res. 2010, 62, 365–383. [Google Scholar] [CrossRef] [PubMed]
- Clerk, A.; Cullingford, T.E.; Fuller, S.J.; Giraldo, A.; Markou, T.; Pikkarainen, S.; Sugden, P.H. Signaling pathways mediating cardiac myocyte gene expression in physiological and stress responses. J. Cell Physiol. 2007, 212, 311–322. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, H.W.; Dekkers, D.H.; Houtsmuller, A.B.; Sharma, H.S.; Lamers, J.M. Differential signaling and hypertrophic responses in cyclically stretched vs endothelin-1 stimulated neonatal rat cardiomyocytes. Cell Biochem. Biophys. 2007, 47, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Haque, Z.K.; Wang, D.Z. How cardiomyocytes sense pathophysiological stresses for cardiac remodeling. Cell. Mol. Life Sci. 2017, 74, 983–1000. [Google Scholar] [CrossRef]
- Hefti, M.A.; Harder, B.A.; Eppenberger, H.M.; Schaub, M.C. Signaling pathways in cardiac myocyte hypertrophy. J. Mol. Cell. Cardiol. 1997, 29, 2873–2892. [Google Scholar] [CrossRef]
- Heineke, J.; Molkentin, J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef]
- Peter, A.K.; Bjerke, M.A.; Leinwand, L.A. Biology of the cardiac myocyte in heart disease. Mol. Biol. Cell 2016, 27, 2149–2160. [Google Scholar] [CrossRef]
- Takano, A.P.C.; Senger, N.; Barreto-Chaves, M.L.M. The endocrinological component and signaling pathways associated to cardiac hypertrophy. Mol. Cell. Endocrinol. 2020, 518, 110972. [Google Scholar] [CrossRef]
- Winkle, A.J.; Nassal, D.M.; Shaheen, R.; Thomas, E.; Mohta, S.; Gratz, D.; Weinberg, S.H.; Hund, T.J. Emerging therapeutic targets for cardiac hypertrophy. Expert Opin. Ther. Targets 2022, 26, 29–40. [Google Scholar] [CrossRef]
- Freire, G.; Ocampo, C.; Ilbawi, N.; Griffin, A.J.; Gupta, M. Overt expression of AP-1 reduces alpha myosin heavy chain expression and contributes to heart failure from chronic volume overload. J. Mol. Cell. Cardiol. 2007, 43, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.Y.; Kinugawa, K.; Vinson, C.; Long, C.S. AFos dissociates cardiac myocyte hypertrophy and expression of the pathological gene program. Circulation 2005, 111, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Montessuit, C.; Thorburn, A. Transcriptional activation of the glucose transporter GLUT1 in ventricular cardiac myocytes by hypertrophic agonists. J. Biol. Chem. 1999, 274, 9006–9012. [Google Scholar] [CrossRef] [Green Version]
- Colella, M.; Grisan, F.; Robert, V.; Turner, J.D.; Thomas, A.P.; Pozzan, T. Ca2+ oscillation frequency decoding in cardiac cell hypertrophy: Role of calcineurin/NFAT as Ca2+ signal integrators. Proc. Natl. Acad. Sci. USA 2008, 105, 2859–2864. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Young, D.; Maitra, R.K.; Gupta, A.; Popovic, Z.B.; Yong, S.L.; Mahajan, A.; Wang, Q.; Sen, S. Prevention of cardiac hypertrophy and heart failure by silencing of NF-kappaB. J. Mol. Biol. 2008, 375, 637–649. [Google Scholar] [CrossRef] [Green Version]
- Robinson, E.L.; Drawnel, F.M.; Mehdi, S.; Archer, C.R.; Liu, W.; Okkenhaug, H.; Alkass, K.; Aronsen, J.M.; Nagaraju, C.K.; Sjaastad, I.; et al. MSK-mediated phosphorylation of histone H3 Ser28 couples MAPK signalling with early gene induction and cardiac hypertrophy. Cells 2022, 11, 604. [Google Scholar] [CrossRef]
- Rosenzweig, A.; Halazonetis, T.D.; Seidman, J.G.; Seidman, C.E. Proximal regulatory domains of rat atrial natriuretic factor gene. Circulation 1991, 84, 1256–1265. [Google Scholar] [CrossRef] [Green Version]
- Saadane, N.; Alpert, L.; Chalifour, L.E. Altered molecular response to adrenoreceptor-induced cardiac hypertrophy in Egr-1-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H796–H805. [Google Scholar] [CrossRef] [Green Version]
- Aoki, H.; Richmond, M.; Izumo, S.; Sadoshima, J. Specific role of the extracellular signal-regulated kinase pathway in angiotensin II-induced cardiac hypertrophy in vitro. Biochem. J. 2000, 347, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Hannan, R.D.; West, A.K. Adrenergic agents, but not triiodo-L-thyronine induce c-fos and c-myc expression in the rat heart. Basic Res. Cardiol. 1991, 86, 154–164. [Google Scholar] [CrossRef]
