
Mitochondrial Dysfunction as a Hallmark of Environmental Injury

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Mitochondria as Crucial Organelles: From Healthy Metabolic Homeostasis to Critical Dysfunction
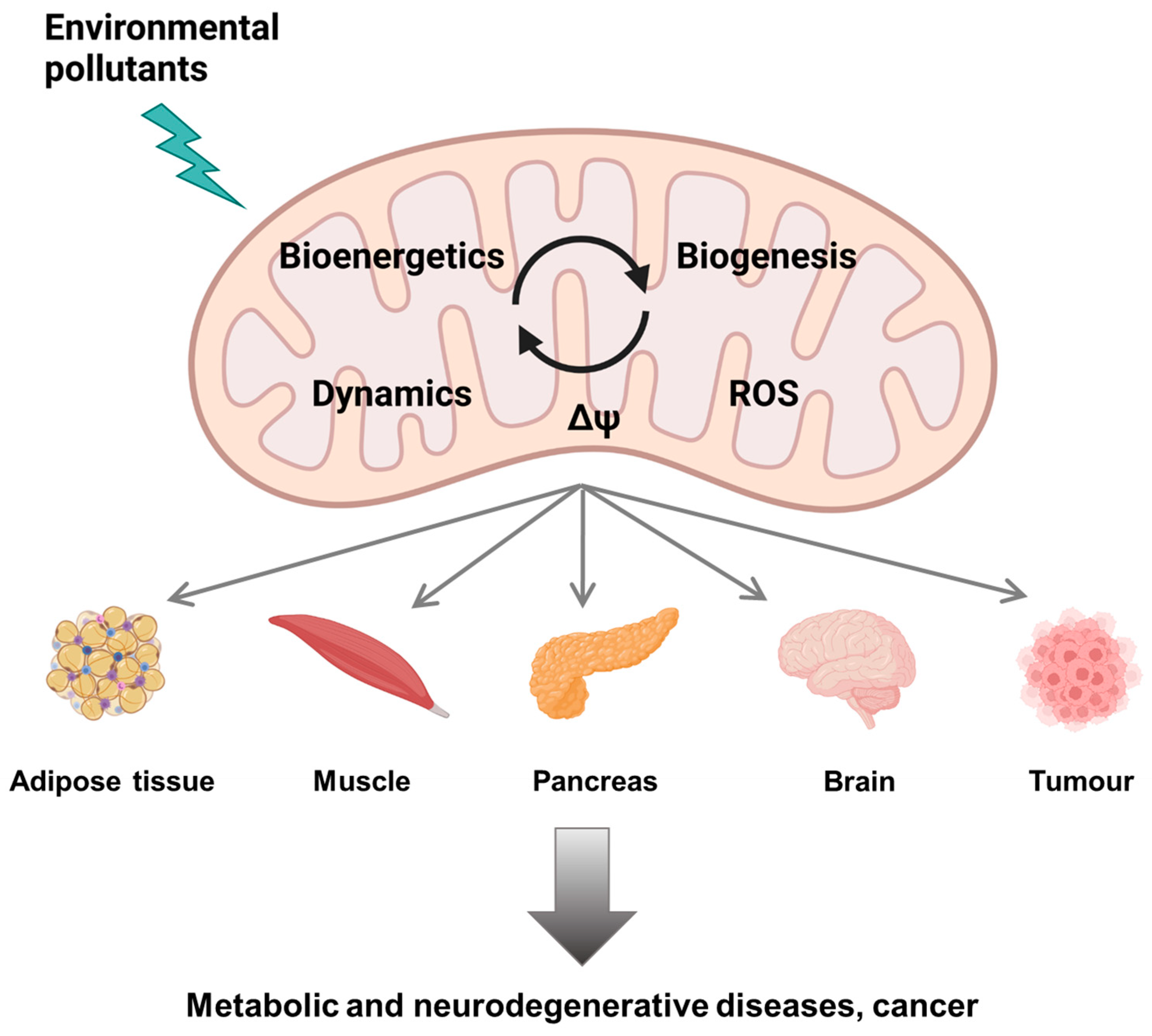
3. Mitochondrial Dysfunction upon Exposure to Environmental Pollutants: Multiple Mechanisms of Toxicity
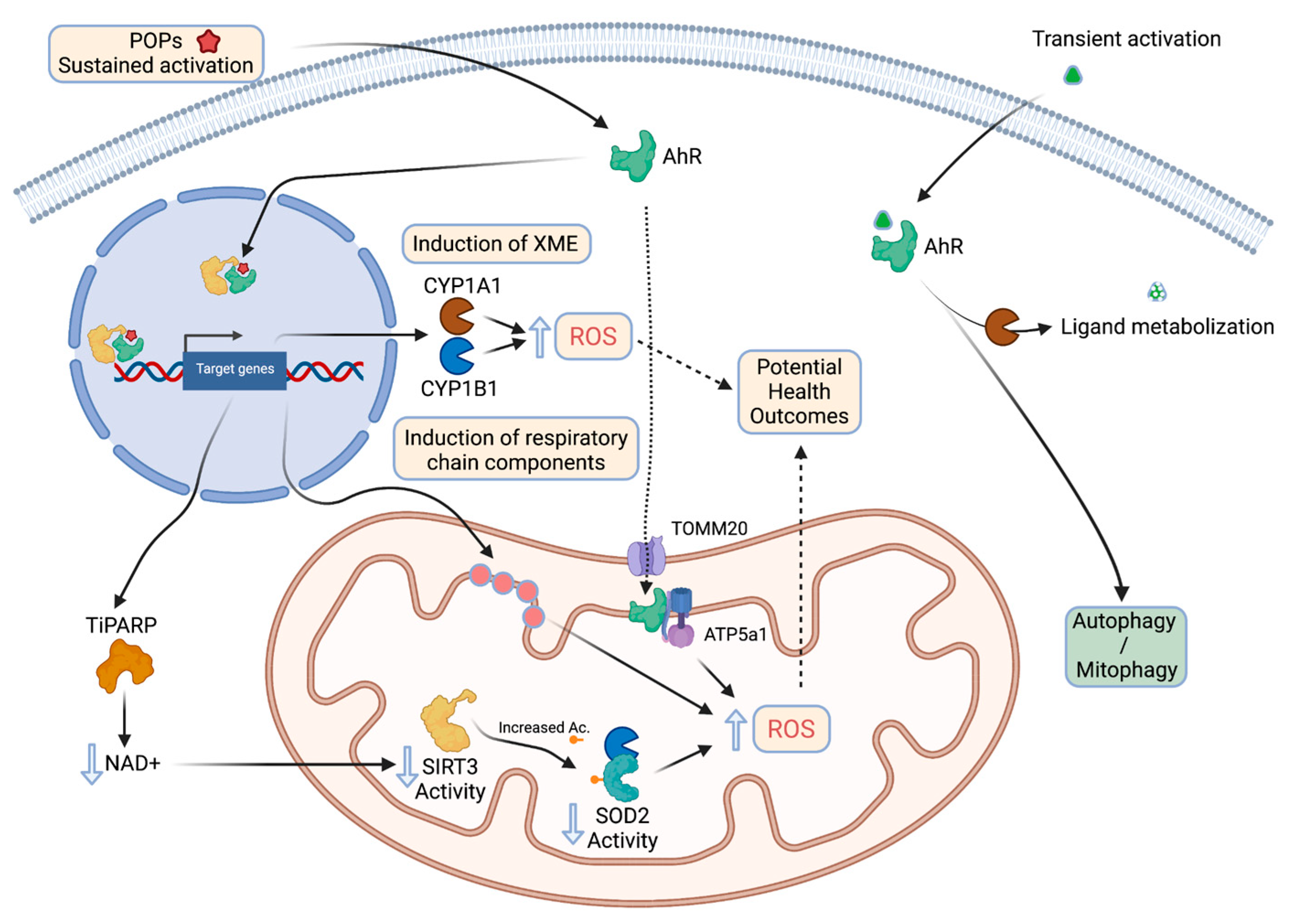
4. AhR as a Crucial Player in Environmental Pollutant-Induced Mitochondrial Dysfunction
5. Concluding Remarks Related to Mitotoxicity and Risk Assessment
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Yang, Y.; He, P.-Y.; Zhang, Y.; Li, N. Natural Products Targeting the Mitochondria in Cancers. Molecules 2020, 26, 92. [Google Scholar] [CrossRef]
- Cole, L.W. The Evolution of Per-Cell Organelle Number. Front. Cell Dev. Biol. 2016, 4, 85. [Google Scholar] [CrossRef] [Green Version]
- Wiemerslage, L.; Lee, D. Quantification of Mitochondrial Morphology in Neurites of Dopaminergic Neurons Using Multiple Parameters. J. Neurosci. Methods 2016, 262, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Sauvanet, C.; Duvezin-Caubet, S.; di Rago, J.-P.; Rojo, M. Energetic Requirements and Bioenergetic Modulation of Mitochondrial Morphology and Dynamics. Semin. Cell Dev. Biol. 2010, 21, 558–565. [Google Scholar] [CrossRef]
- Dard, L.; Blanchard, W.; Hubert, C.; Lacombe, D.; Rossignol, R. Mitochondrial Functions and Rare Diseases. Mol. Asp. Med. 2020, 71, 100842. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial Diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Vafai, S.B.; Mootha, V.K. Mitochondrial Disorders as Windows into an Ancient Organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef]
- Barazzuol, L.; Giamogante, F.; Brini, M.; Calì, T. PINK1/Parkin Mediated Mitophagy, Ca2+ Signalling, and ER-Mitochondria Contacts in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1772. [Google Scholar] [CrossRef] [Green Version]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed Mitochondrial Dynamics and Neurodegenerative Disorders. Nat. Rev. Neurol 2015, 11, 11–24. [Google Scholar] [CrossRef]
- Alavi, M.V.; Fuhrmann, N. Dominant Optic Atrophy, OPA1, and Mitochondrial Quality Control: Understanding Mitochondrial Network Dynamics. Mol. Neurodegener. 2013, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The Role of Mitochondria in Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Duchen, M.R. Roles of Mitochondria in Health and Disease. Diabetes 2004, 53, S96–S102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khacho, M.; Harris, R.; Slack, R.S. Mitochondria as Central Regulators of Neural Stem Cell Fate and Cognitive Function. Nat. Rev. Neurosci. 2019, 20, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of Mitochondria in Human Skeletal Muscle in Type 2 Diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haythorne, E.; Rohm, M.; van de Bunt, M.; Brereton, M.F.; Tarasov, A.I.; Blacker, T.S.; Sachse, G.; Silva Dos Santos, M.; Terron Exposito, R.; Davis, S.; et al. Diabetes Causes Marked Inhibition of Mitochondrial Metabolism in Pancreatic β-Cells. Nat. Commun. 2019, 10, 2474. [Google Scholar] [CrossRef]
