
Neutrophil Functional Heterogeneity and Implications for Viral Infections and Treatments

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Neutrophil Immunobiology
- Primary or azurophil granules that are compacted with MPO, cathepsin G, elastase, proteinase 3, and defensins;
- Secondary or specific granules with lactoferrin as the most important component;
- Tertiary or gelatinase granules consisting of gelatinase proteins, such as matrix metalloproteinase (MMP)-9; and
- Secretory granules that mainly contain serum albumin and pre-formed cytokines [56].
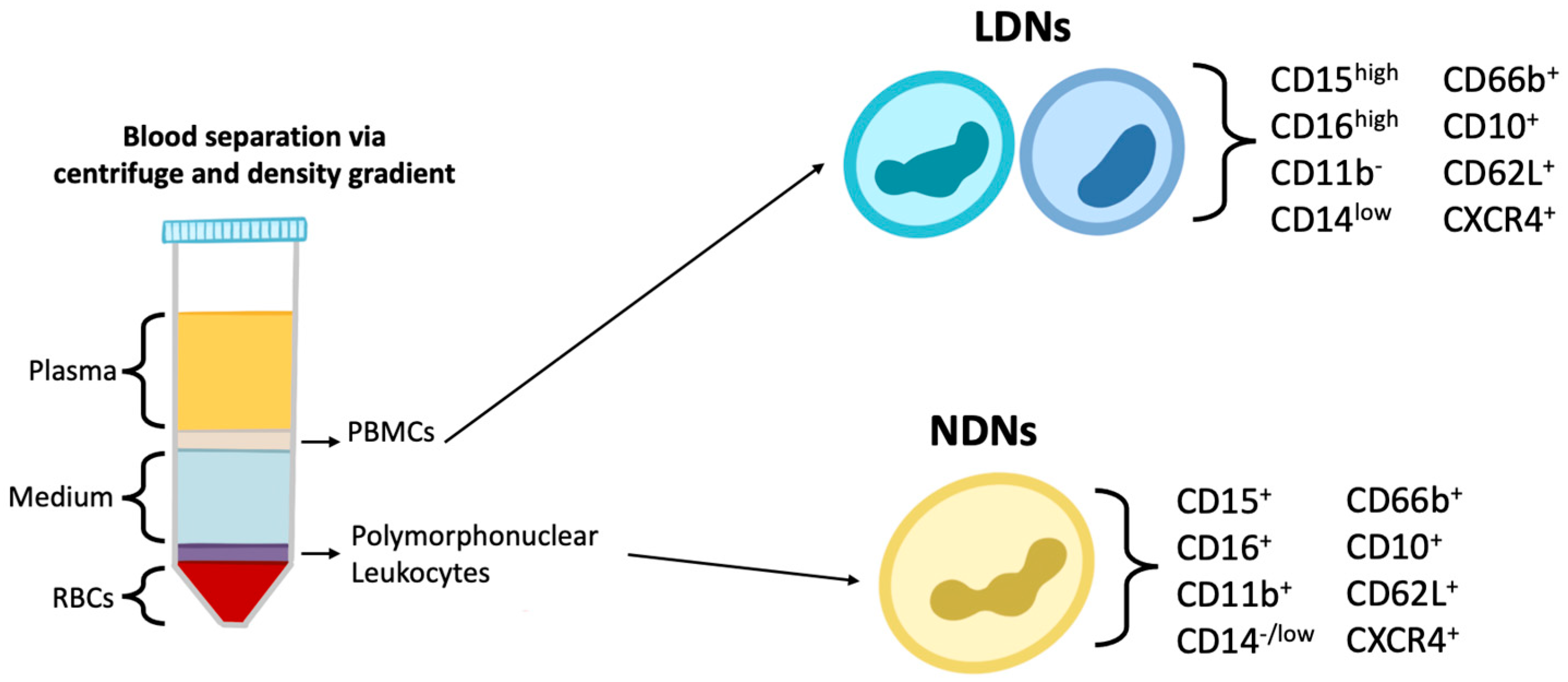
3. Neutrophil Heterogeneity and Plasticity
4. Function of Neutrophil Phenotypes
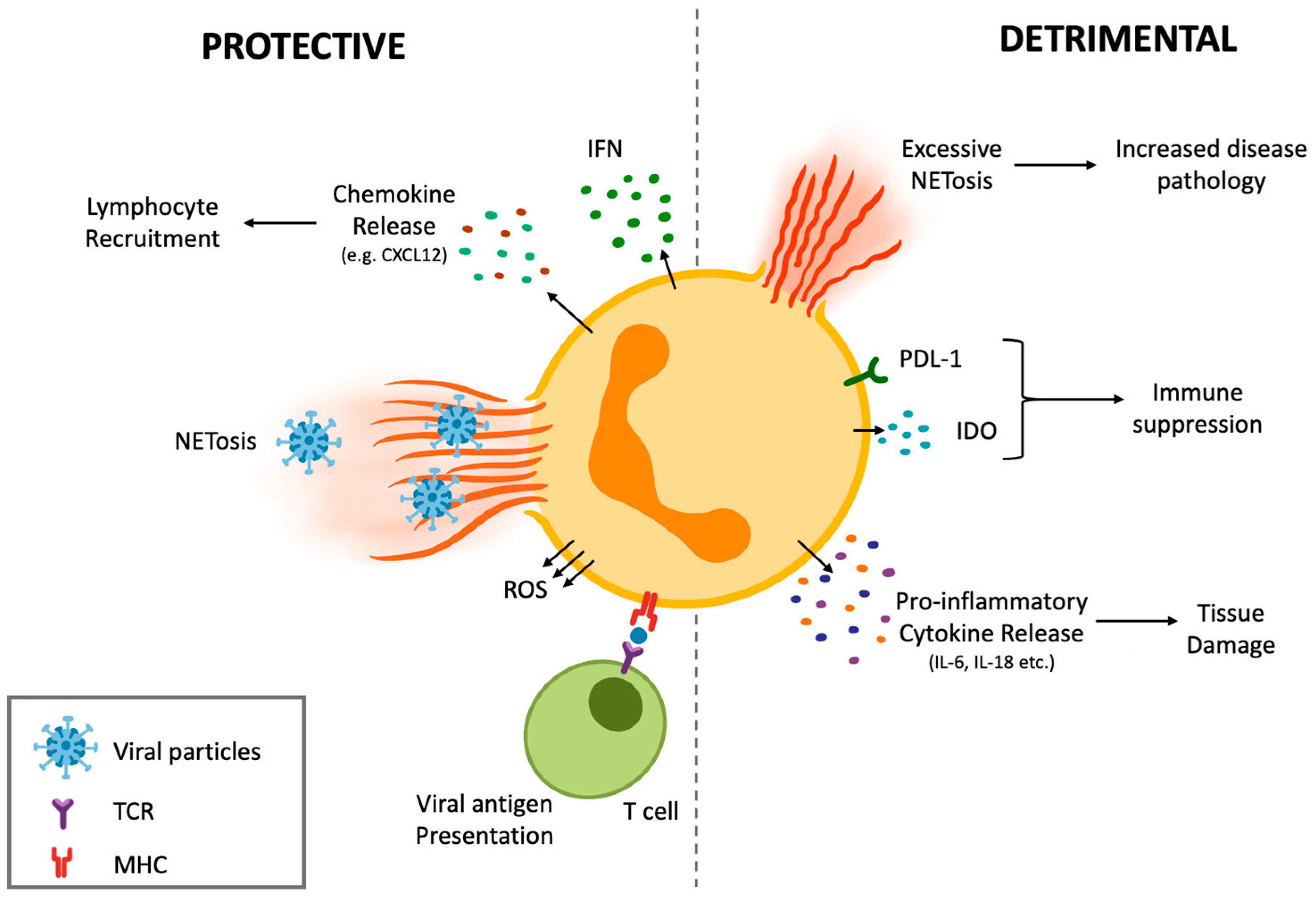
4.1. Protective Effects of Neutrophil Subsets during Anti-Viral Defense
4.2. Detrimental Potential of Different Neutrophil Subsets and Their Promotion of Collateral Damage during Viral Infections
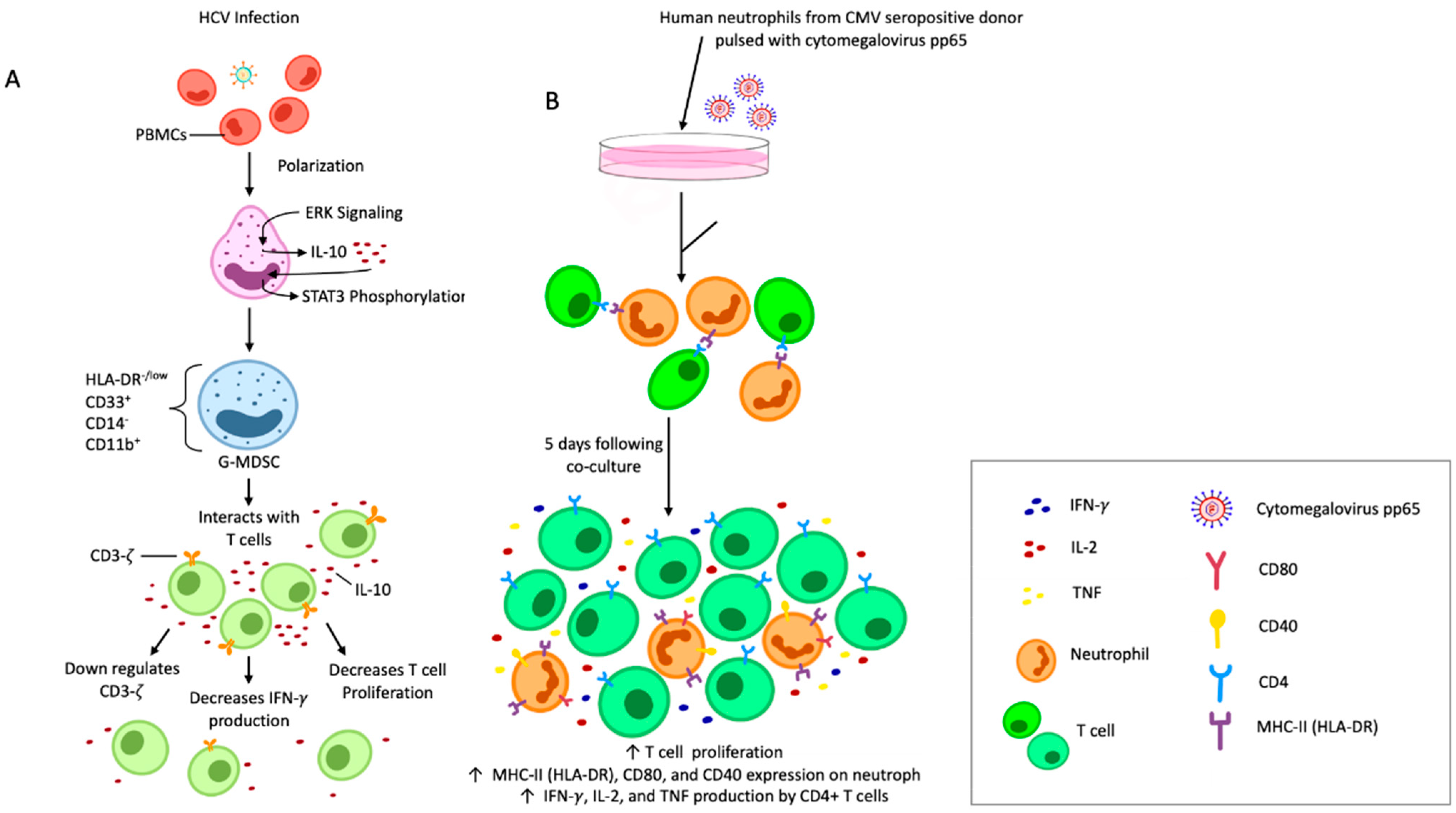
5. Implications of Neutrophil Heterogeneity for the Treatment of Viral Infections
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef] [PubMed]
- Drescher, B.; Bai, F. Neutrophil in viral infections, friend or foe? Virus Res. 2013, 171, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergthaler, A.; Menche, J. The immune system as a social network. Nat. Immunol. 2017, 18, 481–482. [Google Scholar] [CrossRef] [PubMed]
- Stegelmeier, A.A.; Chan, L.; Mehrani, Y.; Petrik, J.J.; Wootton, S.K.; Bridle, B.; Karimi, K. Characterization of the Impact of Oncolytic Vesicular Stomatitis Virus on the Trafficking, Phenotype, and Antigen Presentation Potential of Neutrophils and Their Ability to Acquire a Non-Structural Viral Protein. Int. J. Mol. Sci. 2020, 22, 6347. [Google Scholar] [CrossRef]
- Kruger, P.; Saffarzadeh, M.; Weber, A.N.; Rieber, N.; Radsak, M.; von Bernuth, H.; Benarafa, C.; Roos, D.; Skokowa, J.; Hartl, D. Neutrophils: Between host defence, immune modulation, and tissue injury. PLoS Pathog. 2015, 11, e1004651. [Google Scholar] [CrossRef] [Green Version]
- Conrad, C.; Yildiz, D.; Cleary, S.J.; Margraf, A.; Cook, L.; Schlomann, U.; Panaretou, B.; Bowser, J.L.; Karmouty-Quintana, H.; Li, J.; et al. ADAM8 signaling drives neutrophil migration and ARDS severity. JCI Insight 2022, 7, e149870. [Google Scholar] [CrossRef]
- Effah, C.Y.; Drokow, E.K.; Agboyibor, C.; Ding, L.; He, S.; Liu, S.; Akorli, S.Y.; Nuamah, E.; Sun, T.; Zhou, X.; et al. Neutrophil-Dependent Immunity During Pulmonary Infections and Inflammations. Front. Immunol. 2021, 12, 689866. [Google Scholar] [CrossRef]
- Filep, J.G.; Ariel, A. Neutrophil heterogeneity and fate in inflamed tissues: Implications for the resolution of inflammation. Am. J. Physiol. Cell Physiol. 2020, 319, C510–C532. [Google Scholar] [CrossRef]
- Hellebrekers, P.; Hietbrink, F.; Vrisekoop, N.; Leenen, L.P.H.; Koenderman, L. Neutrophil Functional Heterogeneity: Identification of Competitive Phagocytosis. Front. Immunol. 2017, 8, 1498. [Google Scholar] [CrossRef]
- Ng, L.G.; Ostuni, R.; Hidalgo, A. Heterogeneity of neutrophils. Nat. Rev. Immunol. 2019, 19, 255–265. [Google Scholar] [CrossRef]
- Basu, S.; Hodgson, G.; Katz, M.; Dunn, A.R. Evaluation of role of G-CSF in the production, survival, and release of neutrophils from bone marrow into circulation. Blood J. Am. Soc. Hematol. 2002, 100, 854–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dancey, J.; Deubelbeiss, K.A.; Harker, L.A.; Finch, C.A. Neutrophil kinetics in man. J. Clin. Investig. 1976, 58, 705–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillay, J.; Den Braber, I.; Vrisekoop, N.; Kwast, L.M.; De Boer, R.J.; Borghans, J.A.; Tesselaar, K.; Koenderman, L. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood J. Am. Soc. Hematol. 2010, 116, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Geering, B.; Stoeckle, C.; Conus, S.; Simon, H.-U. Living and dying for inflammation: Neutrophils, eosinophils, basophils. Trends Immunol. 2013, 34, 398–409. [Google Scholar] [CrossRef]
- Nourshargh, S.; Marelli-Berg, F.M. Transmigration through venular walls: A key regulator of leukocyte phenotype and function. Trends Immunol. 2005, 26, 157–165. [Google Scholar] [CrossRef]
- Ross, E.A.; Douglas, M.R.; Wong, S.H.; Ross, E.J.; Curnow, S.J.; Nash, G.B.; Rainger, E.; Scheel-Toellner, D.; Lord, J.M.; Salmon, M. Interaction between integrin α9β1 and vascular cell adhesion molecule-1 (VCAM-1) inhibits neutrophil apoptosis. Blood 2006, 107, 1178–1183. [Google Scholar] [CrossRef] [Green Version]
- Diamond, M.S.; Staunton, D.E.; De Fougerolles, A.R.; Stacker, S.A.; Garcia-Aguilar, J.; Hibbs, M.L.; Springer, T.A. ICAM-1 (CD54): A counter-receptor for Mac-1 (CD11b/CD18). J. Cell Biol. 1990, 111, 3129–3139. [Google Scholar] [CrossRef] [Green Version]
- Diamond, M.S.; Staunton, D.E.; Marlin, S.D.; Springer, T.A. Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell 1991, 65, 961–971. [Google Scholar] [CrossRef]
- Brazil, J.C.; Sumagin, R.; Cummings, R.D.; Louis, N.A.; Parkos, C.A. Targeting of Neutrophil Lewis X Blocks Transepithelial Migration and Increases Phagocytosis and Degranulation. Am. J. Pathol. 2016, 186, 297–311. [Google Scholar] [CrossRef] [Green Version]
- Zen, K.; Cui, L.B.; Zhang, C.Y.; Liu, Y. Critical role of mac-1 sialyl lewis x moieties in regulating neutrophil degranulation and transmigration. J. Mol. Biol. 2007, 374, 54–63. [Google Scholar] [CrossRef]
- Muller, W.A.; Weigl, S.A.; Deng, X.; Phillips, D.M. PECAM-1 is required for transendothelial migration of leukocytes. J. Exp. Med. 1993, 178, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Buckley, C.D.; Ross, E.A.; McGettrick, H.M.; Osborne, C.E.; Haworth, O.; Schmutz, C.; Stone, P.C.; Salmon, M.; Matharu, N.M.; Vohra, R.K. Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J. Leukoc. Biol. 2006, 79, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Chertov, O.; Ueda, H.; Xu, L.L.; Tani, K.; Murphy, W.J.; Wang, J.M.; Howard, O.; Sayers, T.J.; Oppenheim, J.J. Identification of human neutrophil-derived cathepsin G and azurocidin/CAP37 as chemoattractants for mononuclear cells and neutrophils. J. Exp. Med. 1997, 186, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Ethuin, F.; Gérard, B.; Benna, J.E.; Boutten, A.; Gougereot-Pocidalo, M.-A.; Jacob, L.; Chollet-Martin, S. Human neutrophils produce interferon gamma upon stimulation by interleukin-12. Lab. Investig. 2004, 84, 1363–1371. [Google Scholar] [CrossRef] [Green Version]
