
Microfluidic Platforms Designed for Morphological and Photosynthetic Investigations of Chlamydomonas reinhardtii on a Single-Cell Level

 , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
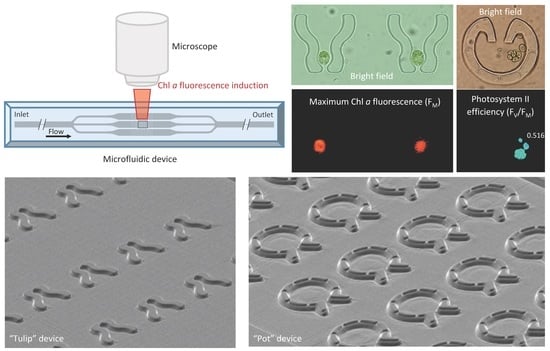
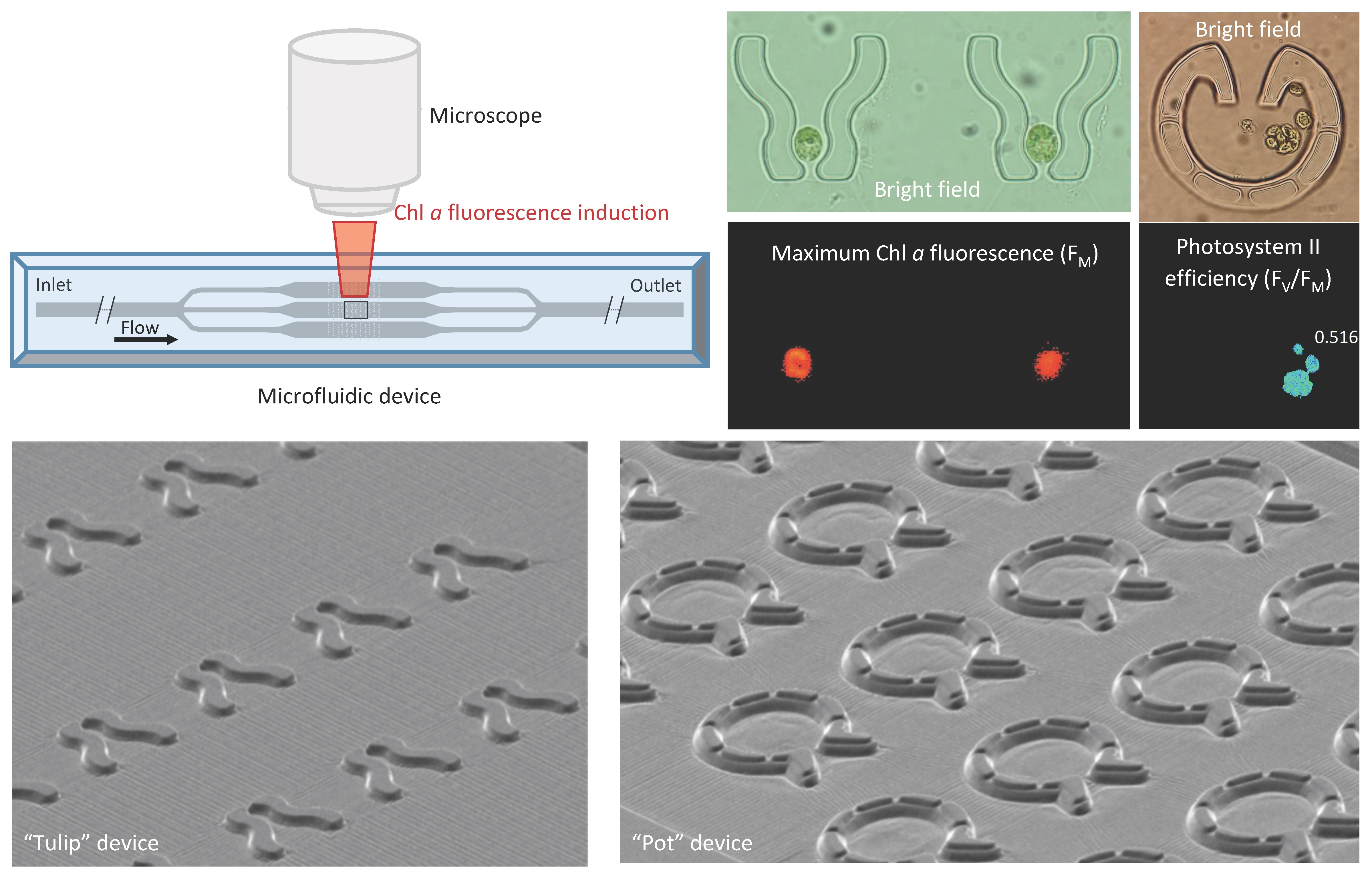
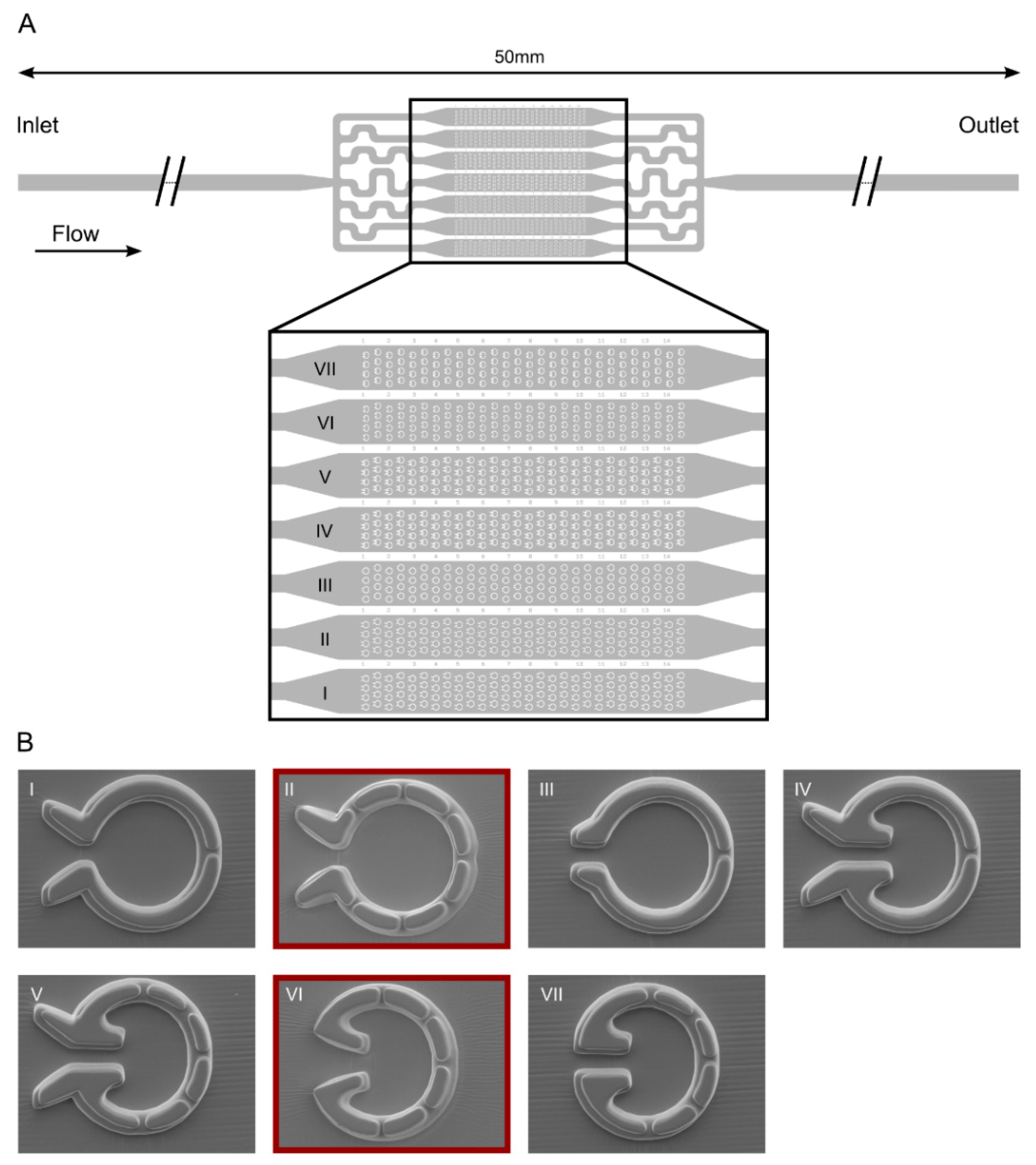
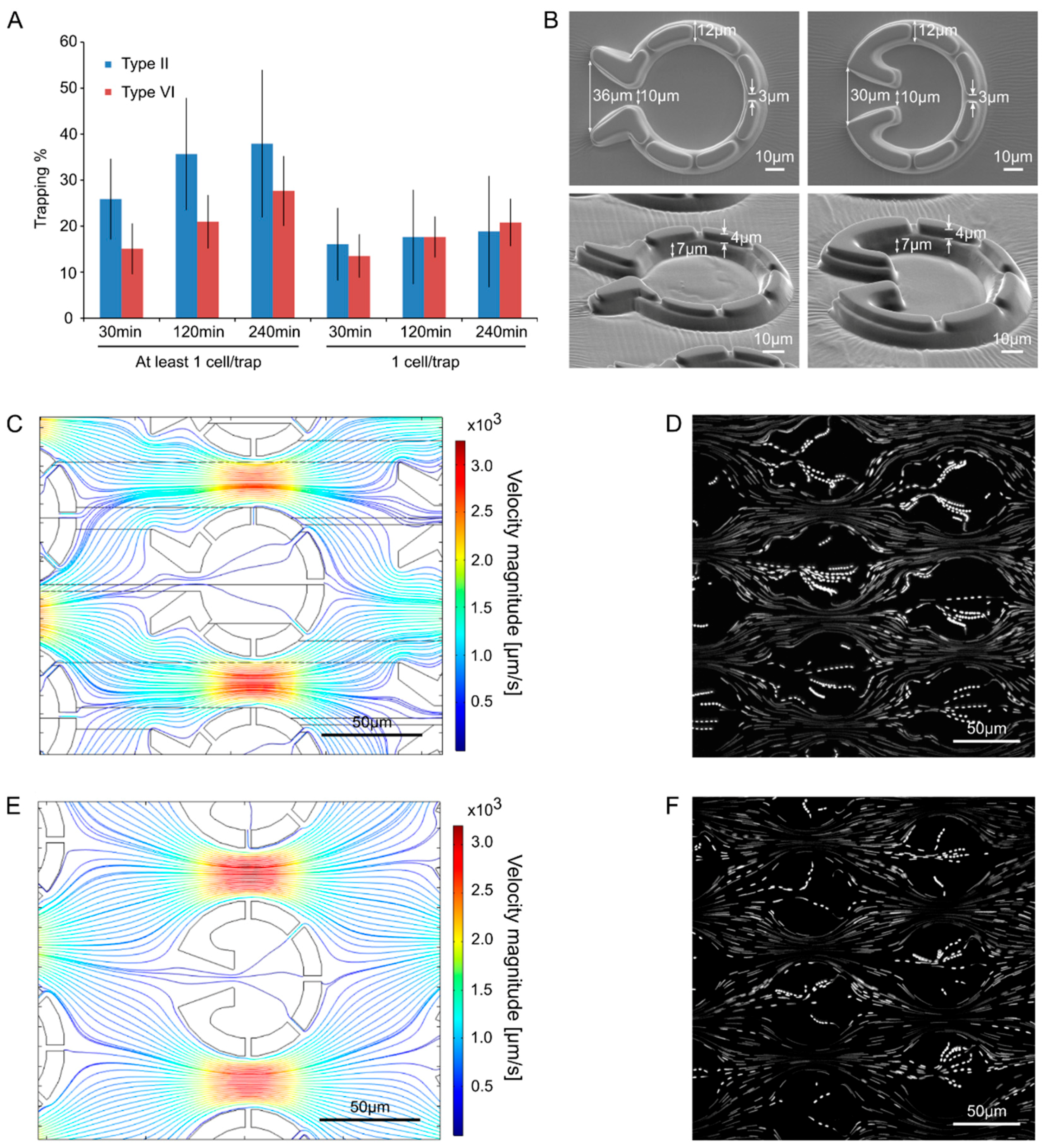
2.1. Design and Construction of Microfluidic Devices for C. reinhardtii
2.2. Characterizing the Properties of the Fluid Flow by Model Calculations and Tracer Particles
2.3. Chlamydomonas reinhardtii Strains and Cell Culture Conditions
2.4. Cell Loading and Culturing in the Microfluidic Devices
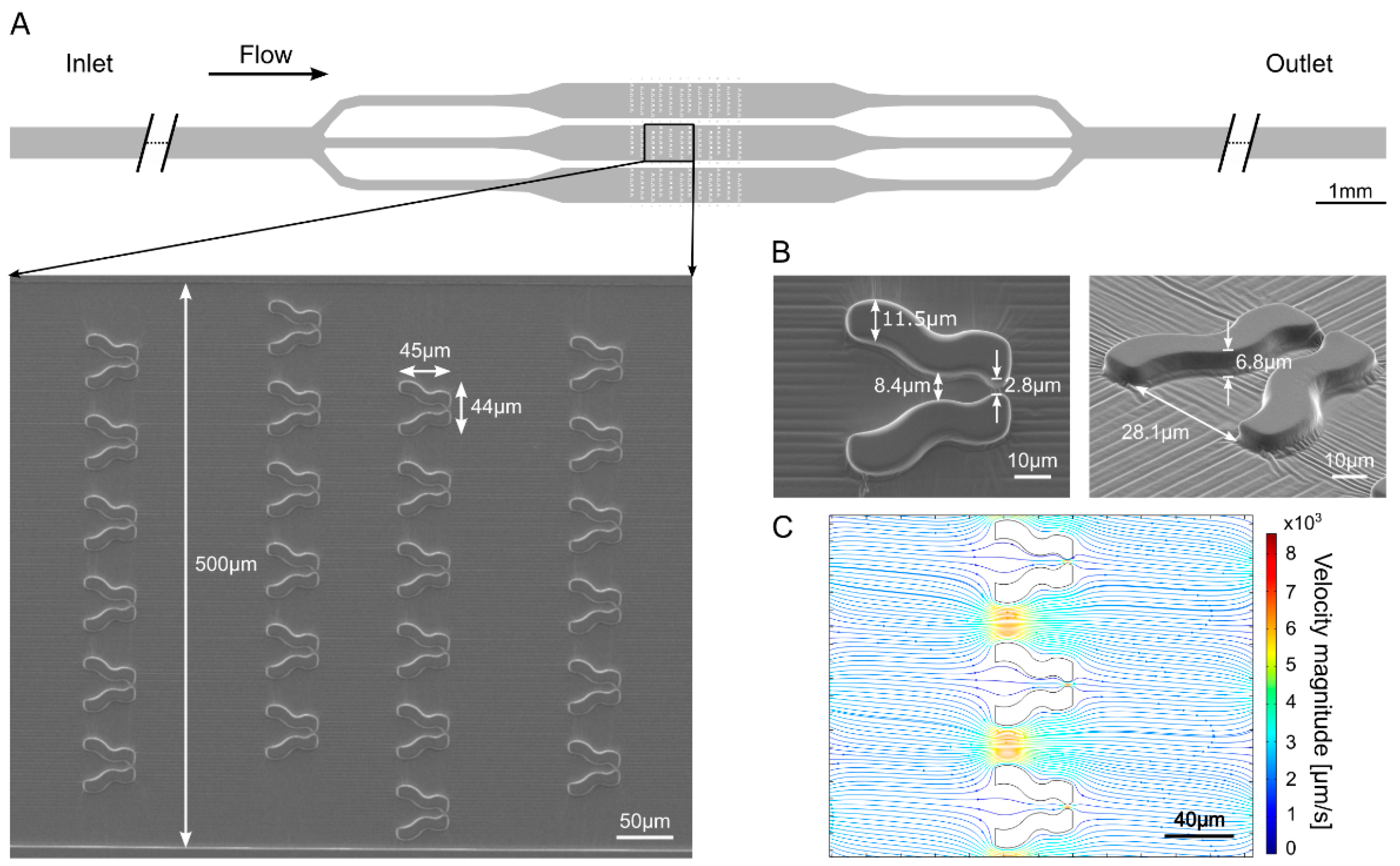
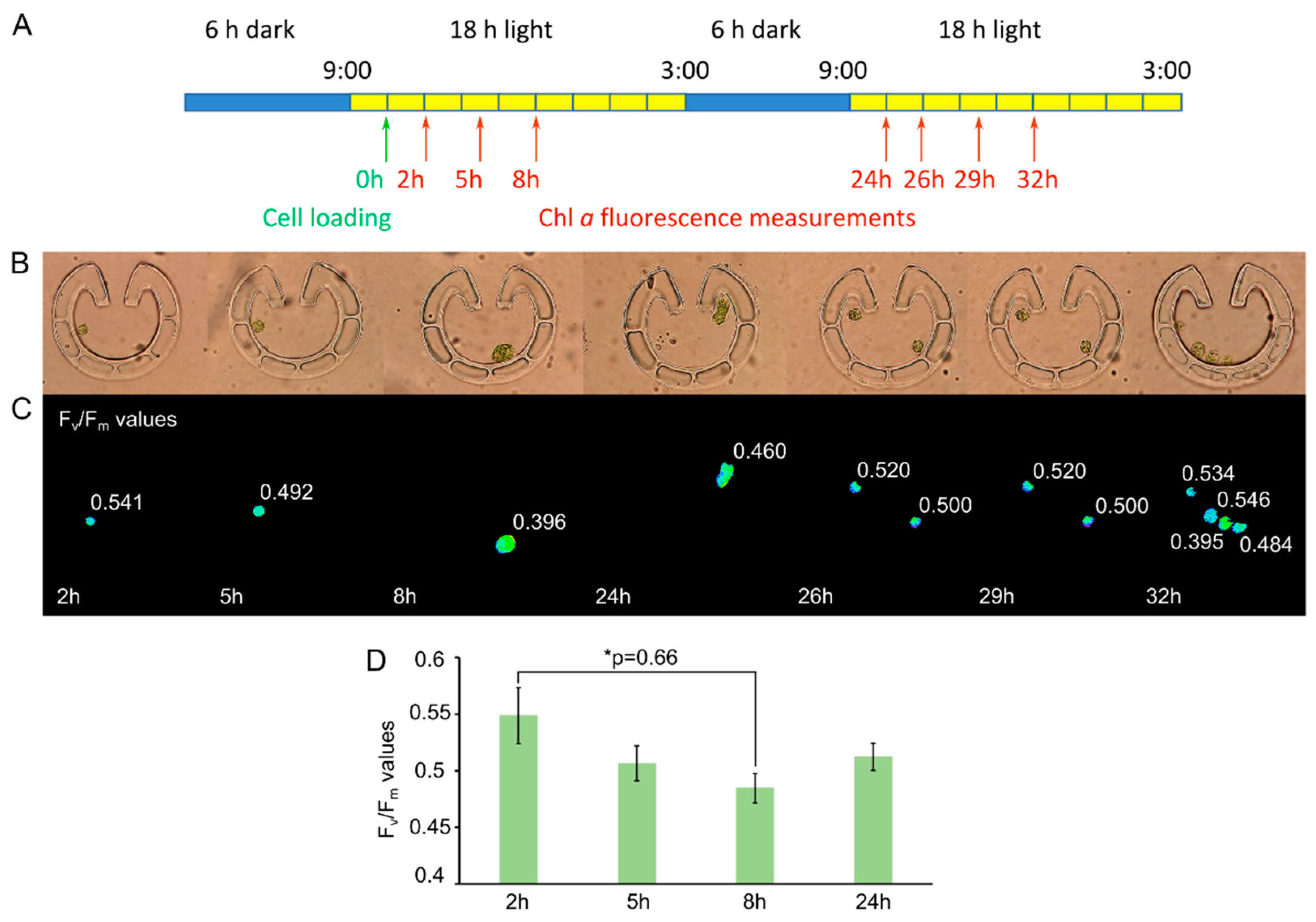
2.5. Microscopy and Chl a Fluorescence Measurements
2.6. Electron Microscopy
2.7. Statistical Analysis
3. Results
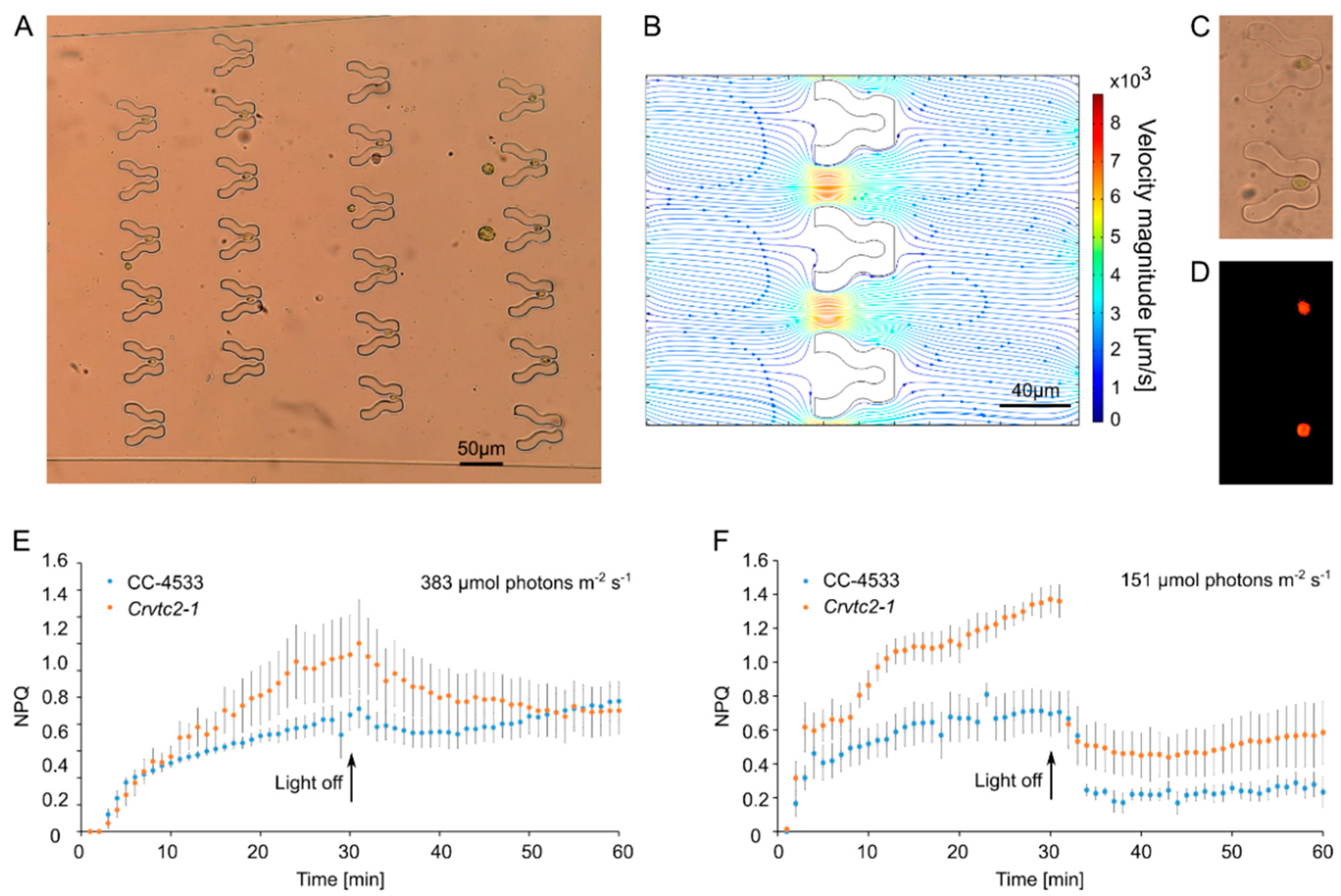
3.1. “Tulip” Microfluidics Platform for Long-Term Chl a Fluorescence Measurements on Single Cells
3.2. “Pot” Microfluidics Platform for Observing Cell Division
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nickelsen, J. Cell biology: The green alga Chlamydomonas reinhardtii—A genetic model organism. In Progress in Botany; Esser, K., Lüttge, U., Beyschlag, W., Murata, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 66, pp. 66–89. [Google Scholar]
- Salomé, P.A.; Merchant, S.S. A series of fortunate events: Introducing Chlamydomonas as a reference organism. Plant Cell 2019, 31, 1682–1707. [Google Scholar] [CrossRef] [Green Version]
- Harris, E.H. Chlamydomonas as a model organism. Ann. Rev. Plant Physiol. Plant Mol. Biol. 2001, 52, 363–406. [Google Scholar] [CrossRef] [Green Version]