- Rocha, M.; Apostolova, N.; Diaz-Rua, R.; Muntane, J.; Victor, V.M. Mitochondria and T2D: Role of Autophagy, ER Stress, and Inflammasome. Trends. Endocrinol. Metab. 2020, 31, 725–741. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Lanza, I.R.; Swain, J.M.; Sarr, M.G.; Nair, K.S.; Jensen, M.D. Adipocyte Mitochondrial Function Is Reduced in Human Obesity Independent of Fat Cell Size. J. Clin. Endocrinol. Metab. 2014, 99, E209–E216. [Google Scholar] [CrossRef] [Green Version]
- Heinonen, S.; Buzkova, J.; Muniandy, M.; Kaksonen, R.; Ollikainen, M.; Ismail, K.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; Vuolteenaho, K.; et al. Impaired Mitochondrial Biogenesis in Adipose Tissue in Acquired Obesity. Diabetes 2015, 64, 3135–3145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A Mitochondria-K+ Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef] [Green Version]
- Christofk, H.R.; Heiden, M.G.V.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 Splice Isoform of Pyruvate Kinase Is Important for Cancer Metabolism and Tumour Growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Saunier, E.; Benelli, C.; Bortoli, S. The Pyruvate Dehydrogenase Complex in Cancer: An Old Metabolic Gatekeeper Regulated by New Pathways and Pharmacological Agents. Int. J. Cancer 2016, 138, 809–817. [Google Scholar] [CrossRef]
- Heiden, M.G.V. Targeting Cancer Metabolism: A Therapeutic Window Opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Rangel, E.; Inzucchi, S.E. Metformin: Clinical Use in Type 2 Diabetes. Diabetologia 2017, 60, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Vial, G.; Detaille, D.; Guigas, B. Role of Mitochondria in the Mechanism(s) of Action of Metformin. Front. Endocrinol. 2019, 10, 294. [Google Scholar] [CrossRef] [Green Version]
- Bárcena, C.; Mayoral, P.; Quirós, P.M. Mitohormesis, an Antiaging Paradigm. Int. Rev. Cell Mol. Biol. 2018, 340, 35–77. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S. Mitochondria as Signaling Organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial Generation of Superoxide and Hydrogen Peroxide as the Source of Mitochondrial Redox Signaling. Free. Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Hwang, M.-S.; Rohlena, J.; Dong, L.-F.; Neuzil, J.; Grimm, S. Powerhouse down: Complex II Dissociation in the Respiratory Chain. Mitochondrion 2014, 19 Pt A, 20–28. [Google Scholar] [CrossRef]