- Hannan, R.D.; Stennard, F.A.; West, A.K. Expression of c-fos and related genes in the rat heart in response to norepinephrine. J. Mol. Cell. Cardiol. 1993, 25, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Schunkert, H.; Jahn, L.; Izumo, S.; Apstein, C.S.; Lorell, B.H. Localization and regulation of c-fos and c-jun protooncogene induction by systolic wall stress in normal and hypertrophied rat hearts. Proc. Natl. Acad. Sci. USA 1991, 88, 11480–11484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, D.J.; Rho, H.W.; Rhee, S.G. CD3 stimulation causes phosphorylation of phospholipase C-γ 1 on serine and tyrosine residues in a human T-cell line. Proc. Natl. Acad. Sci. USA 1991, 88, 5453–5456. [Google Scholar] [CrossRef] [Green Version]
- Schnabel, P.; Mies, F.; Nohr, T.; Geisler, M.; Böhm, M. Differential regulation of phospholipase C-β isozymes in cardiomyocyte hypertrophy. Biochem. Biophys. Res. Commun. 2000, 275, 1–6. [Google Scholar] [CrossRef]
- Katan, M.; Cockcroft, S. Phospholipase C families: Common themes and versatility in physiology and pathology. Prog. Lipid. Res. 2020, 80, 101065. [Google Scholar] [CrossRef]
- Vines, C.M. Phospholipase C. Adv. Exp. Med. Biol. 2012, 740, 235–254. [Google Scholar]
- Bai, H.; Wu, L.L.; Xing, D.Q.; Liu, J.; Zhao, Y.L. Angiotensin II induced upregulation of G αq/11, phospholipase C β3 and extracellular signal-regulated kinase 1/2 via angiotensin II type 1 receptor. Chin. Med. J. 2004, 117, 88–93. [Google Scholar]
- Lamers, J.M.; Eskildsen-Helmond, Y.E.; Resink, A.M.; de Jonge, H.W.; Bezstarosti, K.; Sharma, H.S.; van Heugten, H.A. Endothelin-1-induced phospholipase C-beta and D and protein kinase C isoenzyme signaling leading to hypertrophy in rat cardiomyocytes. J. Cardiovasc. Pharmacol. 1995, 26, S100–S103. [Google Scholar] [CrossRef]
- Ruwhof, C.; Egas, J.M.; van Wamel, A.E.; van der Laarse, A. Signal transduction of mechanical stress in myocytes and fibroblasts derived from neonatal rat ventricles. Neth. Heart J. 2001, 9, 372–378. [Google Scholar]
- Ruwhof, C.; van Wamel, J.T.; Noordzij, L.A.; Aydin, S.; Harper, J.C.; van der Laarse, A. Mechanical stress stimulates phospholipase C activity and intracellular calcium ion levels in neonatal rat cardiomyocytes. Cell Calcium 2001, 29, 73–83. [Google Scholar] [CrossRef]
- Woodcock, E.A.; Grubb, D.R.; Filtz, T.M.; Marasco, S.; Luo, J.; McLeod-Dryden, T.J.; Kaye, D.M.; Sadoshima, J.; Du, X.J.; Wong, C.; et al. Selective activation of the “b” splice variant of phospholipase Cβ1 in chronically dilated human and mouse atria. J. Mol. Cell. Cardiol. 2009, 47, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Wang, K.; Li, P. The role of post-translational modifications in cardiac hypertrophy. J. Cell. Mol. Med. 2019, 23, 3795–3807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.-H.; Zhang, H.-G. Transcription factors involved in the development and prognosis of cardiac remodeling. Front. Pharmacol. 2022, 13, 828549. [Google Scholar] [CrossRef] [PubMed]
- Harden, T.K.; Sondek, J. Regulation of phospholipase C isozymes by ras superfamily GTPases. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 355–379. [Google Scholar] [CrossRef] [Green Version]
- Suh, P.-G.; Ryu, S.O.; Choi, W.C.; Lee, K.Y.; Rhee, S.G. Monoclonal antibodies to three phospholipase C isozymes from bovine brain. J. Biol. Chem. 1988, 263, 14497–14504. [Google Scholar] [CrossRef]
- Gresset, A.; Sondek, J.; Harden, T.K. The phospholipase C isozymes and their regulation. Subcell. Biochem. 2012, 58, 61–94. [Google Scholar]
- Fukami, K.; Inanobe, S.; Kanemaru, K.; Nakamura, Y. Phospholipase C is a key enzyme regulating intracellular calcium and modulating the phosphoinositide balance. Prog. Lipid Res. 2010, 49, 429–437. [Google Scholar] [CrossRef]
- Miao, L.N.; Pan, D.; Shi, J.; Du, J.P.; Chen, P.F.; Gao, J.; Yu, Y.; Shi, D.Z.; Guo, M. Role and mechanism of PKC-δ for cardiovascular disease: Current status and perspective. Front. Cardiovasc. Med. 2022, 9, 816369. [Google Scholar] [CrossRef]
- Singh, R.M.; Cummings, E.; Pantos, C.; Singh, J. Protein kinase C and cardiac dysfunction: A review. Heart Fail. Rev. 2017, 22, 843–859. [Google Scholar] [CrossRef] [Green Version]
