
Correcting Differential Gene Expression Analysis for Cyto—Architectural Alterations in Substantia Nigra of Parkinson’s Disease Patients Reveals Known and Potential Novel Disease—Associated Genes and Pathways

 , ,
, ,  ,
, 
Abstract
:1. Introduction
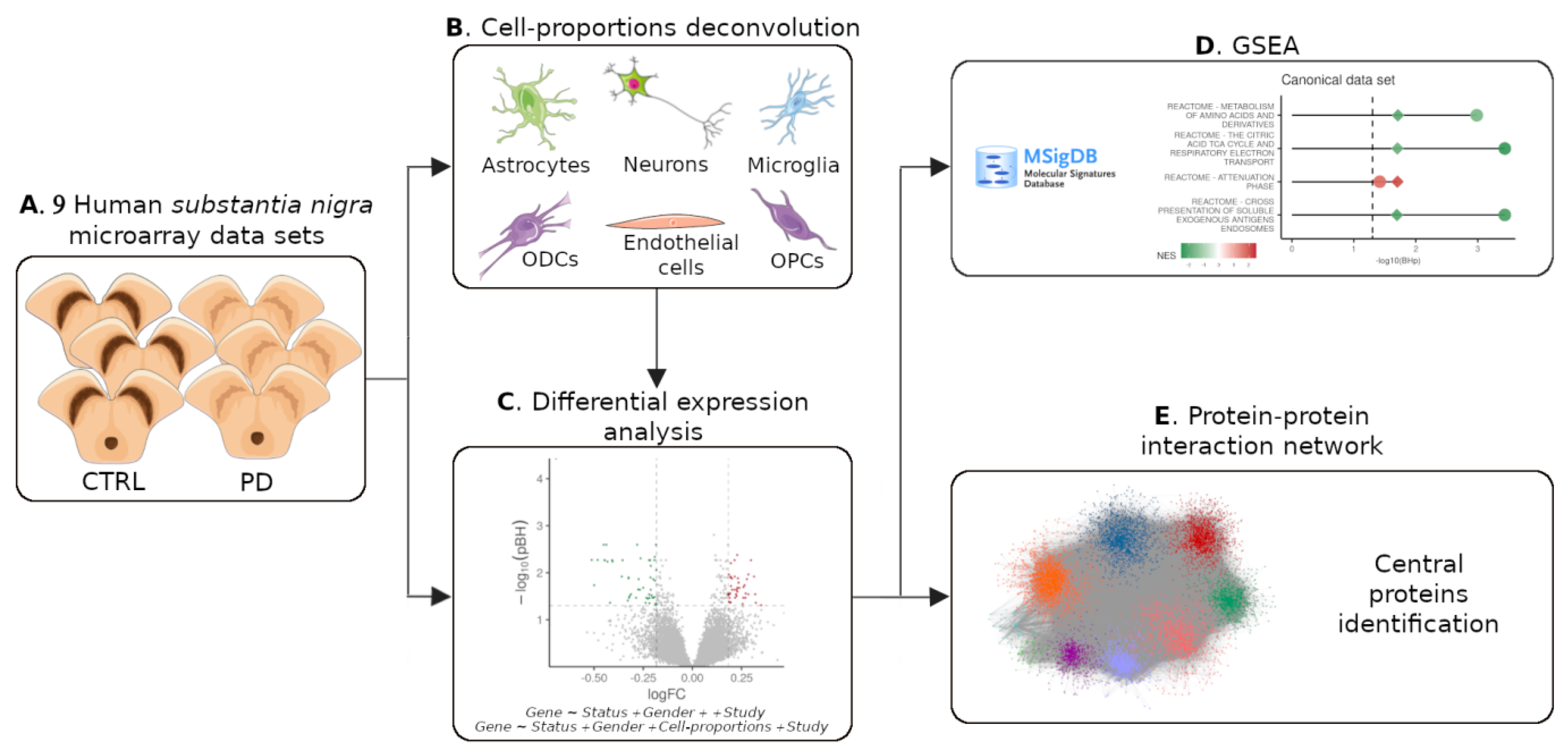
2. Materials and Methods
2.1. Transcriptome Dataset Acquisition, Preprocessing, and Gender Imputation
2.2. DEGs Meta-Analysis
2.3. Processing of Substantia Nigra Single-Nucleus Data
2.4. Cell-Type Proportion Estimation and Marker Selection
2.5. Cell-Type Proportion Comparison and Statistical Analyses
2.6. Gene Set Enrichment Analysis
2.7. Expression-Weighted Cell-Type Enrichment (EWCE)
2.8. Protein-Protein Interaction Network Construction and Analysis
3. Results
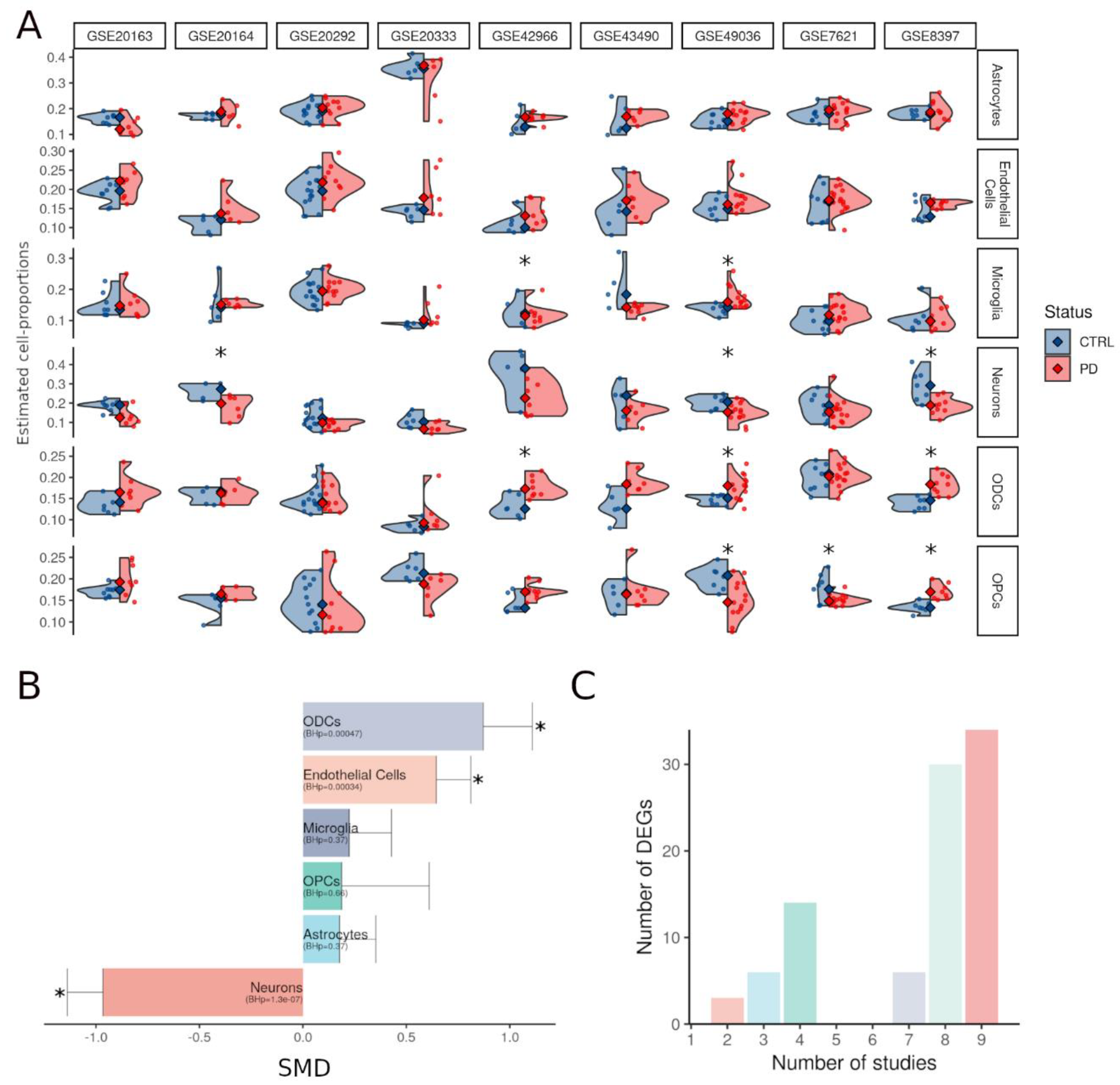
3.1. Alterations in Composition of the Substantia Nigra Are Heterogeneous
3.2. The Cyto-Architectural Alterations Are a Major Confounder in the DEG Identification
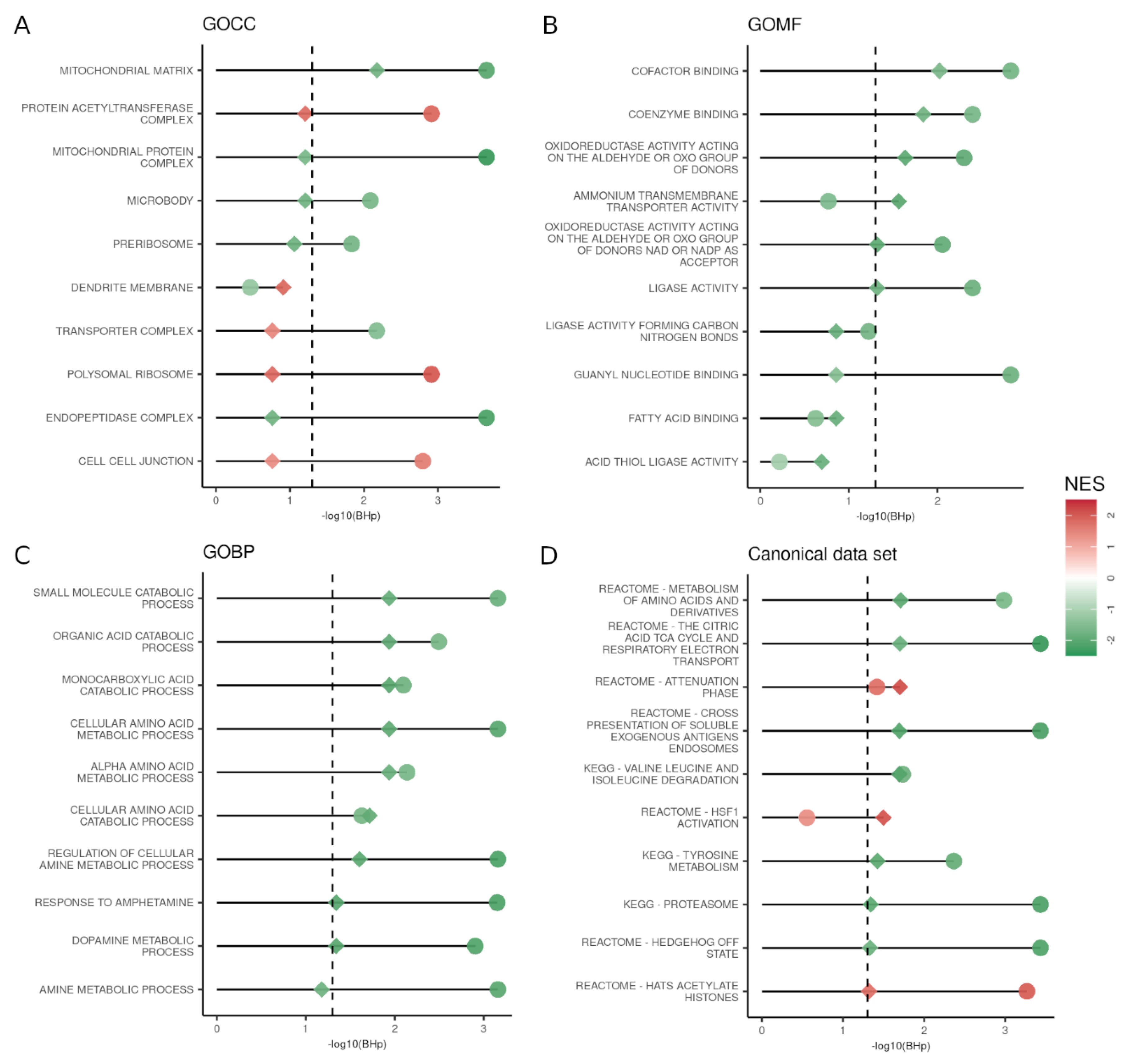
3.3. GSEA Reveals That the Majority of the Altered Pathways in PD Are Downregulated
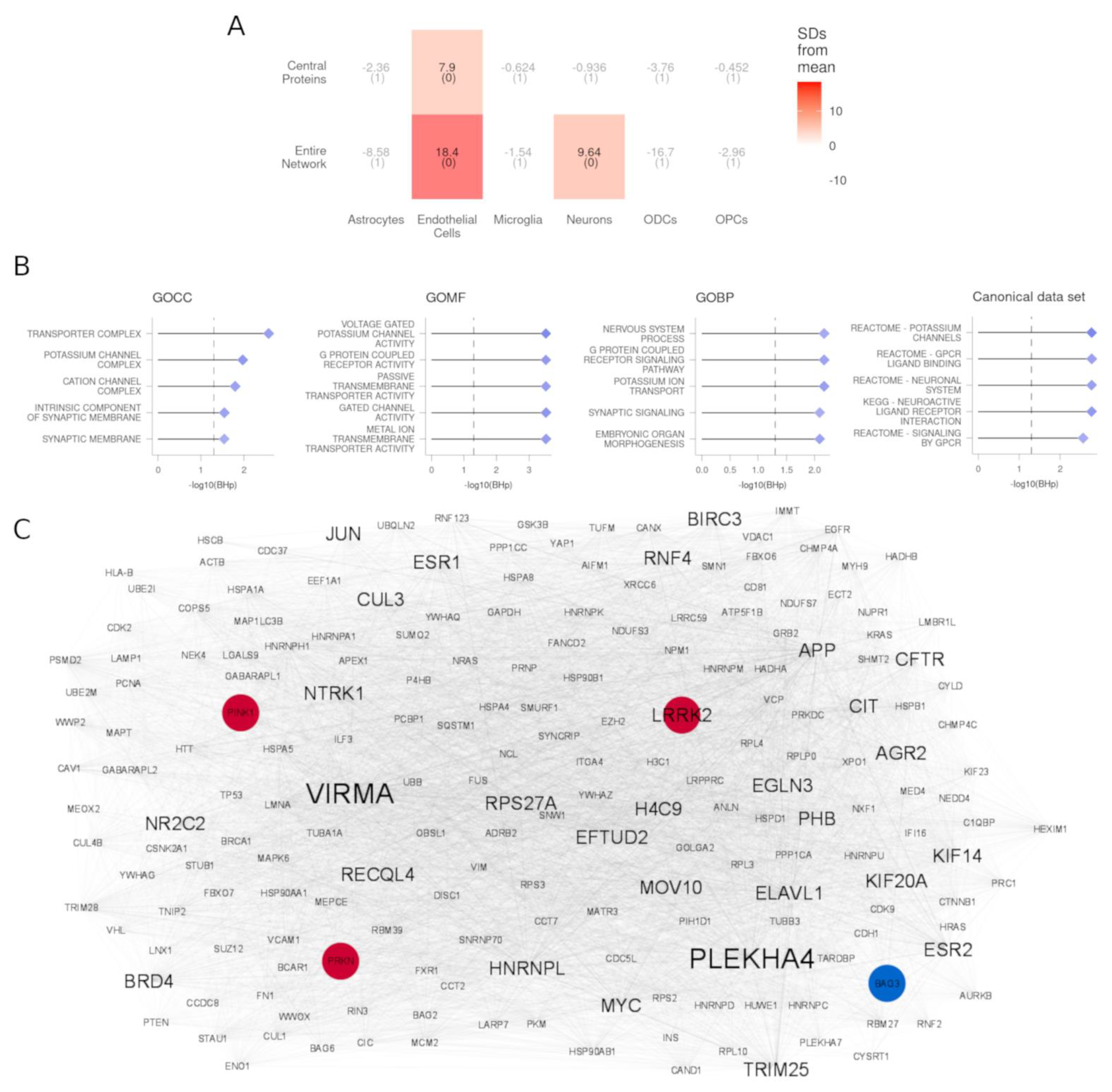
3.4. PPI Partners of DEGs Are Enriched for Genes Expressed in Endothelial Cells and Neurons
3.5. Central Proteins of DEG PPI Partners Are Known PD-Causing Genes and Genes in Proximity to PD Risk Factors