- Kasama, T.; Strieter, R.M.; Lukacs, N.W.; Burdick, M.D.; Kunkel, S.L. Regulation of neutrophil-derived chemokine expression by IL-10. J. Immunol. 1994, 152, 3559–3569. [Google Scholar]
- Kasama, T.; Strieter, R.M.; Standiford, T.; Burdick, M.; Kunkel, S. Expression and regulation of human neutrophil-derived macrophage inflammatory protein 1 alpha. J. Exp. Med. 1993, 178, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Sergejeva, S.; Linden, A. Impact of IL-17 on cells of the monocyte lineage in health and disease. Endocr. Metab. Immune Disord.-Drug Targets (Former. Curr. Drug Targets-Immune Endocr. Metab. Disord.) 2009, 9, 178–186. [Google Scholar] [CrossRef] [Green Version]
- Costantini, C.; Calzetti, F.; Perbellini, O.; Micheletti, A.; Scarponi, C.; Lonardi, S.; Pelletier, M.; Schakel, K.; Pizzolo, G.; Facchetti, F. Human neutrophils interact with both 6-sulfo LacNAc+ DC and NK cells to amplify NK-derived IFNγ: Role of CD18, ICAM-1, and ICAM-3. Blood J. Am. Soc. Hematol. 2011, 117, 1677–1686. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Whyte, M.K.; Haslett, C. Inhibition of apoptosis and prolongation of neutrophil functional longevity by inflammatory mediators. J. Leukoc. Biol. 1993, 54, 283–288. [Google Scholar] [CrossRef]
- Takano, T.; Azuma, N.; Satoh, M.; Toda, A.; Hashida, Y.; Satoh, R.; Hohdatsu, T. Neutrophil survival factors (TNF-alpha, GM-CSF, and G-CSF) produced by macrophages in cats infected with feline infectious peritonitis virus contribute to the pathogenesis of granulomatous lesions. Arch. Virol. 2009, 154, 775–781. [Google Scholar] [CrossRef]
- Bhatnagar, N.; Hong, H.S.; Krishnaswamy, J.K.; Haghikia, A.; Behrens, G.M.; Schmidt, R.E.; Jacobs, R. Cytokine-activated NK cells inhibit PMN apoptosis and preserve their functional capacity. Blood J. Am. Soc. Hematol. 2010, 116, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Colom, B.; Bodkin, J.V.; Beyrau, M.; Woodfin, A.; Ody, C.; Rourke, C.; Chavakis, T.; Brohi, K.; Imhof, B.A.; Nourshargh, S. Leukotriene B4-neutrophil elastase axis drives neutrophil reverse transendothelial cell migration in vivo. Immunity 2015, 42, 1075–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodfin, A.; Voisin, M.-B.; Beyrau, M.; Colom, B.; Caille, D.; Diapouli, F.-M.; Nash, G.B.; Chavakis, T.; Albelda, S.M.; Rainger, G.E. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat. Immunol. 2011, 12, 761–769. [Google Scholar] [CrossRef] [Green Version]
- Duffy, D.; Perrin, H.; Abadie, V.; Benhabiles, N.; Boissonnas, A.; Liard, C.; Descours, B.; Reboulleau, D.; Bonduelle, O.; Verrier, B. Neutrophils transport antigen from the dermis to the bone marrow, initiating a source of memory CD8+ T cells. Immunity 2012, 37, 917–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, A.L.; Holmes, G.R.; Bojarczuk, A.N.; Burgon, J.; Loynes, C.A.; Chimen, M.; Sawtell, A.K.; Hamza, B.; Willson, J.; Walmsley, S.R.; et al. A zebrafish compound screen reveals modulation of neutrophil reverse migration as an anti-inflammatory mechanism. Sci. Transl. Med. 2014, 6, 225ra229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hind, L.E.; Huttenlocher, A. Neutrophil Reverse Migration and a Chemokinetic Resolution. Dev. Cell 2018, 47, 404–405. [Google Scholar] [CrossRef] [Green Version]
- Bennouna, S.; Denkers, E.Y. Microbial antigen triggers rapid mobilization of TNF-α to the surface of mouse neutrophils transforming them into inducers of high-level dendritic cell TNF-α production. J. Immunol. 2005, 174, 4845–4851. [Google Scholar] [CrossRef]
- Yang, D.; de la Rosa, G.; Tewary, P.; Oppenheim, J.J. Alarmins link neutrophils and dendritic cells. Trends Immunol. 2009, 30, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Hampton, H.R.; Bailey, J.; Tomura, M.; Brink, R.; Chtanova, T. Microbe-dependent lymphatic migration of neutrophils modulates lymphocyte proliferation in lymph nodes. Nat. Commun. 2015, 6, 7139. [Google Scholar] [CrossRef]
- Bogoevska, V.; Horst, A.; Klampe, B.; Lucka, L.; Wagener, C.; Nollau, P. CEACAM1, an adhesion molecule of human granulocytes, is fucosylated by fucosyltransferase IX and interacts with DC-SIGN of dendritic cells via Lewis x residues. Glycobiology 2006, 16, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Van Gisbergen, K.P.; Ludwig, I.S.; Geijtenbeek, T.B.; van Kooyk, Y. Interactions of DC-SIGN with Mac-1 and CEACAM1 regulate contact between dendritic cells and neutrophils. FEBS Lett. 2005, 579, 6159–6168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, I.S.; Geijtenbeek, T.B.; van Kooyk, Y. Two way communication between neutrophils and dendritic cells. Curr. Opin. Pharmacol. 2006, 6, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-W.; Unanue, E.R. Neutrophils control the magnitude and spread of the immune response in a thromboxane A2-mediated process. J. Exp. Med. 2013, 210, 375–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotland, R.S.; Stables, M.J.; Madalli, S.; Watson, P.; Gilroy, D.W. Sex differences in resident immune cell phenotype underlie more efficient acute inflammatory responses in female mice. Blood J. Am. Soc. Hematol. 2011, 118, 5918–5927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klink, M.; Bednarska, K.; Blus, E.; Kielbik, M.; Sulowska, Z. Seasonal changes in activities of human neutrophils in vitro. Inflamm. Res. 2012, 61, 11–16. [Google Scholar] [CrossRef] [Green Version]
- NaveenKumar, S.K.; Hemshekhar, M.; Jagadish, S.; Manikanta, K.; Vishalakshi, G.J.; Kemparaju, K.; Girish, K.S. Melatonin restores neutrophil functions and prevents apoptosis amid dysfunctional glutathione redox system. J. Pineal Res. 2020, 69, e12676. [Google Scholar] [CrossRef]