- Barac, Y.D.; Zeevi-Levin, N.; Yaniv, G.; Reiter, I.; Milman, F.; Shilkrut, M.; Coleman, R.; Abassi, Z.; Binah, O. The 1,4,5-inositol trisphosphate pathway is a key component in Fas-mediated hypertrophy in neonatal rat ventricular myocytes. Cardiovasc. Res. 2005, 68, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Kockskämper, J.; Zima, A.V.; Roderick, H.L.; Pieske, B.; Blatter, L.A.; Bootman, M.D. Emerging roles of inositol 1,4,5-trisphosphate signaling in cardiac myocytes. J. Mol. Cell. Cardiol. 2008, 45, 128–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, C.A.; Schroering, A.G.; Robishaw, J.D. Subunit expression of signal transducing G proteins in cardiac tissue: Implications for phospholipase C-β regulation. J. Mol. Cell. Cardiol. 1995, 27, 471–484. [Google Scholar] [CrossRef]
- Schnabel, P.; Gäs, H.; Nohr, T.; Camps, M.; Böhm, M. Identification and characterization of G protein-regulated phospholipase C in human myocardium. J. Mol. Cell. Cardiol. 1996, 28, 2419–2427. [Google Scholar] [CrossRef]
- Smrcka, A.V.; Brown, J.H.; Holz, G.G. Role of phospholipase Cε in physiological phosphoinositide signaling networks. Cell Signal. 2012, 24, 1333–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, P.G.; Ryu, S.H.; Moon, K.H.; Suh, H.W.; Rhee, S.G. Cloning and sequence of multiple forms of phospholipase C. Cell. 1988, 54, 161–169. [Google Scholar] [CrossRef]
- Tappia, P.S.; Liu, S.Y.; Shatadal, S.; Takeda, N.; Dhalla, N.S.; Panagia, V. Changes in sarcolemmal PLC isoenzymes in postinfarct congestive heart failure: Partial correction by imidapril. Am. J. Physiol. 1999, 277, H40–H49. [Google Scholar] [CrossRef]
- Wolf, R.A. Specific expression of phospholipase C-δ1 and γ1 by adult cardiac ventricular Myocytes. Circulation 1993, 88 (Suppl. S1), I-241. [Google Scholar]
- Nakamura, Y.; Fukami, K. Regulation and physiological functions of mammalian phospholipase C. J. Biochem. 2017, 161, 315–321. [Google Scholar] [CrossRef] [Green Version]
- Otaegui, D.; Querejeta, R.; Arrieta, A.; Lazkano, A.; Bidaurrazaga, A.; Arriandiaga, J.R.; Aldazabal, P.; Garro, M.A. Phospholipase Cβ4 isozyme is expressed in human, rat, and murine heart left ventricles and in HL-1 cardiomyocytes. Mol. Cell. Biochem. 2010, 337, 167–173. [Google Scholar] [CrossRef]
- Rhee, S.G. Regulation of phosphoinositide-specific phospholipase C. Annu. Rev. Biochem. 2001, 70, 281–312. [Google Scholar] [CrossRef]
- Lee, C.W.; Lee, K.H.; Lee, S.B.; Park, D.; Rhee, S.G. Regulation of phospholipase C-β4 by ribonucleotides and the alpha subunit of Gq. J. Biol. Chem. 1994, 269, 25335–25338. [Google Scholar] [CrossRef]
- Madukwe, J.C.; Garland-Kuntz, E.E.; Lyon, A.M.; Smrcka, A.V. G protein βγ subunits directly interact with and activate phospholipase Cϵ. J. Biol. Chem. 2018, 293, 6387–6397. [Google Scholar] [CrossRef] [Green Version]
- Cockcroft, S.; Thomas, G.M. Inositol-lipid-specific phospholipase C isoenzymes and their differential regulation by receptors. Biochem. J. 1992, 288, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, F.; Bae, Y.S.; Rhee, S.G. Regulation of phospholipase C isozymes: Activation of phospholipase C-γ in the absence of tyrosine-phosphorylation. Chem. Phys. Lipids. 1999, 98, 3–11. [Google Scholar] [CrossRef]
- Tappia, P.S.; Padua, R.R.; Panagia, V.; Kardami, E. Fibroblast growth factor-2 stimulates phospholipase Cβ in adult cardiomyocytes. Biochem. Cell. Biol. 1999, 77, 569–575. [Google Scholar] [CrossRef]
- Tall, E.; Dormán, G.; Garcia, P.; Runnels, L.; Shah, S.; Chen, J.; Profit, A.; Gu, Q.M.; Chaudhary, A.; Prestwich, G.D.; et al. Phosphoinositide binding specificity among phospholipase C isozymes as determined by photo-cross-linking to novel substrate and product analogs. Biochemistry 1997, 36, 7239–7248. [Google Scholar] [CrossRef]
