
FK506-Binding Protein 11 Is a Novel Plasma Cell-Specific Antibody Folding Catalyst with Increased Expression in Idiopathic Pulmonary Fibrosis

 , , , , , , , , ,
, , , , , , , , ,  add
Show full author list
add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Gene Expression Data
2.3. Cell Culture, Induction of ER Stress and Transfection
2.4. Plasma Cell Differentiation
2.5. RNA Isolation and Real-Time Quantitative Reverse-Transcriptase PCR (qRT-PCR) Analysis
2.6. Protein Isolation and Western Blot Analysis
2.7. Flow Cytometry Analysis
2.8. Immunofluorescent Stainings
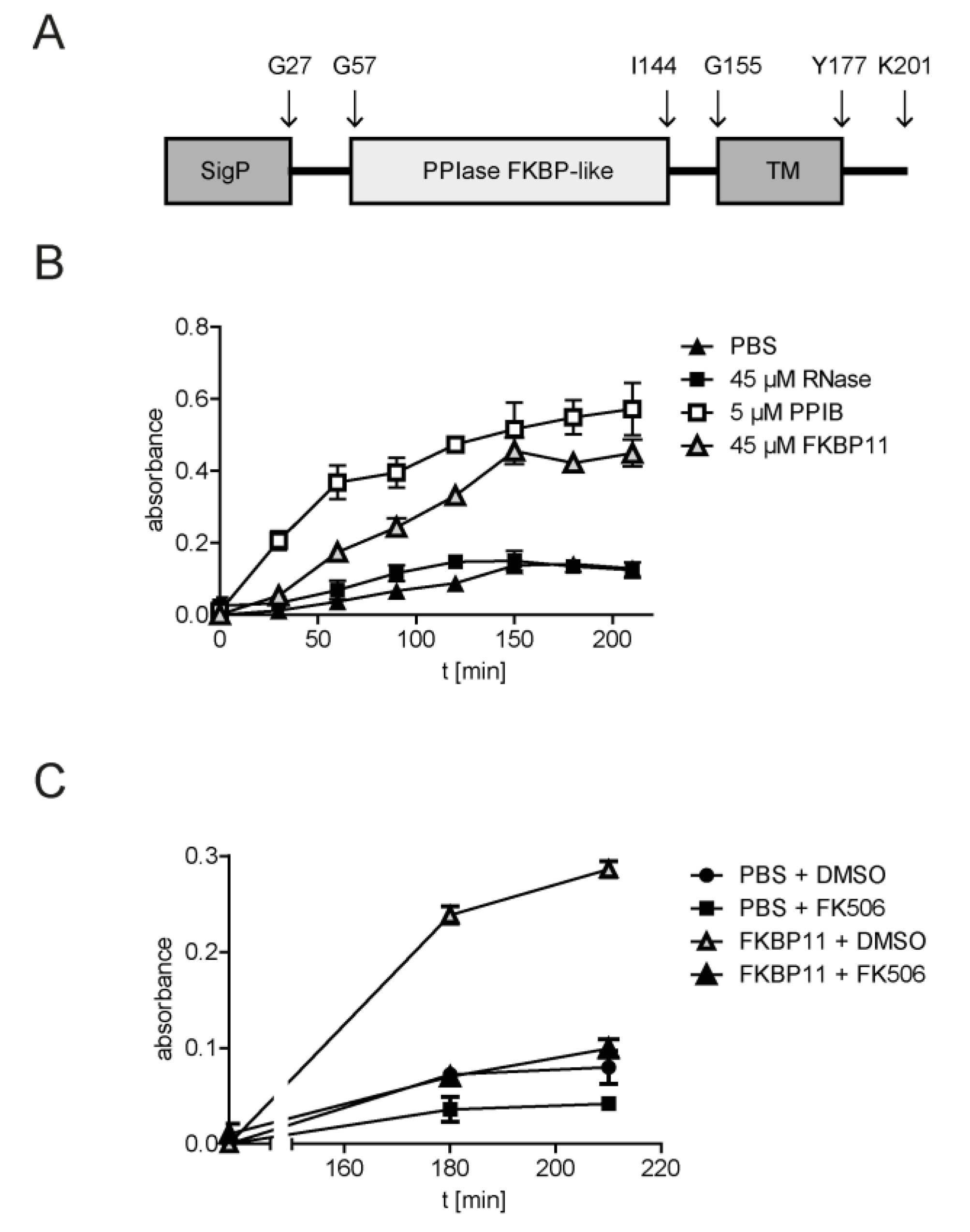
2.9. Unfolding and Refolding of Immunoglobulin G
2.10. ELISA
3. Results
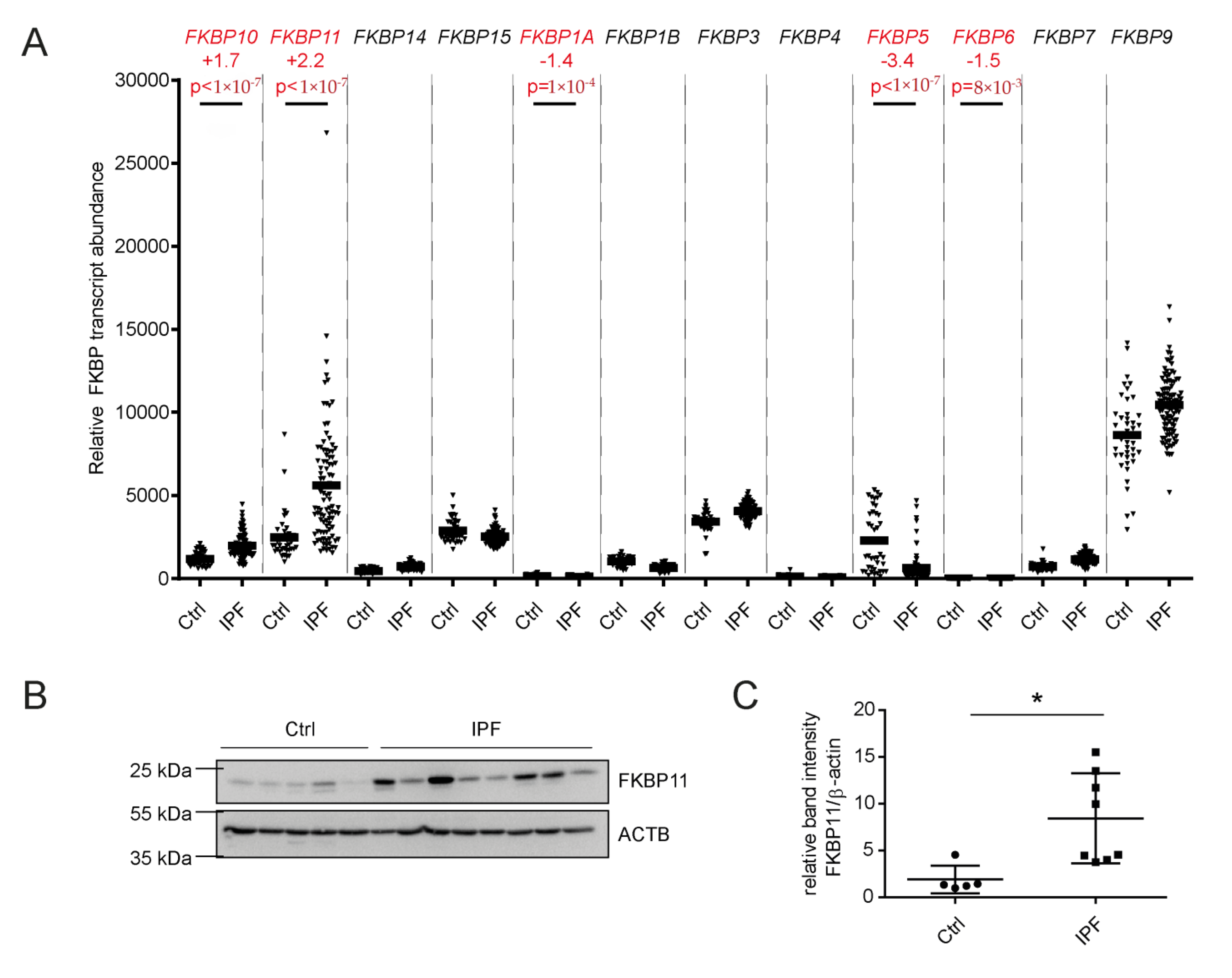
3.1. FKBP11 Expression Is Increased in IPF Lungs
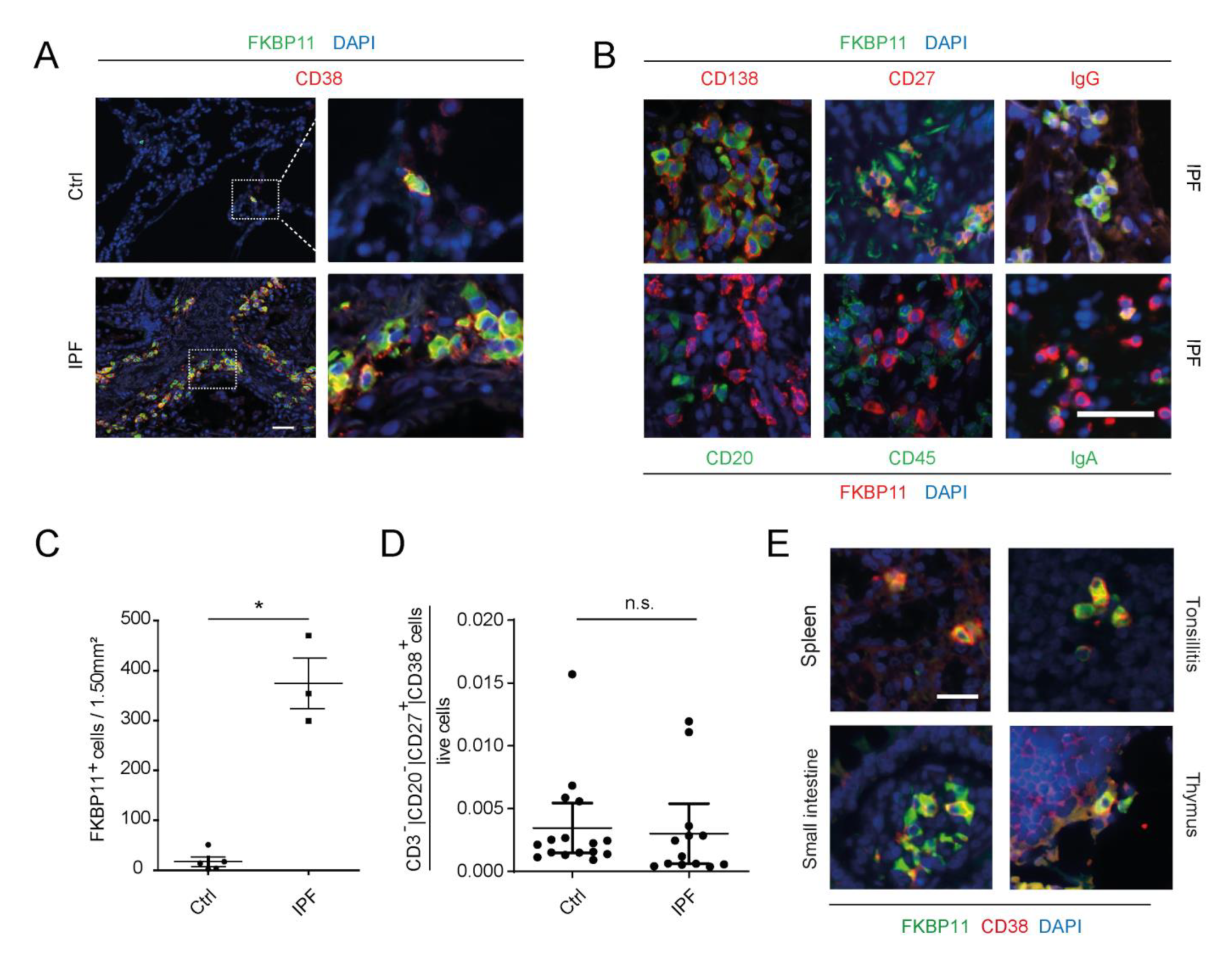
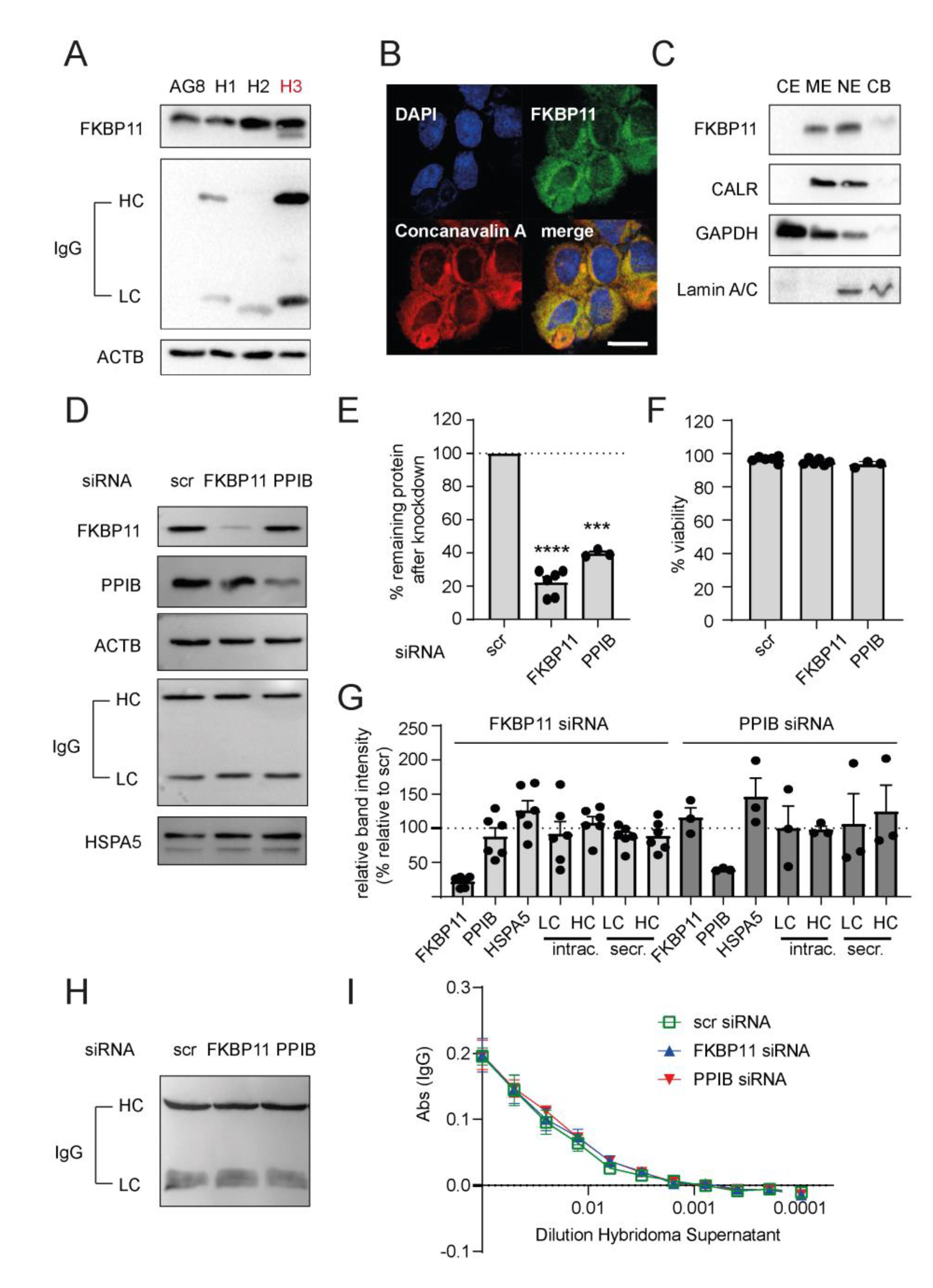
3.2. FKBP11 Localizes Mainly to CD27+/CD38+/CD138+/CD20−/CD45− Plasma Cells
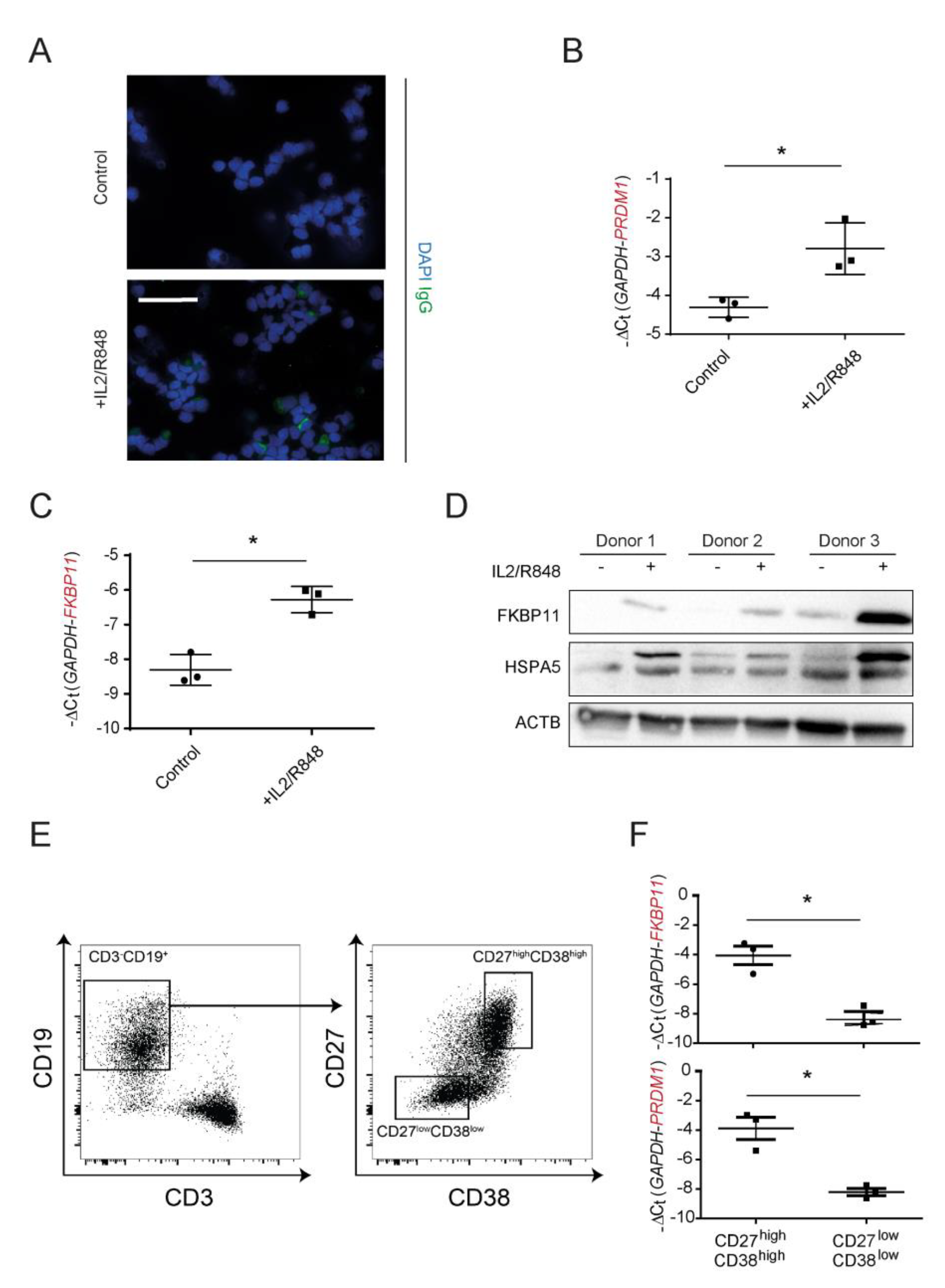
3.3. FKBP11 Is Upregulated during Plasma Cell Transdifferentiation
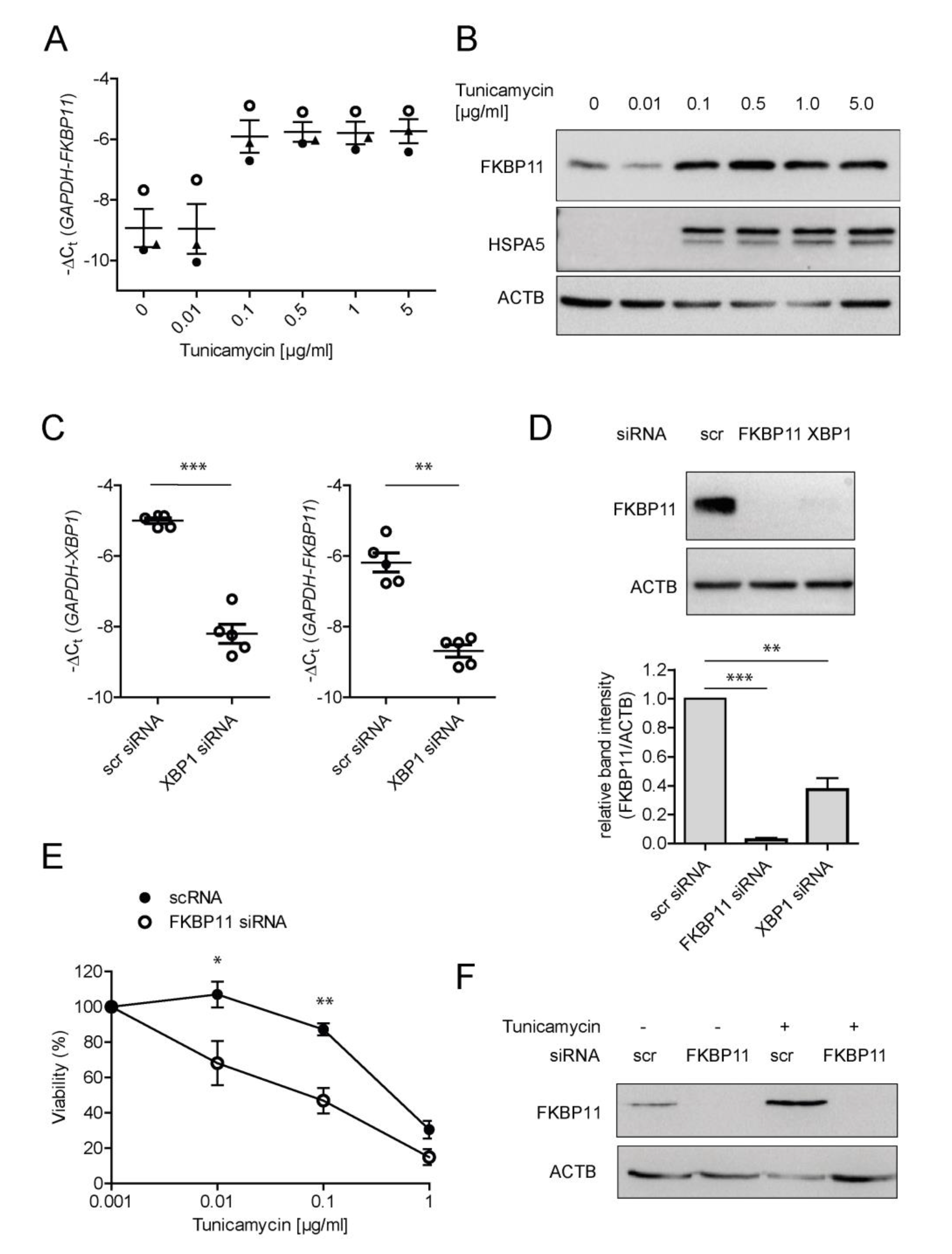
3.4. Expression of FKBP11 Is Induced by the Transcription Factor X-Box Binding Protein 1 (XBP1) and Protects an Alveolar Cell Line from ER-Stress Induced Cell Death
3.5. Recombinant FKBP11 Folds IgG Antibody In Vitro
3.6. Neither Knockdown of FKBP11 Nor Knockdown of Cyclophilin B Affects IgG Yield of an Antibody-Producing Hybridoma Cell Line
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flajnik, M.F.; Kasahara, M. Origin and evolution of the adaptive immune system: Genetic events and selective pressures. Nat. Rev. Genet. 2010, 11, 47–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feige, M.J.; Hendershot, L.M.; Buchner, J. How antibodies fold. Trends Biochem. Sci. 2010, 35, 189–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feige, M.J.; Buchner, J. Principles and engineering of antibody folding and assembly. Biochim. Biophys. Acta 2014, 1844, 2024–2031. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.A.; Hollien, J. The unfolded protein response in secretory cell function. Annu. Rev. Genet. 2012, 46, 165–183. [Google Scholar] [CrossRef]
- Todd, D.J.; Lee, A.H.; Glimcher, L.H. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 2008, 8, 663–674. [Google Scholar] [CrossRef]
- Janssens, S.; Pulendran, B.; Lambrecht, B.N. Emerging functions of the unfolded protein response in immunity. Nat. Immunol. 2014, 15, 910–919. [Google Scholar] [CrossRef] [Green Version]
- Taubenheim, N.; Tarlinton, D.M.; Crawford, S.; Corcoran, L.M.; Hodgkin, P.D.; Nutt, S.L. High rate of antibody secretion is not integral to plasma cell differentiation as revealed by XBP-1 deficiency. J. Immunol. 2012, 189, 3328–3338. [Google Scholar] [CrossRef] [Green Version]
- Todd, D.J.; McHeyzer-Williams, L.J.; Kowal, C.; Lee, A.H.; Volpe, B.T.; Diamond, B.; McHeyzer-Williams, M.G.; Glimcher, L.H. XBP1 governs late events in plasma cell differentiation and is not required for antigen-specific memory B cell development. J. Exp. Med. 2009, 206, 2151–2159. [Google Scholar] [CrossRef] [Green Version]
