
AGEs-Induced and Endoplasmic Reticulum Stress/Inflammation-Mediated Regulation of GLUT4 Expression and Atherogenesis in Diabetes Mellitus



1
Laboratório de Lípides (LIM-10), Hospital das Clínicas (HCFMUSP) da Faculdade de Medicina da Universidade de São Paulo, São Paulo 01246-000, Brazil
2
Programa de Pos-Graduação em Medicina, Universidade Nove de Julho, São Paulo 01525-000, Brazil
3
Department of Physiology and Biophysics, Institute of Biomedical Sciences, University of São Paulo, São Paulo 05508-000, Brazil
*
Author to whom correspondence should be addressed.
Cells 2022, 11(1), 104; https://doi.org/10.3390/cells11010104
Submission received: 8 November 2021
/
Revised: 23 December 2021
/
Accepted: 27 December 2021
/
Published: 29 December 2021
(This article belongs to the Special Issue Unfolded Protein Response in Inflammation and Cancer)
{kind=link}
{kind=link}
Abstract
:In recent decades, complex and exquisite pathways involved in the endoplasmic reticulum (ER) and inflammatory stress responses have been demonstrated to participate in the development and progression of numerous diseases, among them diabetes mellitus (DM). In those pathways, several players participate in both, reflecting a complicated interplay between ER and inflammatory stress. In DM, ER and inflammatory stress are involved in both the pathogenesis of the loss of glycemic control and the development of degenerative complications. Furthermore, hyperglycemia increases the generation of advanced glycation end products (AGEs), which in turn refeed ER and inflammatory stress, contributing to worsening glycemic homeostasis and to accelerating the development of DM complications. In this review, we present the current knowledge regarding AGEs-induced and ER/inflammation-mediated regulation of the expression of GLUT4 (solute carrier family 2, facilitated glucose transporter member 4), as a marker of glycemic homeostasis and of cardiovascular disease (CVD) development/progression, as a leading cause of morbidity and mortality in DM.
1. Introduction
Diabetes mellitus (DM) is a global public health burden, as the number of affected adults is expected to rise to 630 million by 2045 [1]. DM is a metabolic disorder characterized by inappropriate hyperglycemia and can be classified into type 1 DM (T1D), which primarily results from the lack of insulin secretion, and type 2 DM (T2D), which primarily results from insulin resistance (IR) [2]. However, whereas T2D subjects develop impaired insulin secretion, T1D subjects may also develop IR, as a complex result of interactions among age, sex, pregnancy, ethnicity, duration of disease, increased life expectancy, genetic predisposition, epigenetic changes, glucotoxicity, lipotoxicity, overweight and intensive insulin therapy [2,3,4,5]. In addition to glycemic disorder, chronic hyperglycemia injures various organic systems, leading to the development of several complications, prompting a 2- to 4-fold increase in the risk of cardiovascular disease (CVD) [6,7].
In DM, several metabolic mechanisms, notably hyperglycemia, lead to the activation of intracellular pathways that increase the generation of advanced glycation end products (AGEs); thus, increased plasma level of AGEs has become a hallmark of uncontrolled DM [8]. In turn, AGEs contribute to worsening IR and glycemic control [9], and participate in the development of chronic diabetic complications, including CVD [10]. The receptor for AGEs—AGER (advanced glycation end product-specific receptor, formerly RAGE)—mediates several biological effects of AGEs, including the activation of endoplasmic reticulum stress (ER stress) and inflammatory stress pathways [10].
Considering that AGEs can participate in both the impairment of glycemic homeostasis and the development/progression of chronic complications of DM, and considering that these effects involve ER stress- and inflammatory-mediated pathways, the present manuscript reviews these regulations upon the expression of solute carrier family 2, facilitated glucose transporter member 4 (GLUT4) expression, as a marker of altered glycemic homeostasis [11,12,13] and, upon atherogenesis, as an important inducer of the development/progression of CVD [7].
Names and symbols of genes and proteins referred to in this manuscript are in accordance with the HGNC (https://www.genenames.org/, accessed on 2 November 2021) and the UNIPROT (https://www.uniprot.org/, accessed on 2 November 2021) databases. Whenever necessary, alias symbols will be provided when they first appear in the text.
2. Pathogenesis of DM and Its Complications
2.1. Insulin Resistance (IR)
Insulin resistance involves complex and variable disturbances in the insulin signaling pathway, eventually culminating in impaired glucose utilization by muscle and adipose tissues, contributing to the impairment of glycemic control [12,14]. Furthermore, IR aggravates hyperglycemia by favoring the output of glucose from the liver by increasing gluconeogenesis. Compensatory hyperglycemia, in turn, is not able to neutralize hepatic glucose production, whereas, in a non-resistant pathway, induces an increase in lipogenesis mediated by the overactivation of sterol regulatory element-binding protein 1 (SREBP1), which aggravates peripheral insulin resistance and favors a pro-atherogenic state [15]. Then, plasma glucose levels are dictated by an interplay of mechanisms triggered by IR and hyperinsulinemia in one or more different organs.
Glucose uptake in muscle and adipose tissues depends on the insulin-responsive glucose transporter GLUT4, codified by the SLC2A4 gene (solute carrier family 2 member 4), which was cloned 30 years ago [16]. Since then, it has been clear that GLUT4 plays a fundamental role in plasma glucose clearance and glycemia homeostasis as well [11,12]. Furthermore, alterations in GLUT4 expression have been related to alterations in glycemic control, with reduced GLUT4 content participating in the hyperglycemia prone. Because of that, in the present manuscript, muscle and adipose tissues regulation of SLC2A4/GLUT4 is reviewed as a marker of glycemic control in DM.
On the other hand, combinations of IR and compensatory hyperinsulinemia (in T2D) or with hyperinsulinemia induced by insulin therapy (in T1D) have also been reported as capable of causing damage to various organ systems. This leads to the development of disabling and life-threatening health complications, most of which are microvascular (retinopathy, nephropathy and neuropathy) and macrovascular complications (CVD, cerebrovascular disease and peripheral artery disease) [7].
Insulin resistance and hyperinsulinemia have been associated with polycystic ovary syndrome, nonalcoholic fatty liver disease, certain forms of cancer, sleep apnea and, especially, with hypertension and CVD [17,18]. Related to that, several disorders that course with IR have been considered as risk factors for the development of hypertension and CVD, including a high atherogenic lipoprotein profile (in its complexity), elevated plasma concentrations of plasminogen activator inhibitor-1, increased sympathetic nervous system activity and endothelial disfunction [17,18]. Along with these risk factors, a pro-inflammatory profile and the activation of reticulum endoplasmic stress and oxidative stress are observed, in a complex relationship, creating a dangerous vicious circle [19,20]. Because of that, in this manuscript, CVD (with special focus on atherogenesis) was chosen to be reviewed as the most important complication of DM.
2.2. GLUT4 Expression and Glycemic Control
The glucose transporter protein GLUT4 belongs to a family of proteins responsible for the glucose facilitative diffusion across the plasma membrane (PM) (for a review, see [21]). GLUT4 is the only insulin-sensitive glucose transporter, which is mainly expressed in the skeletal muscle and adipose tissue, where it is responsible for the insulin-induced glucose uptake. In myocytes and adipocytes, the binding of insulin in its receptor triggers the activation of an exquisite intracellular sorting of signals that, eventually, culminates in a GLUT4 storage vesicle translocation to the PM. After docking and fusing events, the amount of GLUT4 in PM increases, enhancing the glucose influx. Since intracellular consumption of glucose is high in these cells, the diffusion gradient continuously favors the influx of the substrate. Disruption of the insulin stimulus leads to the internalization of GLUT4, restoring the glucose uptake to basal levels (for a review, see [16]). The GLUT4-mediated increase in glucose uptake in muscle and adipose tissues is a fundamental mechanism involved in the blood glucose clearance, especially in the postprandial state. Thus, several research groups have investigated the regulation of SLC2A4 gene expression, which codifies the GLUT4 protein, for better understanding the insulin-mediated plasma glucose clearance [13,22,23].
Impairment of insulin signaling transduction can compromise GLUT4 translocation to the PM, notably in acute situations, in which the total cellular GLUT4 content is unchanged. However, under chronic insulin-resistant states, a decreased total GLUT4 content is currently observed, and that certainly contributes to decreased GLUT4 at the PM in response to insulin. Even considering a preserved translocation system, when the intracellular GLUT4 content is reduced, the final amount of GLUT4 at the PM will be reduced [24]. This emphasizes the great relevance of the repression of Slc2a4 gene expression, and consequent reduction in GLUT4 protein, in the IR and DM states. Besides, the role of Slc2a4/GLUT4 expression in glycemic control has been reinforced by studies with transgenic mice. It is well recognized that Slc2a4 knockout induces hyperglycemia, whereas the overexpression of Slc2a4 improves glycemic control even in DM mice [25,26], regulations that were directly associated with the amount of GLUT4 at the PM, independently of changes in insulin signaling. These data reinforce the importance of the regulation of SLC2A4 gene expression in glycemic control and qualify this gene as a promising target for the pharmacogenomics of IR/DM [12].
At present, several transcriptional factors are described as involved in the expression of SLC2A4/Slc2a4 (human/murine genes, respectively), most of them acting as enhancers and a few as repressors (for a review, see [22,23]). Importantly, some of these transcription factors have been related to the inflammatory and/or ER stress activity, characterizing these routes as important modulators of GLUT4 expression, as will be commented on next.
2.3. Cellular Cholesterol Homeostasis and Cardiovascular Disease (CVD) Development
Cardiovascular disease is the most prevalent cause of disability and morbimortality in DM [27]. Although CVD is prevalent in both T1D and T2D, it is especially of greater concern in T2D, which comprises about 91% of all DM cases. Insulin resistance and compensatory hyperinsulinemia, obesity, chronic inflammation and derangements in lipid metabolism that contribute to lipid accumulation in the arterial wall favor the clinical burden of CVD complications. Moreover, the presence of other CVD risk factors commonly associated with metabolic syndrome increases atherogenesis [28].
In T2D, quantitative changes of plasma lipoproteins occur as a primary consequence of the insulin-resistant state [29]. Hyperinsulinemia increases hepatic triglycerides synthesis by overactivating the SREBP1. This transcriptional factor resides in the ER membrane and its transference to the Golgi compartment is controlled by hormonal and nutritional signaling. Then, the small fragment containing the leucine zipper, released after two consecutive proteolytic cleavages, enters the nucleus, where it induces the transactivation of genes involved in the de novo synthesis of fatty acids. Moreover, increased lipolysis favors the free fatty acids flow from the adipose tissue to the liver, allowing more substrate for triglycerides synthesis. These events contribute to a greater secretion of very-low-density lipoprotein (VLDL), which in the blood is less metabolized by the lipoprotein lipase, contributing to hypertriglyceridemia. The impairment of triglyceride-rich lipoprotein lipolysis reduces the generation of high-density lipoprotein (HDL). Furthermore, small and dense low-density lipoprotein (LDL), generated according to the increase in plasma triglycerides levels, characterizes a more atherogenic particle that has more access to the arterial wall, being susceptible to oxidation. Monocyte-derived macrophages take up oxidized LDL, contributing to the first stage of atherosclerotic plaque formation. The increased generation of oxysterols accompanies the intracellular accumulation of cholesterol, eliciting inflammation [30].
The balance between cholesterol supply to arterial wall cells (mainly via modified LDL) and lipid removal (by HDL) dictates the amount of accumulated lipids that modulates atherogenesis. Reverse cholesterol transport is the centripetal flux of cholesterol that allows its excretion into bile and feces, characterizing the major driving force of cholesterol out of the arterial wall compartment. Lipid-free apolipoprotein A-1 (APOA1) interacts with the phospholipid-transporting ATPase ABCA1 (ABCA1), a putative receptor for APOA1 and nascent HDL (pre-beta HDL), which drives excess cholesterol outside cells by obtaining energy from the hydrolysis of two ATP molecules. After esterification by the phosphatidylcholine-sterol acyltransferase (alias lecithin cholesterol acyltransferase), esterified cholesterol becomes part of the growing hydrophobic core of HDL, namely HDL3, and subsequently HDL2. Larger HDL interacts with the ATP-binding cassette transporter G-1 (ABCG1), which exports excess cholesterol and some oxysterols. Then, HDL can discharge esterified cholesterol directly into the liver by interacting with the scavenger receptor class B member 1 or indirectly by transferring esterified cholesterol to LDL, VLDL or chylomicrons by the action of the cholesteryl ester transfer protein. In the latter pathway, cholesterol removal is mediated by the uptake of APOB-containing lipoproteins by hepatocytes [29].