- Yagisawa, H.; Sakuma, K.; Paterson, H.F.; Cheung, R.; Allen, V.; Hirata, H.; Watanabe, Y.; Hirata, M.; Williams, R.L.; Katan, M. Replacements of single basic amino acids in the pleckstrin homology domain of phospholipase C-δ1 alter the ligand binding, phospholipase activity, and interaction with the plasma membrane. J. Biol. Chem. 1998, 273, 417–424. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, M.; Houdeau, E.; Mhaouty-Kodja, S. Increased potency of α1-adrenergic receptors to induce inositol phosphates production correlates with the up-regulation of α1d/Gh α/phospholipase C δ1 signaling pathway in term rat myometrium. Reproduction 2008, 135, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.F.; Yeh, T.S.; Chen, S.F.; Tsai, Y.H.; Chou, C.M.; Yang, Y.Y.; Huang, H.M. Nonmuscle myosin IIA (myosin heavy polypeptide 9): A novel class of signal transducer mediating the activation of Gαh/phospholipase C-δ1 pathway. Endocrinology 2010, 151, 876–885. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, H.; Sano, H.; Iizuka, K.; Okada, H.; Kudo, T.; Kageyama, K.; Muramoto, S.; Murakami, T.; Okamoto, H.; Mochizuki, N. Phosphatidylinositol metabolism in hypertrophic rat heart. Circ. Res. 1993, 72, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Shoki, M.; Kawaguchi, H.; Okamoto, H.; Sano, H.; Sawa, H.; Kudo, T.; Hirao, N.; Sakata, Y.; Yasuda, H. Phosphatidylinositol and inositolphosphatide metabolism in hypertrophied rat heart. Jpn. Circ. J. 1992, 56, 142–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dent, M.R.; Aroutiounova, N.; Dhalla, N.S.; Tappia, P.S. Losartan attenuates phospholipase C isozyme gene expression in hypertrophied hearts due to volume overload. J. Cell. Mol. Med. 2006, 10, 470–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dent, M.R.; Dhalla, N.S.; Tappia, P.S. Phospholipase C gene expression, protein content and activities in cardiac hypertrophy and heart failure due to volume overload. Am. J. Physiol. 2004, 282, H719–H727. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zhang, C.; Liu, C.; Zhang, A.; Li, A.; Zhang, J.; Zhang, H. Aortic constriction induces hypertension and cardiac hypertrophy via (pro)renin receptor activation and the PLC-β3 signaling pathway. Mol. Med. Rep. 2019, 19, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalili, T.; Takeishi, Y.; Song, G.; Ball, N.A.; Howles, G.; Walsh, R.A. PKC translocation without changes in Gαq and PLC-β protein abundance in cardiac hypertrophy and failure. Am. J. Physiol. 1999, 277, H2298–H2304. [Google Scholar] [PubMed]
- Wang, H.; Oestreich, E.A.; Maekawa, N.; Bullard, T.A.; Vikstrom, K.L.; Dirksen, R.T.; Kelley, G.G.; Blaxall, B.C.; Smrcka, A.V. Phospholipase C ε modulates β-adrenergic receptor-dependent cardiac contraction and inhibits cardiac hypertrophy. Circ. Res. 2005, 97, 1305–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganguly, P.K.; Lee, S.L.; Beamish, R.E.; Dhalla, N.S. Altered sympathetic system and adrenoceptors during the development of cardiac hypertrophy. Am. Heart J. 1989, 118, 520–525. [Google Scholar] [CrossRef]
- Adams, J.W.; Sakata, Y.; Davis, M.G.; Sah, V.P.; Wang, Y.; Liggett, S.B.; Chien, K.R.; Brown, J.H.; Dorn, G.W., 2nd. Enhanced Gαq signaling: A common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc. Natl. Acad. Sci. USA 1998, 95, 10140–10145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, D.D.; Sakata, Y.; Lorenz, J.N.; Boivin, G.P.; Walsh, R.A.; Liggett, S.B.; Dorn, G.W., 2nd. Transgenic Gαq overexpression induces cardiac contractile failure in mice. Proc. Natl. Acad. Sci. USA 1997, 94, 8121–8126. [Google Scholar] [CrossRef] [Green Version]
- Sakata, Y.; Hoit, B.D.; Liggett, S.B.; Walsh, R.A.; Dorn, G.W., 2nd. Decompensation of pressure-overload hypertrophy in Gαq-overexpressing mice. Circulation 1998, 97, 1488–1495. [Google Scholar] [CrossRef] [Green Version]
- Sussman, M.A.; Welch, S.; Walker, A.; Klevitsky, R.; Hewett, T.E.; Price, R.L.; Schaefer, E.; Yager, K. Altered focal adhesion regulation correlates with cardiomyopathy in mice expressing constitutively active rac1. J. Clin. Investig. 2000, 105, 875–886. [Google Scholar] [CrossRef] [Green Version]