- Harikishore, A.; Yoon, H.S. Immunophilins: Structures, Mechanisms and Ligands. Curr. Mol. Pharmacol. 2015, 9, 37–47. [Google Scholar] [CrossRef]
- Meunier, L.; Usherwood, Y.K.; Chung, K.T.; Hendershot, L.M. A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol. Biol. Cell 2002, 13, 4456–4469. [Google Scholar] [CrossRef]
- Feige, M.J.; Groscurth, S.; Marcinowski, M.; Shimizu, Y.; Kessler, H.; Hendershot, L.M.; Buchner, J. An unfolded CH1 domain controls the assembly and secretion of IgG antibodies. Mol. Cell 2009, 34, 569–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Choi, T.G.; Ha, J.; Kim, S.S. Cyclosporine A suppresses immunoglobulin G biosynthesis via inhibition of cyclophilin B in murine hybridomas and B cells. Int. Immunopharmacol. 2012, 12, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Jansen, G.; Maattanen, P.; Denisov, A.Y.; Scarffe, L.; Schade, B.; Balghi, H.; Dejgaard, K.; Chen, L.Y.; Muller, W.J.; Gehring, K.; et al. An interaction map of endoplasmic reticulum chaperones and foldases. Mol. Cell. Proteom. 2012, 11, 710–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lilie, H.; Lang, K.; Rudolph, R.; Buchner, J. Prolyl isomerases catalyze antibody folding in vitro. Protein Sci. 1993, 2, 1490–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, M.; Jiang, Y. FK506-Binding Proteins and Their Diverse Functions. Curr. Mol. Pharmacol. 2015, 9, 48–65. [Google Scholar] [CrossRef] [PubMed]
- Rulten, S.L.; Kinloch, R.A.; Tateossian, H.; Robinson, C.; Gettins, L.; Kay, J.E. The human FK506-binding proteins: Characterization of human FKBP19. Mamm. Genome 2006, 17, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Bensellam, M.; Chan, J.Y.; Lee, K.; Joglekar, M.V.; Hardikar, A.A.; Loudovaris, T.; Thomas, H.E.; Jonas, J.C.; Laybutt, D.R. Phlda3 regulates beta cell survival during stress. Sci. Rep. 2019, 9, 12827. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.Y.; Lee, K.; Maxwell, E.L.; Liang, C.; Laybutt, D.R. Macrophage alterations in islets of obese mice linked to beta cell disruption in diabetes. Diabetologia 2019, 62, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Bensellam, M.; Maxwell, E.L.; Chan, J.Y.; Luzuriaga, J.; West, P.K.; Jonas, J.C.; Gunton, J.E.; Laybutt, D.R. Hypoxia reduces ER-to-Golgi protein trafficking and increases cell death by inhibiting the adaptive unfolded protein response in mouse beta cells. Diabetologia 2016, 59, 1492–1502. [Google Scholar] [CrossRef] [Green Version]
- Hanagata, N.; Li, X. Osteoblast-enriched membrane protein IFITM5 regulates the association of CD9 with an FKBP11-CD81-FPRP complex and stimulates expression of interferon-induced genes. Biochem. Biophys. Res. Commun. 2011, 409, 378–384. [Google Scholar] [CrossRef]