- Mettler, T.; Mühlhaus, T.; Hemme, D.; Schöttler, M.-A.; Rupprecht, J.; Idoine, A.; Veyel, D.; Kumar Pal, S.; Yaneva-Roder, L.; Winck, F.V.; et al. Systems analysis of the response of photosynthesis, metabolism, and growth to an increase in irradiance in the photosynthetic model organism Chlamydomonas reinhardtii. Plant Cell 2014, 26, 2310–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim., L.; Toh, Y.; Voldman, J.; Yu, H. A practical guide to microfluidic perfusion culture of adherent mammalian cells. Lab Chip 2007, 7, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Barber, R.; Emerson, D. Optimal design of microfluidic networks using biologically inspired principles. Microfluid. Nanofluid. 2007, 4, 179–191. [Google Scholar] [CrossRef]
- Song, K.; Li, G.; Zu, X.; Du, Z.; Liu, L.; Hu, Z. The fabrication and application mechanism of microfluidic systems for high throughput biomedical screening: A review. Micromachines 2020, 11, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitesides, G.M.; Ostuni, E.; Takayama, S.; Jiang, X.; Ingber, D.E. Soft lithography in biology and chemistry. Annu. Rev. Biomed. Eng. 2001, 3, 335–373. [Google Scholar] [CrossRef] [Green Version]
- Qin, D.; Xia, Y.; Whitesides, G.M. Soft lithography for micro- and nanoscale patterning. Nat. Protoc. 2010, 5, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Vidal-Meireles, A.; Tóth, D.; Kovács, L.; Neupert, J.; Tóth, S.Z. Ascorbate deficiency does not limit nonphotochemical quenching in Chlamydomonas reinhardtii. Plant Physiol. 2020, 182, 597–611. [Google Scholar] [CrossRef] [Green Version]