- Mende, U.; Kagen, A.; Cohen, A.; Aramburu, J.; Schoen, F.J.; Neer, E.J. Transient cardiac expression of constitutively active Gαq leads to hypertrophy and dilated cardiomyopathy by calcineurin-dependent and independent pathways. Proc. Natl. Acad. Sci. USA 1998, 95, 13893–13898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mende, U.; Semsarian, C.; Martins, D.C.; Kagen, A.; Duffy, C.; Schoen, F.J.; Neer, E.J. Dilated cardiomyopathy in two transgenic mouse lines expressing activated G protein αq: Lack of correlation between phospholipase C activation and the phenotype. J. Mol. Cell. Cardiol. 2001, 33, 1477–14791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Malik, S.; Kelley, G.G.; Kapiloff, M.S.; Smrcka, A.V. Phospholipase C ε scaffolds to muscle-specific A kinase anchoring protein (mAKAPβ) and integrates multiple hypertrophic stimuli in cardiac myocytes. J. Biol. Chem. 2011, 286, 23012–23021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, J.F.; Matkovich, S.J.; Mitchell, C.J.; Biden, T.J.; Woodcock, E.A. Evidence for selective coupling of α1-adrenergic receptors to phospholipase C-β1 in rat neonatal cardiomyocytes. J. Biol. Chem. 2001, 276, 37341–37346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubb, D.R.; Vasilevski, O.; Huynh, H.; Woodcock, E.A. The extreme C-terminal region of phospholipase Cβ1 determines subcellular localization and function; the “b” splice variant mediates alpha1-adrenergic receptor responses in cardiomyocytes. FASEB J. 2008, 22, 2768–2774. [Google Scholar] [CrossRef]
- Filtz, T.M.; Grubb, D.R.; McLeod-Dryden, T.J.; Luo, J.; Woodcock, E.A. Gq-initiated cardiomyocyte hypertrophy is mediated by phospholipase Cβ1b. FASEB J. 2009, 23, 3564–3570. [Google Scholar] [CrossRef]
- Singal, T.; Dhalla, N.S.; Tappia, P.S. Regulation of c-Fos and c-Jun gene expression by phospholipase C activity in adult cardiomyocytes. Mol. Cell. Biochem. 2009, 327, 229–239. [Google Scholar] [CrossRef]
- Singal, T.; Dhalla, N.S.; Tappia, P.S. Reciprocal regulation of transcription factors and PLC isozyme gene expression in adult cardiomyocytes. J. Cell. Mol. Med. 2010, 14, 1824–1835. [Google Scholar] [CrossRef] [Green Version]
- Singal, T.; Dhalla, N.S.; Tappia, P.S. Norepinephrine-induced changes in gene expression of phospholipase C in cardiomyocytes. J. Mol. Cell. Cardiol. 2006, 41, 126–137. [Google Scholar] [CrossRef]
- Giles, T.D.; Sander, G.E.; Thomas, M.G.; Quiroz, A.C. α-adrenergic mechanisms in the pathophysiology of left ventricular heart failure-An analysis of their role in systolic and diastolic dysfunction. J. Mol. Cell. Cardiol. 1996, 18, 33–43. [Google Scholar] [CrossRef]
- Motz, W.; Klepzig, M.; Strauer, B.E. Regression of cardiac hypertrophy: Experimental and clinical results. J. Cardiovasc. Pharmacol. 1987, 10, S148–S152. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; O’Neil, C.L.; Bharadwaj, B. Effect of prolonged prazosin treatment on hemodynamic and biochemical changes in the dog heart due to chronic pressure overload. Jpn. Heart J. 1984, 25, 461–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauer, B.E.; Bayer, F.; Brecht, H.M.; Motz, W. The influence of sympathetic nervous activity on regression of cardiac hypertrophy. J. Hypertens. Suppl. 1985, 3, S39–S44. [Google Scholar]
- Strauer, B.E. Progression and regression of heart hypertrophy in arterial hypertension: Pathophysiology and clinical aspects. Z. Kardiol. 1995, 74, 171–178. [Google Scholar]
- Babick, A.; Elimban, V.; Dhalla, N.S. Reversal of cardiac remodeling and subcellular defects by prazosin in heart failure due to myocardial infarction. J. Clin. Exp. Cardiol. 2012, 5, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Babick, A.; Elimban, V.; Zieroth, S.; Dhalla, N.S. Reversal of cardiac dysfunction and subcellular alterations by metoprolol in heart failure due to myocardial infarction. J. Cell Physiol. 2013, 228, 2063–2070. [Google Scholar] [CrossRef]
- Massart, P.E.; Donckier, J.; Kyselovic, J.; Godfraind, T.; Heyndrickx, G.R.; Wibo, M. Carvedilol and lacidipine prevent cardiac hypertrophy and endothelin-1 gene expression after aortic banding. Hypertension 1999, 34, 1197–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosendorff, C. Beta-blocking agents with vasodilator activity. J. Hypertens. Suppl. 1993, 11, S37–S40. [Google Scholar] [CrossRef] [PubMed]
- Antani, J.A.; Antani, N.J.; Nanivadekar, A.S. Prazosin in chronic congestive heart failure due to ischemic heart disease. Clin. Cardiol. 1991, 14, 495–500. [Google Scholar] [CrossRef]
- Colucci, W.S.; Wynne, J.; Holman, B.L.; Braunwald, E. Long-term therapy of heart failure with prazosin: A randomized double blind trial. Am. J. Cardiol. 1980, 45, 337–344. [Google Scholar] [CrossRef]