- Hanagata, N.; Li, X.; Morita, H.; Takemura, T.; Li, J.; Minowa, T. Characterization of the osteoblast-specific transmembrane protein IFITM5 and analysis of IFITM5-deficient mice. J. Bone Mineral. Metab. 2011, 29, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, T.; Li, X.; Morita, H.; Minowa, T.; Aizawa, T.; Hanagata, N.; Demura, M. Role of S-palmitoylation on IFITM5 for the interaction with FKBP11 in osteoblast cells. PLoS ONE 2013, 8, e75831. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Q.; Huang, G.; Lin, B.Y.; Lin, D.; Ma, Y.; Zhang, Z.; Chen, T.; Zhou, J. Tandem Mass Tag-Based Proteomic Analysis of Potential Biomarkers for Hepatocellular Carcinoma Differentiation. Onco Targets Ther. 2021, 14, 1007–1020. [Google Scholar] [CrossRef] [PubMed]
- Lin, I.Y.; Yen, C.H.; Liao, Y.J.; Lin, S.E.; Ma, H.P.; Chan, Y.J.; Chen, Y.M. Identification of FKBP11 as a biomarker for hepatocellular carcinoma. Anticancer Res. 2013, 33, 2763–2769. [Google Scholar] [PubMed]
- Shi, W.; Liao, Y.; Willis, S.N.; Taubenheim, N.; Inouye, M.; Tarlinton, D.M.; Smyth, G.K.; Hodgkin, P.D.; Nutt, S.L.; Corcoran, L.M. Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nat. Immunol. 2015, 16, 663–673. [Google Scholar] [CrossRef]
- Tellier, J.; Shi, W.; Minnich, M.; Liao, Y.; Crawford, S.; Smyth, G.K.; Kallies, A.; Busslinger, M.; Nutt, S.L. Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat. Immunol. 2016, 17, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Ruer-Laventie, J.; Simoni, L.; Schickel, J.N.; Soley, A.; Duval, M.; Knapp, A.M.; Marcellin, L.; Lamon, D.; Korganow, A.S.; Martin, T.; et al. Overexpression of Fkbp11, a feature of lupus B cells, leads to B cell tolerance breakdown and initiates plasma cell differentiation. Immun. Inflamm. Dis. 2015, 3, 265–279. [Google Scholar] [CrossRef]
- Hofmann, K.; Clauder, A.K.; Manz, R.A. Targeting B Cells and Plasma Cells in Autoimmune Diseases. Front. Immunol. 2018, 9, 835. [Google Scholar] [CrossRef]
- Lee, D.S.W.; Rojas, O.L.; Gommerman, J.L. B cell depletion therapies in autoimmune disease: Advances and mechanistic insights. Nat. Rev. Drug Discov. 2021, 20, 179–199. [Google Scholar] [CrossRef]
- Kao, D.; Lux, A.; Schwab, I.; Nimmerjahn, F. Targeting B cells and autoantibodies in the therapy of autoimmune diseases. Semin. Immunopathol. 2014, 36, 289–299. [Google Scholar] [CrossRef]
- Hutchinson, J.; Fogarty, A.; Hubbard, R.; McKeever, T. Global incidence and mortality of idiopathic pulmonary fibrosis: A systematic review. Eur. Respir. J. 2015, 46, 795–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet 2012, 380, 680–688. [Google Scholar] [CrossRef]
- Chakraborty, A.; Mastalerz, M.; Ansari, M.; Schiller, H.B.; Staab-Weijnitz, C.A. Emerging Roles of Airway Epithelial Cells in Idiopathic Pulmonary Fibrosis. Cells 2022, 11, 1050. [Google Scholar] [CrossRef]
- Wells, A.U.; Denton, C.P. Interstitial lung disease in connective tissue disease--mechanisms and management. Nat. Rev. Rheumatol. 2014, 10, 728–739. [Google Scholar] [CrossRef]