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nature reviews. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Balestrino, R.; Schapira, A. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Fares, M.B.; Jagannath, S.; Lashuel, H.A. Reverse engineering Lewy bodies: How far have we come and how far can we go? Nature reviews. Neuroscience 2021, 22, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Parkinson, J. An Essay on the Shaking Palsy. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Borrageiro, G.; Haylett, W.; Seedat, S.; Kuivaniemi, H.; Bardien, S. A review of genome-wide transcriptomics studies in Parkinson’s disease. Eur. J. Neurosci. 2018, 47, 1–16. [Google Scholar] [CrossRef]
- Keo, A.; Mahfouz, A.; Ingrassia, A.; Meneboo, J.P.; Villenet, C.; Mutez, E.; Comptdaer, T.; Lelieveldt, B.; Figeac, M.; Chartier-Harlin, M.C.; et al. Transcriptomic signatures of brain regional vulnerability to Parkinson’s disease. Commun. Biol. 2020, 3, 101. [Google Scholar] [CrossRef] [Green Version]
- Oerton, E.; Bender, A. Concordance analysis of microarray studies identifies representative gene expression changes in Parkinson’s disease: A comparison of 33 human and animal studies. BMC Neurol. 2017, 17, 58. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, Y.; Wang, M.; Song, W.M.; Shen, Q.; McKenzie, A.; Choi, I.; Zhou, X.; Pan, P.Y.; Yue, Z.; et al. The landscape of multiscale transcriptomic networks and key regulators in Parkinson’s disease. Nat. Commun. 2019, 10, 5234. [Google Scholar] [CrossRef]