- Goldman, S.A.; Johnson, L.L.; Escala, E.; Cannon, P.J.; Weiss, M.B. Improved exercise ejection fraction with long-term prazosin therapy in patients with heart failure. Am. J. Med. 1980, 68, 36–42. [Google Scholar] [CrossRef]
- Gomes, M.E.; Mulder, A.; Bellersen, L.; Verheugt, F.W.; Smits, P.; Tack, C.J. α-receptor blockade improves muscle perfusion and glucose uptake in heart failure. Eur. J. Heart Fail. 2010, 12, 1061–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukin, M.L.; Kalman, J.; Mannino, M.; Freudenberger, R.; Buchholz, C.; Ocampo, O. Combined α-β blockade (doxazosin plus metoprolol) compared with beta blockade alone in chronic congestive heart failure. Am. J. Cardiol. 1996, 77, 486–491. [Google Scholar] [CrossRef]
- Poole-Wilson, P.A.; Swedberg, K.; Cleland, J.G.; Di Lenarda, A.; Hanrath, P.; Komajda, M.; Lubsen, J.; Lutiger, B.; Metra, M.; Remme, W.J.; et al. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol or Metoprolol European Trial (COMET): Randomised controlled trial. Lancet 2003, 362, 7–13. [Google Scholar] [CrossRef] [Green Version]
- ALLHAT Collaborative Research Group. Major cardiovascular events in hypertensive patients randomized to doxazosin vs. chlorthalidone: The antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT). JAMA 2000, 283, 1967–1975. [Google Scholar] [CrossRef] [Green Version]
- Dhaliwal, A.S.; Habib, G.; Deswal, A.; Verduzco, M.; Souchek, J.; Ramasubbu, K.; Aguilar, D.; Ma, T.S.; Jneid, H.M.; Bolos, M.; et al. Impact of α1-adrenergic antagonist use for benign prostatic hypertrophy on outcomes in patients with heart failure. Am. J. Cardiol. 2009, 104, 270–275. [Google Scholar] [CrossRef]
- Kokoz, Y.M.; Evdokimovskii, E.V.; Maltsev, A.V.; Nenov, M.N.; Nakipova, O.V.; Averin, A.S.; Pimenov, O.Y.; Teplov, I.Y.; Berezhnov, A.V.; Reyes, S.; et al. Sarcolemmal α2-adrenoceptors control protective cardiomyocyte-delimited sympathoadrenal response. J. Mol. Cell. Cardiol. 2016, 100, 9–20. [Google Scholar] [CrossRef]
- Alekseev, A.E.; Park, S.; Pimenov, O.Y.; Reyes, S.; Terzic, A. Sarcolemmal α2-adrenoceptors in feedback control of myocardial response to sympathetic challenge. Pharmacol. Ther. 2019, 197, 179–190. [Google Scholar] [CrossRef]
- Maltsev, A.V.; Kokoz, Y.M.; Evdokimovskii, E.V.; Pimenov, O.Y.; Reyes, S.; Alekseev, A.E. Alpha-2 adrenoceptors and imidazoline receptors in cardiomyocytes mediate counterbalancing effect of agmatine on NO synthesis and intracellular calcium handling. J. Mol. Cell. Cardiol. 2014, 68, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Xu, Y.J.; Sheu, S.S.; Tappia, P.S.; Panagia, V. Phosphatidic acid: A potential signal transducer for cardiac hypertrophy. J. Mol. Cell. Cardiol. 1997, 29, 2865–2871. [Google Scholar] [CrossRef] [PubMed]
- Tappia, P.S. Phospholipid-mediated signaling systems as novel targets for treatment of heart disease. Can. J. Physiol. Pharmacol. 2007, 85, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Tappia, P.S.; Asemu, G.; Rodriguez-Leyva, D. Phospholipase C as a potential target for cardioprotection during oxidative stress. Can. J. Physiol. Pharmacol. 2010, 88, 249–263. [Google Scholar] [CrossRef]
- Tappia, P.S.; Dent, M.R.; Dhalla, N.S. Oxidative stress and redox regulation of phospholipase D in myocardial disease. Free Radic. Biol. Med. 2006, 41, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Tappia, P.S.; Dhalla, N.S. Mechanisms for the defects in phospholipid signal transduction in diabetic cardiomyopathy. Indian J. Biochem. Biophys. 2014, 51, 431–440. [Google Scholar] [PubMed]
- Tappia, P.S.; Singal, T. Phospholipid-mediated signaling and heart disease. Subcell. Biochem. 2008, 49, 299–324. [Google Scholar] [PubMed]
- Tappia, P.S.; Elimban, V.; Dhalla, N.S. Involvement of phospholipase C in the norepinephrine—Induced hypertrophic response in cardiomyocytes. Scr. Med. 2022, 53, 149–157. [Google Scholar] [CrossRef]
- Tappia, P.S.; Ramjiawan, B.; Dhalla, N.S. Role of phospholipase C in catecholamine-induced increase in myocardial protein synthesis. Can. J. Physiol. Pharmacol. 2022; in press. [Google Scholar] [CrossRef]
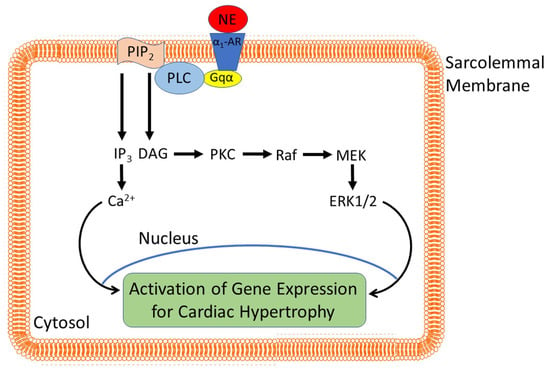
Figure 1.