- Komura, K.; Yanaba, K.; Horikawa, M.; Ogawa, F.; Fujimoto, M.; Tedder, T.F.; Sato, S. CD19 regulates the development of bleomycin-induced pulmonary fibrosis in a mouse model. Arthritis Rheum. 2008, 58, 3574–3584. [Google Scholar] [CrossRef]
- Vittal, R.; Mickler, E.A.; Fisher, A.J.; Zhang, C.; Rothhaar, K.; Gu, H.; Brown, K.M.; Emtiazdjoo, A.; Lott, J.M.; Frye, S.B.; et al. Type V collagen induced tolerance suppresses collagen deposition, TGF-beta and associated transcripts in pulmonary fibrosis. PLoS ONE 2013, 8, e76451. [Google Scholar] [CrossRef]
- Shum, A.K.; Alimohammadi, M.; Tan, C.L.; Cheng, M.H.; Metzger, T.C.; Law, C.S.; Lwin, W.; Perheentupa, J.; Bour-Jordan, H.; Carel, J.C.; et al. BPIFB1 is a lung-specific autoantigen associated with interstitial lung disease. Sci. Transl. Med. 2013, 5, 206ra139. [Google Scholar] [CrossRef] [Green Version]
- Mehta, H.; Goulet, P.O.; Nguyen, V.; Perez, G.; Koenig, M.; Senecal, J.L.; Sarfati, M. Topoisomerase I peptide-loaded dendritic cells induce autoantibody response as well as skin and lung fibrosis. Autoimmunity 2016, 49, 503–513. [Google Scholar] [CrossRef]
- Yoshizaki, A.; Iwata, Y.; Komura, K.; Ogawa, F.; Hara, T.; Muroi, E.; Takenaka, M.; Shimizu, K.; Hasegawa, M.; Fujimoto, M.; et al. CD19 regulates skin and lung fibrosis via Toll-like receptor signaling in a model of bleomycin-induced scleroderma. Am. J. Pathol. 2008, 172, 1650–1663. [Google Scholar] [CrossRef] [Green Version]
- Francois, A.; Gombault, A.; Villeret, B.; Alsaleh, G.; Fanny, M.; Gasse, P.; Adam, S.M.; Crestani, B.; Sibilia, J.; Schneider, P.; et al. B cell activating factor is central to bleomycin- and IL-17-mediated experimental pulmonary fibrosis. J. Autoimmun. 2015, 56, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, T.; Kobayashi, T.; Mizumaki, K.; Kano, M.; Sawada, T.; Tennichi, M.; Okamura, A.; Hamaguchi, Y.; Iwakura, Y.; Hasegawa, M.; et al. BAFF inhibition attenuates fibrosis in scleroderma by modulating the regulatory and effector B cell balance. Sci. Adv. 2018, 4, eaas9944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiller, H.B.; Mayr, C.H.; Leuschner, G.; Strunz, M.; Staab-Weijnitz, C.; Preisendorfer, S.; Eckes, B.; Moinzadeh, P.; Krieg, T.; Schwartz, D.A.; et al. Deep Proteome Profiling Reveals Common Prevalence of MZB1-Positive Plasma B Cells in Human Lung and Skin Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 1298–1310. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Kass, D.J.; Bon, J.; Vuga, L.; Tan, J.; Csizmadia, E.; Otterbein, L.; Soejima, M.; Levesque, M.C.; Gibson, K.F.; et al. Plasma B lymphocyte stimulator and B cell differentiation in idiopathic pulmonary fibrosis patients. J. Immunol. 2013, 191, 2089–2095. [Google Scholar] [CrossRef]
- Hoyne, G.F.; Elliott, H.; Mutsaers, S.E.; Prele, C.M. Idiopathic pulmonary fibrosis and a role for autoimmunity. Immunol. Cell Biol. 2017, 95, 577–583. [Google Scholar] [CrossRef]