- Ren, D.L.; Li, Y.J.; Hu, B.B.; Wang, H.; Hu, B. Melatonin regulates the rhythmic migration of neutrophils in live zebrafish. J. Pineal Res. 2015, 58, 452–460. [Google Scholar] [CrossRef]
- Liew, P.X.; Kubes, P. The Neutrophil’s Role During Health and Disease. Physiol. Rev. 2019, 99, 1223–1248. [Google Scholar] [CrossRef]
- Roberts, R.E.; Hallett, M.B. Neutrophil Cell Shape Change: Mechanism and Signalling during Cell Spreading and Phagocytosis. Int. J. Mol. Sci. 2019, 20, 1383. [Google Scholar] [CrossRef] [Green Version]
- Segal, A.W. How neutrophils kill microbes. Annu. Rev. Immunol. 2005, 23, 197–223. [Google Scholar] [CrossRef] [Green Version]
- Laarman, A.; Milder, F.; van Strijp, J.; Rooijakkers, S. Complement inhibition by gram-positive pathogens: Molecular mechanisms and therapeutic implications. J. Mol. Med. 2010, 88, 115–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Kessel, K.P.; Bestebroer, J.; van Strijp, J.A. Neutrophil-Mediated Phagocytosis of Staphylococcus aureus. Front. Immunol. 2014, 5, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellebrekers, P.; Vrisekoop, N.; Koenderman, L. Neutrophil phenotypes in health and disease. Eur. J. Clin. Investig. 2018, 48 (Suppl. S2), e12943. [Google Scholar] [CrossRef] [PubMed]
- El-Benna, J.; Hurtado-Nedelec, M.; Marzaioli, V.; Marie, J.C.; Gougerot-Pocidalo, M.A.; Dang, P.M. Priming of the neutrophil respiratory burst: Role in host defense and inflammation. Immunol. Rev. 2016, 273, 180–193. [Google Scholar] [CrossRef]
- Odobasic, D.; Kitching, A.R.; Holdsworth, S.R. Neutrophil-Mediated Regulation of Innate and Adaptive Immunity: The Role of Myeloperoxidase. J. Immunol. Res. 2016, 2016, 2349817. [Google Scholar] [CrossRef] [Green Version]
- Sheshachalam, A.; Srivastava, N.; Mitchell, T.; Lacy, P.; Eitzen, G. Granule protein processing and regulated secretion in neutrophils. Front. Immunol. 2014, 5, 448. [Google Scholar] [CrossRef] [Green Version]
- Lacy, P. Mechanisms of degranulation in neutrophils. Allergy Asthma Clin. Immunol. 2006, 2, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Stacey, H.D.; Golubeva, D.; Posca, A.; Ang, J.C.; Novakowski, K.E.; Zahoor, M.A.; Kaushic, C.; Cairns, E.; Bowdish, D.M.E.; Mullarkey, C.E.; et al. IgA potentiates NETosis in response to viral infection. Proc. Natl. Acad. Sci. USA 2021, 118, e2101497118. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.; Surette, M.G.; Sugai, M.; et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, B.; Urrutia, R.; Yipp, B.G.; Jenne, C.N.; Kubes, P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 2012, 12, 324–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manfredi, A.A.; Ramirez, G.A.; Rovere-Querini, P.; Maugeri, N. The Neutrophil’s Choice: Phagocytose vs. Make Neutrophil Extracellular Traps. Front. Immunol. 2018, 9, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akgul, C.; Edwards, S. Regulation of neutrophil apoptosis via death receptors. Cell. Mol. Life Sci. CMLS 2003, 60, 2402–2408. [Google Scholar] [CrossRef]
- Milot, E.; Filep, J.G. Regulation of neutrophil survival/apoptosis by Mcl-1. Sci. World J. 2011, 11, 1948–1962. [Google Scholar] [CrossRef] [Green Version]
- Capucetti, A.; Albano, F.; Bonecchi, R. Multiple Roles for Chemokines in Neutrophil Biology. Front. Immunol. 2020, 11, 1259. [Google Scholar] [CrossRef]
- Suratt, B.T.; Young, S.K.; Lieber, J.; Nick, J.A.; Henson, P.M.; Worthen, G.S. Neutrophil maturation and activation determine anatomic site of clearance from circulation. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2001, 281, L913–L921. [Google Scholar] [CrossRef] [Green Version]
- Weisel, K.C.; Bautz, F.; Seitz, G.; Yildirim, S.; Kanz, L.; Möhle, R. Modulation of CXC chemokine receptor expression and function in human neutrophils during aging in vitro suggests a role in their clearance from circulation. Mediat. Inflamm. 2009, 2009, 790174. [Google Scholar] [CrossRef]
- Martin, C.; Burdon, P.C.; Bridger, G.; Gutierrez-Ramos, J.C.; Williams, T.J.; Rankin, S.M. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity 2003, 19, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Wolach, B.; van der Laan, L.J.; Maianski, N.A.; Tool, A.T.; van Bruggen, R.; Roos, D.; Kuijpers, T.W. Growth factors G-CSF and GM-CSF differentially preserve chemotaxis of neutrophils aging in vitro. Exp. Hematol. 2007, 35, 541–550. [Google Scholar] [CrossRef]
- Nagase, H.; Miyamasu, M.; Yamaguchi, M.; Imanishi, M.; Tsuno, N.H.; Matsushima, K.; Yamamoto, K.; Morita, Y.; Hirai, K. Cytokine-mediated regulation of CXCR4 expression in human neutrophils. J. Leukoc. Biol. 2002, 71, 711–717. [Google Scholar] [PubMed]
- Tortorella, C.; Ottolenghi, A.; Pugliese, P.; Jirillo, E.; Antonaci, S. Relationship between respiratory burst and adhesiveness capacity in elderly polymorphonuclear cells. Mech. Ageing Dev. 1993, 69, 53–63. [Google Scholar] [CrossRef]
- Butcher, S.; Chahal, H.; Nayak, L.; Sinclair, A.; Henriquez, N.; Sapey, E.; O’mahony, D.; Lord, J. Senescence in innate immune responses: Reduced neutrophil phagocytic capacity and CD16 expression in elderly humans. J. Leukoc. Biol. 2001, 70, 881–886. [Google Scholar] [PubMed]
- Fülöp, T., Jr.; Fouquet, C.; Allaire, P.; Perrin, N.; Lacombe, G.; Stankova, J.; Rola-Pleszczynski, M.; Gagne, D.; Wagner, J.; Khalil, A. Changes in apoptosis of human polymorphonuclear granulocytes with aging. Mech. Ageing Dev. 1997, 96, 15–34. [Google Scholar] [CrossRef]
- Peters, T.; Weiss, J.M.; Sindrilaru, A.; Wang, H.; Oreshkova, T.; Wlaschek, M.; Maity, P.; Reimann, J.; Scharffetter-Kochanek, K. Reactive oxygen intermediate-induced pathomechanisms contribute to immunosenescence, chronic inflammation and autoimmunity. Mech. Ageing Dev. 2009, 130, 564–587. [Google Scholar] [CrossRef]