2.4. Advanced Glycation End Products (AGEs)
Hyperglycemia induces intracellular oxidative stress that is considered a link for all DM-related complications. Increased cellular glucose influx favors its metabolism through glycolytic pathway enhancing the electron flow in the mitochondrial respiratory chain, which relates to the generation of reactive oxygen species (ROS), particularly superoxide anion. The consequent increased expression and activity of poly (ADP-ribose) polymerase, as an adaptive mechanism that protects against DNA damage, leads to the modification of the structure of glyceraldehyde-3-phosphate dehydrogenase by poly-ADP-ribosylation. The impairment of glycolysis flow deviates glyceraldehyde 3-phosphate and dihydroxyacetone phosphate to the formation of methylglyoxal (MGO), a very reactive oxoaldehyde that interacts with proteins, phospholipids and nucleic acids, leading to the irreversible formation of AGEs [31]. Usually, under normoglycemic conditions, MGO formation occurs in very low rates, with around 0.05–0.1% of the triosephosphates degrading to MGO. In addition, its cellular concentration is low due to the action of the glyoxalase system consisting of glyoxalases 1 and 2, which convert MGO into, respectively, S-D-lactoylgluthatione and D-lactate (for a review, see [32]). In the setting of hyperglycemia, AGEs are further increased in some cells by the coactivation of the polyol pathway. Aldose reductase catalyzes glucose conversion into sorbitol, which is converted into fructose by the sorbitol dehydrogenase activity. Fructose is very reactive with proteins leading to the rapid formation of AGEs; in addition, by depleting NADPH, the polyol pathway impairs the glutathione resynthesis, favoring oxidative stress that increases the generation of AGEs [31].
In the blood circulation, excess glucose nonenzymatically interacts with the amino terminal portion of lysine and arginine residues in proteins, or with amino residues of phospholipids, leading to the formation of an unstable Schiff base. According to the maintenance of hyperglycemia, an Amadori product is generated, and after inter- and intramolecular rearrangements, AGEs are formed, including very heterogeneous compounds. Oxoaldehydes, including MGO, glyoxal, 3-deoxyglucosone and glycolaldehyde, are intermediates of this process; dicarbonyl sugars and other oxoaldehydes are more reactive than glucose and increase not only in DM, but also in chronic kidney disease, inflammation, and disorders associated with oxidative stress, leading to the rapid generation of AGEs. Moreover, oxoaldehydes increase in the postprandial period favoring a rapid modification of circulating proteins. As a consequence, there is a change in the intracellular and extracellular protein structure and functionality, affecting the extracellular matrix, cellular interactions, receptor-mediated cell responses and DNA structure. Some AGEs are intense fluorophores, allowing their measurement by fluorimetry and may induce covalent crosslinks [33]. Carboxymethyllysine (CML), carboxyethyllysine, pentosidine, pyrraline, MGO dimers, glyoxal dimers, MGO-derived hydroimidazolones and glucosepane are major species of AGEs that have been associated with long-term complications of DM [34]. AGEs, together with other metabolic pathways altered during hyperglycemia, including the polyol pathway, activation of protein kinase C and hexosamine pathways, constitute the molecular basis for cellular damage in DM (for a review, see [28]).
Advanced glycation end products interact with the receptor AGER, a multiligand receptor that activates NADPH oxidase, inducing ROS generation. This leads to the NFKB nuclear factor kappa-B (NFKB) activation and transactivation of several genes, including AGER, which contributes to a deleterious vicious circle. In addition, AGER overlaps with toll-like receptor signaling and can bind to other ligands, including calgranulins, serum amyloid and high-mobility group protein 1 (alias amphoterin), increasing inflammatory stress. The localization of AGER in the cell surface as well as in intracellular organelles enables signaling triggered by different ligands and both intra- and extracellularly formed AGEs [35]. Furthermore, soluble isoforms of AGER, lacking the intracellular domain of the native receptor, can bind AGEs without triggering intracellular signaling; thus, the amount of soluble AGER in blood might be a protective biomarker of DM complications risk [36].
On the other hand, the AGE receptor DDOST (dolichyl-diphosphooligosaccharide-protein glycosyltransferase, alias AGER1) antagonizes AGER signaling by inducing antioxidant genes attenuating the effects of AGEs. In T1D, a lower expression of DDOST in peripheral blood mononuclear cells was found in comparison to healthy controls after adjustment for sex, age, use of statins, angiotensin-converting enzyme inhibitors and angiotensin receptor blockers [37]. In this sense, the balance between AGER and DDOST expression may be detrimental when analyzing the contribution of AGEs to tissue damage in DM.
Advanced glycation impairs lipid metabolism, contributing to qualitative and quantitative alterations in plasma lipoproteins. AGEs modify both phospholipids and apolipoproteins in the lipoprotein structure, disturbing its recognition by cellular receptors and its metabolism by enzymes and proteins in the plasmatic and lymphatic compartments. VLDL and chylomicron glycation damages the activity of lipoprotein lipase; glycation of APOB increases its half-life, allowing the entrance of LDL in the arterial wall. Glycated LDL is also more oxidized and immunogenic, and for this reason, is more rapidly captured by arterial macrophages [38]. In DM, HDL modified by MGO is increased and severely compromises HDL generation, half-life and functionality, affecting cholesterol flow along the reverse cholesterol transport as well as other antiatherogenic properties of HDL [39].
3. Endoplasmic Reticulum Stress (ER Stress) and Inflammation in DM
Thirty years ago, the study of ER stress was initiated by investigating the cell reaction to heat shock, including the rapid induction of heat shock proteins (HSPs), such as the heat shock 70 kDa protein (HSP70) [40]. After the initial description of the HSP participation in cellular homeostasis and immune function, an exquisite ER stress pathway was rapidly characterized and pleiotropic roles have been proposed, including participation in the development of DM (for a review, see [41]).
In DM, ER stress was first associated to a protective effect of pancreatic beta cells. Despite the normal development of endocrine pancreas in eukaryotic translation initiation factor 2-alpha kinase 3 (EIF2AK3, alias PERK)-deficient mice, postnatal activation of ER stress is accompanied by increased cell death and leads to a progressive development of DM [42]. Later, it was demonstrated that inositol-requiring enzyme 1 (IRE1)-deficient mice exhibited mild hypoinsulinemia with hyperglycemia, especially after a glucose challenge, and despite an unaltered histological analysis of the pancreatic islets, a reduction in pancreas mass was described [43]. Furthermore, during the development of DM in Akita mice, overexpression of the ER-stress-related proteins BIP (endoplasmic reticulum chaperone BIP; alias GRP78) and DDIT3 (DNA damage-inducible transcript 3 protein; alias CHOP10/GADD153) was observed, and targeted disruption of the Ddit3 gene delayed the onset of DM, confirming the participation of ER stress in beta cell damage [44].
Additionally, ER stress has also been related to the pathogenesis of IR and T2D. The development of high-fat diet-induced obesity and T2D decreases in Hspa5 (heat shock protein family A member 5)-null mice (knockout for BIP) [45]. Repression of many UPR (unfolded protein response)-related genes also has been observed in beta cell exposed to a high concentration of glucose [46]. Indeed, multiple studies have demonstrated the involvement of UPR in the development and progression of metabolic diseases (for a review, see [47]). Thus, nowadays, it is known that ER stress participates in the impairment of insulin secretion and action; furthermore, ER stress has also been related to the development of degenerative complications (for a review, see [48,49,50]).
It was in the early 2000s that metabolic disturbances were proposed to be associated to increased inflammatory activity, due to evolutionary reasons (for a review, see [51,52]). It was demonstrated that the development of obesity was accompanied by increased production of inflammatory cytokines by the adipose tissue, defining obesity as a disease of a subclinical inflammatory activity [53]. Rapidly, it was verified that pro-inflammatory activity would be spread to other territories, participating in the pathogenesis of T2D [50,51,52].
As the molecular mechanisms triggered by inflammation were being characterized, it was becoming clear that many of them were shared with the UPR pathway, revealing an interplay between these two processes. For instance, the activation of some UPR components, such as BIP, IRE1 and TRAF2 (TNF receptor-associated factor 2), leads to IKKB/A (inhibitor of nuclear factor kappa-B kinase subunits beta/alpha) phosphorylation, triggering the activation of NFKB, an important mediator of the inflammatory activity [54]. Nowadays, it is clear that UPR is a central proteostatic pathway that can modulate both immunity and inflammation (for a review, see [55,56]).
3.1. GLUT4 Expression and ER Stress/Inflammation
After the suggestion that the activation of ER stress plays an important role in the pathophysiology of T2D, ER stress began to be investigated in the regulation of GLUT4 expression in adipocytes. Later, it was reported that ER stress response decreased Slc2a4 expression at the gene transcription level, along with increased expression of the Ddit3 gene. It is worth recalling that the DDIT3 protein is an inhibitor of the activity and expression of the Cebpa gene (CCAAT enhancer binding protein alpha), and the CEBPA transcription factor is a potent activator of Slc2a4/GLUT4 expression [57,58].
Studies in the muscle started in insulin-resistant mouse C2C12 myotubes, in which a mixture of black soybean peptides increased the glucose transport and GLUT4 translocation, in parallel to the inhibition of the ER stress response [59]. Further, in rat myotubes, induction of IR by glucosamine treatment was reported to activate some ER stress markers, such as BIP and XBP1(X-box-binding protein 1), and to increase the expression of the activating transcription factor 6 (Atf6) gene [60]. ATF6 overproduction inhibits the expression of some important enhancers of Slc2a4 expression, such as MEF2A (myocyte enhancer factor 2A) and PRGC1A (peroxisome proliferator-activated receptor gamma coactivator 1-alpha, alias PGC1A), thus explaining the repression of Slc2a4/GLUT4. Finally, it was confirmed that Atf6 silencing (with small interfering RNA) was sufficient to completely avoid these glucosamine-induced effects. These data elegantly demonstrate that ER stress causes IR in myotubes by impairing GLUT4 expression via an ATF6-mediated pathway [60].
After the first indication that inflammation could inhibit the expression of the GLUT4 [53], we started investigations focusing on demonstrating this regulation. First, it was suggested (in soleus muscle) that in vivo metabolic conditions (such as fasting) and in vitro treatments (such as muscle contraction and incubation with insulin) alter Slc2a4 mRNA expression, conversely to the Nfkb mRNA expression [61]. Electrophoretic mobility shift assay (EMSA) also revealed that the NFKB-binding activity to a consensus NFKB-binding site changes in parallel to the Nfkb mRNA expression [61]. These data strongly suggest that the inflammatory-induced repression of Slc2a4 gene expression involved a NFKB-mediated transcriptional effect.
Participation of the NFKB-mediated inflammatory activity in Slc2a4 mRNA expression was further confirmed by (1) the anti-inflammatory effect of atorvastatin in adipose tissue of T2D mice [62]; (2) the insulin sensitizer effect of inhibition of cannabinoid receptor 1 in 3T3-L1 adipocyte [63]; (3) the dose-dependent insulin-induced enhancement of the Slc2a4 expression in soleus muscle [64]; and (4) the oleic and linoleic fatty acids-induced repression of the Slc2a4 expression in L6 muscle cell [65]. All these studies revealed a converse regulation between the Slc2a4 expression and the NFKB expression/activity, indicating that the NFKB transcription factor has a repressor effect upon the Slc2a4 gene; that is very unusual considering that NFKB is currently known as an enhancer of many genes.
Importantly, the promoter region of the Slc2a4 gene does not exhibit the consensus kappa-B-binding site; thus, we investigated a homologous sequence located at the -134/-113 region of the mouse Slc2a4 gene. We confirmed that both the p50 and p65 subunits of NFKB bind into this region in vitro (EMSA) and in vivo (chromatin immunoprecipitation assay); moreover, a reporter gene assay confirmed that this region is responsible for inhibiting the Slc2a4 transcription [62]. In fact, in most of our studies commented on above concerning inflammatory-induced regulation of the Slc2a4 expression, we have confirmed the binding activity of both p50 and p65 NFKB subunits into this -143/-113 Slc2a4 promoter region [62,63,64,65,66].