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial Mutations and Mitoepigenetics: Focus on Regulation of Oxidative Stress-Induced Responses in Breast Cancers. Semin. Cancer Biol. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P.H. Oxidative Stress in Ischemic Brain Damage: Mechanisms of Cell Death and Potential Molecular Targets for Neuroprotection. Antioxid Redox Signal 2011, 14, 1505–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, P.H. Reactive Oxygen Radicals in Signaling and Damage in the Ischemic Brain. J. Cereb. Blood Flow. Metab. 2001, 21, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F.; Stoccoro, A. Mitoepigenetics and Neurodegenerative Diseases. Front. Endocrinol. 2019, 10, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R.; Van Houten, B. SnapShot: Mitochondrial Quality Control. Cell 2011, 147, 950.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Khacho, M.; Slack, R.S. Mitochondrial and Reactive Oxygen Species Signaling Coordinate Stem Cell Fate Decisions and Life Long Maintenance. Antioxid Redox Signal 2018, 28, 1090–1101. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab. 2017, 26, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serasinghe, M.N.; Wieder, S.Y.; Renault, T.T.; Elkholi, R.; Asciolla, J.J.; Yao, J.L.; Jabado, O.; Hoehn, K.; Kageyama, Y.; Sesaki, H.; et al. Mitochondrial Division Is Requisite to RAS-Induced Transformation and Targeted by Oncogenic MAPK Pathway Inhibitors. Mol. Cell 2015, 57, 521–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, E.; Altman, B.J.; Ho Seo, J.; Bertolini, I.; Ghosh, J.C.; Kaur, A.; Kossenkov, A.V.; Languino, L.R.; Gabrilovich, D.I.; Speicher, D.W.; et al. Myc Regulation of a Mitochondrial Trafficking Network Mediates Tumor Cell Invasion and Metastasis. Mol. Cell Biol. 2019, 39, e00109-19. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial Dynamics Regulates Migration and Invasion of Breast Cancer Cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef]
- Peters, A.; Nawrot, T.S.; Baccarelli, A.A. Hallmarks of Environmental Insults. Cell 2021, 184, 1455–1468. [Google Scholar] [CrossRef]
- Marroqui, L.; Tudurí, E.; Alonso-Magdalena, P.; Quesada, I.; Nadal, Á.; Dos Santos, R.S. Mitochondria as Target of Endocrine-Disrupting Chemicals: Implications for Type 2 Diabetes. J. Endocrinol. 2018, 239, R27–R45. [Google Scholar] [CrossRef]
- Meyer, J.N.; Leung, M.C.K.; Rooney, J.P.; Sendoel, A.; Hengartner, M.O.; Kisby, G.E.; Bess, A.S. Mitochondria as a Target of Environmental Toxicants. Toxicol. Sci. 2013, 134, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Huang, X.; Liu, L.; Shi, N. Deltamethrin Induces Mitochondrial Membrane Permeability and Altered Expression of Cytochrome C in Rat Brain. J. Appl. Toxicol. 2007, 27, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Singh, A.; Tripathi, P.; Mishra, M.; Singh, P.K.; Singh, M.P. Cypermethrin-Induced Nigrostriatal Dopaminergic Neurodegeneration Alters the Mitochondrial Function: A Proteomics Study. Mol. Neurobiol. 2015, 51, 448–465. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Kong, A.P.S.; Cai, Z.; Chung, A.C.K. Persistent Organic Pollutants as Risk Factors for Obesity and Diabetes. Curr. Diab. Rep. 2017, 17, 132. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Ahn, S.Y.; Song, I.C.; Chung, M.H.; Jang, H.C.; Park, K.S.; Lee, K.-U.; Pak, Y.K.; Lee, H.K. Chronic Exposure to the Herbicide, Atrazine, Causes Mitochondrial Dysfunction and Insulin Resistance. PLoS ONE 2009, 4, e5186. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.; Galloway, C.A.; Jhun, B.S.; Yu, T. Mitochondrial Dynamics in Diabetes. Antioxid Redox Signal 2011, 14, 439–457. [Google Scholar] [CrossRef] [PubMed]