- Jaffe, A.E.; Tao, R.; Norris, A.L.; Kealhofer, M.; Nellore, A.; Shin, J.H.; Kim, D.; Jia, Y.; Hyde, T.M.; Kleinman, J.E.; et al. qSVA framework for RNA quality correction in differential expression analysis. Proc. Natl. Acad. Sci. USA 2017, 114, 7130–7135. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, K.; Friedman, B.A.; Larson, J.L.; Lauffer, B.E.; Goldstein, L.D.; Appling, L.L.; Borneo, J.; Poon, C.; Ho, T.; Cai, F.; et al. Untangling the brain’s neuroinflammatory and neurodegenerative transcriptional responses. Nat. Commun. 2016, 7, 11295. [Google Scholar] [CrossRef]
- Smajić, S.; Prada-Medina, C.A.; Landoulsi, Z.; Dietrich, C.; Jarazo, J.; Henck, J.; Balachandran, S.; Pachchek, S.; Morris, C.M.; Antony, P.; et al. Single-cell sequencing of the human midbrain reveals glial activation and a neuronal state specific to Parkinson’s disease. medRxiv 2021. [Google Scholar] [CrossRef]
- Shen-Orr, S.S.; Tibshirani, R.; Khatri, P.; Bodian, D.L.; Staedtler, F.; Perry, N.M.; Hastie, T.; Sarwal, M.M.; Davis, M.M.; Butt, A.J. Cell type-specific gene expression differences in complex tissues. Nat. Methods 2010, 7, 287–289. [Google Scholar] [CrossRef] [PubMed]
- Mancarci, B.O.; Toker, L.; Tripathy, S.J.; Li, B.; Rocco, B.; Sibille, E.; Pavlidis, P. Cross-Laboratory Analysis of Brain Cell Type Transcriptomes with Applications to Interpretation of Bulk Tissue Data. eNeuro 2017, 4, ENEURO.0212-17. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, A.T.; Wang, M.; Hauberg, M.E.; Fullard, J.F.; Kozlenkov, A.; Keenan, A.; Hurd, Y.L.; Dracheva, S.; Casaccia, P.; Roussos, P.; et al. Brain Cell Type Specific Gene Expression and Co-expression Network Architectures. Sci. Rep. 2018, 8, 8868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Wang, X.; Park, J.; Susztak, K.; Zhang, N.R.; Li, M. Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat. Commun. 2019, 10, 380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaitsev, K.; Bambouskova, M.; Swain, A.; Artyomov, M.N. Complete deconvolution of cellular mixtures based on linearity of transcriptional signatures. Nat. Commun. 2019, 10, 2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, M.; Thennavan, A.; Urrutia, E.; Li, Y.; Perou, C.M.; Zou, F.; Jiang, Y. SCDC: Bulk gene expression deconvolution by multiple single-cell RNA sequencing references. Brief. Bioinform. 2021, 22, 416–427. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Guo, Z.; Cheng, Y.; Jin, P.; Wu, H. Robust partial reference-free cell composition estimation from tissue expression. Bioinformatics 2020, 36, 3431–3438. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, G.T.; Matigian, N.A.; Chalk, A.M.; Anderson, M.J.; Silburn, P.A.; Mackay-Sim, A.; Wells, C.A.; Mellick, G.D. A cross-study transcriptional analysis of Parkinson’s disease. PLoS ONE 2009, 4, e4955. [Google Scholar] [CrossRef] [Green Version]