- Fortin, C.F.; Larbi, A.; Lesur, O.; Douziech, N.; Fulop, T., Jr. Impairment of SHP-1 down-regulation in the lipid rafts of human neutrophils under GM-CSF stimulation contributes to their age-related, altered functions. J. Leukoc. Biol. 2006, 79, 1061–1072. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Douziech, N.; Fortin, C.; Guérard, K.P.; Lesur, O.; Khalil, A.; Dupuis, G. Signal transduction and functional changes in neutrophils with aging. Aging Cell 2004, 3, 217–226. [Google Scholar] [CrossRef]
- Sato, N.; Sakamaki, K.; Terada, N.; Arai, K.-i.; Miyajima, A. Signal transduction by the high-affinity GM-CSF receptor: Two distinct cytoplasmic regions of the common beta subunit responsible for different signaling. EMBO J. 1993, 12, 4181–4189. [Google Scholar] [CrossRef]
- Watanabe, S.; Itoh, T.; Arai, K.-i. Roles of JAK kinase in human GM-CSF receptor signals. Leukemia 1997, 11, 76–78. [Google Scholar]
- Biasi, D.; Carletto, A.; Dell’Agnola, C.; Caramaschi, P.; Montesanti, F.; Zavateri, G.; Zeminian, S.; Bellavite, P.; Bambara, L. Neutrophil migration, oxidative metabolism, and adhesion in elderly and young subjects. Inflammation 1996, 20, 673–681. [Google Scholar] [CrossRef]
- Rao, K.M.K.; Currie, M.S.; Padmanabhan, J.; Cohen, H.J. Age-related alterations in actin cytoskeleton and receptor expression in human leukocytes. J. Gerontol. 1992, 47, B37–B44. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T., Jr.; Larbi, A.; Linteau, A.; Desgeorges, S.; Douziech, N. The role of Mcl-1 and Bax expression alteration in the decreased rescue of human neutrophils from apoptosis by GM-CSF with aging. Ann. N. Y. Acad. Sci. 2002, 973, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Postnikov, Y.V.; Li, Y.; Tewary, P.; de la Rosa, G.; Wei, F.; Klinman, D.; Gioannini, T.; Weiss, J.P.; Furusawa, T. High-mobility group nucleosome-binding protein 1 acts as an alarmin and is critical for lipopolysaccharide-induced immune responses. J. Exp. Med. 2012, 209, 157–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weksler, M.E. Changes in the B-cell repertoire with age. Vaccine 2000, 18, 1624–1628. [Google Scholar] [CrossRef]
- MacGregor, R.R.; Shalit, M. Neutrophil function in healthy elderly subjects. J. Gerontol. 1990, 45, M55–M60. [Google Scholar] [CrossRef]
- Cakman, I.; Rohwer, J.; Schütz, R.-M.; Kirchner, H.; Rink, L. Dysregulation between TH1 and TH2 T cell subpopulations in the elderly. Mech. Ageing Dev. 1996, 87, 197–209. [Google Scholar] [CrossRef]
- Morrissey, S.M.; Geller, A.E.; Hu, X.; Tieri, D.; Ding, C.; Klaes, C.K.; Cooke, E.A.; Woeste, M.R.; Martin, Z.C.; Chen, O.; et al. A specific low-density neutrophil population correlates with hypercoagulation and disease severity in hospitalized COVID-19 patients. JCI Insight 2021, 6, e148435. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Fridlender, Z.G.; Glogauer, M.; Scapini, P. Neutrophil Diversity in Health and Disease. Trends Immunol. 2019, 40, 565–583. [Google Scholar] [CrossRef]
- Scapini, P.; Marini, O.; Tecchio, C.; Cassatella, M.A. Human neutrophils in the saga of cellular heterogeneity: Insights and open questions. Immunol. Rev. 2016, 273, 48–60. [Google Scholar] [CrossRef]
- Denny, M.F.; Yalavarthi, S.; Zhao, W.; Thacker, S.G.; Anderson, M.; Sandy, A.R.; McCune, W.J.; Kaplan, M.J. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J. Immunol. 2010, 184, 3284–3297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Camarillo, C.; Alemán, O.R.; Rosales, C. Low-Density Neutrophils in Healthy Individuals Display a Mature Primed Phenotype. Front. Immunol. 2021, 12, 672520. [Google Scholar] [CrossRef] [PubMed]
- Lourda, M.; Dzidic, M.; Hertwig, L.; Bergsten, H.; Palma Medina, L.M.; Sinha, I.; Kvedaraite, E.; Chen, P.; Muvva, J.R.; Gorin, J.B.; et al. High-dimensional profiling reveals phenotypic heterogeneity and disease-specific alterations of granulocytes in COVID-19. Proc. Natl. Acad. Sci. USA 2021, 118, e2109123118. [Google Scholar] [CrossRef]
- Marini, O.; Costa, S.; Bevilacqua, D.; Calzetti, F.; Tamassia, N.; Spina, C.; De Sabata, D.; Tinazzi, E.; Lunardi, C.; Scupoli, M.T. Mature CD10+ and immature CD10− neutrophils present in G-CSF–treated donors display opposite effects on T cells. Blood J. Am. Soc. Hematol. 2017, 129, 1343–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Bergenfelz, C.; Leandersson, K. The Generation and Identity of Human Myeloid-Derived Suppressor Cells. Front. Oncol. 2020, 10, 109. [Google Scholar] [CrossRef] [Green Version]
- Lang, S.; Bruderek, K.; Kaspar, C.; Höing, B.; Kanaan, O.; Dominas, N.; Hussain, T.; Droege, F.; Eyth, C.; Hadaschik, B.; et al. Clinical Relevance and Suppressive Capacity of Human Myeloid-Derived Suppressor Cell Subsets. Clin. Cancer Res. 2018, 24, 4834–4844. [Google Scholar] [CrossRef] [Green Version]
- Filipazzi, P.; Huber, V.; Rivoltini, L. Phenotype, function and clinical implications of myeloid-derived suppressor cells in cancer patients. Cancer Immunol. Immunother. 2012, 61, 255–263. [Google Scholar] [CrossRef]
- Almand, B.; Clark, J.I.; Nikitina, E.; van Beynen, J.; English, N.R.; Knight, S.C.; Carbone, D.P.; Gabrilovich, D.I. Increased production of immature myeloid cells in cancer patients: A mechanism of immunosuppression in cancer. J. Immunol. 2001, 166, 678–689. [Google Scholar] [CrossRef] [Green Version]
- Kusmartsev, S.; Nefedova, Y.; Yoder, D.; Gabrilovich, D.I. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol. 2004, 172, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.Y.; Fuchsberger, M.; Xiang, D.S.; Apostolopoulos, V.; Plebanski, M. Myeloid derived suppressor cells and their role in diseases. Curr. Med. Chem. 2013, 20, 1437–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, E.; Zhang, L.; Li, C. LOX-1+ PMN-MDSC enhances immune suppression which promotes glioblastoma multiforme progression. Cancer Manag. Res. 2019, 11, 7307–7315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moses, K.; Brandau, S. Human neutrophils: Their role in cancer and relation to myeloid-derived suppressor cells. In Proceedings of Seminars in Immunology; Elsevier: Amsterdam, The Netherlands; pp. 187–196.