- Novo, P.; Chu, V.; Conde, J.P. Integrated fluorescence detection of labeled biomolecules using a prism-like PDMS microfluidic chip and lateral light excitation. Lab Chip 2014, 14, 1991–1995. [Google Scholar] [CrossRef]
- Tian, R.; Li, K.; Shi, W.; Ding, C.; Lu, C. In situ visualization of hydrophilic spatial heterogeneity inside microfluidic chips by fluorescence microscopy. Lab Chip 2019, 19, 934–940. [Google Scholar] [CrossRef]
- Wang, J.; Sun, J.; Song, Y.; Xu, Y.; Pan, X.; Sun, Y.; Li, D. A label-free microfluidic biosensor for activity detection of single microalgae cells based on chlorophyll fluorescence. Sensors 2013, 13, 16075–16089. [Google Scholar] [CrossRef]
- Juang, Y.-J.; Chang, J.-S. Applications of microfluidics in microalgae biotechnology: A review. Biotechnol. J. 2016, 11, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, O.H.; Kramarsky-Winter, E.; Gavish, A.R.; Stocker, R.; Vardi, A. A coral-on-a-chip microfluidic platform enabling live-imaging microscopy of reef-building corals. Nat. Commun. 2016, 7, 10860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, F.R.; Umen, J.G. The Chlamydomonas cell cycle. Plant J. 2015, 82, 370–392. [Google Scholar] [CrossRef]
- Heldt, F.S.; Tyson, J.J.; Cross, F.R.; Novák, B. A single light-responsive sizer can control multiple-fission cycles in Chlamydomonas. Curr. Biol. 2020, 30, 634–644. [Google Scholar] [CrossRef]
- Kropat, J.; Hong-Hermesdorf, A.; Casero, D.; Ent, P.; Castruita, M.; Pellegrini, M.; Merchant, S.S.; Malasarn, D. A revised mineral nutrient supplement increases biomass and growth rate in Chlamydomonas reinhardtii. Plant J. 2011, 66, 