- Duke, D.C.; Moran, L.B.; Pearce, R.K.; Graeber, M.B. The medial and lateral substantia nigra in Parkinson’s disease: mRNA profiles associated with higher brain tissue vulnerability. Neurogenetics 2007, 8, 83–94. [Google Scholar] [CrossRef]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.A.; Cai, C.; Langfelder, P.; Geschwind, D.H.; Kurian, S.M.; Salomon, D.R.; Horvath, S. Strategies for aggregating gene expression data: The collapseRows R function. BMC Bioinform. 2011, 12, 322. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Buckberry, S.; Bent, S.J.; Bianco-Miotto, T.; Roberts, C.T. massiR: A method for predicting the sex of samples in gene expression microarray datasets. Bioinformatics 2014, 30, 2084–2085. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, D.; Sandor, C.; Volpato, V.; Caffrey, T.M.; Monzón-Sandoval, J.; Bowden, R.; Alegre-Abarrategui, J.; Wade-Martins, R.; Webber, C. A single-cell atlas of the human substantia nigra reveals cell-specific pathways associated with neurological disorders. Nat. Commun. 2020, 11, 4183. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wu, H. TOAST: Improving reference-free cell composition estimation by cross-cell type differential analysis. Genome Biol. 2019, 20, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viechtbauer, W. Conducting meta-analyses in R with the metafor package. J. Stat. Softw. 2010, 36, 1–48. Available online: https://www.jstatsoft.org/v36/i03/.