- Rovira-Llopis, S.; Bañuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial Dynamics in Type 2 Diabetes: Pathophysiological Implications. Redox. Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Pi, H.; Chen, Y.; Zhang, N.; Guo, P.; Lu, Y.; He, M.; Xie, J.; Zhong, M.; Zhang, Y.; et al. Cadmium Induced Drp1-Dependent Mitochondrial Fragmentation by Disturbing Calcium Homeostasis in Its Hepatotoxicity. Cell Death Dis. 2013, 4, e540. [Google Scholar] [CrossRef] [Green Version]
- Hardonnière, K.; Saunier, E.; Lemarié, A.; Fernier, M.; Gallais, I.; Héliès-Toussaint, C.; Mograbi, B.; Antonio, S.; Bénit, P.; Rustin, P.; et al. The Environmental Carcinogen Benzo[a]Pyrene Induces a Warburg-like Metabolic Reprogramming Dependent on NHE1 and Associated with Cell Survival. Sci. Rep. 2016, 6, 30776. [Google Scholar] [CrossRef]
- Holme, J.A.; Gorria, M.; Arlt, V.M.; Ovrebø, S.; Solhaug, A.; Tekpli, X.; Landvik, N.E.; Huc, L.; Fardel, O.; Lagadic-Gossmann, D. Different Mechanisms Involved in Apoptosis Following Exposure to Benzo[a]Pyrene in F258 and Hepa1c1c7 Cells. Chem. Biol. Interact. 2007, 167, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Hardonnière, K.; Fernier, M.; Gallais, I.; Mograbi, B.; Podechard, N.; Le Ferrec, E.; Grova, N.; Appenzeller, B.; Burel, A.; Chevanne, M.; et al. Role for the ATPase Inhibitory Factor 1 in the Environmental Carcinogen-Induced Warburg Phenotype. Sci. Rep. 2017, 7, 195. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C.; Fan, W. Energetics, Epigenetics, Mitochondrial Genetics. Mitochondrion 2010, 10, 12–31. [Google Scholar] [CrossRef] [Green Version]
- Morin, A.; Letouzé, E.; Gimenez-Roqueplo, A.-P.; Favier, J. Oncometabolites-Driven Tumorigenesis: From Genetics to Targeted Therapy. Int. J. Cancer 2014, 135, 2237–2248. [Google Scholar] [CrossRef]
- Alonso-Magdalena, P.; Rivera, F.J.; Guerrero-Bosagna, C. Bisphenol-A and Metabolic Diseases: Epigenetic, Developmental and Transgenerational Basis. Env. Epigenet 2016, 2, dvw022. [Google Scholar] [CrossRef] [PubMed]
- García-Arevalo, M.; Alonso-Magdalena, P.; Rebelo Dos Santos, J.; Quesada, I.; Carneiro, E.M.; Nadal, A. Exposure to Bisphenol-A during Pregnancy Partially Mimics the Effects of a High-Fat Diet Altering Glucose Homeostasis and Gene Expression in Adult Male Mice. PLoS ONE 2014, 9, e100214. [Google Scholar] [CrossRef] [Green Version]
- Kirchner, S.; Kieu, T.; Chow, C.; Casey, S.; Blumberg, B. Prenatal Exposure to the Environmental Obesogen Tributyltin Predisposes Multipotent Stem Cells to Become Adipocytes. Mol. Endocrinol. 2010, 24, 526–539. [Google Scholar] [CrossRef]