- Rieber, N.; Singh, A.; Öz, H.; Carevic, M.; Bouzani, M.; Amich, J.; Ost, M.; Ye, Z.; Ballbach, M.; Schäfer, I. Pathogenic fungi regulate immunity by inducing neutrophilic myeloid-derived suppressor cells. Cell Host Microbe 2015, 17, 507–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Lelis, F.; Braig, S.; Schäfer, I.; Hartl, D.; Rieber, N. Differential regulation of myeloid-derived suppressor cells by Candida species. Front. Microbiol. 2016, 7, 1624. [Google Scholar] [CrossRef]
- Heim, C.E.; Vidlak, D.; Scherr, T.D.; Kozel, J.A.; Holzapfel, M.; Muirhead, D.E.; Kielian, T. Myeloid-derived suppressor cells contribute to Staphylococcus aureus orthopedic biofilm infection. J. Immunol. 2014, 192, 3778–3792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poe, S.L.; Arora, M.; Oriss, T.B.; Yarlagadda, M.; Isse, K.; Khare, A.; Levy, D.E.; Lee, J.S.; Mallampalli, R.; Chan, Y. STAT1-regulated lung MDSC-like cells produce IL-10 and efferocytose apoptotic neutrophils with relevance in resolution of bacterial pneumonia. Mucosal Immunol. 2013, 6, 189–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, G.; Jung, B.; Chiu, P.Y.; Aslam, R.; Palacios, F.; Mazzarello, A.N.; Vergani, S.; Bagnara, D.; Chen, S.-S.; Yancopoulos, S.; et al. Myeloid-derived suppressor cell subtypes differentially influence T-cell function, T-helper subset differentiation, and clinical course in CLL. Leukemia 2021, 35, 3163–3175. [Google Scholar] [CrossRef]
- Nagaraj, S.; Gabrilovich, D.I. Regulation of suppressive function of myeloid-derived suppressor cells by CD4+ T cells. Semin. Cancer Biol. 2012, 22, 282–288. [Google Scholar] [CrossRef] [Green Version]
- Bertoletti, A.; Tan, A.T.; Koh, S. T-cell therapy for chronic viral hepatitis. Cytotherapy 2017, 19, 1317–1324. [Google Scholar] [CrossRef]
- Wang, M.; Ping, Y.; Li, Z.; Li, J.; Zhang, Z.; Yue, D.; Chen, X.; Wang, L.; Huang, L.; Huang, J. Polarization of granulocytic myeloid-derived suppressor cells by hepatitis C core protein is mediated via IL-10/STAT3 signalling. J. Viral Hepat. 2019, 26, 246–257. [Google Scholar] [CrossRef]
- Li, X.-K.; Lu, Q.-B.; Chen, W.-W.; Xu, W.; Liu, R.; Zhang, S.-F.; Du, J.; Li, H.; Yao, K.; Zhai, D. Arginine deficiency is involved in thrombocytopenia and immunosuppression in severe fever with thrombocytopenia syndrome. Sci. Transl. Med. 2018, 10, eaat4162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Q.-L.; Yang, B.; Sun, H.-Q.; Feng, G.-H.; Jin, L.; Zou, Z.-S.; Zhang, Z.; Zhang, J.-Y.; Wang, F.-S. Myeloid-derived suppressor cells are associated with viral persistence and downregulation of TCR ζ chain expression on CD8+ T cells in chronic hepatitis C patients. Mol. Cells 2014, 37, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoermer, K.A.; Burrack, A.; Oko, L.; Montgomery, S.A.; Borst, L.B.; Gill, R.G.; Morrison, T.E. Genetic ablation of arginase 1 in macrophages and neutrophils enhances clearance of an arthritogenic alphavirus. J. Immunol. 2012, 189, 4047–4059. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Spector, S.A. HIV type 1 gp120–induced expansion of myeloid derived suppressor cells is dependent on interleukin 6 and suppresses immunity. J. Infect. Dis. 2014, 209, 441–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vollbrecht, T.; Stirner, R.; Tufman, A.; Roider, J.; Huber, R.M.; Bogner, J.R.; Lechner, A.; Bourquin, C.; Draenert, R. Chronic progressive HIV-1 infection is associated with elevated levels of myeloid-derived suppressor cells. Aids 2012, 26, F31–F37. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-N.; Yi, N.; Zhang, T.-W.; Zhang, L.-L.; Wu, X.; Liu, M.; Fu, Y.-J.; He, S.-J.; Jiang, Y.-J.; Ding, H.-B. Myeloid-derived suppressor cells associated with disease progression in primary HIV infection: PD-L1 blockade attenuates inhibition. JAIDS J. Acquir. Immune Defic. Syndr. 2017, 76, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Tumino, N.; Turchi, F.; Meschi, S.; Lalle, E.; Bordoni, V.; Casetti, R.; Agrati, C.; Cimini, E.; Montesano, C.; Colizzi, V. In HIV-positive patients, myeloid-derived suppressor cells induce T-cell anergy by suppressing CD3ζ expression through ELF-1 inhibition. Aids 2015, 29, 2397–2407. [Google Scholar] [CrossRef]
- Chen, S.; Akbar, S.; Abe, M.; Hiasa, Y.; Onji, M. Immunosuppressive functions of hepatic myeloid-derived suppressor cells of normal mice and in a murine model of chronic hepatitis B virus. Clin. Exp. Immunol. 2011, 166, 134–142. [Google Scholar] [CrossRef]
- Norris, B.A.; Uebelhoer, L.S.; Nakaya, H.I.; Price, A.A.; Grakoui, A.; Pulendran, B. Chronic but not acute virus infection induces sustained expansion of myeloid suppressor cell numbers that inhibit viral-specific T cell immunity. Immunity 2013, 38, 309–321. [Google Scholar] [CrossRef] [Green Version]
- He, Y.-M.; Li, X.; Perego, M.; Nefedova, Y.; Kossenkov, A.V.; Jensen, E.A.; Kagan, V.; Liu, Y.-F.; Fu, S.-Y.; Ye, Q.-J. Transitory presence of myeloid-derived suppressor cells in neonates is critical for control of inflammation. Nat. Med. 2018, 24, 224–231. [Google Scholar] [CrossRef]
- Condamine, T.; Dominguez, G.A.; Youn, J.-I.; Kossenkov, A.V.; Mony, S.; Alicea-Torres, K.; Tcyganov, E.; Hashimoto, A.; Nefedova, Y.; Lin, C. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 2016, 1, aaf8943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nefedova, Y.; Huang, M.; Kusmartsev, S.; Bhattacharya, R.; Cheng, P.; Salup, R.; Jove, R.; Gabrilovich, D. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J. Immunol. 2004, 172, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Rébé, C.; Végran, F.; Berger, H.; Ghiringhelli, F. STAT3 activation: A key factor in tumor immunoescape. Jak-stat 2013, 2, e23010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Cheng, P.; Condamine, T.; Mony, S.; Languino, L.R.; McCaffrey, J.C.; Hockstein, N.; Guarino, M.; Masters, G.; Penman, E. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity 2016, 44, 303–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Zhang, N.; Qi, L.; Yuan, J.; Wang, K.; Wang, K.; Ma, S.; Wang, H.; Lou, W.; Hu, P. Myeloid-derived suppressor cells inhibit T follicular helper cell immune response in Japanese encephalitis virus infection. J. Immunol. 2017, 199, 3094–3105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Huang, X.; Yang, Y. Myeloid-derived suppressor cells regulate natural killer cell response to adenovirus-mediated gene transfer. J. Virol. 2012, 86, 13689–13696. [Google Scholar] [CrossRef] [Green Version]
- Lukens, M.V.; van de Pol, A.C.; Coenjaerts, F.E.; Jansen, N.J.; Kamp, V.M.; Kimpen, J.L.; Rossen, J.W.; Ulfman, L.H.; Tacke, C.E.; Viveen, M.C. A systemic neutrophil response precedes robust CD8+ T-cell activation during natural respiratory syncytial virus infection in infants. J. Virol. 2010, 84, 2374–2383. [Google Scholar] [CrossRef] [Green Version]
- Cortjens, B.; Ingelse, S.A.; Calis, J.C.; Vlaar, A.P.; Koenderman, L.; Bem, R.A.; van Woensel, J.B. Neutrophil subset responses in infants with severe viral respiratory infection. Clin. Immunol. 2017, 176, 100–106. [Google Scholar] [CrossRef]
- Pillay, J.; Kamp, V.M.; van Hoffen, E.; Visser, T.; Tak, T.; Lammers, J.-W.; Ulfman, L.H.; Leenen, L.P.; Pickkers, P.; Koenderman, L. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J. Clin. Investig. 2012, 122, 327–336. [Google Scholar] [CrossRef]
- Hiroki, C.H.; Toller-Kawahisa, J.E.; Fumagalli, M.J.; Colon, D.F.; Figueiredo, L.T.M.; Fonseca, B.A.L.D.; Franca, R.F.O.; Cunha, F.Q. Neutrophil Extracellular Traps Effectively Control Acute Chikungunya Virus Infection. Front. Immunol. 2020, 10, 3108. [Google Scholar] [CrossRef] [Green Version]