- Xue, J.; Gochuico, B.R.; Alawad, A.S.; Feghali-Bostwick, C.A.; Noth, I.; Nathan, S.D.; Rosen, G.D.; Rosas, I.O.; Dacic, S.; Ocak, I.; et al. The HLA class II Allele DRB1*1501 is over-represented in patients with idiopathic pulmonary fibrosis. PLoS ONE 2011, 6, e14715. [Google Scholar] [CrossRef] [Green Version]
- Fingerlin, T.E.; Zhang, W.; Yang, I.V.; Ainsworth, H.C.; Russell, P.H.; Blumhagen, R.Z.; Schwarz, M.I.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; et al. Genome-wide imputation study identifies novel HLA locus for pulmonary fibrosis and potential role for auto-immunity in fibrotic idiopathic interstitial pneumonia. BMC Genet. 2016, 17, 74. [Google Scholar] [CrossRef] [Green Version]
- Preisendörfer, S.; Ishikawa, Y.; Knüppel, L.; Fernandez, I.; Binzenhofer, L.; Juan-Guardela, B.; Hennen, E.; Ruppert, C.; Guenther, A.; Kneidinger, N.; et al. FK506-binding protein 11, a novel plasma cell specific antibody folding catalyst, is increased in idiopathic pulmonary fibrosis. Eur. Respir. J. 2020, 56, 2293. [Google Scholar] [CrossRef]
- Preisendörfer, S.; Knüppel, L.; Ishikawa, Y.; Binzenhofer, L.; Fernandez, I.E.; Juan-Guardela, B.M.; Ruppert, C.; Guenther, A.; Hatz, R.; Behr, J.; et al. FK506 Binding Protein 11, a Plasma Cell-Specific Antibody Folding Catalyst, Is Increased in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 197, A2204. [Google Scholar]
- Bauer, Y.; Tedrow, J.; de Bernard, S.; Birker-Robaczewska, M.; Gibson, K.F.; Guardela, B.J.; Hess, P.; Klenk, A.; Lindell, K.O.; Poirey, S.; et al. A novel genomic signature with translational significance for human idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 52, 217–231. [Google Scholar] [CrossRef] [Green Version]
- Yang, I.V.; Pedersen, B.S.; Rabinovich, E.; Hennessy, C.E.; Davidson, E.J.; Murphy, E.; Guardela, B.J.; Tedrow, J.R.; Zhang, Y.; Singh, M.K.; et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Herazo-Maya, J.D.; Noth, I.; Duncan, S.R.; Kim, S.; Ma, S.F.; Tseng, G.C.; Feingold, E.; Juan-Guardela, B.M.; Richards, T.J.; Lussier, Y.; et al. Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci. Transl. Med. 2013, 5, 205ra136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbrunn, T.; Chatterjee, M.; Bargou, R.C.; Stuhmer, T. Efficient transient transfection of human multiple myeloma cells by electroporation--an appraisal. PLoS ONE 2014, 9, e97443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinna, D.; Corti, D.; Jarrossay, D.; Sallusto, F.; Lanzavecchia, A. Clonal dissection of the human memory B-cell repertoire following infection and vaccination. Eur. J. Immunol. 2009, 39, 1260–1270. [Google Scholar] [CrossRef]
- Laurent, S.A.; Hoffmann, F.S.; Kuhn, P.H.; Cheng, Q.; Chu, Y.; Schmidt-Supprian, M.; Hauck, S.M.; Schuh, E.; Krumbholz, M.; Rubsamen, H.; et al. gamma-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat. Commun. 2015, 6, 7333. [Google Scholar] [CrossRef]