- Korotkevich, G.; Sukhov, V.; Sergushichev, A.; Budin, N.; Shpak, B.; Artyomov, M.N. Fast gene set enrichment analysis. BioRxiv 2019, 060012. [Google Scholar] [CrossRef] [Green Version]
- Zito, A.; Lualdi, M.; Granata, P.; Cocciadiferro, D.; Novelli, A.; Alberio, T.; Casalone, R.; Fasano, M. Gene Set Enrichment Analysis of Interaction Networks Weighted by Node Centrality. Front. Genet. 2021, 12, 577623. [Google Scholar] [CrossRef] [PubMed]
- Skene, N.G.; Grant, S.G. Identification of Vulnerable Cell Types in Major Brain Disorders Using Single Cell Transcriptomes and Expression Weighted Cell Type Enrichment. Front. Neurosci. 2016, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Luck, K.; Kim, D.-K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B. A reference map of the human binary protein interactome. Nature 2020, 580, 402–408. [Google Scholar] [CrossRef]
- Oughtred, R.; Stark, C.; Breitkreutz, B.J.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O’Donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019, 47, D529–D541. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Kotlyar, M.; Pastrello, C.; Malik, Z.; Jurisica, I. IID 2018 update: Context-specific physical protein-protein interactions in human, model organisms and domesticated species. Nucleic Acids Res. 2019, 47, D581–D589. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Bruckner, R.J.; Navarete-Perea, J.; Cannon, J.R.; Baltier, K.; Gebreab, F.; Gygi, M.P.; Thornock, A.; Zarraga, G.; Tam, S.; et al. Dual Proteome-scale Networks Reveal Cell-specific Remodeling of the Human Interactome. Cell 2020, 184, 3022–3040.e28. [Google Scholar] [CrossRef]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJ. Complex. Syst. 2006, 1695, 1–9. Available online: https://igraph.org.
- Lesnick, T.G.; Papapetropoulos, S.; Mash, D.C.; Ffrench-Mullen, J.; Shehadeh, L.; de Andrade, M.; Henley, J.R.; Rocca, W.A.; Ahlskog, J.E.; Maraganor, D.M. A genomic pathway approach to a complex disease: Axon guidance and Parkinson disease. PLoS Genet. 2007, 3, e98. [Google Scholar] [CrossRef]
- Moran, L.B.; Duke, D.C.; Deprez, M.; Dexter, D.T.; Pearce, R.K.; Graeber, M.B. Whole genome expression profiling of the medial and lateral substantia nigra in Parkinson’s disease. Neurogenetics 2006, 7, 1–11. [Google Scholar] [CrossRef]
- Grunblatt, E.; Mandel, S.; Jacob-Hirsch, J.; Zeligson, S.; Amariglo, N.; Rechavi, G.; Li, J.; Ravid, R.; Roggendorf, W.; Riederer, P.; et al. Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J. Neural. Transm. 2004, 111, 1543–1573. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; James, M.; Middleton, F.A.; Davis, R.L. Transcriptional analysis of multiple brain regions in Parkinson’s disease supports the involvement of specific protein processing, energy metabolism, and signaling pathways, and suggests novel disease mechanisms. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 137, 5–16. [Google Scholar] [CrossRef]
- Miller, R.M.; Kiser, G.L.; Kaysser-Kranich, T.M.; Lockner, R.J.; Palaniappan, C.; Federoff, H.J. Robust dysregulation of gene expression in substantia nigra and striatum in Parkinson’s disease. Neurobiol. Dis. 2006, 21, 305–313. [Google Scholar] [CrossRef]
- Hauser, M.A.; Li, Y.J.; Xu, H.; Noureddine, M.A.; Shao, Y.S.; Gullans, S.R.; Scherzer, C.R.; Jensen, R.V.; McLaurin, A.C.; Gibson, J.R.; et al. Expression profiling of substantia nigra in Parkinson disease, progressive supranuclear palsy, and frontotemporal dementia with parkinsonism. Arch. Neurol. 2005, 62, 917–921. [Google Scholar] [CrossRef] [Green Version]
- Dijkstra, A.A.; Ingrassia, A.; de Menezes, R.X.; van Kesteren, R.E.; Rozemuller, A.J.; Heutink, P.; van de Berg, W.D. Evidence for Immune Response, Axonal Dysfunction and Reduced Endocytosis in the Substantia Nigra in Early Stage Parkinson’s Disease. PLoS ONE 2015, 10, e0128651. [Google Scholar] [CrossRef] [Green Version]