Norepinephrine-induced signal transduction resulting in cardiomyocyte hypertrophy. α1-AR, α1-adrenoceptors; Gαq, Gq protein alpha subunit; PLC, phospholipase C. There are several proteins that are phosphorylated such as ERK1/2, PKC, Ca2+/Calmodulin-dependent kinase, and JNK [32], which in turn phosphorylate and activate transcription factors such as c-Fos and c-Jun leading to the gene expression of PLC as well as the expression of fetal genes that are characteristic of cardiac hypertrophy [9,33].
Figure 1.
Norepinephrine-induced signal transduction resulting in cardiomyocyte hypertrophy. α1-AR, α1-adrenoceptors; Gαq, Gq protein alpha subunit; PLC, phospholipase C. There are several proteins that are phosphorylated such as ERK1/2, PKC, Ca2+/Calmodulin-dependent kinase, and JNK [32], which in turn phosphorylate and activate transcription factors such as c-Fos and c-Jun leading to the gene expression of PLC as well as the expression of fetal genes that are characteristic of cardiac hypertrophy [9,33].

Figure 2.
Role of phospholipase C in the perpetuation of cardiomyocyte growth response to norepinephrine. DAG = sn-1,2-diacylglycerol; IP3, inositol-1,4,5-trisphosphate. The response to increased levels of norepinephrine (input) is a cyclical process that produces an increase in the expression of PLC isozyme genes and subsequent higher generation of DAG and IP3 and stimulation of the signal for cardiac hypertrophy (output). The activation and amplification of PLC isozymes ensure continuation of this cycle of events for continuation of abnormal hypertrophic growth due sustained exposure to high levels of norepinephrine.
Figure 2.
Role of phospholipase C in the perpetuation of cardiomyocyte growth response to norepinephrine. DAG = sn-1,2-diacylglycerol; IP3, inositol-1,4,5-trisphosphate. The response to increased levels of norepinephrine (input) is a cyclical process that produces an increase in the expression of PLC isozyme genes and subsequent higher generation of DAG and IP3 and stimulation of the signal for cardiac hypertrophy (output). The activation and amplification of PLC isozymes ensure continuation of this cycle of events for continuation of abnormal hypertrophic growth due sustained exposure to high levels of norepinephrine.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Specific alterations in transcription factor gene expression in cardiomyocytes treated with norepinephrine and phenylephrine.
Table 1.
Specific alterations in transcription factor gene expression in cardiomyocytes treated with norepinephrine and phenylephrine.
| Transcription Factor mRNA Level (% of Control) | ||||||
|---|---|---|---|---|---|---|
| NFAT3 (99 bp) | NFκB (124 bp) | MEF2C (92 bp) | MEF2D (105 bp) | c-Fos (74 bp) | c-Jun (163 bp) | |
| Agonist | ||||||
| NE | 110 ± 8 | 112 ± 9 | 121 ± 11 | 113 ± 6 | 268 ± 12 * | 217 ± 6 * |
| PhE | 125 ± 11 | 109 ± 8 | 127 ± 8 | 130 ± 13 | 261 ± 7 * | 225 ± 8 * |
* Significantly different (p < 0.05) vs. control value. Information presented is based on the data in our paper [78].
Table 2.
Inhibition of norepinephrine the increases in PLC isozyme gene expression (A) and activities (B) in response to norepinephrine in cardiomyocytes transfected with c-Fos and c-Jun siRNA.
Table 2.
Inhibition of norepinephrine the increases in PLC isozyme gene expression (A) and activities (B) in response to norepinephrine in cardiomyocytes transfected with c-Fos and c-Jun siRNA.
| A: | PLC mRNA Levels (% of Control) | |||
| β1 (114 bp) | β3 (230 bp) | γ1 (123 bp) | δ1 (190 bp) | |
| NE | 219 ± 11 * | 182 ± 18 * | 168 ± 18 * | 221 ± 18 * |
| NE + cFos siRNA | 80 ± 21 # | 89 ± 11 # | 160 ± 15 # | 218 ± 17 # |
| NE + cJun siRNA | 75 ± 19 # | 91 ± 9 # | 170 ± 14 # | 74 ± 8 # |
| B: | Inositol Phosphates (pmol/min/mg Protein) | |||
| PLC β1 | PLC β3 | PLC δ1 | ||
| Control | 2.8 ± 0.6 | 4.1 ± 0.8 | 10.0 ± 1.4 | |
| NE | 6.8 ± 1.0 * | 7.0 ± 1.3 * | 17.1 ± 3.0 * | |
| NE + cFos siRNA | 3.5 ± 0.7 # | 4.5 ± 0.8 # | 16.6 ± 2.4 | |
| NE + cJun siRNA | 3.0 ± 0.8 # | 3.9 ± 0.9 # | 11.8 ± 2.6 # | |
* Significantly different (p < 0.05) vs. control; # significantly different (p < 0.05) vs. NE. NE, norepinephrine; siRNA = small interfering RNA. Information presented is based on the data in our paper [79].