Finally, we have demonstrated the involvement of inflammation and ER stress in the repression of Slc2a4/GLUT4 expression in muscle cells exposed to palmitate [67]. Acute treatment increased the BIP, EIF2A (eukaryotic translation initiation factor 2A), EIF2AK3, IRE1 and TRAF2 protein content, and EIF2AK3 phosphorylation, but did not elicit EIF2A and IKK phosphorylation or increased XBP1 nuclear content; additionally, acute and chronic treatments increased the NFKB p65 nuclear content and the NFKB-binding activity in the Slc2a4 gene promoter. All these data reveal that palmitate treatment induces the activation of the initial components of UPR, such as the formation of a IRE1–TRAF2–IKK complex, converging to a NFKB-mediated repression of Slc2a4/GLUT4, and revealing a link between ER stress and inflammation in IR [67].
Endoplasmic reticulum and inflammatory stress (with reduced GLUT4 expression) have been observed in insulin-resistant skeletal muscle from women with gestational DM, and suppression of ER stress by tauroursodeoxycholic acid (TUDCA) or siRNA knockdown of IRE1A and BIP protein significantly downregulated the activation of ER and inflammatory stress, and increased the GLUT4 expression and glucose uptake [68]. Interestingly, it was demonstrated that prolonged preoperative fasting in rats induced postoperative ER stress (the activation of IRE1A) and the repression of muscle GLUT4, leading to IR and hyperglycemia [69]. Finally, a recent study in the brain hippocampus of DM rats has associated the activation of ER stress (BIP, DDIT3 and ATF4) and inflammatory stress (tumor necrosis factor (TNF) and interleukin-6 (IL6)) with reduced GLUT4 expression; furthermore, metformin/donepezil treatment was demonstrated as efficacious to reverse these alterations, becoming a promising way to manage DM-associated dementia [70].
3.2. Atherogenesis/CVD and ER Stress/Inflammation
Hyperglycemia is known as a detrimental factor for the development of long-term complications of DM. In the association of CVD, DM, obesity and other metabolic diseases, altered lipid metabolism is an important etiopathogenic mechanism. The modification in lipid metabolism is both a biomarker, and a pathological factor, for CVD, T2D, obesity and other metabolic diseases; under these conditions, impaired cellular homeostasis hinders the proper functioning of the ER, and thus ER stress as well as inflammation play a fundamental role in the pathogenicity of these diseases (for a review, see [71]).
The scavenger receptor A, the platelet glycoprotein 4 (alias FAT and SCARB3), and the oxidized low-density lipoprotein receptor 1 are directly linked to ER stress, and their inhibition is a target to prevent the development of atherosclerosis [72,73,74]. Inflammatory and ER stress markers driven by the accumulation of free cholesterol and 7-ketocholesterol are increased in atherosclerotic lesion areas prone to rupture [30,75].
The chronic activation of ER stress is closely associated with endothelial dysfunction and atherosclerosis (for a review, see [76]). The accumulation of free cholesterol in cells, inflammation, and other associated CV risk factors (hyperhomocysteinemia, saturated fatty acids, modified lipoproteins and DM) trigger or worsen ER stress (for a review, see [77]). Conceivably, cholesterol is esterified by the sterol O-acyltransferase (alias ACAT), allowing the storage of esterified cholesterol in a relatively inert form in the cytosol and the rapid bioavailability of free cholesterol. In addition, it avoids the unspecific flux of free cholesterol among cell organelles that is especially toxic to the ER. The analysis of the transcriptome of aortic tissues in high-fat-diet-fed Apoe-knockout mice demonstrated the enhancement of three major UPR pathways, as well as 50 overlapping genes involved in UPR signaling, adaptation and apoptosis [78]. Furthermore, in DM Apoe-deficient mice, supplemented with glucosamine, larger and more advanced atherosclerotic lesions at the aortic root, displaying increased immunoreactivity for BIP and ENPL (endoplasmin, alias GRP94) at the lesion tissue, was observed [79]
Excess cholesterol and crystals of cholesterol also trigger the activation of the inflammasome, which together with the activation of ER stress are in an intricate connection that underlies the development and progression of atherosclerosis [80]. Interestingly, one of the mediators of UPR, the ER-resident ATF6, is similar to SREBP, activated by two proteolytic cleavages in the Golgi compartment [81]. It has been shown that ATF6, although inhibiting the activation of SREBP, is itself capable of inducing lipogenesis, establishing a link between UPR and dyslipidemia [82,83].
The activation of liver X receptor (LXR), by enhancing the expression of ABCA1 and ABCG1, improves the cholesterol efflux, alleviates ER stress and reduces apoptosis and defective efferocytosis in atherosclerotic lesion [84,85]. The pharmacological inhibition of cholesterol esterification and the stimulus to cholesterol efflux after treating with APOA1 mimetics, or overexpressing APOA1, abrogate ER and inflammatory stress [86,87]. Besides, by inhibiting oxidation and inflammation, and stimulating excess cholesterol efflux, HDL contributes to preventing ER stress [87]. Treatment of vascular smooth cells with a high-glucose medium induces the expression of CD36, cytokines and markers of calcification and ER stress; a condition that is aggravated by adding oxidized LDL. Those results were reinforced by findings in carotid plaques obtained from DM subjects in comparison to non-diabetic individuals [88].
4. AGEs-Induced and ER Stress/Inflammation-Mediated Effects in DM
Inflammation, oxidative stress and sterol accumulation, induced by AGE, trigger ER stress characterized by the enhanced expression of BIP, ENPL, EIF2A and ATF6A (cyclic AMP-dependent transcription factor ATF-6 alpha). Furthermore, glycation per se is a contributing factor to protein misfolding, eliciting UPR as an intracellular protein quality control system [89,90]. Glycation changes the electrostatic interactions and the hydrophobicity of polypeptide residues that trigger ubiquitination and degradation [91]. In fact, AGEs and ER stress often chronically coexist under many biological conditions, evincing their integrated action in the progression of DM- and other carbonyl stress-related complications. In addition, the propagation of this intricate mechanism is strengthened by noticing that the expression of AGER and its ligand—calgranulin S100 B—increases in neurological disorders, marked by increased ER stress activation [92].
The proteome of endothelial cells incubated with high-glucose medium identified the abundance of 331 proteins differentially expressed in comparison to the normoglycemic medium. Apart from the proteins directly involved in glycolysis and gluconeogenesis, the heat shock proteins related to UPR and protein refolding, the ubiquitin E2 ligases involved in protein degradation by the proteasomal and lysosomal system increased upon toxic glycoxidative stress mediated by MGO generation [93]. Recently, the plasma concentration of the tribbles homolog 3 protein (TRB3) (which is enhanced by hyperglycemia and ER stress) was found increased in subjects with T2D together with AGEs, BIP and TNF; moreover, it is positively correlated with fasting plasma glucose and AGEs [94].
4.1. AGEs and GLUT4 Expression Regulation
Advanced glycation has been extensively related to the development and progression of DM-induced degenerative complications; however, little is known about their participation in the glycemic homeostasis since, classically, they emerge only in situations of hyperglycemia. However, more recently, it has become evident that AGEs can be formed under other conditions of carbonyl stress, as well as obtained through exogenous sources, and that has led AGEs to occupy a key role in insulin sensitivity/GLUT4/glycemic homeostasis, closing a vicious circle. In fact, there is considerable evidence regarding the role of exogenous AGEs, mainly from dietary sources, in the induction of IR and DM in animal models and humans (for a review, see [95]).
In humans, long-term intervention trials with a high-AGE-containing diet are not used for ethical issues; nevertheless, studies with a low-AGE containing diet revealed amelioration of IR, which is, in part, ascribed to the enhanced expression of DDOST and SIRTUIN 1, due to their antioxidant and anti-inflammatory actions [96,97].
In vitro studies have contributed to demonstrating the direct effect of AGEs upon tissue insulin sensitivity, especially in GLUT4 regulation and inflammatory activity. AGER overexpression in 3T3-L1 preadipocytes accelerates adipocyte hypertrophy, whereas inhibition of AGER by small interfering RNA decreases adipocyte hypertrophy; furthermore, AGER-induced adipocyte hypertrophy was associated with the reduction of insulin signaling and glucose uptake, downregulation of GLUT4 and upregulation of toll-like receptor [98]. Additionally, in cultured human preadipocytes, adipogenesis was associated with increased levels of CML and AGER, and CML was seen to induce an AGER-dependent dysregulation of inflammatory adipokines [99]. Furthermore, a recent study confirmed that 3T3-L1 adipocytes cultured in a proglycating medium (with MGO or MGO-modified bovine serum albumin) increase Ager and reduces the Slc2a4 gene expression; besides, bovine serum albumin-MGO reduces glucose uptake [100].
Interestingly, epicardial adipose tissue (EAT), a visceral fat surrounding the myocardium, which is potentially involved in the onset/progression of coronary artery disease, has also been investigated in subjects undergoing open-heart surgery. Increased AGER expression in EAT was observed to correlate with increased EAT thickness, reduced expression of GLUT4, adiponectin and lactoylglutathione lyase (alias glyoxalase 1), and increased expression of the high-mobility group protein B1, toll-like receptor and MYD88 (myeloid differentiation primary response protein MyD88). These data indicate that, in subjects with coronary disease, AGER may be involved in promoting EAT adiposity and metabolic dysfunction, and that is related to the decreased GLUT4 expression [101].
The first indication of a primary in vivo effect of AGE upon glycemic homeostasis and GLUT4 expression was reported in experimental healthy Sprague-Dawley rats subjected to chronic administration of MGO. In this model, increased plasma glucose, reduced GLUT4 expression and glucose uptake (adipose tissue) and severe beta cell damage were observed, evincing a classic T2D profile [102]. Later, in healthy Wistar rats, chronic administration of AGE-albumin (3 months) was reported to increase adiposity and body weight as well as to induce whole-body IR, associated with an increased expression of inflammatory markers and decreased expression of Scl2a4/Glut4 in periepididymal adipose tissue [103].
Finally, we have investigated the participation of ER stress and inflammation in the in vivo and in vitro effects of AGEs in the Slc2a4/GLUT4 expression in skeletal soleus muscle of healthy rats [9]. For in vivo analysis, rats were treated (12 weeks) with AGE-albumin, and developed whole-body IR, with decreased Slc2a4/GLUT4 expression, increased nuclear content of NFKB (p50) and increased cellular content of BIP [8]. For in vitro analysis, muscles from healthy rats were incubated (2.5 to 7.5 h) with AGE-albumin, and displayed a decreased Slc2a4/GLUT4 content, increased BIP and ENPL contents, as well as increased phosphorylation of IKKA and IKKB and nuclear NFKB (p50 and p65) content; furthermore, EMSA analysis revealed an increase in nuclear protein binding in the NFKB-binding site of the Slc2a4 promoter [9].