- Winklmeier, S.; Schluter, M.; Spadaro, M.; Thaler, F.S.; Vural, A.; Gerhards, R.; Macrini, C.; Mader, S.; Kurne, A.; Inan, B.; et al. Identification of circulating MOG-specific B cells in patients with MOG antibodies. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, 625. [Google Scholar] [CrossRef] [Green Version]
- Lilie, H. Folding of the Fab fragment within the intact antibody. FEBS Lett. 1997, 417, 239–242. [Google Scholar] [CrossRef] [Green Version]
- Reinhardt, D.P.; Keene, D.R.; Corson, G.M.; Poschl, E.; Bachinger, H.P.; Gambee, J.E.; Sakai, L.Y. Fibrillin-1: Organization in microfibrils and structural properties. J. Mol. Biol. 1996, 258, 104–116. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Mizuno, K.; Bächinger, H.P. Ziploc-ing the structure 2.0: Endoplasmic reticulum-resident peptidyl prolyl isomerases show different activities toward hydroxyproline. J. Biol. Chem. 2017, 292, 9273–9282. [Google Scholar] [CrossRef] [Green Version]
- Krämer, P.M.; Weber, C.M.; Forster, S.; Rauch, P.; Kremmer, E. Analysis of DDT isomers with enzyme-linked immunosorbent assay and optical immunosensor based on rat monoclonal antibodies as biological recognition elements. J. AOAC Int. 2010, 93, 44–58. [Google Scholar] [CrossRef] [Green Version]
- Staab-Weijnitz, C.A.; Fernandez, I.E.; Knüppel, L.; Maul, J.; Heinzelmann, K.; Juan-Guardela, B.M.; Hennen, E.; Preissler, G.; Winter, H.; Neurohr, C.; et al. FK506-Binding Protein 10, a Potential Novel Drug Target for Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 455–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Herazo-Maya, J.D.; Kang, D.D.; Juan-Guardela, B.M.; Tedrow, J.; Martinez, F.J.; Sciurba, F.C.; Tseng, G.C.; Kaminski, N. Integrative phenotyping framework (iPF): Integrative clustering of multiple omics data identifies novel lung disease subphenotypes. BMC Genom. 2015, 16, 924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelin-Duclos, C.; Cattoretti, G.; Lin, K.I.; Calame, K. Commitment of B lymphocytes to a plasma cell fate is associated with Blimp-1 expression in vivo. J. Immunol. 2000, 165, 5462–5471. [Google Scholar] [CrossRef]
- Angelidis, I.; Simon, L.M.; Fernandez, I.E.; Strunz, M.; Mayr, C.H.; Greiffo, F.R.; Tsitsiridis, G.; Ansari, M.; Graf, E.; Strom, T.M.; et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat. Commun. 2019, 10, 963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gass, J.N.; Gunn, K.E.; Sriburi, R.; Brewer, J.W. Stressed-out B cells? Plasma-cell differentiation and the unfolded protein response. Trends Immunol. 2004, 25, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Iwakoshi, N.N.; Lee, A.H.; Glimcher, L.H. The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol. Rev. 2003, 194, 29–38. [Google Scholar] [CrossRef]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [Green Version]