- Palha, N.; Guivel-Benhassine, F.; Briolat, V.; Lutfalla, G.; Sourisseau, M.; Ellett, F.; Wang, C.-H.; Lieschke, G.J.; Herbomel, P.; Schwartz, O.; et al. Real-Time Whole-Body Visualization of Chikungunya Virus Infection and Host Interferon Response in Zebrafish. PLoS Pathog. 2013, 9, e1003619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opasawatchai, A.; Amornsupawat, P.; Jiravejchakul, N.; Chan-In, W.; Spoerk, N.J.; Manopwisedjaroen, K.; Singhasivanon, P.; Yingtaweesak, T.; Suraamornkul, S.; Mongkolsapaya, J.; et al. Neutrophil Activation and Early Features of NET Formation Are Associated With Dengue Virus Infection in Human. Front. Immunol. 2018, 9, 3007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzagalli, D.U.; Latino, I.; Pulfer, A.; Palomino-Segura, M.; Virgilio, T.; Farsakoglu, Y.; Krause, R.; Gonzalez, S.F. Characterization of the Dynamic Behavior of Neutrophils Following Influenza Vaccination. Front. Immunol. 2019, 10, 2621. [Google Scholar] [CrossRef] [Green Version]
- Peiró, T.; Patel, D.F.; Akthar, S.; Gregory, L.G.; Pyle, C.J.; Harker, J.A.; Birrell, M.A.; Lloyd, C.M.; Snelgrove, R.J. Neutrophils drive alveolar macrophage IL-1β release during respiratory viral infection. Thorax 2018, 73, 546–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, K.; Hyun, Y.-M.; Lambert-Emo, K.; Capece, T.; Bae, S.; Miller, R.; Topham, D.J.; Kim, M. Neutrophil trails guide influenza-specific CD8(+) T cells in the airways. Science 2015, 349, aaa4352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Pilato, M.; Mejías-Pérez, E.; Zonca, M.; Perdiguero, B.; Gómez, C.E.; Trakala, M.; Nieto, J.; Nájera, J.L.; Sorzano, C.O.S.; Combadière, C.; et al. NFκB activation by modified vaccinia virus as a novel strategy to enhance neutrophil migration and HIV-specific T-cell responses. Proc. Natl. Acad. Sci. USA 2015, 112, E1333–E1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Pilato, M.; Palomino-Segura, M.; Mejías-Pérez, E.; Gómez, C.E.; Rubio-Ponce, A.; D’Antuono, R.; Pizzagalli, D.U.; Pérez, P.; Kfuri-Rubens, R.; Benguría, A.; et al. Neutrophil subtypes shape HIV-specific CD8 T-cell responses after vaccinia virus infection. Npj Vaccines 2021, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Vono, M.; Lin, A.; Norrby-Teglund, A.; Koup, R.A.; Liang, F.; Loré, K. Neutrophils acquire the capacity for antigen presentation to memory CD4+ T cells in vitro and ex vivo. Blood 2017, 129, 1991–2001. [Google Scholar] [CrossRef] [Green Version]
- Puga, I.; Cols, M.; Barra, C.M.; He, B.; Cassis, L.; Gentile, M.; Comerma, L.; Chorny, A.; Shan, M.; Xu, W.; et al. B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat. Immunol. 2012, 13, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Tak, T.; Rygiel, T.P.; Karnam, G.; Bastian, O.W.; Boon, L.; Viveen, M.; Coenjaerts, F.E.; Meyaard, L.; Koenderman, L.; Pillay, J. Neutrophil-mediated Suppression of Influenza-induced Pathology Requires CD11b/CD18 (MAC-1). Am. J. Respir. Cell Mol. Biol. 2018, 58, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Agrati, C.; Sacchi, A.; Bordoni, V.; Cimini, E.; Notari, S.; Grassi, G.; Casetti, R.; Tartaglia, E.; Lalle, E.; D’Abramo, A.; et al. Expansion of myeloid-derived suppressor cells in patients with severe coronavirus disease (COVID-19). Cell Death Differ. 2020, 27, 3196–3207. [Google Scholar] [CrossRef] [PubMed]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.-A.; Phoon, M.C.; van Rooijen, N.; Chow, V.T. Excessive Neutrophils and Neutrophil Extracellular Traps Contribute to Acute Lung Injury of Influenza Pneumonitis. Am. J. Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.M.D.P.; Message, S.D.M.D.P.; Qiu, Y.M.D.P.; Mallia, P.M.D.P.; Kebadze, T.M.D.; Contoli, M.M.D.; Ward, C.K.P.; Barnathan, E.S.M.D.; Mascelli, M.A.P.; Kon, O.M.M.D.; et al. Airway Inflammation and Illness Severity in Response to Experimental Rhinovirus Infection in Asthma. Chest 2014, 145, 1219–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Message, S.D.; Laza-Stanca, V.; Mallia, P.; Parker, H.L.; Zhu, J.; Kebadze, T.; Contoli, M.; Sanderson, G.; Kon, O.M.; Papi, A.; et al. Rhinovirus-Induced Lower Respiratory Illness Is Increased in Asthma and Related to Virus Load and Th1/2 Cytokine and IL-10 Production. Proc. Natl. Acad. Sci. USA 2008, 105, 13562–13567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishna, C.; Cantin, E.M. IFNγ inhibits G-CSF induced neutrophil expansion and invasion of the CNS to prevent viral encephalitis. PLoS Pathog. 2018, 14, e1006822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmat, N.; Derakhshani, A.; Bannazadeh Baghi, H.; Silvestris, N.; Baradaran, B.; De Summa, S. Neutrophils, Crucial, or Harmful Immune Cells Involved in Coronavirus Infection: A Bioinformatics Study. Front. Genet. 2020, 11, 641. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, M.; Chen, X.; Montaner, L.J. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: Review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J. Leukoc. Biol. 2020, 108, 17–41. [Google Scholar] [CrossRef]
- Singh, K.; Mittal, S.; Gollapudi, S.; Butzmann, A.; Kumar, J.; Ohgami, R.S. A meta-analysis of SARS-CoV-2 patients identifies the combinatorial significance of D-dimer, C-reactive protein, lymphocyte, and neutrophil values as a predictor of disease severity. Int. J. Lab. Hematol. 2021, 43, 324–328. [Google Scholar] [CrossRef]
- Guan, J.; Wei, X.; Qin, S.; Liu, X.; Jiang, Y.; Chen, Y.; Chen, Y.; Lu, H.; Qian, J.; Wang, Z.; et al. Continuous tracking of COVID-19 patients’ immune status. Int. Immunopharmacol. 2020, 89, 107034. [Google Scholar] [CrossRef]
- Liu, J.; Li, S.; Liu, J.; Liang, B.; Wang, X.; Wang, H.; Li, W.; Tong, Q.; Yi, J.; Zhao, L.; et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine 2020, 55, 102763. [Google Scholar] [CrossRef] [PubMed]
- Kiani, A.; Roesch, R.; Wendtner, C.M.; Kullmann, F.; Kubin, T.; Südhoff, T.; Augustin, M.; Schaich, M.; Müller-Naendrup, C.; Illerhaus, G.; et al. Preinfection laboratory parameters may predict COVID-19 severity in tumor patients. Cancer Med. 2021, 10, 4424–4436. [Google Scholar] [CrossRef] [PubMed]
- McNamara, P.S.; Ritson, P.; Selby, A.; Hart, C.A.; Smyth, R.L. Bronchoalveolar lavage cellularity in infants with severe respiratory syncytial virus bronchiolitis. Arch. Dis. Child. 2003, 88, 922–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emboriadou, M.; Hatzistilianou, M.; Magnisali, C.; Sakelaropoulou, A.; Exintari, M.; Conti, P.; Aivazis, V. Human neutrophil elastase in RSV bronchiolitis. Ann. Clin. Lab. Sci. 2007, 37, 79–84. [Google Scholar]
- Kirsebom, F.; Michalaki, C.; Agueda-Oyarzabal, M.; Johansson, C. Neutrophils do not impact viral load or the peak of disease severity during RSV infection. Sci. Rep. 2020, 10, 1110. [Google Scholar] [CrossRef] [Green Version]
- Geerdink, R.J.M.; Pillay, J.M.D.P.; Meyaard, L.P.; Bont, L.M.D.P. Neutrophils in respiratory syncytial virus infection: A target for asthma prevention. J. Allergy Clin. Immunol. 2015, 136, 838–847. [Google Scholar] [CrossRef]
- Seda, S.; Lohann, D.; Irem, K.; Antoine, G.; Adam, L.; Loïc, G.; Thomas, B.; Mustapha, S.-T.; Sylvain, M.-A.; Dieter, E.J.; et al. Proteinase release from activated neutrophils in mechanically ventilated patients with non-COVID-19 and COVID-19 pneumonia. Eur. Respir. J. 2021, 57, 2003755. [Google Scholar] [CrossRef]
- Komaravelli, N.; Casola, A. Respiratory Viral Infections and Subversion of Cellular Antioxidant Defenses. J. Pharm. Pharm. 2014, 5, 1000141. [Google Scholar] [CrossRef]
- Laforge, M.; Elbim, C.; Frere, C.; Hemadi, M.; Massaad, C.; Nuss, P.; Benoliel, J.-J.; Becker, C. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat. Rev. Immunol. 2020, 20, 515–516. [Google Scholar] [CrossRef]