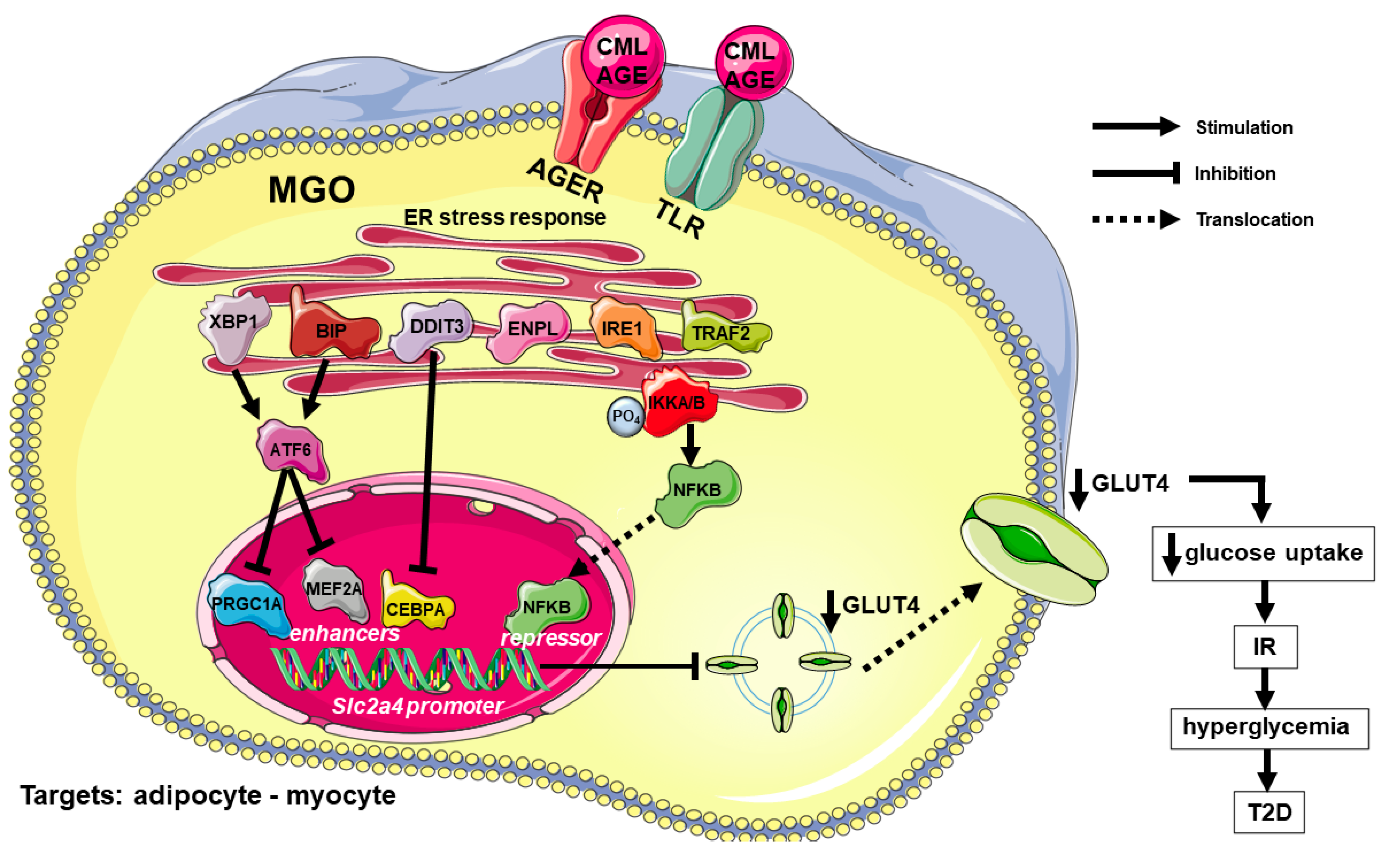
These studies reveal that high AGE concentrations impair glucose homeostasis through a mechanism that involves ER stress/inflammation-mediated repression of the Slc2a4/GLUT4 expression. Figure 1 summarizes the main mechanisms involved in the AGE-induced and ER stress-mediated repression of GLUT4 expression and the impairment of glycemic homeostasis.
4.2. AGEs and Atherogenesis/CVD Development
Inflammation has an important role in the development of CVD, which can be initially observed by its role in the macrophage homeostasis. Albumin, the most abundant protein in circulation, has enhanced susceptibility to glycation, and AGE-albumin increases the generation of ROS in macrophages, priming these cells to the lipopolysaccharide-induced secretion of inflammatory cytokines [104], and inducing the accumulation of oxysterols [105].
In non-diabetic dyslipidemic mice (Apoe-knockout mouse), treatment with AGE-albumin increased lipid deposition in the aortic arch, enhanced the expression of Ager, Tnf (tumor necrosis factor) and Nox4 (NADPH oxidase 4) genes, and increased the marker of lipid peroxidation (4-hydroxynonenal) and CML, in comparison with animals receiving control-albumin. Interestingly, treatment with losartan (antagonist of the angiotensin 2 receptor) abrogated lipid accumulation and expression of the AGE–AGER axis in the aortic arch of animals treated with AGE-albumin [106].
The participation of ER stress in the development of CVD has been firstly characterized by its role in macrophage homeostasis, a field we have contributed to in the last decade. Oxidative stress induces a reduction in the ABCA1 protein level and cholesterol efflux to APOA1; both events are avoided by incubating cells with aminoguanidine, an antiglycation and antioxidation compound that reduces ROS generation [107]. Besides, the use of a chemical chaperone (4-phenylbutyric acid), which alleviates ER protein misfolding, recovers the ABCA-1 levels [108]. The ER-associated degradation (ERAD) pathway is involved in the degradation of ABCA1, together with the endosomal sorting complex required for transport pathway and the proteolysis mediated by surface calpains. [109].
Macrophages treated with AGE-albumin (produced in vitro and isolated from subjects with poorly controlled DM) have a faster intracellular degradation of ABCA1, due to the activity of ubiquitin-proteasomal and lysosomal systems, which are independent of surface calpains [110]. The effects on disturbing the APOA1-mediated cholesterol removal and inducing inflammation persisted even after a long period of cell resting in the absence of AGE-albumin. In addition, the impairment of cholesterol efflux was restored after the improvement of the glycemic control, reflected by diminished levels of AGEs [111]. In the absence of AGER in macrophages, attained by AGER silencing or using peritoneal macrophages from Ager knockout mice, the deleterious effects of AGE-albumin produced in vitro or isolated from subjects with DM were abrogated. Moreover, the altered expression of genes involved in lipid efflux was restored in the absence of AGE/AGER signaling [112]. Considering the ability of many other receptors to interact with AGEs, further studies are necessary to better understand the blockage of AGE signaling in the improvement of cholesterol balance in the arterial wall.
Diabetic kidney disease is a prevalent complication of DM that increases cardiovascular mortality. AGEs are also prevalent in chronic kidney disease due to the failure in the detoxification of the Maillard reaction intermediates. Besides, carbamoylation occurs because of urea dissociation into thiocyanate that reacts with proteins. In an animal model of uremia, carbamoylated albumin disturbed cholesterol efflux, which was associated with ER stress [113]. In humans with diabetic kidney disease, albumin carbamoylation enhances according to the reduction in estimated glomerular filtration rate and diminishes HDL-mediated cholesterol efflux from macrophages [114]. Regarding the potential mechanisms involved, it has been reported that AGE-albumin increases the expression of BIP and induces apoptosis dependent of the intracellular calcium increase in kidney podocytes; furthermore, TUDCA, which impairs ER stress by acting as a chaperone, prevents the cell from dying [115]. Furthermore, MGO-derived AGEs were confirmed to induce ER stress (increased BIP, ATF4 and DDIT3) in kidney cells, associated with inflammation, apoptosis and an imbalance in mitochondrial function [116].
In animal model of DM, the activation of EIF2AK3 was observed as involved in the AGE-induced coronary dysfunction [117]; furthermore, AGE-induced activation of ER stress was also related to post-myocardial infarction ventricular arrhythmias [118]. Glycated LDL induces intense ER stress in bovine aortic endothelial cells, activating BIP, EIF2AK3 and ATF6, which was accompanied by impaired endothelium-dependent vasodilation [119]. Moreover, in human vascular endothelial cells, the reduction in the antioxidant enzyme paraoxonase 2 elicited by AGEs is associated with increased oxidative, inflammatory and ER stress [120]. Finally, a beneficial effect of lowering the AGEs levels for CVD in humans was suggested in the recent CORDIOPREV study, in which the reduction in circulating AGEs achieved by the Mediterranean diet, expressed by the MGO levels, was associated with a greater probability of T2D remission in subjects with CVD [121]. Figure 2 summarizes the main mechanisms involved in the AGE-induced and ER stress-mediated atherogenesis and CVD development.
5. AGEs, ER Stress and Inflammation in Non-Alcoholic Fatty Liver Disease (NAFLD)
As the knowledge of ER stress and inflammation advances, their involvement in several chronic diseases, such as DM, CVD, neurodegenerative disorders, fatty liver disease, inflammatory bowel and cancer, among others, become increasingly evident (for a review, see [122]). NAFLD has received considerable attention as a complication of DM, because of its increasing incidence and severity, and ER and inflammatory stress have been demonstrated to be involved in this condition.
Diabetes mellitus Apoe-knockout mice, supplemented with glucosamine, developed intracellular lipid accumulation, indicating the development of NAFLD, and that was associated with increased content of the UPR markers BIP, ENPL, DDIT3 and protein disulfide-isomerase in liver tissue [79]. On the other hand, we have reported that T2D mice develop histological signals of non-alcoholic steatohepatitis (NASH, according to NASH-CRN Pathology Committee System), associated with impaired glucose metabolism in the liver, and increased expression of the inflammatory markers TNF, IL6 and NFKB, as well as increased binding activity of nuclear proteins in NFKB-binding site target genes, as measured in the Slc2a2 (solute carrier family 2 member 2) gene [123].
The P2Y purinergic receptor 12, an adenosine diphosphate responsive G protein-coupled receptor expressed on the surface of platelets (required for normal platelets aggregation), is also expressed by macrophages in the liver from mice with cirrhosis and hepatocellular carcinoma. Ticagrelor-induced inhibition of P2Y purinergic receptor 12 enhances tumor cell phagocytosis by macrophages and induces an anti-tumoral phenotype, which was associated with the increased expression of several actors of the ER stress pathways, indicating activation of the UPR [124]. Besides, the inhibition of the UPR with TUDCA diminishes the pro-phagocytotic effect of ticagrelor, confirming that P2Y purinergic receptor 12 mediates macrophage function through the activation of ER stress [124]. This seems to be relevant in the pathogenesis of chronic liver disease and cancer.
Increased consumption of sugars, especially fructose, and AGE-enriched diets are related to the generation of intracellular AGEs that induce lipogenesis and inflammation in crosstalk with ER stress [125]. Besides, the extravasation of AGEs and its interaction with RAGE is associated with the development and evolution of several chronic diseases, including steatosis and CVD [126]. A recent meta-analysis found an association among elevated levels of several types of AGEs with NAFLD [127] and the determination of fluorescent AGE has been considered as a potential biomarker for NAFLD stratification [128].
6. Concluding Remarks
Over the last two decades, extensive knowledge of the molecular players in the pathways of ER and inflammatory stress has been gained; at the same time, their participation in the etiopathogenesis and pathophysiology of innumerous diseases has also grown. Interestingly, the sharing of some players through the two pathways has been increasingly demonstrated, evincing a very complicate and intricate interplay between ER and inflammatory stress.
Endoplasmic reticulum and inflammatory stress are recurrently found to be involved in the pathophysiology of DM, whether in mechanisms related to glycemic control (insulin secretion and action) or in the development/progression of DM complications. Furthermore, hyperglycemia increases the generation of AGEs, which in turn refeed ER and inflammatory stress, contributing to the impairment of glycemic homeostasis and the acceleration of DM complications.
In this article, we have reviewed the molecular mechanisms involved in the AGEs-induced activation of ER and inflammatory stress pathways, leading to the repression of the Slc2a4 gene and consequently to the decrease in GLUT4 content in adipose and muscle tissue. This event reduces plasma glucose clearance and contributes to hyperglycemia, establishing a dangerous vicious circle in DM. Additionally, AGEs also induce, by an ER stress/inflammation-mediated way, the development of atherogenesis, contributing to the progression of CVD.
To date, in DM, it is clear that the impairment of glycemic homeostasis and the development of degenerative complications have important AGEs-induced and ER stress/inflammation-mediated components. Experimentally, the blockage of some ER and inflammatory stress players in specific tissues has exhibited excellent results for the improvement of glycemic control or the inhibition of CVD development/progression. Thus, based on ER and inflammatory stress inhibition, several potential targets for the prevention and/or treatment of DM and its complications can be developed in the future.
Author Contributions
M.P. and U.F.M. have equally participated in the conceptualization of this review. M.P. is expert in diabetes and atherogenesis; U.F.M. is expert in diabetes and insulin resistance/GLUT4. All authors have read and agreed to the published version of the manuscript.
Funding
Research articles from the authors cited in this manuscript were funded by FAPESP grant numbers: #2002/07384-4; #2002/07945-6; #2007/50554-1; #2008/51094-7; #2007/57745-7; #2009/53869-9; #2011/08570-5; #2012/04831-1; #2012/19755-9; #2015/21072-5; #2016/15603-0; 2019/18431-4. The authors thank Conselho Nacional de Desenvolvimento Científico, CNPq, Brazil, for receiving a research award.