770–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, D.; López-Paz, C.; Olson, B.J.S.C.; Umen, J.G. A new class of cyclin dependent kinase in Chlamydomonas is required for coupling cell size to cell division. eLife 2016, 5, e10767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; Hsu, S.C.; Han, S.I.; Thapa, H.R.; Guzman, A.R.; Browne, D.R.; Tatli, M.; Devarenne, T.P.; Stern, D.B.; Han, A. High-throughput droplet microfluidics screening platform for selecting fast-growing and high lipid-producing microalgae from a mutant library. Plant Direct 2017, 1, e00011. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Devarenne, T.P.; Han, A. Microfluidic systems for microalgal biotechnology: A review. Algal Res. 2018, 30, 149–161. [Google Scholar] [CrossRef]
- Benazzi, G.; Holmes, D.; Sun, T.; Mowlem, M.C.; Morgan, H. Discrimination and analysis of phytoplankton using a microfluidic cytometer. IET Nanobiotechnol. 2008, 1, 94–101. [Google Scholar] [CrossRef]
- Schaap, A.; Dumon, J.; den Toonder, J. Sorting algal cells by morphology in spiral microchannels using inertial microfluidics. Microfluid. Nanofluid. 2016, 20, 125. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.H.; Kwak, H.S.; Sung, Y.J.; Choi, H.I.; Hong, M.E.; Lim, H.S.; Lee, J.H.; Lee, S.Y.; Sim, S.J. Microfluidic high-throughput selection of microalgal strains with superior photosynthetic productivity using competitive phototaxis. Sci. Rep. 2016, 6, 21155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, Y.J.; Kim, J.Y.H.; Choi, H.I.; Kwak, H.S.; Sim, S.J. Magnetophoretic sorting of microdroplets with different microalgal cell densities for rapid isolation of