- Kamstra, J.H.; Hruba, E.; Blumberg, B.; Janesick, A.; Mandrup, S.; Hamers, T.; Legler, J. Transcriptional and Epigenetic Mechanisms Underlying Enhanced in Vitro Adipocyte Differentiation by the Brominated Flame Retardant BDE-47. Environ. Sci Technol. 2014, 48, 4110–4119. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, S.; Khalili, N.P.; Khan, F.; Hassani, S.; Ghafour-Boroujerdi, E.; Abdollahi, M. Bisphenol A: What Lies beneath Its Induced Diabetes and the Epigenetic Modulation? Life Sci. 2018, 214, 136–144. [Google Scholar] [CrossRef]
- Manikkam, M.; Tracey, R.; Guerrero-Bosagna, C.; Skinner, M.K. Plastics Derived Endocrine Disruptors (BPA, DEHP and DBP) Induce Epigenetic Transgenerational Inheritance of Obesity, Reproductive Disease and Sperm Epimutations. PLoS ONE 2013, 8, e55387. [Google Scholar] [CrossRef]
- Chamorro-Garcia, R.; Diaz-Castillo, C.; Shoucri, B.M.; Käch, H.; Leavitt, R.; Shioda, T.; Blumberg, B. Ancestral Perinatal Obesogen Exposure Results in a Transgenerational Thrifty Phenotype in Mice. Nat. Commun. 2017, 8, 2012. [Google Scholar] [CrossRef] [Green Version]
- Janssen, B.G.; Byun, H.-M.; Gyselaers, W.; Lefebvre, W.; Baccarelli, A.A.; Nawrot, T.S. Placental Mitochondrial Methylation and Exposure to Airborne Particulate Matter in the Early Life Environment: An ENVIRONAGE Birth Cohort Study. Epigenetics 2015, 10, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Byun, H.-M.; Panni, T.; Motta, V.; Hou, L.; Nordio, F.; Apostoli, P.; Bertazzi, P.A.; Baccarelli, A.A. Effects of Airborne Pollutants on Mitochondrial DNA Methylation. Part. Fibre. Toxicol. 2013, 10, 18. [Google Scholar] [CrossRef] [Green Version]
- Byun, H.-M.; Colicino, E.; Trevisi, L.; Fan, T.; Christiani, D.C.; Baccarelli, A.A. Effects of Air Pollution and Blood Mitochondrial DNA Methylation on Markers of Heart Rate Variability. J. Am. Heart Assoc. 2016, 5, e003218. [Google Scholar] [CrossRef] [Green Version]
- Byun, H.-M.; Benachour, N.; Zalko, D.; Frisardi, M.C.; Colicino, E.; Takser, L.; Baccarelli, A.A. Epigenetic Effects of Low Perinatal Doses of Flame Retardant BDE-47 on Mitochondrial and Nuclear Genes in Rat Offspring. Toxicology 2015, 328, 152–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tappenden, D.M.; Lynn, S.G.; Crawford, R.B.; Lee, K.; Vengellur, A.; Kaminski, N.E.; Thomas, R.S.; LaPres, J.J. The Aryl Hydrocarbon Receptor Interacts with ATP5α1, a Subunit of the ATP Synthase Complex, and Modulates Mitochondrial Function. Toxicol. Appl. Pharmacol. 2011, 254, 299–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Wang, X.; Li, F.; Wang, Y.; Yang, L.; Zhen, X.; Tan, W. Mitochondrial Activity and Oxidative Stress Functions Are Influenced by the Activation of AhR-Induced CYP1A1 Overexpression in Cardiomyocytes. Mol. Med. Rep. 2017, 16, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Senft, A.P.; Dalton, T.P.; Nebert, D.W.; Genter, M.B.; Puga, A.; Hutchinson, R.J.; Kerzee, J.K.; Uno, S.; Shertzer, H.G. Mitochondrial Reactive Oxygen Production Is Dependent on the Aromatic Hydrocarbon Receptor. Free Radic. Biol. Med. 2002, 33, 1268–1278. [Google Scholar] [CrossRef]