- Corradini, B.Z.; Iamashita, P.; Tampellini, E.; Farfel, J.M.; Grinberg, L.T.; Moreira-Filho, C.A. Complex network-driven view of genomic mechanisms underlying Parkinson’s disease: Analyses in dorsal motor vagal nucleus, locus coeruleus, and substantia nigra. BioMed. Res. Int. 2014, 2014, 543673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bando, S.Y.; Iamashita, P.; Fujita, A.; Tampellini, E.; Farfel, J.M.; Grinberg, L.T.; Moreira-Filho, C.A. Transcriptional and Interactome Network Analyses of Substantia Nigra in Progressive Stages of Parkinson’s Disease; Faculdade de Medicina da Universidade de Sao Paulo: Sao Paulo, Brazil, 2018. [Google Scholar]
- Reynolds, R.H.; Botía, J.; Nalls, M.A.; International Parkinson’s Disease Genomics Consortium; System Genomics of Parkinson’s Disease; Hardy, J.; Gagliano, T.S.A.; Ryten, M. Moving beyond neurons: The role of cell type-specific gene regulation in Parkinson’s disease heritability. NPJ Parkinson’s Dis. 2019, 5, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryois, J.; Skene, N.G.; Hansen, T.F.; Kogelman, L.; Watson, H.J.; Liu, Z.; Eating Disorders Working Group of the Psychiatric Genomics Consortium; International Headache Genetics Consortium; 23andMe Research Team; Brueggeman, L.; et al. Genetic identification of cell types underlying brain complex traits yields insights into the etiology of Parkinson’s disease. Nat. Genet. 2020, 52, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Faucheux, B.A.; Bonnet, A.M.; Agid, Y.; Hirsch, E.C. Blood vessels change in the mesencephalon of patients with Parkinson’s disease. Lancet 1999, 353, 981–982. [Google Scholar] [CrossRef]
- Desai Bradaric, B.; Patel, A.; Schneider, J.A.; Carvey, P.M.; Hendey, B. Evidence for angiogenesis in Parkinson’s disease, incidental Lewy body disease, and progressive supranuclear palsy. J. Neural Transm. 2012, 119, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Naldini, A.; Carraro, F. Role of inflammatory mediators in angiogenesis. Curr. Drug Targets Inflamm. Allergy 2005, 4, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Wada, K.; Arai, H.; Takanashi, M.; Fukae, J.; Oizumi, H.; Yasuda, T.; Mizuno, Y.; Mochizuki, H. Expression levels of vascular endothelial growth factor and its receptors in Parkinson’s disease. Neuroreport 2006, 17, 705–709. [Google Scholar] [CrossRef]
- Barcia, C.; Emborg, M.E.; Hirsch, E.C.; Herrero, M.T. Blood vessels and parkinsonism. Front. Biosci. J. Virtual Libr. 2004, 9, 277–282. [Google Scholar] [CrossRef] [Green Version]
- Vila, M.; Jackson-Lewis, V.; Guégan, C.; Wu, D.C.; Teismann, P.; Choi, D.K.; Tieu, K.; Przedborski, S. The role of glial cells in Parkinson’s disease. Curr. Opin. Neurol. 2001, 14, 483–489. [Google Scholar] [CrossRef]
- Tong, J.; Ang, L.C.; Williams, B.; Furukawa, Y.; Fitzmaurice, P.; Guttman, M.; Boileau, I.; Hornykiewicz, O.; Kish, S.J. Low levels of astroglial markers in Parkinson’s disease: Relationship to α-synuclein accumulation. Neurobiol. Dis. 2015, 82, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Masato, A.; Plotegher, N.; Boassa, D.; Bubacco, L. Impaired dopamine metabolism in Parkinson’s disease pathogenesis. Mol. Neurodegener. 2019, 14, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNaught, K.S.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C.W. Altered proteasomal function in sporadic Parkinson’s disease. Exp. Neurol. 2003, 179, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nature reviews. Mol. Cell Biol. 2018, 19, 4–19. [Google Scholar] [CrossRef]
- Gonzalez-Reyes, L.E.; Verbitsky, M.; Blesa, J.; Jackson-Lewis, V.; Paredes, D.; Tillack, K.; Phani, S.; Kramer, E.R.; Przedborski, S.; Kottmann, A.H. Sonic hedgehog maintains cellular and neurochemical homeostasis in the adult nigrostriatal circuit. Neuron 2012, 75, 306–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adlimoghaddam, A.; Sabbir, M.G.; Albensi, B.C. Ammonia as a Potential Neurotoxic Factor in Alzheimer’s Disease. Front. Mol. Neurosci. 2016, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.J.; Plum, F. Biochemistry and physiology of brain ammonia. Physiol. Rev. 1987, 67, 440–519. [Google Scholar] [CrossRef]