fast growing strains. Sci. Rep. 2017, 7, 10390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, M.S.; Rafeie, M.; Vandamme, D.; Asadnia, M.; Henderson, R.; Taylor, R.A.; Warkiani, M.E. Selective separation of microalgae cells using inertial microfluidics. Bioresour. Technol. 2018, 252, 91–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, D.; Zhao, Q.; Yan, S.; Tang, S.Y.; Zhang, Y.; Yun, G.; Nguyen, N.T.; Zhang, J.; Li, M.; Li, W. Sheathless separation of microalgae from bacteria using a simple straight channel based on viscoelastic microfluidics. Lab Chip 2019, 19, 2811–2821. [Google Scholar] [CrossRef]
- Korensky, G.; Chen, X.; Bao, M.; Miller, A.; Lapizco-Encinas, B.; Park, M.; Du, K. Single Chlamydomonas reinhardtii cell separation from bacterial cells and auto-fluorescence tracking with a nanosieve device. Electrophoresis 2020, 42, 95–102. [Google Scholar] [CrossRef]
- Kim, H.S.; Devarenne, T.P.; Han, A. A high-throughput microfluidic single-cell screening platform capable of selective cell extraction. Lab Chip 2015, 15, 2467–2475. [Google Scholar] [CrossRef]
- Tahirbegi, I.B.; Ehgartner, J.; Sulzer, P.; Zieger, S.; Kasjanow, A.; Paradiso, M.; Strobl, M.; Bouwes, D.; Mayr, T. Fast pesticide detection inside microfluidic device with integrated optical pH, oxygen sensors and algal fluorescence. Biosens. Bioelectron. 2017, 88, 188–195. [Google Scholar] [CrossRef]
- Lefèvre, F.; Chalifour, A.; Yu, L.; Chodavarapu, V.; Juneau, P.; Izquierdo, R. Algal fluorescence sensor integrated into a microfluidic chip for water pollutant detection. Lab Chip 2012, 12, 787. [Google Scholar] [CrossRef] [PubMed]