- Ghosh, J.; Chowdhury, A.R.; Srinivasan, S.; Chattopadhyay, M.; Bose, M.; Bhattacharya, S.; Raza, H.; Fuchs, S.Y.; Rustgi, A.K.; Gonzalez, F.J.; et al. Cigarette Smoke Toxins-Induced Mitochondrial Dysfunction and Pancreatitis Involves Aryl Hydrocarbon Receptor Mediated Cyp1 Gene Expression: Protective Effects of Resveratrol. Toxicol. Sci. 2018, 166, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.J.; Dornbos, P.; Steidemann, M.; Dunivin, T.K.; Rizzo, M.; LaPres, J.J. Mitochondrial-Targeted Aryl Hydrocarbon Receptor and the Impact of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin on Cellular Respiration and the Mitochondrial Proteome. Toxicol. Appl. Pharmacol. 2016, 304, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forgacs, A.L.; Burgoon, L.D.; Lynn, S.G.; LaPres, J.J.; Zacharewski, T. Effects of TCDD on the Expression of Nuclear Encoded Mitochondrial Genes. Toxicol. Appl. Pharmacol. 2010, 246, 58–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Hu, B.; Shi, X.; Weidert, E.R.; Lu, P.; Xu, M.; Huang, M.; Kelley, E.E.; Xie, W. Activation of the Aryl Hydrocarbon Receptor Sensitizes Mice to Nonalcoholic Steatohepatitis by Deactivating Mitochondrial Sirtuin Deacetylase Sirt3. Mol. Cell Biol. 2013, 33, 2047–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucher, S.; Le Guillou, D.; Allard, J.; Pinon, G.; Begriche, K.; Tête, A.; Sergent, O.; Lagadic-Gossmann, D.; Fromenty, B. Possible Involvement of Mitochondrial Dysfunction and Oxidative Stress in a Cellular Model of NAFLD Progression Induced by Benzo[a]Pyrene/Ethanol CoExposure. Oxid. Med. Cell Longev. 2018, 2018, 4396403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Zhang, J.; Tao, Y.; Ji, C.; Aniagu, S.; Jiang, Y.; Chen, T. AHR/ROS-Mediated Mitochondria Apoptosis Contributes to Benzo[a]Pyrene-Induced Heart Defects and the Protective Effects of Resveratrol. Toxicology 2021, 462, 152965. [Google Scholar] [CrossRef]
- Wang, B.; Xu, A. Aryl Hydrocarbon Receptor Pathway Participates in Myocardial Ischemia Reperfusion Injury by Regulating Mitochondrial Apoptosis. Med. Hypotheses 2019, 123, 2–5. [Google Scholar] [CrossRef]
- Fisher, M.T.; Nagarkatti, M.; Nagarkatti, P.S. Aryl Hydrocarbon Receptor-Dependent Induction of Loss of Mitochondrial Membrane Potential in Epididydimal Spermatozoa by 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD). Toxicol. Lett. 2005, 157, 99–107. [Google Scholar] [CrossRef]
- de Souza, A.R.; Zago, M.; Pollock, S.J.; Sime, P.J.; Phipps, R.P.; Baglole, C.J. Genetic Ablation of the Aryl Hydrocarbon Receptor Causes Cigarette Smoke-Induced Mitochondrial Dysfunction and Apoptosis. J. Biol. Chem. 2011, 286, 43214–43228. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Shi, Z.; Lin, Y.; Zhang, M.; Liu, J.; Zhu, L.; Chen, Q.; Bi, J.; Li, S.; Ni, Z.; et al. Benzo(a)Pyrene Induces MUC5AC Expression through the AhR/Mitochondrial ROS/ERK Pathway in Airway Epithelial Cells. Ecotoxicol. Environ. Saf. 2021, 210, 111857. [Google Scholar] [CrossRef]