- Michael, B.D.; Bricio-Moreno, L.; Sorensen, E.W.; Miyabe, Y.; Lian, J.; Solomon, T.; Kurt-Jones, E.A.; Luster, A.D. Astrocyte- and Neuron-Derived CXCL1 Drives Neutrophil Transmigration and Blood-Brain Barrier Permeability in Viral Encephalitis. Cell Rep. 2020, 32, 108150. [Google Scholar] [CrossRef]
- Cortjens, B.; de Boer, O.J.; de Jong, R.; Antonis, A.F.G.; Sabogal Piñeros, Y.S.; Lutter, R.; van Woensel, J.B.M.; Bem, R.A. Neutrophil extracellular traps cause airway obstruction during respiratory syncytial virus disease. J. Pathol. 2016, 238, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Jenne, D.E.; Krumbholz, M.; Schönermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Gröne, H.-J.; Brinkmann, V. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Liu, L.; Zhang, Y.; Pu, L.; Liu, J.; Li, X.; Chen, Z.; Hao, Y.; Wang, B.; Han, J.; et al. High Level of Neutrophil Extracellular Traps Correlates with Poor Prognosis of Severe Influenza A Infection. J. Infect. Dis. 2018, 217, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Lefrançais, E.; Mallavia, B.; Zhuo, H.; Calfee, C.S.; Looney, M.R. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight 2018, 3, e98178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toussaint, M.; Jackson, D.J.; Swieboda, D.; Guedán, A.; Tsourouktsoglou, T.-D.; Ching, Y.M.; Radermecker, C.; Makrinioti, H.; Aniscenko, J.; Bartlett, N.W.; et al. Host DNA released by NETosis promotes rhinovirus-induced type-2 allergic asthma exacerbation. Nat. Med. 2017, 23, 681–691. [Google Scholar] [CrossRef] [Green Version]
- Radermecker, C.; Detrembleur, N.; Guiot, J.; Cavalier, E.; Henket, M.; d’Emal, C.; Vanwinge, C.; Cataldo, D.; Oury, C.; Delvenne, P.; et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J. Exp. Med. 2020, 217, e20201012. [Google Scholar] [CrossRef]
- Laridan, E.; Martinod, K.; De Meyer, S.F. Neutrophil Extracellular Traps in Arterial and Venous Thrombosis. Semin. Thromb. Hemost. 2019, 45, 086–093. [Google Scholar] [CrossRef]
- Saitoh, T.; Komano, J.; Saitoh, Y.; Misawa, T.; Takahama, M.; Kozaki, T.; Uehata, T.; Iwasaki, H.; Omori, H.; Yamaoka, S.; et al. Neutrophil Extracellular Traps Mediate a Host Defense Response to Human Immunodeficiency Virus-1. Cell Host Microbe 2012, 12, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Sung, P.-S.; Huang, T.-F.; Hsieh, S.-L. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat. Commun. 2019, 10, 2402. [Google Scholar] [CrossRef] [Green Version]
- Tumino, N.; Bilotta, M.T.; Pinnetti, C.; Ammassari, A.; Antinori, A.; Turchi, F.; Agrati, C.; Casetti, R.; Bordoni, V.; Cimini, E.; et al. Granulocytic Myeloid–Derived Suppressor Cells Increased in Early Phases of Primary HIV Infection Depending on TRAIL Plasma Level. J. Acquir. Immune Defic. Syndr. 2017, 74, 575–582. [Google Scholar] [CrossRef] [Green Version]
- Qin, A.; Cai, W.; Pan, T.; Wu, K.; Yang, Q.; Wang, N.; Liu, Y.; Yan, D.; Hu, F.; Guo, P.; et al. Expansion of Monocytic Myeloid-Derived Suppressor Cells Dampens T Cell Function in HIV-1-Seropositive Individuals. J. Virol. 2013, 87, 1477–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallett, L.J.; Gill, U.S.; Quaglia, A.; Sinclair, L.V.; Jover-Cobos, M.; Schurich, A.; Singh, K.P.; Thomas, N.; Das, A.; Chen, A.; et al. Metabolic regulation of hepatitis B immunopathology by myeloid-derived suppressor cells. Nat. Med. 2015, 21, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Fang, Z.; Li, J.; Yu, X.; Zhang, D.; Ren, G.; Shi, B.; Wang, C.; Kosinska, A.D.; Wang, S.; Zhou, X.; et al. Polarization of Monocytic Myeloid-Derived Suppressor Cells by Hepatitis B Surface Antigen Is Mediated via ERK/IL-6/STAT3 Signaling Feedback and Restrains the Activation of T Cells in Chronic Hepatitis B Virus Infection. J. Immunol. 2015, 195, 4873–4883. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.; Zhang, B.; Yan, W.; Wang, B.; Wei, H.; Zhang, F.; Wu, L.; Fan, K.; Guo, Y. Myeloid-derived suppressor cells regulate immune response in patients with chronic hepatitis B virus infection through PD-1-induced IL-10. J. Immunol. 2014, 193, 5461–5469. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.; Cui, M.; Lv, Z.; Lu, J.; Zhang, X.; Zhao, Z.; Wang, Y.; Gao, L.; Tsuji, N.M.; Yan, H. Expression and significance of peripheral myeloid-derived suppressor cells in chronic hepatitis B patients. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Ning, G.; She, L.; Lu, L.; Liu, Y.; Zeng, Y.; Yan, Y.; Lin, C. Analysis of Monocytic and Granulocytic Myeloid-Derived Suppressor Cells Subsets in Patients with Hepatitis C Virus Infection and Their Clinical Significance. BioMed Res. Int. 2015, 2015, 385378. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Shimosegawa, T. Significant roles of regulatory T cells and myeloid derived suppressor cells in hepatitis B virus persistent infection and hepatitis B virus-related HCCs. Int. J. Mol. Sci. 2015, 16, 3307–3322. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Zhang, X.; Lv, Z.; Gao, L.; Yan, H. Increased Expression of Myeloid-Derived Suppressor Cells in Patients with HBV-Related Hepatocellular Carcinoma. BioMed Res. Int. 2020, 2020, 6527192–6527198. [Google Scholar] [CrossRef] [Green Version]
- Hetta, H.F.; Zahran, A.M.; Mansor, S.G.; Abdel-Malek, M.O.; Mekky, M.A.; Abbas, W.A. Frequency and Implications of myeloid-derived suppressor cells and lymphocyte subsets in Egyptian patients with hepatitis C virus-related hepatocellular carcinoma. J. Med. Virol. 2019, 91, 1319–1328. [Google Scholar] [CrossRef]
- Reizine, F.; Lesouhaitier, M.; Gregoire, M.; Pinceaux, K.; Gacouin, A.; Maamar, A.; Painvin, B.; Camus, C.; Le Tulzo, Y.; Tattevin, P.; et al. SARS-CoV-2-Induced ARDS Associates with MDSC Expansion, Lymphocyte Dysfunction, and Arginine Shortage. J. Clin. Immunol. 2021, 41, 515–525. [Google Scholar] [CrossRef]
- Hou, A.; Hou, K.; Huang, Q.; Lei, Y.; Chen, W. Targeting Myeloid-Derived Suppressor Cell, a Promising Strategy to Overcome Resistance to Immune Checkpoint Inhibitors. Front. Immunol. 2020, 11, 783. [Google Scholar] [CrossRef] [PubMed]
- Drabczyk-Pluta, M.; Werner, T.; Hoffmann, D.; Leng, Q.; Chen, L.; Dittmer, U.; Zelinskyy, G. Granulocytic myeloid-derived suppressor cells suppress virus-specific CD8 + T cell responses during acute Friend retrovirus infection. Retrovirology 2017, 14, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Yuan, W.; Deng, J.; Wang, D.; Zhang, T.; Peng, L.; Tian, H.; Wang, Z.; Ma, J. Granulocyte colony stimulating factor (G-CSF) regulates neutrophils infiltration and periodontal tissue destruction in an experimental periodontitis. Mol. Immunol. 2020, 117, 110–121. [Google Scholar] [CrossRef]
- Campbell, I.K.; Leong, D.; Edwards, K.M.; Rayzman, V.; Ng, M.; Goldberg, G.L.; Wilson, N.J.; Scalzo-Inguanti, K.; Mackenzie-Kludas, C.; Lawlor, K.E.; et al. Therapeutic Targeting of the G-CSF Receptor Reduces Neutrophil Trafficking and Joint Inflammation in Antibody-Mediated Inflammatory Arthritis. J. Immunol. 2016, 197, 4392–4402. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Shukla, S.; Pandey, A.D.; Goswami, S.; Bandyopadhyay, B.; Ramachandran, V.; Das, S.; Malhotra, A.; Agarwal, A.; Adhikari, S.; et al. RNA-Seq analysis of peripheral blood mononuclear cells reveals unique transcriptional signatures associated with disease progression in dengue patients. Transl. Res. 2017, 186, 62–78.e69. [Google Scholar] [CrossRef] [PubMed]
- Cloke, T.; Munder, M.; Taylor, G.; Müller, I.; Kropf, P. Characterization of a novel population of low-density granulocytes associated with disease severity in HIV-1 infection. PLoS ONE 2012, 7, e48939. [Google Scholar] [CrossRef] [Green Version]