- Gosset, A.; Durrieu, C.; Renaud, L.; Deman, A.L.; Barbe, P.; Bayard, R.; Chateaux, J.F. Xurography-based microfluidic algal biosensor and dedicated portable measurement station for online monitoring of urban polluted samples. Biosens. Bioelectron. 2018, 117, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, J.; Zhou, C.; Ding, G.; Chen, M.; Zou, J.; Wang, G.; Kang, Y.; Pan, X. A microfluidic prototype system towards microalgae cell separation, treatment and viability characterization. Sensors 2019, 19, 4940. [Google Scholar] [CrossRef] [Green Version]
- Westerwalbesloh, C.; Brehl, C.; Weber, S.; Probst, C.; Widzgowski, J.; Grünberger, A.; Pfaff, C.; Nedbal, L.; Kohlheyer, D. A microfluidic photobioreactor for simultaneous observation and cultivation of single microalgal cells or cell aggregates. PLoS ONE 2019, 14, e0216093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhao, H.; Liu, X.; Lin, W.; Jiang, Y.; Li, J.; Zhang, Q.; Zheng, G. An integrated digital microfluidic bioreactor for fully automatic screening of microalgal growth and stress-induced lipid accumulation. Biotechnol. Bioeng. 2020, 118, 294–304. [Google Scholar] [CrossRef]
- Behrendt, L.; Salek, M.M.; Trampe, E.L.; Fernandez, V.I.; Lee, K.S.; Kühl, M.; Stocker, R. PhenoChip: A single-cell phenomic platform for highthroughput photophysiological analyses of microalgae. Sci. Adv. 2020, 6, eabb2754. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Woong Kim, C.; Seob Choi, J.; Yang, J.-W.; Seo, T.S. An integrated microfluidic device for the high-throughput screening of microalgal cell culture conditions that induce high growth rate and lipid content. Anal. Bioanal. Chem. 2013, 405, 9365–9374. [Google Scholar] [CrossRef]