Acknowledgments
The authors are thankful to Adauri Brezolin for English revision of the manuscript.
Conflicts of Interest
The authors declare that they have no conflict of interest.
List of Abbreviations and Acronyms
Names and symbols of genes and proteins referred to in this manuscript are in accordance with the HGNC (https://www.genenames.org/, accessed on 2 November 2021) and the UNIPROT (https://www.uniprot.org/, accessed on 2 November 2021) databases.
| ABCA1 | the phospholipid-transporting ATPase ABCA1 |
| ABCG1 | ATP binding cassette transporter G-1 |
| AGE | advanced glycation end products |
| AGER | advanced glycation end product-specific receptor, (alias RAGE) |
| APOA1 | apolipoprotein A-1 |
| APOB | apolipoprotein B |
| Apoe | apolipoprotein E gene (rat/mouse) |
| ATF4 | cyclic AMP-dependent transcription factor ATF-4 |
| ATF6 | cyclic AMP-dependent transcription factor ATF-6 |
| Atf6 | activating transcription factor 6 gene (rat/mouse) |
| ATF6A | cyclic AMP-dependent transcription factor ATF-6 alpha |
| BIP | endoplasmic reticulum chaperone BIP (alias GRP78) |
| CD36 | platelet glycoprotein 4 (alias FAT/SCARB3) |
| CEBPA | CCAAT enhancer binding protein alpha |
| Cebpa | CCAAT enhancer binding protein alpha gene (rat/mouse) |
| CML | carboxymethyllysine |
| CVD | cardiovascular disease |
| DDIT3 | DNA damage-inducible transcript 3 protein (alias CHOP10/GADD153) |
| Ddit3 | DNA damage-inducible transcript 3 gene (rat/mouse) |
| DDOST | dolichyl-diphosphooligosaccharide-protein glycosyltransferase, alias AGER1 |
| DDOST | dolichyl-diphosphooligosaccharide-protein glycosyltransferase gene (human) |
| DM | diabetes mellitus |
| EAT | epicardial adipose tissue |
| EIF2A | eukaryotic translation initiation factor 2A |
| EIF2AK3 | eukaryotic translation initiation factor 2-alpha kinase 3 (alias PERK) |
| EMSA | electrophoretic mobility shift assay |
| ENPL | endoplasmin (alias GRP94) |
| ER | endoplasmic reticulum |
| GLUT4 | solute carrier family 2, facilitated glucose transporter member 4 |
| HDL | high-density lipoprotein |
| HSP | heat shock protein |
| HSP70 | heat shock 70 kDa protein |
| Hspa5 | heat shock protein family A member 5 gene (rat-mouse) |
| IKKA | inhibitor of nuclear factor kappa-B kinase subunits alpha |
| IKKB | inhibitor of nuclear factor kappa-B kinase subunits beta |
| IL6 | interleukin-6 |
| IR | insulin resistance |
| IRE1 | inositol requiring enzyme 1 |
| LDL | low-density lipoprotein |
| LXR | liver X receptor |
| MEF2A | myocyte enhancer factor 2A |
| MGO | methylglyoxal |
| MYD88 | myeloid differentiation primary response protein MyD88 |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| NFKB | NFKB nuclear factor-kappa-B |
| Nfkb | nuclear factor-kappa-B gene (rat/mouse) |
| Nox4 | NADPH oxidase 4 gene (rat/mouse) |
| PM | plasma membrane |
| PRGC1A | peroxisome proliferator-activated receptor gamma coactivator 1-alpha (alias PGC1A) |
| ROS | reactive oxygen species |
| Slc2a2 | solute carrier family 2 member 2 gene (rat/mouse) |
| Slc2a4 | solute carrier family 2 member 4 gene (rat/mouse) |
| SLC2A4 | solute carrier family 2 member 4 gene (human) |
| SREBP1 | sterol regulatory element-binding protein |
| T1D | type 1 diabetes mellitus |
| T2D | type 2 diabetes mellitus |
| TRAF2 | TNF receptor-associated factor 2 |
| TRB3 | tribbles homolog 3 protein |
| TNF | tumor necrosis factor |
| Tnf | tumor necrosis factor gene (rat/mouse) |
| TUDCA | tauroursodeoxycholic acid |
| UPR | unfolded protein response |
| VLDL | very low-density lipoprotein |
References
- Forouhi, N.G.; Wareham, N.J. Epidemiology of Diabetes. Medicine 2019, 47, 22–27. [Google Scholar] [CrossRef]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2011, 34, S62–S69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, M.M.; Anhê, G.F.; Sabino-Silva, R.; Marques, M.F.; Freitas, H.S.; Mori, R.C.; Melo, K.F.; Machado, U.F. Intensive insulin treatment induces insulin resistance in diabetic rats by impairing glucose metabolism-related mechanisms in muscle and liver. J. Endocrinol. 2011, 211, 55–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donga, E.; Dekkers, O.M.; Corssmit, E.P.M.; Johannes, A.; Romijn, J.A. Insulin resistance in patients with type 1 diabetes assessed by glucose clamp studies: Systematic review and meta-analysis. Eur. J. Endocrinol. 2015, 173, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolosowics, M.; Lukaszuk, B.; Chabowski, A. The causes of insulin resistance in type 1 diabetes mellitus: Is there a place for quaternary prevention? Int. J. Environ. Res. Public Health 2020, 17, 8651. [Google Scholar] [CrossRef]
- Nathan, D.M. Realising the long-term promise of insulin therapy: The DCCT/EDIC study. Diabetologia 2021, 64, 1049–1058. [Google Scholar] [CrossRef]
- Artime, E.; Romera, I.; Díaz-Cerezo, S.; Delgado, E. Epidemiology and Economic Burden of Cardiovascular Disease in Patients with Type 2 Diabetes Mellitus in Spain: A Systematic Review. Diabetes Ther. 2021, 12, 1631–1659. [Google Scholar] [CrossRef]
- Lester, E. The clinical value of glycated haemoglobin and glycated plasma proteins. Ann. Clin. Biochem. 1989, 26, 213–219. [Google Scholar] [CrossRef]
- Pinto-Junior, D.C.; Silva, K.S.; Michalani, M.L.; Yonamine, C.Y.; Esteves, J.V.; Fabre, N.T.; Thieme, K.; Catanozi, S.; Okamoto, M.M.; Seraphim, P.M.; et al. Advanced glycation end products-induced insulin resistance involves repression of skeletal muscle GLUT4 expression. Sci. Rep. 2018, 8, 8109. [Google Scholar] [CrossRef]
- Pinto, R.S.; Machado, U.F.; Passarelli, M. Advanced glycation end products as biomarkers for cardiovascular disease: Browning clarifying atherogenesis. Biomark. Med. 2020, 14, 611–614. [Google Scholar] [CrossRef]
- Klip, A.; Sun, Y.; Chiu, T.T.; Foley, K.P. Signal transduction meets vesicle traffic: The software and hardware of GLUT4 translocation. Am. J. Physiol. Cell Physiol. 2014, 306, C879–C886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klip, A.; McGraw, T.E.; James, D.E. Thirty sweet years of GLUT4. J. Biol. Chem. 2019, 294, 11369–11381. [Google Scholar] [CrossRef] [Green Version]
- Corrêa-Giannella, M.L.; Machado, U.F. SLC2A4 gene: A promising target for pharmacogenomics of insulin resistance. Pharmacogenomics 2013, 14, 847–850. [Google Scholar] [CrossRef] [Green Version]
- Frendo-Cumbo, S.; Tokarz, V.L.; Bilan, P.J.; Brumell, J.H.; Klip, A. Communication Between Autophagy and Insulin Action: At the Crux of Insulin Action-Insulin Resistance? Front. Cell Dev. Biol. 2021, 9, 708431. [Google Scholar] [CrossRef]
- Sajan, M.P.; Lee, M.C.; Foufelle, F.; Sajan, J.; Cleland, C.; Farese, R.V. Coordinated regulation of hepatic FoxO1, PGC-1α, and SREBP-1c facilitates insulin action and resistance. Cell. Signal. 2018, 43, 62–70. [Google Scholar] [CrossRef]
- James, D.E.; Strube, M.; Mueckler, M. Molecular cloning and characterization of an insulin-regulatable glucose transporter. Nature 1989, 338, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef]
- Salazar, M.R.; Carbajal, H.A.; Espeche, W.G.; Aizpurúa, M.; Leiva Sisnieguez, C.E.; Leiva Sisnieguez, B.C.; Stavile, R.N.; March, C.E.; Reaven, G.M. Insulin resistance: The linchpin between prediabetes and cardiovascular disease. Diab. Vasc. Dis. Res. 2016, 13, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Villalobos-Labra, R.; Subiabre, M.; Toledo, F.; Pardo, F.; Sobrevia, I. Endoplasmic reticulum stress and development of insulin resistance in adipose, skeletal, liver, and foetoplacental tissue in diabesity. Mol. Asp. Med. 2019, 66, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Tirosh, A.; Tuncman, G.; Calay, E.S.; Rathaus, M.; Ron, I.; Tirosh, A.; Yalcin, A.; Lee, Y.G.; Livne, R.; Ron, S.; et al. Intercellular transmission of hepatic ER stress in obesity disrupts systemic metabolism. Cell Metab. 2021, 33, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Asp. Med. 2013, 34, 121–138. [Google Scholar] [CrossRef] [Green Version]
- Karnieli, E.; Armoni, M. Transcriptional regulation of the insulin-responsive glucose transporter GLUT4 gene: From physiology to pathology. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E38–E45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Im, S.S.; Kwon, S.K.; Kim, T.H.; Kim, H.-I.; Ahn, Y.H. Regulation of glucose transporter type 4 isoform gene expression in muscle and adipocytes. IUBMB Life 2007, 59, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Machado, U.F.; Shimizu, I.; Saito, M. Reduced content and preserved translocation of glucose transporter (GLUT 4) in white adipose tissue of obese mice. Physiol. Behav. 1994, 55, 621–625. [Google Scholar] [CrossRef]
- Gibbs, E.M.; Stock, J.L.; McCoid, S.C.; Stukenbrok, H.A.; Pessin, J.E.; Stevenson, R.W.; Milici, A.J.; McNeish, J.D. Glycemic improvement in diabetic db/db mice by overexpression of the human insulin-regulatable glucose transporter (GLUT4). J. Clin. Investig. 1995, 95, 1512–1518. [Google Scholar] [CrossRef] [Green Version]
- Zisman, A.; Peroni, O.D.; Abel, E.D.; Michael, M.D.; Mauvais-Jarvis, F.; Lowell, B.B.; Wojtaszewski, J.F.P.; Hirshman, M.F.; Virkamaki, A.; Goodyear, L.J.; et al. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat. Med. 2000, 6, 924–928. [Google Scholar] [CrossRef]
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Prevalence of cardiovascular disease in type 2 diabetes: A systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovasc. Diabetol. 2018, 17, 83. [Google Scholar] [CrossRef] [Green Version]
- Eckel, R.H.; Bornfeldt, K.E.; Goldberg, I.J. Cardiovascular disease in diabetes, beyond glucose. Cell Metab. 2021, 33, 1519–1545. [Google Scholar] [CrossRef]
- Thambiah, S.C.; Lai, L.C. Diabetic dyslipidaemia. Pract. Lab. Med. 2021, 26, e00248. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [Green Version]
- Myoishi, M.; Hao, H.; Minamino, T.; Watanabe, K.; Nishihira, K.; Hatakeyama, K.; Asada, Y.; Okada, K.I.; Ishibashi-Ueda, H.; Gabbiani, G.; et al. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation 2007, 116, 1226–1233. [Google Scholar] [CrossRef] [Green Version]
- Morgenstern, J.; Campos Campos, M.; Nawroth, P.; Fleming, T. The Glyoxalase System-New Insights into an Ancient Metabolism. Antioxidants 2020, 9, 939. [Google Scholar] [CrossRef]