- Brinkmann, V.; Ale-Agha, N.; Haendeler, J.; Ventura, N. The Aryl Hydrocarbon Receptor (AhR) in the Aging Process: Another Puzzling Role for This Highly Conserved Transcription Factor. Front. Physiol. 2019, 10, 1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, D.N.; Naik, P.P.; Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Meher, B.R.; Bhutia, S.K. Elimination of Dysfunctional Mitochondria through Mitophagy Suppresses Benzo[a]Pyrene-Induced Apoptosis. Free Radic. Biol. Med. 2017, 112, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Ferecatu, I.; Borot, M.-C.; Bossard, C.; Leroux, M.; Boggetto, N.; Marano, F.; Baeza-Squiban, A.; Andreau, K. Polycyclic Aromatic Hydrocarbon Components Contribute to the Mitochondria-Antiapoptotic Effect of Fine Particulate Matter on Human Bronchial Epithelial Cells via the Aryl Hydrocarbon Receptor. Part. Fibre. Toxicol. 2010, 7, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, S.; Liu, L.; Jian, Z.; Cui, T.; Yang, Y.; Guo, S.; Yi, X.; Wang, G.; Li, C.; et al. Role of the Aryl Hydrocarbon Receptor Signaling Pathway in Promoting Mitochondrial Biogenesis against Oxidative Damage in Human Melanocytes. J. Derm. Sci. 2019, 96, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Gan, M.; Ding, H.; Chen, G. 6-Formylindolo[3,2-b]Carbazole Reduces Apoptosis Induced by Benzo[a]Pyrene in a Mitochondrial-Dependent Manner. Cell Biol. Int. 2020, 44, 2427–2437. [Google Scholar] [CrossRef] [PubMed]
- Sahebnasagh, A.; Hashemi, J.; Khoshi, A.; Saghafi, F.; Avan, R.; Faramarzi, F.; Azimi, S.; Habtemariam, S.; Sureda, A.; Khayatkashani, M.; et al. Aromatic Hydrocarbon Receptors in Mitochondrial Biogenesis and Function. Mitochondrion 2021, 61, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Xu, M.; Meng, X.; Li, S.; Liu, Q.; Bai, M.; You, R.; Huang, S.; Yang, L.; Zhang, Y.; et al. Nuclear Receptor PXR Targets AKR1B7 to Protect Mitochondrial Metabolism and Renal Function in AKI. Sci. Transl. Med. 2020, 12, eaay7591. [Google Scholar] [CrossRef] [PubMed]
- van der Mark, V.A.; Adam, A.A.A.; Chang, J.-C.; Oude Elferink, R.P.; Chamuleau, R.A.F.M.; Hoekstra, R. Overexpression of the Constitutive Androstane Receptor and Shaken 3D-Culturing Increase Biotransformation and Oxidative Phosphorylation and Sensitivity to Mitochondrial Amiodarone Toxicity of HepaRG Cells. Toxicol. Appl. Pharm. 2020, 399, 115055. [Google Scholar] [CrossRef]
- Liao, T.-L.; Tzeng, C.-R.; Yu, C.-L.; Wang, Y.-P.; Kao, S.-H. Estrogen Receptor-β in Mitochondria: Implications for Mitochondrial Bioenergetics and Tumorigenesis. Ann. N. Y. Acad. Sci. 2015, 1350, 52–60. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Key Features of the Mitochondria | Consequences Regarding the Exposure to Xenobiotics |
|---|---|