- Matsumura, K.; Yagi, T.; Yasuda, K. Role of timer and sizer in regulation of Chlamydomonas cell cycle. Biochem. Biophys. Res. Commun. 2003, 306, 1042–1049. [Google Scholar] [CrossRef]
- Folcik, A.M.; Haire, T.; Cutshaw, K.; Riddle, M.; Shola, C.; Nassani, S.; Rice, P.; Richardson, B.; Shah, P.; Nazamoddini-Kachouie, N.; et al. Computer-assisted tracking of Chlamydomonas species. Front. Plant Sci. 2020, 10, 1616. [Google Scholar] [CrossRef]
- Pokora, W.; Aksmann, A.; Bascik-Remisiewicz, A.; Dettlaff-Pokora, A.; Rykaczewski, M.; Gappa, M.; Tukaj, Z. Changes in nitric oxide/hydrogen peroxide content and cell cycle progression: Study with synchronized cultures of green alga Chlamydomonas reinhardtii. J. Plant Physiol. 2017, 208, 84–93. [Google Scholar] [CrossRef]
- Tulin, F.; Cross, F.R. Cyclin-dependent kinase regulation of diurnal transcription in Chlamydomonas. Plant Cell 2015, 27, 2727–2742. [Google Scholar] [CrossRef]
- Strenkert, D.; Schmollinger, S.; Gallaher, S.D.; Salomé, P.A.; Purvine, S.O.; Nicora, C.D.; Mettler-Altmann, T.; Soubeyrand, E.; Weber, A.P.M.; Lipton, M.S.; et al. Multiomics resolution of molecular events during a day in the life of Chlamydomonas. Proc. Natl. Acad. Sci. USA 2019, 116, 2374–2383. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.; Piccinini, F.; Balassa, T.; Koos, K.; Danka, T.; Azizpour, H.; Horvath, P. Phenotypic image analysis software tools for exploring and understanding big image data from cell-based assays. Cell Syst. 2018, 6, 636–653. [Google Scholar] [CrossRef] [PubMed]