- Sell, D.R.; Sun, W.; Gao, X.; Strauch, C.; Lachin, J.M.; Cleary, P.A.; Genuth, S.; DCCT/EDIC Research Group; Monnier, V.M. Skin collagen fluorophore LW-1 versus skin fluorescence as markers for the long-term progression of subclinical macrovascular disease in type 1 diabetes. Cardiovasc. Diabetol. 2016, 15, 30. [Google Scholar] [CrossRef] [Green Version]
- Genuth, S.; Sun, W.; Cleary, P.; Gao, X.; Sell, D.R.; Lachin, J.; DCCT/EDIC Research Group; Monnier, V.M. Skin advanced glycation end products glucosepane and methylglyoxal hydroimidazolone are independently associated with long-term microvascular complication progression of type 1 diabetes. Diabetes 2015, 64, 266–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akirav, E.M.; Preston-Hurlburt, P.; Garyu, J.; Henegariu, O.; Clynes, R.; Schmidt, A.M.; Herold, K.C. RAGE expression in human T cells: A link between environmental factors and adaptive immune responses. PLoS ONE 2012, 7, e34698. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K. Low levels of serum soluble receptors for advanced glycation end products, biomarkers for disease state: Myth or reality. Int. J. Angiol. 2014, 23, 11–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Bezerra, D.P.; Machado-Lima, A.; Monteiro, M.B.; Admoni, S.N.; Perez, R.V.; Machado, C.G.; Shimizu, M.H.; Cavaleiro, A.M.; Thieme, K.; Queiroz, M.S.; et al. Dietary advanced glycated end-products and medicines influence the expression of SIRT1 and DDOST in peripheral mononuclear cells from long-term type 1 diabetes patients. Diab. Vasc. Dis. Res. 2018, 15, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Godfrey, L.; Xue, M.; Shahe, F.; Geoffrion, M.; Milne, R.; Thornalley, P.J. Glycation of LDL by methylglyoxal increases arterial atherogenicity: A possible contributor to increased risk of cardiovascular disease in diabetes. Diabetes 2011, 60, 1973–1980. [Google Scholar] [CrossRef] [Green Version]
- Godfrey, L.; Yamada-Fowler, N.; Smith, J.; Thornalley, P.J.; Rabbani, N. Arginine-directed glycation and decreased HDL plasma con;centration and functionality. Nutr. Diabetes 2014, 4, e134. [Google Scholar] [CrossRef] [Green Version]
- Leung, P.S.; Gershwin, M.E. The immunobiology of heat shock proteins. J. Investig. Allergol. Clin. Immunol. 1991, 1, 23–30. [Google Scholar]
- Schröder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zeng, H.; Zhang, Y.; Jungries, R.; Chung, P.; Plesken, H.; Sabatini, D.D.; Ron, D. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol. Cell 2001, 7, 1153–1163. [Google Scholar] [CrossRef]
- Iwawaki, T.; Akai, R.; Kohno, K. IRE1a Disruption Causes Histological Abnormality of Exocrine Tissues, Increase of Blood Glucose Level, and Decrease of Serum Immunoglobulin Level. PLoS ONE 2010, 5, e13052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyadomari, S.; Koizumi, A.; Takeda, K.; Gotoh, T.; Akira, S.; Araki, E.; Mori, M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Investig. 2002, 109, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.; Jung, D.Y.; Jun, J.Y.; Li, J.; Luo, S.; Ko, H.J.; Kim, J.K.; Lee, A. Grp78 heterozygosity promotes adaptive unfolded protein response and attenuates diet-induced obesity and insulin resistance. Diabetes 2010, 59, 6–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, S.T.; Luzuriaga, J.; Chan, J.Y.; Grey, S.T.; Laybutt, D.R. Influence of chronic hyperglycemia on the loss of the unfolded protein response in transplanted islets. J. Mol. Endocrinol. 2013, 51, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Pandey, V.K.; Mathur, A.; Kakkar, P. Emerging role of Unfolded Protein Response (UPR) mediated proteotoxic apoptosis in diabetes. Life Sci. 2019, 216, 246–258. [Google Scholar] [CrossRef]
- Yilmaz, E. Endoplasmic Reticulum Stress and Obesity. Adv. Exp. Med. Biol. 2017, 960, 261–276. [Google Scholar]
- Mustapha, S.; Mohammed, M.; Azemi, A.K.; Jatau, A.I.; Shehu, A.; Mustapha, L.; Aliyu, I.M.; Danraka, R.N.; Amin, A.; Bala, A.A.; et al. Current Status of Endoplasmic Reticulum Stress in Type II Diabetes. Molecules 2021, 26, 4362. [Google Scholar] [CrossRef]
- Mukherjee, N.; Lin, L.; Contreras, C.J.; Templin, A.T. Β-cell death in diabetes: Past discoveries, present understanding, and potential future advances. Metabolites 2021, 11, 796. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Investig. 2005, 115, 1111–1119. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Ruan, H.; Hacohen, N.; Golub, T.R.; Van Parijs, L.; Lodish, H.F. Tumor necrosis factor-alpha suppresses adipocyte-specific genes and activates expression of preadipocyte genes in 3T3-L1 adipocytes: Nuclear factor-kappaB activation by TNF-alpha is obligatory. Diabetes 2002, 51, 1319–1336. [Google Scholar] [CrossRef] [Green Version]
- Tam, A.B.; Mercado, E.L.; Hoffmann, A.; Niwa, M. ER Stress Activates NF-κB by Integrating Functions of Basal IKK Activity, IRE1 and PERK. PLoS ONE 2012, 7, e45078. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S. Inflammation and endoplasmic reticulum stress in obesity and diabetes. Int. J. Obes. 2008, 32, S52–S54. [Google Scholar] [CrossRef] [Green Version]
- Fernández, D.; Geisse, A.; Bernales, J.I.; Lira, A.; Osorio, F. The Unfolded Protein Response in Immune Cells as an Emerging Regulator of Neuroinflammation. Front. Aging Neurosci. 2021, 13, 682633. [Google Scholar] [CrossRef]
- Fatima, L.A.; Campello, R.S.; Barreto-Andrade, J.N.; Passarelli, M.; Santos, R.S.; Clegg, D.J.; Machado, U.F. Estradiol stimulates adipogenesis and Slc2a4/GLUT4 expression via ESR1-mediated activation of CEBPA. Mol. Cell. Endocrinol. 2019, 498, 110447. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.S.; Diaczok, D.; Cooke, D.W. Repression of GLUT4 expression by the endoplasmic reticulum stress response in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 2007, 362, 188–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, E.H.; Ko, J.H.; Ahn, C.W.; Lee, H.H.; Shin, J.K.; Chang, S.J.; Park, C.S.; Kang, J.H. In vivo and in vitro application of black soybean peptides in the amelioration of endoplasmic reticulum stress and improvement of insulin resistance. Life Sci. 2010, 86, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Raciti, G.A.; Iadicicco, C.; Ulianich, L.; Vind, B.F.; Gaster, M.; Andreozzi, F.; Longo, M.; Teperino, R.; Ungaro, P.; Di Jeso, B.; et al. Glucosamine-induced endoplasmic reticulum stress affects GLUT4 expression via activating transcription factor 6 in rat and human skeletal muscle cells. Diabetologia 2010, 53, 955–965. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.L.; Giannocco, G.; Furuya, D.T.; Lima, G.A.; Moraes, P.A.; Nachef, S.; Bordin, S.; Britto, L.R.; Nunes, M.T.; Machado, U.F. NF-kappaB, MEF2A, MEF2D and HIF1-a involvement on insulin- and contraction-induced regulation of GLUT4 gene expression in soleus muscle. Mol. Cell. Endocrinol. 2005, 240, 82–93. [Google Scholar] [CrossRef]
- Furuya, D.T.; Poletto, A.C.; Favaro, R.R.; Martins, J.O.; Zorn, T.M.; Machado, U.F. Anti-inflammatory effect of atorvastatin ameliorates insulin resistance in monosodium glutamate-treated obese mice. Metabolism 2010, 59, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Furuya, D.T.; Poletto, A.C.; Freitas, H.S.; Machado, U.F. Inhibition of cannabinoid CB1 receptor upregulates Slc2a4 expression via nuclear factor-κB and sterol regulatory element-binding protein-1 in adipocytes. J. Mol. Endocrinol. 2012, 49, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moraes, P.A.; Yonamine, C.Y.; Pinto Junior, D.C.; Esteves, J.V.D.C.; Machado, U.F.; Mori, R.C. Insulin acutely triggers transcription of Slc2a4 gene: Participation of the AT-rich, E-box and NFKB-binding sites. Life Sci. 2014, 114, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Poletto, A.C.; Furuya, D.T.; David-Silva, A.; Ebersbach-Silva, P.; Santos, C.L.; Corrêa-Giannella, M.L.; Passarelli, M.; Machado, U.F. Oleic and linoleic fatty acids downregulate Slc2a4/GLUT4 expression via NFKB and SREBP1 in skeletal muscle cells. Mol. Cell. Endocrinol. 2015, 401, 65–72. [Google Scholar] [CrossRef]
- Furuya, D.T.; Neri, E.A.; Poletto, A.C.; Anhê, G.F.; Freitas, H.S.; Campello, R.S.; Rebouças, N.A.; Machado, U.F. Identification of nuclear factor-κB sites in the Slc2a4 gene promoter. Mol. Cell. Endocrinol. 2013, 370, 87–95. [Google Scholar] [CrossRef]
- Ebersbach-Silva, P.; Poletto, A.C.; David-Silva, A.; Seraphim, P.M.; Anhê, G.F.; Passarelli, M.; Furuya, D.T.; Machado, U.F. Palmitate-induced Slc2a4/GLUT4 downregulation in L6 muscle cells: Evidence of inflammatory and endoplasmic reticulum stress involvement. Lipids Health Dis. 2018, 17, 64. [Google Scholar] [CrossRef] [Green Version]
- Liong, S.; Lappas, M. Endoplasmic reticulum stress regulates inflammation and insulin resistance in skeletal muscle from pregnant women. Mol. Cell. Endocrinol. 2016, 425, 11–25. [Google Scholar] [CrossRef]
- Lin, M.W.; Chen, C.I.; Cheng, T.T.; Huang, C.C.; Tsai, J.W.; Feng, G.M.; Hwang, T.Z.; Lam, C.F. Prolonged preoperative fasting induces postoperative insulin resistance by ER-stress mediated Glut4 down-regulation in skeletal muscles. Int. J. Med. Sci. 2021, 18, 1189–1197. [Google Scholar] [CrossRef]
- Obafemi, T.O.; Olasehinde, O.R.; Olaoye, O.A.; Jaiyesimi, K.F.; Adewumi, F.D.; Adewale, O.B.; Afolabi, B.A. Metformin/Donepezil combination modulates brain antioxidant status and hippocampal endoplasmic reticulum stress in type 2 diabetic rats. J. Diabetes Metab. Disord. 2020, 19, 499–510. [Google Scholar] [CrossRef]
- Varghese, D.S.; Ali, B.R. Pathological Crosstalk Between Oxidized LDL and ER Stress in Human Diseases: A Comprehensive Review. Front. Cell Dev. Biol. 2021, 9, 674103. [Google Scholar] [CrossRef]
- Devries-Seimon, T.; Li, Y.; Yao, P.M.; Stone, E.; Wang, Y.; Davis, R.J.; Flavell, R.; Tabas, I. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. J. Cell Biol. 2005, 171, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Seimon, T.A.; Nadolski, M.J.; Liao, X.; Magallon, J.; Nguyen, M.; Feric, N.T.; Koschinsky, M.L.; Harkewicz, R.; Witztum, J.L.; Tsimikas, S.; et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010, 12, 467–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, D.; Bai, Y.P.; Gao, H.C.; Wang, X.; Li, L.F.; Zhang, G.G.; Hu, C.P. Ox-LDL induces endothelial cell apoptosis via the LOX-1-dependent endoplasmic reticulum stress pathway. Atherosclerosis 2014, 235, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Tsukano, H.; Gotoh, T.; Endo, M.; Miyata, K.; Tazume, H.; Kadomatsu, T.; Yano, M.; Iwawaki, T.; Kohno, K.; Araki, K. The endoplas-mic reticulum stress-C/EBP homologous protein pathway-mediated apoptosisin macrophages contributes to the instability of atherosclerosis plaques. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1925–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Bi, Y.; Sowers, J.R.; Hetz, C.; Zhang, Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat. Rev. Cardiol. 2021, 18, 499–521. [Google Scholar] [CrossRef]