- Kelly, T.; Rose, C.R. Ammonium influx pathways into astrocytes and neurones of hippocampal slices. J. Neurochem. 2010, 115, 1123–1136. [Google Scholar] [CrossRef]
- Nido, G.S.; Dick, F.; Toker, L.; Petersen, K.; Alves, G.; Tysnes, O.B.; Jonassen, I.; Haugarvoll, K.; Tzoulis, C. Common gene expression signatures in Parkinson’s disease are driven by changes in cell composition. Acta Neuropathol. Commun. 2020, 8, 55. [Google Scholar] [CrossRef] [Green Version]
- Raghunathan, R.; Hogan, J.D.; Labadorf, A.; Myers, R.H.; Zaia, J. A glycomics and proteomics study of aging and Parkinson’s disease in human brain. Sci. Rep. 2020, 10, 12804. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shojaee, S.; Sina, F.; Banihosseini, S.S.; Kazemi, M.H.; Kalhor, R.; Shahidi, G.A.; Fakhrai-Rad, H.; Ronaghi, M.; Elah, E. Genome-wide linkage analysis of a Parkinsonian-pyramidal syndrome pedigree by 500 K SNP arrays. Am J. Hum. Genet. 2008, 82, 1375–1384. [Google Scholar] [CrossRef] [Green Version]
- Chan, N.; Le, C.; Shieh, P.; Mozaffar, T.; Khare, M.; Bronstein, J.; Kimonis, V. Valosin-containing protein mutation and Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18, 107–109. [Google Scholar] [CrossRef]
- Wang, H.Y.; Juo, L.I.; Lin, Y.T.; Hsiao, M.; Lin, J.T.; Tsai, C.H.; Tzeng, Y.H.; Chuang, Y.C.; Chang, N.S.; Yang, C.N.; et al. WW domain-containing oxidoreductase promotes neuronal differentiation via negative regulation of glycogen synthase kinase 3β. Cell Death Differ. 2012, 19, 1049–1059. [Google Scholar] [CrossRef] [Green Version]
- Pagano, G.; Ferrara, N.; Brooks, D.J.; Pavese, N. Age at onset and Parkinson disease phenotype. Neurology 2016, 86, 1400–1407. [Google Scholar] [CrossRef]
- Fereshtehnejad, S.M.; Zeighami, Y.; Dagher, A.; Postuma, R.B. Clinical criteria for subtyping Parkinson’s disease: Biomarkers and longitudinal progression. Brain J. Neurol. 2017, 140, 1959–1976. [Google Scholar] [CrossRef]
- Koros, C.; Simitsi, A.; Stefanis, L. Genetics of Parkinson’s Disease: Genotype-Phenotype Correlations. Int. Rev. Neurobiol. 2017, 132, 197–231. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO ID | Tissue | #CTRL (F:M) | #PD (F:M) | Platform | #n Probes | F:M | Average Age of Death | Reference |
|---|---|---|---|---|---|---|---|---|
| GSE7621 | SN | 9 (5:4) | 16 (3:13) | GPL570 | 54,675 | 8:17 | 78 | [45] |
| GSE8397 | LatSN | 7 (2:5) | 9 (4:5) | GPL96 | 22,283 | 6:10 | 76 | [46] |
| GSE20333 | SN | 6 (1:5) | 6 (5:1) | GPL201 | 8793 | 6:6 | 78 | [47] |
| GSE20292 | SN | 15 (5:10) | 11 6:5) | GPL96 | 22,283 | 11:15 | 73 | [48] |
| GSE20163 | SN | 9 (1:8) | 8 (4:4) | GPL96 | 22,283 | 5:12 | 74 | [49] |
| GSE20164 | SN | 5 (4:1) | 6 (2:4) | GPL96 | 22,283 | 6:5 | 82 | [50] |
| GSE49036 | SN | 8 (5:3) | 15 (7:8) | GPL570 | 54,675 | 12:11 | 78 | [51] |
| GSE43490 | SN | 5 (2:3) | 8 (0:8) | GPL6480 | 41,108 | 2:11 | 74 | [52] |
| GSE42966 | SN | 6 (2:4) | 9 (4:5) | GPL4133 | 45,220 | 6:9 | 73 | [53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferraro, F.; Fevga, C.; Bonifati, V.; Mandemakers, W.; Mahfouz, A.; Reinders, M. Correcting Differential Gene Expression Analysis for Cyto—Architectural Alterations in Substantia Nigra of Parkinson’s Disease Patients Reveals Known and Potential Novel Disease—Associated Genes and Pathways. Cells 2022, 11, 198. https://doi.org/10.3390/cells11020198
Ferraro F, Fevga C, Bonifati V, Mandemakers W, Mahfouz A, Reinders M. Correcting Differential Gene Expression Analysis for Cyto—Architectural Alterations in Substantia Nigra of Parkinson’s Disease Patients Reveals Known and Potential Novel Disease—Associated Genes and Pathways. Cells. 2022; 11(2):198. https://doi.org/10.3390/cells11020198
Chicago/Turabian StyleFerraro, Federico, Christina Fevga, Vincenzo Bonifati, Wim Mandemakers, Ahmed Mahfouz, and Marcel Reinders. 2022. "Correcting Differential Gene Expression Analysis for Cyto—Architectural Alterations in Substantia Nigra of Parkinson’s Disease Patients Reveals Known and Potential Novel Disease—Associated Genes and Pathways" Cells 11, no. 2: 198. https://doi.org/10.3390/cells11020198