- McCord, R.P.; Yukich, J.N.; Bernd, K.K. Analysis of force generation during flagellar assembly through optical trapping of free-swimming Chlamydomonas reinhardtii. Cell Mot. Cytoskeleton 2005, 61, 137–144. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CC-4533 | Crvtc2-1 | ||||
|---|---|---|---|---|---|
| TAP | HSM | TAP | HSM | ||
| FV/FM | Batch culture | 0.467 ± 0.016 (n = 9) | 0.429 ± 0.024 (n = 4) | 0.500 ± 0.024 (n = 8) | 0.426 ± 0.024 (n = 4) |
| Single cell | 0.473 ± 0.016 (n = 9) | 0.448 ± 0.026 (n = 4) | 0.481 ± 0.017 (n = 8) | 0.469 ± 0.026 (n = 4) | |
| NPQ | 0.962 ± 0.105 (n = 9) | 0.705 ± 0.119 (n = 4) | 1.868 ± 0.392 * (n = 5) | 1.360 ± 0.096 * (n = 4) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Széles, E.; Nagy, K.; Ábrahám, Á.; Kovács, S.; Podmaniczki, A.; Nagy, V.; Kovács, L.; Galajda, P.; Tóth, S.Z. Microfluidic Platforms Designed for Morphological and Photosynthetic Investigations of Chlamydomonas reinhardtii on a Single-Cell Level. Cells 2022, 11, 285. https://doi.org/10.3390/cells11020285
Széles E, Nagy K, Ábrahám Á, Kovács S, Podmaniczki A, Nagy V, Kovács L, Galajda P, Tóth SZ. Microfluidic Platforms Designed for Morphological and Photosynthetic Investigations of Chlamydomonas reinhardtii on a Single-Cell Level. Cells. 2022; 11(2):285. https://doi.org/10.3390/cells11020285
Chicago/Turabian StyleSzéles, Eszter, Krisztina Nagy, Ágnes Ábrahám, Sándor Kovács, Anna Podmaniczki, Valéria Nagy, László Kovács, Péter Galajda, and Szilvia Z. Tóth. 2022. "Microfluidic Platforms Designed for Morphological and Photosynthetic Investigations of Chlamydomonas reinhardtii on a Single-Cell Level" Cells 11, no. 2: 285. https://doi.org/10.3390/cells11020285