- Schulte-Schrepping, J.; Reusch, N.; Paclik, D.; Baßler, K.; Schlickeiser, S.; Zhang, B.; Krämer, B.; Krammer, T.; Brumhard, S.; Bonaguro, L.; et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell 2020, 182, 1419–1440.e1423. [Google Scholar] [CrossRef]
- Li, Y.; Li, H.; Wang, H.; Pan, H.; Zhao, H.; Jin, H.; Jie, S. The proportion, origin and pro-inflammation roles of low density neutrophils in SFTS disease. BMC Infect. Dis. 2019, 19, 109. [Google Scholar] [CrossRef]
- Siemińska, I.; Węglarczyk, K.; Surmiak, M.; Kurowska-Baran, D.; Sanak, M.; Siedlar, M.; Baran, J. Mild and Asymptomatic COVID-19 Convalescents Present Long-Term Endotype of Immunosuppression Associated With Neutrophil Subsets Possessing Regulatory Functions. Front. Immunol. 2021, 12, 748097. [Google Scholar] [CrossRef]
- Villanueva, E.; Yalavarthi, S.; Berthier, C.C.; Hodgin, J.B.; Khandpur, R.; Lin, A.M.; Rubin, C.J.; Zhao, W.; Olsen, S.H.; Klinker, M.; et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 2011, 187, 538–552. [Google Scholar] [CrossRef] [Green Version]
- Mauracher, L.M.; Krall, M.; Roiß, J.; Hell, L.; Koder, S.; Hofbauer, T.M.; Gebhart, J.; Hayden, H.; Brostjan, C.; Ay, C.; et al. Neutrophil subpopulations and their activation potential in patients with antiphospholipid syndrome and healthy individuals. Rheumatology 2021, 60, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, N.L.; Helton, E.S.; Huijbregts, R.P.; Goepfert, P.A.; Heath, S.L.; Hel, Z. Immune suppression by neutrophils in HIV-1 infection: Role of PD-L1/PD-1 pathway. PLoS Pathog. 2014, 10, e1003993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Huang, H.H.; Yang, T.; Jiao, Y.M.; Zhang, C.; Song, J.W.; Zhang, J.Y.; Zhou, C.B.; Yuan, J.H.; Cao, W.J.; et al. Increased Neutrophil Aging Contributes to T Cell Immune Suppression by PD-L1 and Arginase-1 in HIV-1 Treatment Naïve Patients. Front. Immunol. 2021, 12, 670616. [Google Scholar] [CrossRef]
- Xie, X.; Shi, Q.; Wu, P.; Zhang, X.; Kambara, H.; Su, J.; Yu, H.; Park, S.Y.; Guo, R.; Ren, Q.; et al. Single-cell transcriptome profiling reveals neutrophil heterogeneity in homeostasis and infection. Nat. Immunol. 2020, 21, 1119–1133. [Google Scholar] [CrossRef]
- Sinha, S.; Rosin, N.L.; Arora, R.; Labit, E.; Jaffer, A.; Cao, L.; Farias, R.; Nguyen, A.P.; de Almeida, L.G.N.; Dufour, A.; et al. Dexamethasone modulates immature neutrophils and interferon programming in severe COVID-19. Nat. Med. 2022, 28, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Rudd, J.M.; Pulavendran, S.; Ashar, H.K.; Ritchey, J.W.; Snider, T.A.; Malayer, J.R.; Marie, M.; Chow, V.T.K.; Narasaraju, T. Neutrophils Induce a Novel Chemokine Receptors Repertoire During Influenza Pneumonia. Front. Cell. Infect. Microbiol. 2019, 9, 108. [Google Scholar] [CrossRef] [Green Version]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [Green Version]
- Silva, C.M.S.; Wanderley, C.W.S.; Veras, F.P.; Sonego, F.; Nascimento, D.C.; Gonçalves, A.V.; Martins, T.V.; Cólon, D.F.; Borges, V.F.; Brauer, V.S.; et al. Gasdermin D inhibition prevents multiple organ dysfunction during sepsis by blocking NET formation. Blood 2021, 138, 2702–2713. [Google Scholar] [CrossRef]
- Adrover, J.M.; Carrau, L.; Daßler-Plenker, J.; Bram, Y.; Chandar, V.; Houghton, S.; Redmond, D.; Merrill, J.R.; Shevik, M.; tenOever, B.R.; et al. Disulfiram inhibits neutrophil extracellular trap formation and protects rodents from acute lung injury and SARS-CoV-2 infection. JCI Insight 2022, 7, e157342. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.L.; Pattemore, P.K.; Sanderson, G.; Smith, S.; Campbell, M.J.; Josephs, L.K.; Cunningham, A.; Robinson, B.S.; Myint, S.H.; Ward, M.E. The relationship between upper respiratory infections and hospital admissions for asthma: A time-trend analysis. Am. J. Respir. Crit. Care Med. 1996, 154, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Sigurs, N. Epidemiologic and clinical evidence of a respiratory syncytial virus–reactive airway disease link. Am. J. Respir. Crit. Care Med. 2001, 163, S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Sigurs, N. A cohort of children hospitalised with acute RSV bronchiolitis: Impact on later respiratory disease. Paediatr. Respir. Rev. 2002, 3, 177–183. [Google Scholar] [CrossRef]
- Cheung, D.S.; Ehlenbach, S.J.; Kitchens, R.T.; Riley, D.A.; Thomas, L.L.; Holtzman, M.J.; Grayson, M.H. Cutting edge: CD49d+ neutrophils induce FcεRI expression on lung dendritic cells in a mouse model of postviral asthma. J. Immunol. 2010, 185, 4983–4987. [Google Scholar] [CrossRef] [Green Version]
- Walter, M.J.; Morton, J.D.; Kajiwara, N.; Agapov, E.; Holtzman, M.J. Viral induction of a chronic asthma phenotype and genetic segregation from the acute response. J. Clin. Investig. 2002, 110, 165–175. [Google Scholar] [CrossRef]
- Simões, E.A.; Carbonell-Estrany, X.; Rieger, C.H.; Mitchell, I.; Fredrick, L.; Groothuis, J.R.; Palivizumab Long-Term Respiratory Outcomes Study Group. The effect of respiratory syncytial virus on subsequent recurrent wheezing in atopic and nonatopic children. J. Allergy Clin. Immunol. 2010, 126, 256–262. [Google Scholar] [CrossRef]
- Cheung, D.S.; Sigua, J.A.; Simpson, P.M.; Yan, K.; Hussain, S.-R.A.; Santoro, J.L.; Buell, E.J.; Hunter, D.A.; Rohlfing, M.; Patadia, D. Cysteinyl leukotriene receptor 1 expression identifies a subset of neutrophils during the antiviral response that contributes to postviral atopic airway disease. J. Allergy Clin. Immunol. 2018, 142, 1206–1217.e1205. [Google Scholar] [CrossRef] [Green Version]
- Massena, S.; Christoffersson, G.; Vågesjö, E.; Seignez, C.; Gustafsson, K.; Binet, F.; Herrera Hidalgo, C.; Giraud, A.; Lomei, J.; Weström, S.; et al. Identification and characterization of VEGF-A-responsive neutrophils expressing CD49d, VEGFR1, and CXCR4 in mice and humans. Blood 2015, 126, 2016–2026. [Google Scholar] [CrossRef]
- Rawat, S.; Vrati, S.; Banerjee, A. Neutrophils at the crossroads of acute viral infections and severity. Mol. Aspects Med. 2021, 81, 100996. [Google Scholar] [CrossRef]
- Chan, L.; Alizadeh, K.; Alizadeh, K.; Fazel, F.; Kakish, J.E.; Karimi, N.; Knapp, J.P.; Mehrani, Y.; Minott, J.A.; Morovati, S.; et al. Review of Influenza Virus Vaccines: The Qualitative Nature of Immune Responses to Infection and Vaccination Is a Critical Consideration. Vaccines 2021, 9, 979. [Google Scholar] [CrossRef] [PubMed]
- Hafezi, B.; Chan, L.; Knapp, J.P.; Karimi, N.; Alizadeh, K.; Mehrani, Y.; Bridle, B.W.; Karimi, K. Cytokine Storm Syndrome in SARS-CoV-2 Infections: A Functional Role of Mast Cells. Cells 2021, 10, 1761. [Google Scholar] [CrossRef] [PubMed]
- Speth, C.; Brodde, M.F.; Hagleitner, M.; Rambach, G.; Van Aken, H.; Dierich, M.; Kehrel, B.E. Neutrophils Turn Plasma Proteins into Weapons against HIV-1. PLoS ONE 2013, 8, e66073. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, L.; Morovati, S.; Karimi, N.; Alizadeh, K.; Vanderkamp, S.; Kakish, J.E.; Bridle, B.W.; Karimi, K. Neutrophil Functional Heterogeneity and Implications for Viral Infections and Treatments. Cells 2022, 11, 1322. https://doi.org/10.3390/cells11081322
Chan L, Morovati S, Karimi N, Alizadeh K, Vanderkamp S, Kakish JE, Bridle BW, Karimi K. Neutrophil Functional Heterogeneity and Implications for Viral Infections and Treatments. Cells. 2022; 11(8):1322. https://doi.org/10.3390/cells11081322
Chicago/Turabian StyleChan, Lily, Solmaz Morovati, Negar Karimi, Kasra Alizadeh, Sierra Vanderkamp, Julia E. Kakish, Byram W. Bridle, and Khalil Karimi. 2022. "Neutrophil Functional Heterogeneity and Implications for Viral Infections and Treatments" Cells 11, no. 8: 1322. https://doi.org/10.3390/cells11081322