- Zhou, Y.; Murugan, D.D.; Khan, H.; Huang, Y.; Cheang, W.S. Roles and Therapeutic Implications of Endoplasmic Reticulum Stress and Oxidative Stress in Cardiovascular Diseases. Antioxidants 2021, 10, 1167. [Google Scholar] [CrossRef]
- Zhou, Y.; Wan, X.; Seidel, K.; Zhang, M.; Goodman, J.B.; Seta, F.; Hamburg, N.; Han, J. Aging and Hypercholesterolemia Differentially Affect the Unfolded Protein Response in the Vasculature of ApoE−/− Mice. J. Am. Heart Assoc. 2021, 10, e020441. [Google Scholar] [CrossRef]
- Beriault, D.R.; Sharma, S.; Shi, Y.; Khan, M.I.; Werstuck, G.H. Glucosamine-supplementation promotes endoplasmic reticulum stress, hepatic steatosis and accelerated atherogenesis in apoE-/- mice. Atherosclerosis 2011, 219, 134–140. [Google Scholar] [CrossRef]
- Chong, W.C.; Shastri, M.D.; Peterson, G.M.; Patel, R.P.; Pathinayake, P.S.; Dua, K.; Hansbro, N.G.; Hsu, A.C.; Wark, P.A.; Shukla, S.D.; et al. The complex interplay between endoplasmic reticulum stress and the NLRP3 inflammasome: A potential therapeutic target for inflammatory disorders. Clin. Transl. Immunol. 2021, 10, e1247. [Google Scholar] [CrossRef]
- Zeng, L.; Lu, M.; Mori, K.; Luo, S.; Lee, A.S.; Zhu, Y.; Shyy, J.Y. ATF6 modulates SREBP2-mediated lipogenesis. EMBO J. 2004, 23, 950–958. [Google Scholar] [CrossRef] [Green Version]
- Howarth, D.L.; Lindtner, C.; Vacaru, A.M.; Sachidanandam, R.; Tsedensodnom, O.; Vasilkova, T.; Buettner, C.; Sadler, K.C. Activating transcription factor 6 is necessary and sufficient for alcoholic fatty liver disease in zebrafish. PLoS Genet. 2014, 10, e1004335. [Google Scholar] [CrossRef]
- Xu, X.; Lei, T.; Li, W.; Ou, H. Enhanced cellular cholesterol efflux by naringenin is mediated through inhibiting endoplasmic reticulum stress—ATF6 activity in macrophages. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1472–1482. [Google Scholar] [CrossRef]
- Wang, T.; Zhao, Y.; You, Z.; Li, X.; Xiong, M.; Li, H.; Yan, N. Endoplasmic Reticulum Stress Affects Cholesterol Homeostasis by Inhibiting LXRα Expression in Hepatocytes and Macrophages. Nutrients 2020, 12, 3088. [Google Scholar] [CrossRef]
- Che, X.; Xiao, Q.; Song, W.; Zhang, H.; Sun, B.; Geng, N.; Tao, Z.; Shao, Q.; Pu, J. Protective Functions of Liver X Receptor α in Established Vulnerable Plaques: Involvement of Regulating Endoplasmic Reticulum-Mediated Macrophage Apoptosis and Efferocytosis. J. Am. Heart Assoc. 2021, 10, e018455. [Google Scholar] [CrossRef]
- Amengual, J.; Ogando, Y.; Nikain, C.; Quezada, A.; Qian, K.; Vaisar, T.; Fisher, E.A. Short-Term Acyl-CoA:Cholesterol Acyltransferase Inhibition, Combined with Apoprotein A1 Overexpression, Promotes Atherosclerosis Inflammation Resolution in Mice. Mol. Pharmacol. 2021, 99, 175–183. [Google Scholar] [CrossRef]
- Song, G.; Wu, X.; Zhang, P.; Yu, Y.; Yang, M.; Jiao, P.; Wang, N.; Song, H.; Wu, Y.; Zhang, X.; et al. High-density lipoprotein inhibits ox-LDL-induced adipokine secretion by upregulating SR-BI expression and suppressing ER Stress pathway. Sci. Rep. 2016, 6, 30889. [Google Scholar] [CrossRef] [Green Version]
- Navas-Madroñal, M.; Castelblanco, E.; Camacho, M.; Consegal, M.; Ramirez-Morros, A.; Sarrias, M.R.; Perez, P.; Alonso, N.; Galán, M.; Mauricio, D. Role of the Scavenger Receptor CD36 in Accelerated Diabetic Atherosclerosis. Int. J. Mol. Sci. 2020, 21, 7360. [Google Scholar] [CrossRef]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Dicarbonyl stress, protein glycation and the unfolded protein response. Glycoconj. J. 2021, 38, 331–340. [Google Scholar] [CrossRef]
- Fredrickson, E.K.; Rosenbaum, J.C.; Locke, M.N.; Milac, T.I.; Gardner, R.G. Exposed hydrophobicity is a key determinant of nuclear quality control degradation. Mol. Biol. Cell 2011, 22, 2384–2395. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Kim, S.J.; Zhang, Z.; Lee, Y.C.; Sarkar, C.; Tsai, P.C.; Mukherjee, A.B. RAGE signaling contributes to neuroinflammation in infantile neuronal ceroid lipofuscinosis. FEBS Lett. 2008, 582, 3823–3831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irshad, Z.; Xue, M.; Ashour, A.; Larkin, J.R.; Thornalley, P.J.; Rabbani, N. Activation of the unfolded protein response in high glucose treated endothelial cells is mediated by methylglyoxal. Sci. Rep. 2019, 9, 7889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nourbakhsh, M.; Sharifi, R.; Heydari, N.; Nourbakhsh, M.; Ezzati-Mobasser, S.; Zarrinnahad, H. Circulating TRB3 and GRP78 levels in type 2 diabetes patients: Crosstalk between glucose homeostasis and endoplasmic reticulum stress. J. Endocrinol. Investig. 2021. [Google Scholar] [CrossRef]
- Garay-Sevilla, M.E.; Rojas, A.; Portero-Otin, M.; Uribarri, J. Dietary AGEs as Exogenous Boosters of Inflammation. Nutrients 2021, 13, 2802. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Cai, W.; Pyzik, R.; Goodman, S.; Chen, X.; Zhu, L.; Ramdas, M.; Striker, G.E.; Vlassara, H. Suppression of native defense mechanisms, SIRT1 and PPARγ, by dietary glycoxidants precedes disease in adult humans; relevance to lifestyle-engendered chronic diseases. Amino Acids 2014, 46, 301–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uribarri, J.; Cai, W.; Ramdas, M.; Goodman, S.; Pyzik, R.; Chen, X.; Zhu, L.; Striker, G.E.; Vlassara, H. Restriction of advanced glycation end products improves insulin resistance in human type 2 diabetes: Potential role of AGER1 and SIRT1. Diabetes Care 2011, 34, 1610–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monden, M.; Koyama, H.; Otsuka, Y.; Morioka, T.; Mori, K.; Shoji, T.; Mima, Y.; Motoyama, K.; Fukumoto, S.; Shioi, A.; et al. Receptor for advanced glycation end products regulates adipocyte hypertrophy and insulin sensitivity in mice: Involvement of toll-like receptor 2. Diabetes 2013, 62, 478–489. [Google Scholar] [CrossRef] [Green Version]
- Gaens, K.H.J.; Goossens, G.H.; Niessen, P.M.; van Greevenbroek, M.M.; van der Kallen, C.J.H.; Niessen, H.W.; Rensen, S.S.; Buurman, W.A.; Greve, J.W.M.; Blaak, E.E.; et al. Nε-(carboxymethyl)lysine-receptor for advanced glycation end product axis is a key modulator of obesity-induced dysregulation of adipokine expression and insulin resistance. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1199–1208. [Google Scholar] [CrossRef] [Green Version]
- Chilelli, N.C.; Faggian, A.; Favaretto, F.; Milan, G.; Compagnin, C.; Dassie, F.; Bettini, S.; Roverso, M.; Seraglia, R.; Lapolla, A.; et al. In vitro chronic glycation induces AGEs accumulation reducing insulin-stimulated glucose uptake and increasing GLP1R in adipocytes. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E976–E988. [Google Scholar] [CrossRef]
- Dozio, E.; Vianello, E.; Briganti, S.; Lamont, J.; Tacchini, L.; Schmitz, G.; Romanelli, M.M.C. Expression of the Receptor for Advanced Glycation End Products in Epicardial Fat: Link with Tissue Thickness and Local Insulin Resistance in Coronary Artery Disease. J. Diabetes Res. 2016, 2016, 2327341. [Google Scholar] [CrossRef]
- Dhar, A.; Dhar, I.; Jiang, B.; Desai, K.M.; Wu, L. Chronic methylglyoxal infusion by minipump causes pancreatic beta-cell dysfunction and induces type 2 diabetes in Sprague-Dawley rats. Diabetes 2011, 60, 899–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, K.S.; Pinto, P.R.; Fabre, N.T.; Gomes, D.J.; Thieme, K.; Okuda, L.S.; Iborra, R.T.; Freitas, V.G.; Shimizu, M.H.M.; Teodoro, W.R.; et al. N-acetylcysteine Counteracts Adipose Tissue Macrophage Infiltration and Insulin Resistance Elicited by Advanced Glycated Albumin in Healthy Rats. Front. Physiol. 2017, 8, 723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuda, L.S.; Castilho, G.; Rocco, D.D.; Nakandakare, E.R.; Catanozi, S.; Passarelli, M. Advanced glycated albumin impairs HDL anti-inflammatory activity and primes macrophages for inflammatory response that reduces reverse cholesterol transport. Biochim. Biophys. Acta 2012, 1821, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Iborra, R.T.; Machado-Lima, A.; Castilho, G.; Nunes, V.S.; Abdalla, D.S.; Nakandakare, E.R.; Passarelli, M. Advanced glycation in macrophages induces intracellular accumulation of 7-ketocholesterol and total sterols by decreasing the expression of ABCA-1 and ABCG-1. Lipids Health Dis. 2011, 10, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, D.J.; Velosa, A.P.; Okuda, L.S.; Fusco, F.B.; da Silva, K.S.; Pinto, P.R.; Nakandakare, E.R.; Correa-Giannella, M.L.; Woods, T.; Brimble, M.A.; et al. Glycated albumin induces lipid infiltration in mice aorta independently of DM and RAS local modulation by inducing lipid peroxidation and inflammation. J. Diabetes Complicat. 2016, 30, 1614–1621. [Google Scholar] [CrossRef]
- de Souza, P.R.; Castilho, G.; Paim, B.A.; Machado-Lima, A.; Inada, N.M.; Nakandakare, E.R.; Vercesi, A.E.; Passarelli, M. Inhibition of macrophage oxidative stress prevents the reduction of ABCA-1 transporter induced by advanced glycated albumin. Lipids 2012, 47, 443–450. [Google Scholar] [CrossRef]
- Castilho, G.; Okuda, L.S.; Pinto, R.S.; Iborra, R.T.; Nakandakare, E.R.; Santos, C.X.; Laurindo, F.R.; Passarelli, M. ER stress is associated with reduced ABCA-1 protein levels in macrophages treated with advanced glycated albumin—reversal by a chemical chaperone. Int. J. Biochem. Cell Biol. 2012, 44, 1078–1086. [Google Scholar] [CrossRef]
- Mizuno, T.; Hayashi, H.; Naoi, S.; Sugiyama, Y. Ubiquitination is associated with lysosomal degradation of cell surface-resident ATP-binding cassette transporter A1 (ABCA1) through the endosomal sorting complex required for transport (ESCRT) pathway. Hepatology 2011, 54, 631–643. [Google Scholar] [CrossRef]
- Iborra, R.T.; Machado-Lima, A.; Okuda, L.S.; Pinto, P.R.; Nakandakare, E.R.; Machado, U.F.; Correa-Giannella, M.L.; Pickford, R.; Woods, T.; Brimble, M.A.; et al. AGE-albumin enhances ABCA1 degradation by ubiquitin-proteasome and lysosomal pathways in macrophages. J. Diabetes Complicat. 2018, 32, 1–10. [Google Scholar] [CrossRef]