| Mitochondria are complex organelles whose functions are governed by multiple and diverse proteins | As a result, a wide variety of xenobiotics can target the mitochondria such as polycyclic aromatic hydrocarbons, pesticides (including rotenone, pyrethroids, atrazine), metals (cadmium). |
| Mitochondria are involved in the production of ATP which is coupled with the activity of the respiratory chain | Both processes (ATP production and respiration) are governed by complexes of the inner membrane which are targeted by natural and foreign compounds. Disruption of the respiratory chain can lead to the misuse of O2 and production of reactive oxygen species. It can also promote the Warburg Effect. |
| Mitochondria regulate multiple cellular processes including cell proliferation, differentiation, apoptosis and mitophagy | Exposure to xenobiotics targeting the mitochondria can lead to disruption of such processes, leading to various outcomes (increased apoptosis and neurodegeneration; increased survival in cancer). |
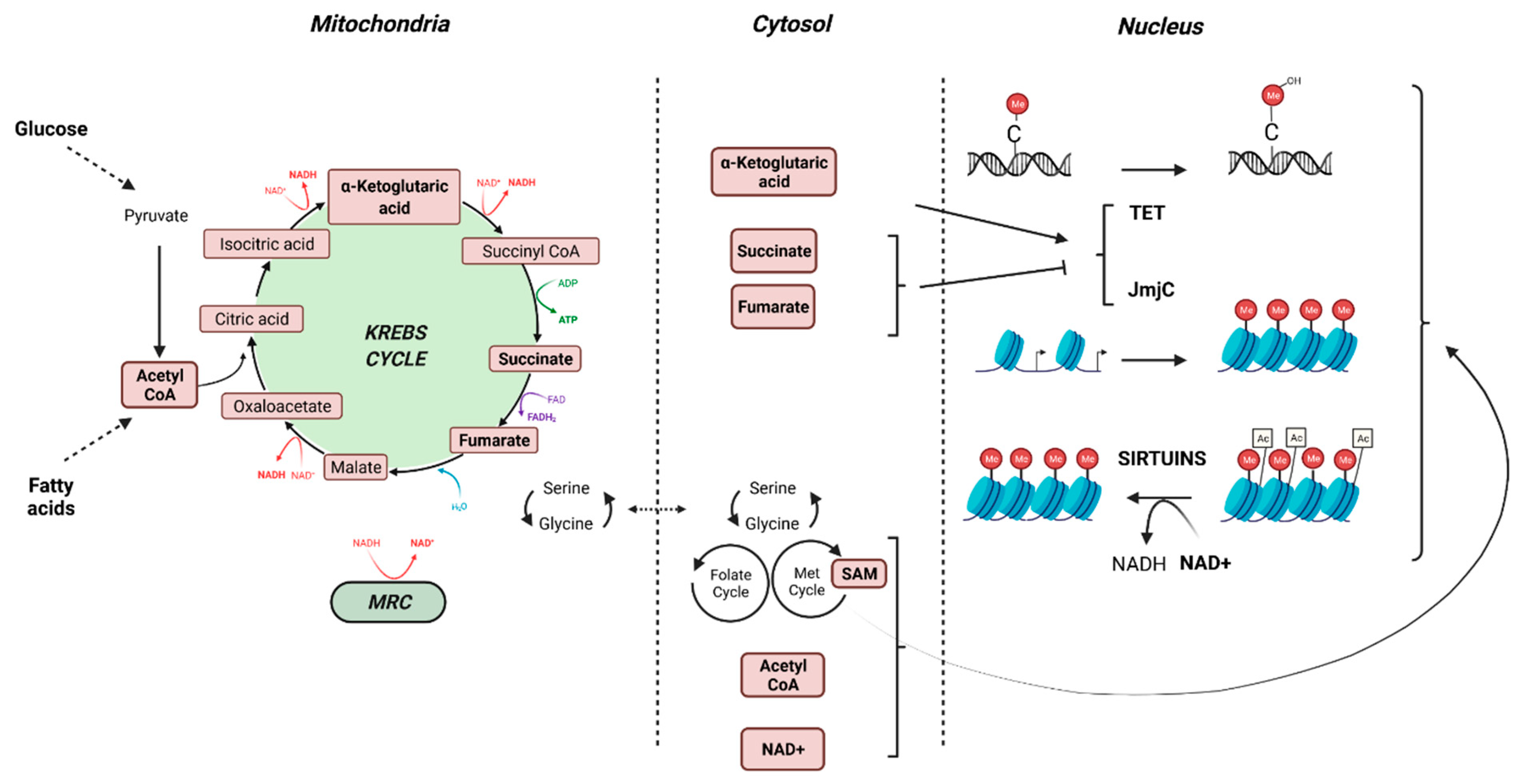
| Mitochondria crosstalk with epigenetics by regulating the levels of NAD+, S-adenosylmethionine, α-ketoglutarate, succinate and fumarate which impact epigenetic marks. | Several pollutants impact epigenetic marks through the deregulation of mitochondria-related metabolites and/or co-factors production, such as DDE, hexachlorobenzene, tributyltin, bisphenol A, phthalates or organochlorines |
| Mitochondria contain transcriptional factors that regulate some of the functions mentioned earlier. | Binding of ligands to such transcriptional factors may lead to relocalization of these proteins to mitochondria and alteration of their properties (i.e., AhR and the Seveso dioxin) due for example to increased ROS production |
| Some pollutants are non-persistent while other are persistent (with a long-half life in the body) | The nature of each xenobiotic is crucial to determine their long-term effects on the mitochondria: endogenous ligands or low doses-exogenous ligands can then be opposed to persistent exogenous ligands regarding the potential health outcomes they would induce |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duarte-Hospital, C.; Tête, A.; Brial, F.; Benoit, L.; Koual, M.; Tomkiewicz, C.; Kim, M.J.; Blanc, E.B.; Coumoul, X.; Bortoli, S. Mitochondrial Dysfunction as a Hallmark of Environmental Injury. Cells 2022, 11, 110. https://doi.org/10.3390/cells11010110
Duarte-Hospital C, Tête A, Brial F, Benoit L, Koual M, Tomkiewicz C, Kim MJ, Blanc EB, Coumoul X, Bortoli S. Mitochondrial Dysfunction as a Hallmark of Environmental Injury. Cells. 2022; 11(1):110. https://doi.org/10.3390/cells11010110
Chicago/Turabian StyleDuarte-Hospital, Carolina, Arnaud Tête, François Brial, Louise Benoit, Meriem Koual, Céline Tomkiewicz, Min Ji Kim, Etienne B. Blanc, Xavier Coumoul, and Sylvie Bortoli. 2022. "Mitochondrial Dysfunction as a Hallmark of Environmental Injury" Cells 11, no. 1: 110. https://doi.org/10.3390/cells11010110