- Kay, J.E. Structure-function relationships in the FK506-binding protein (FKBP) family of peptidylprolyl cis-trans isomerases. Biochem. J. 1996, 314 Pt 2, 361–385. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, Y.; Bachinger, H.P. A molecular ensemble in the rER for procollagen maturation. Biochim. Biophys. Acta 2013, 1833, 2479–2491. [Google Scholar] [CrossRef] [Green Version]
- Lecca, M.R.; Wagner, U.; Patrignani, A.; Berger, E.G.; Hennet, T. Genome-wide analysis of the unfolded protein response in fibroblasts from congenital disorders of glycosylation type-I patients. FASEB J. 2005, 19, 240–242. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Shapiro-Shelef, M.; Iwakoshi, N.N.; Lee, A.H.; Qian, S.B.; Zhao, H.; Yu, X.; Yang, L.; Tan, B.K.; Rosenwald, A.; et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 2004, 21, 81–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonner, J.M.; Boulianne, G.L. Diverse structures, functions and uses of FK506 binding proteins. Cell. Signal. 2017, 38, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Lilie, H.; Rudolph, R.; Buchner, J. Association of antibody chains at different stages of folding: Prolyl isomerization occurs after formation of quaternary structure. J. Mol. Biol. 1995, 248, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.; Jang, E.; Choi, S.S.; Ji, C.; Lee, K.; Youn, J. The Function of FK506-Binding Protein 13 in Protein Quality Control Protects Plasma Cells from Endoplasmic Reticulum Stress-Associated Apoptosis. Front. Immunol. 2017, 8, 222. [Google Scholar] [CrossRef] [Green Version]
- Tat, V.; Ayaub, E.A.; Ayoub, A.; Vierhout, M.; Naiel, S.; Padwal, M.K.; Abed, S.; Mekhael, O.; Tandon, K.; Revill, S.D.; et al. FK506-Binding Protein 13 Expression Is Upregulated in Interstitial Lung Disease and Correlated with Clinical Severity. A Potentially Protective Role. Am. J. Respir. Cell Mol. Biol. 2021, 64, 235–246. [Google Scholar] [CrossRef]
- Calame, K.L. Plasma cells: Finding new light at the end of B cell development. Nat. Immunol. 2001, 2, 1103–1108. [Google Scholar] [CrossRef]
- Caraux, A.; Klein, B.; Paiva, B.; Bret, C.; Schmitz, A.; Fuhler, G.M.; Bos, N.A.; Johnsen, H.E.; Orfao, A.; Perez-Andres, M.; et al. Circulating human B and plasma cells. Age-associated changes in counts and detailed characterization of circulating normal CD138- and CD138+ plasma cells. Haematologica 2010, 95, 1016–1020. [Google Scholar] [CrossRef] [Green Version]
- Feghali-Bostwick, C.A.; Wilkes, D.S. Autoimmunity in idiopathic pulmonary fibrosis: Are circulating autoantibodies pathogenic or epiphenomena? Am. J. Respir. Crit. Care Med. 2011, 183, 692–693. [Google Scholar] [CrossRef]
- Donahoe, M.; Valentine, V.G.; Chien, N.; Gibson, K.F.; Raval, J.S.; Saul, M.; Xue, J.; Zhang, Y.; Duncan, S.R. Autoantibody-Targeted Treatments for Acute Exacerbations of Idiopathic Pulmonary Fibrosis. PLoS ONE 2015, 10, e0127771. [Google Scholar] [CrossRef]