Table 3.
Pharmacological and gene silencing interventions for the inhibition of increases in c-Fos and c-Jun gene expression levels in response to norepinephrine.
Table 3.
Pharmacological and gene silencing interventions for the inhibition of increases in c-Fos and c-Jun gene expression levels in response to norepinephrine.
| Condition | c-Fos mRNA Expression Levels | c-Jun mRNA Expression Levels |
|---|---|---|
| NE | 214 ± 23 * | 198 ± 20 * |
| +Prazosin | 114 ± 11# | 100 ± 8 # |
| +U73122 | 83 ± 8 # | 95 ± 9 # |
| +PLC β1 siRNA | 110 ± 8 # | 85 ± 8 # |
| +PLC β3 siRNA | 105 ± 7 # | 95 ± 11 # |
| +PLC γ1 siRNA | 175 ± 25 * | 180 ± 13 * |
| +PLC δ1 siRNA | 108 ± 7 # | 80 ± 7 # |
Table 4.
Attenuation of the increases in phospholipase C gene expression due to norepinephrine by pharmacological and gene silencing interventions.
Table 4.
Attenuation of the increases in phospholipase C gene expression due to norepinephrine by pharmacological and gene silencing interventions.
| PLC Isozyme mRNA Levels (% of Control) | ||||
|---|---|---|---|---|
| Condition | β1 | β3 | γ1 | δ1 |
| NE | 201 ± 9 * | 188 ± 8 * | 181 ± 9 * | 159 ± 8 * |
| +Prazosin | 99 ± 11 # | 102 ± 4 # | 120 ± 5 # | 90 ± 4 # |
| +U73122 | 68 ± 5 # | 80 ± 4 # | 103 ± 11 # | 67 ± 12 # |
| +PLC β1 siRNA | 90 ± 8 # | - | - | - |
| +PLC β3 siRNA | - | 80 ± 9 # | - | - |
| +PLC γ1 siRNA | - | - | 61 ± 7 * | - |
| +PLC δ1 siRNA | - | - | - | 60 ± 8 # |
Table 5.
PLC isozyme gene expression in adult rat cardiomyocytes treated with phorbol 12-myristate 13-acetate, bisindolylmaleimide, or PD98509.
Table 5.
PLC isozyme gene expression in adult rat cardiomyocytes treated with phorbol 12-myristate 13-acetate, bisindolylmaleimide, or PD98509.
| PLC mRNA Levels (% of Control) | ||||
|---|---|---|---|---|
| Treatment | β1 | β3 | γ1 | δ1 |
| PMA (µM) | ||||
| 10.1 | 118 ± 11 | 131 ± 8 | 110 ± 10 | 108 ± 10 |
| 1.0 | 190 ± 12 * | 183 ± 9 * | 176 ± 13 * | 161 ± 15 * |
| 10.0 | 175 ± 11 * | 171 ± 10 * | 130 ± 12 * | 123 ± 12 * |
| NE (µM) | ||||
| 5.0 | 223 ± 11 * | 192 ± 19 * | 186 ± 15 * | 193 ± 15 * |
| +Bis-1 (nM) | ||||
| 50 | 95 ± 10 # | 96 ± 9 # | 111 ± 14 # | 100 ± 8 # |
| 100 | 83 ± 8 # | 80 ± 12 # | 100 ± 10 # | 82 ± 9 # |
| 200 | 82 ± 9 # | 75 ± 6 # | 82 ± 9 # | 77 ± 9 # |
| +PD98059 (nM) | ||||
| 2 | 107 ± 11 # | 90 ± 9 # | 117 ± 14 # | 120 ± 11 # |
| 10 | 84 ± 9 # | 87 ± 11 # | 103 ± 9 # | 94 ± 8 # |
| 25 | 77 ± 8 # | 82 ± 7 # | 82 ± 11 # | 90 ± 12 # |
* Significantly different (p < 0.05) vs. control; # significantly different (p < 0.05) vs. NE. NE, norepinephrine; PMA, phorbol myristate acetate; Bis-1, bisindolylmaleimide Information presented is based on the data in our paper [80].
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tappia, P.S.; Dhalla, N.S. Upregulation of Phospholipase C Gene Expression Due to Norepinephrine-Induced Hypertrophic Response. Cells 2022, 11, 2488. https://doi.org/10.3390/cells11162488
AMA Style
Tappia PS, Dhalla NS. Upregulation of Phospholipase C Gene Expression Due to Norepinephrine-Induced Hypertrophic Response. Cells. 2022; 11(16):2488. https://doi.org/10.3390/cells11162488
Chicago/Turabian StyleTappia, Paramjit S., and Naranjan S. Dhalla. 2022. "Upregulation of Phospholipase C Gene Expression Due to Norepinephrine-Induced Hypertrophic Response" Cells 11, no. 16: 2488. https://doi.org/10.3390/cells11162488
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.