- van de Donk, N.W.; Janmaat, M.L.; Mutis, T.; Lammerts van Bueren, J.J.; Ahmadi, T.; Sasser, A.K.; Lokhorst, H.M.; Parren, P.W. Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunol. Rev. 2016, 270, 95–112. [Google Scholar] [CrossRef] [Green Version]
- Shih, T.; De, S.; Barnes, B.J. RNAi Transfection Optimized in Primary Naive B Cells for the Targeted Analysis of Human Plasma Cell Differentiation. Front. Immunol. 2019, 10, 1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, B.; Amador, G.; Viswanatha, R.; Zirin, J.; Mohr, S.E.; Perrimon, N. CRISPR-based engineering of gene knockout cells by homology-directed insertion in polyploid Drosophila S2R+ cells. Nat. Protoc. 2020, 15, 3478–3498. [Google Scholar] [CrossRef]
- Korfei, M.; Ruppert, C.; Mahavadi, P.; Henneke, I.; Markart, P.; Koch, M.; Lang, G.; Fink, L.; Bohle, R.M.; Seeger, W.; et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 838–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, W.E.; Crossno, P.F.; Polosukhin, V.V.; Roldan, J.; Cheng, D.S.; Lane, K.B.; Blackwell, T.R.; Xu, C.; Markin, C.; Ware, L.B.; et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: Association with altered surfactant protein processing and herpesvirus infection. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L1119–L1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanjore, H.; Lawson, W.E.; Blackwell, T.S. Endoplasmic reticulum stress as a pro-fibrotic stimulus. Biochim. Biophys. Acta 2013, 1832, 940–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghartey-Kwansah, G.; Li, Z.; Feng, R.; Wang, L.; Zhou, X.; Chen, F.Z.; Xu, M.M.; Jones, O.; Mu, Y.; Chen, S.; et al. Comparative analysis of FKBP family protein: Evaluation, structure, and function in mammals and Drosophila melanogaster. BMC Dev. Biol. 2018, 18, 7. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Preisendörfer, S.; Ishikawa, Y.; Hennen, E.; Winklmeier, S.; Schupp, J.C.; Knüppel, L.; Fernandez, I.E.; Binzenhöfer, L.; Flatley, A.; Juan-Guardela, B.M.; et al. FK506-Binding Protein 11 Is a Novel Plasma Cell-Specific Antibody Folding Catalyst with Increased Expression in Idiopathic Pulmonary Fibrosis. Cells 2022, 11, 1341. https://doi.org/10.3390/cells11081341
Preisendörfer S, Ishikawa Y, Hennen E, Winklmeier S, Schupp JC, Knüppel L, Fernandez IE, Binzenhöfer L, Flatley A, Juan-Guardela BM, et al. FK506-Binding Protein 11 Is a Novel Plasma Cell-Specific Antibody Folding Catalyst with Increased Expression in Idiopathic Pulmonary Fibrosis. Cells. 2022; 11(8):1341. https://doi.org/10.3390/cells11081341
Chicago/Turabian StylePreisendörfer, Stefan, Yoshihiro Ishikawa, Elisabeth Hennen, Stephan Winklmeier, Jonas C. Schupp, Larissa Knüppel, Isis E. Fernandez, Leonhard Binzenhöfer, Andrew Flatley, Brenda M. Juan-Guardela, and et al. 2022. "FK506-Binding Protein 11 Is a Novel Plasma Cell-Specific Antibody Folding Catalyst with Increased Expression in Idiopathic Pulmonary Fibrosis" Cells 11, no. 8: 1341. https://doi.org/10.3390/cells11081341