- Minanni, C.A.; Machado-Lima, A.; Iborra, R.T.; Okuda, L.S.; de Souza Pinto, R.; Santana, M.F.M.; Lira, A.L.A.; Nakandakare, E.R.; Côrrea-Giannella, M.L.C.; Passarelli, M. Persistent Effect of Advanced Glycated Albumin Driving Inflammation and Disturbances in Cholesterol Efflux in Macrophages. Nutrients 2021, 13, 3633. [Google Scholar] [CrossRef] [PubMed]
- Machado-Lima, A.; López-Díez, R.; Iborra, R.T.; Pinto, R.S.; Daffu, G.; Shen, X.; Nakandakare, E.R.; Machado, U.F.; Corrêa-Giannella, M.L.C.; Schmidt, A.M.; et al. RAGE Mediates Cholesterol Efflux Impairment in Macrophages Caused by Human Advanced Glycated Albumin. Int. J. Mol. Sci. 2020, 21, 7265. [Google Scholar] [CrossRef]
- Machado, J.T.; Iborra, R.T.; Fusco, F.B.; Castilho, G.; Pinto, R.S.; Machado-Lima, A.; Nakandakare, E.R.; Seguro, A.C.; Shimizu, M.H.; Catanozi, S.; et al. N-acetylcysteine prevents endoplasmic reticulum stress elicited in macrophages by serum albumin drawn from chronic kidney disease rats and selectively affects lipid transporters, ABCA-1 and ABCG-1. Atherosclerosis 2014, 237, 343–352. [Google Scholar] [CrossRef]
- de Araújo Lira, A.L.; de Fátima Mello Santana, M.; de Souza Pinto, R.; Minanni, C.A.; Iborra, R.T.; de Lima, A.M.S.; Correa-Giannella, M.L.; Passarelli, M.; Queiroz, M.S. Serum albumin modified by carbamoylation impairs macrophage cholesterol efflux in diabetic kidney disease. J. Diabetes Complicat. 2021, 35, 107969. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, C.P.; Xu, K.F.; Mao, X.D.; Lu, Y.B.; Fang, L.; Yang, J.W.; Liu, C. Effect of taurine-conjugated ursodeoxycholic acid on endoplasmic reticulum stress and apoptosis induced by advanced glycation end products in cultured mouse podocytes. Am. J. Nephrol. 2008, 28, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.R.; Lee, K.W. Methylglyoxal-Derived Advanced Glycation End Product (AGE4)-Induced Apoptosis Leads to Mitochondrial Dysfunction and Endoplasmic Reticulum Stress through the RAGE/JNK Pathway in Kidney Cells. Int. J. Mol. Sci. 2021, 22, 6530. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Y.; Pan, S.; Qiu, C.; Jia, H.; Wang, Y.; Zhu, H. Activation of RAGE-dependent endoplasmic reticulum stress associates with exacerbated postmyocardial infarction ventricular arrhythmias in diabetes. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E539–E550. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhu, H.; Ma, Y.; Tang, Z.; Zhao, N.; Wang, Y.; Pan, S. AGEs exacerbates coronary microvascular dysfunction in NoCAD by activating endoplasmic reticulum stress-mediated PERK signaling pathway. Metabolism 2021, 117, 154710. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, M.; Wang, S.; Liang, B.; Zhao, Z.; Liu, C.; Wu, M.; Choi, H.C.; Lyons, T.J.; Zou, M.H. Activation of AMP-activated protein kinase inhibits oxidized LDL-triggered endoplasmic reticulum stress in vivo. Diabetes 2010, 59, 1386–1396. [Google Scholar] [CrossRef] [Green Version]
- Ravi, R.; Ragavachetty Nagaraj, N.; Subramaniam Rajesh, B. Effect of advanced glycation end product on paraoxonase 2 expression: Its impact on endoplasmic reticulum stress and inflammation in HUVECs. Life Sci. 2020, 246, 117397. [Google Scholar] [CrossRef]
- Gutierrez-Mariscal, F.M.; Cardelo, M.P.; de la Cruz, S.; Alcala-Diaz, J.F.; Roncero-Ramos, I.; Guler, I.; Vals-Delgado, C.; López-Moreno, A.; Luque, R.M.; Delgado-Lista, J.; et al. Reduction in Circulating Advanced Glycation End Products by Mediterranean Diet Is Associated with Increased Likelihood of Type 2 Diabetes Remission in Patients with Coronary Heart Disease: From the Cordioprev Study. Mol. Nutr. Food Res. 2021, 65, e1901290. [Google Scholar] [CrossRef] [PubMed]
- Koksal, A.R.; Verne, G.N.; Zhou, Q. Endoplasmic reticulum stress in biological processing and disease. J. Investig. Med. 2021, 69, 309–315. [Google Scholar] [CrossRef] [PubMed]
- David-Silva, A.; Esteves, J.V.; Morais, M.R.P.T.; Freitas, H.S.; Zorn, T.M.; Correa-Giannella, M.L.; Machado, U.F. Dual SGLT1/SGLT2 Inhibitor Phlorizin Ameliorates Non-Alcoholic Fatty Liver Disease and Hepatic Glucose Production in Type 2 Diabetic Mice. Diabetes Metab. Syndr. Obes. 2020, 13, 739–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlović, N.; Kopsida, M.; Gerwins, P.; Heindryckx, F. Inhibiting P2Y12 in Macrophages Induces Endoplasmic Reticulum Stress and Promotes an Anti-Tumoral Phenotype. Int. J. Mol. Sci. 2020, 21, 8177. [Google Scholar] [CrossRef]
- Mastrocola, R.; Nigro, D.; Chiazza, F.; Medana, C.; Dal Bello, F.; Boccuzzi, G.; Collino, M.; Aragno, M. Fructose-derived advanced glycation end-products drive lipogenesis and skeletal muscle reprogramming via SREBP-1c dysregulation in mice. Free Radic. Biol. Med. 2016, 91, 224–235. [Google Scholar] [CrossRef]
- Takeuchi, M.; Sakasai-Sakai, A.; Takata, T.; Takino, J.I.; Koriyama, Y.; Kikuchi, C.; Furukawa, A.; Nagamine, K.; Hori, T.; Matsunaga, T. Intracellular Toxic AGEs (TAGE) Triggers Numerous Types of Cell Damage. Biomolecules 2021, 11, 387. [Google Scholar] [CrossRef]
- Litwinowicz, K.; Waszczuk, E.; Gamian, A. Advanced Glycation End-Products in Common Non-Infectious Liver Diseases: Systematic Review and Meta-Analysis. Nutrients 2021, 13, 3370. [Google Scholar] [CrossRef]
- Pereira, E.N.G.D.S.; Paula, D.P.; de Araujo, B.P.; da Fonseca, M.J.M.; Diniz, M.F.H.S.; Daliry, A.; Griep, R.H. Advanced glycation end product: A potential biomarker for risk stratification of non-alcoholic fatty liver disease in ELSA-Brasil study. World J. Gastroenterol. 2021, 27, 4913–4928. [Google Scholar] [CrossRef]
- Ziolkowska, S.; Binienda, A.; Jabłkowski, M.; Szemraj, J.; Czarny, P. The Interplay between Insulin Resistance, Inflammation, Oxidative Stress, Base Excision Repair and Metabolic Syndrome in Nonalcoholic Fatty Liver Disease. J. Mol. Sci. 2021, 22, 11128. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Liu, S.; Klionsky, D.J.; Lip, G.Y.H.; Tuomilehto, J.; Kavalakatt, S.; Pereira, D.M.; Samali, A.; Ren, J. ER stress in obesity pathogenesis and management. Trends Pharmacol. Sci. 2021, 7, S0165-6147(21)00228-5. [Google Scholar] [CrossRef]
Figure 1.
Advanced glycation end products (AGE)-induced regulation of glycemic homeostasis and glycemic homeostasis: participation of inflammatory and endoplasmic reticulum stress. In adipose and muscle tissues, AGE/CML binding to AGER and TLR activate the ER-stress response, including increased expression and/or activation of BIP, ENPL, XBP1, DDIT3, IRE1 and TRAF2. XBP1 and BIP activate ATF6, which is a repressor of the transcription factor PRGC1A and MEF2A (enhancers of Slc2a4 gene), thus decreasing Slc2a4 expression. On the other, IRE1/TRAF2 activation overlap the inflammatory pathway phosphorylating IKKA/B, and promoting NFKB dissociation and migration into the nucleus, where it plays a potent repressor effect upon Slc2a4 gene expression. Besides, intracellular MGO can contribute to the increase in the activity of proinflammatory cytokines, reinforcing this pathway. These effects on the Slc2a4 gene expression decreases GLUT4 synthesis and plasma membrane content, eventually decreasing insulin-induced glucose uptake, and establishing IR. In turn, IR leads to hyperglycemia, which can cause T2D or worsen preexisting DM. Symbols are: AGE, advanced glycated end product; AGER, advanced glycation end product-specific receptor, alias RAGE; ATF6, cyclic AMP-dependent transcription factor ATF; BIP, endoplasmic reticulum chaperone BIP, alias GRP78; CEBPA, CCAAT enhancer binding protein alpha; CML, carboxymethyl-lysine; DM, diabetes mellitus; DDIT3, DNA damage-inducible transcript 3 protein, alias CHOP10/GADD153; DM, diabetes mellitus; ENPL, endoplasmin, alias GRP94; ER, endoplasmic reticulum; GLUT4, solute carrier family 2, facilitated glucose transporter member 4; IKKA/B, inhibitor of nuclear factor kappa-B kinase subunits alpha/beta; IR, insulin resistance; IRE1, inositol requiring enzyme-1; MGO, methylglyoxal; MEF2A, myocyte enhancer factor 2A; NFKB, nuclear factor NF-kappa-B; PRGC1A, peroxisome proliferator-activated receptor gamma coactivator 1-alpha, alias PGC1A; Slc2a4, solute carrier family 2 member 4 gene; T2D, type 2 diabetes mellitus; TLR, toll-like receptors; TRAF2, TNF receptor-associated factor 2; XBP1, X-box-binding protein 1. Names and symbols of proteins are in accordance with the UNIPROT database (https://www.uniprot.org/, accessed on 2 November 2021). Parts of the figure were drawn from Servier Medical Art (https://smart.servier.com/, accessed on 2 November 2021).
Figure 1.
Advanced glycation end products (AGE)-induced regulation of glycemic homeostasis and glycemic homeostasis: participation of inflammatory and endoplasmic reticulum stress. In adipose and muscle tissues, AGE/CML binding to AGER and TLR activate the ER-stress response, including increased expression and/or activation of BIP, ENPL, XBP1, DDIT3, IRE1 and TRAF2. XBP1 and BIP activate ATF6, which is a repressor of the transcription factor PRGC1A and MEF2A (enhancers of Slc2a4 gene), thus decreasing Slc2a4 expression. On the other, IRE1/TRAF2 activation overlap the inflammatory pathway phosphorylating IKKA/B, and promoting NFKB dissociation and migration into the nucleus, where it plays a potent repressor effect upon Slc2a4 gene expression. Besides, intracellular MGO can contribute to the increase in the activity of proinflammatory cytokines, reinforcing this pathway. These effects on the Slc2a4 gene expression decreases GLUT4 synthesis and plasma membrane content, eventually decreasing insulin-induced glucose uptake, and establishing IR. In turn, IR leads to hyperglycemia, which can cause T2D or worsen preexisting DM. Symbols are: AGE, advanced glycated end product; AGER, advanced glycation end product-specific receptor, alias RAGE; ATF6, cyclic AMP-dependent transcription factor ATF; BIP, endoplasmic reticulum chaperone BIP, alias GRP78; CEBPA, CCAAT enhancer binding protein alpha; CML, carboxymethyl-lysine; DM, diabetes mellitus; DDIT3, DNA damage-inducible transcript 3 protein, alias CHOP10/GADD153; DM, diabetes mellitus; ENPL, endoplasmin, alias GRP94; ER, endoplasmic reticulum; GLUT4, solute carrier family 2, facilitated glucose transporter member 4; IKKA/B, inhibitor of nuclear factor kappa-B kinase subunits alpha/beta; IR, insulin resistance; IRE1, inositol requiring enzyme-1; MGO, methylglyoxal; MEF2A, myocyte enhancer factor 2A; NFKB, nuclear factor NF-kappa-B; PRGC1A, peroxisome proliferator-activated receptor gamma coactivator 1-alpha, alias PGC1A; Slc2a4, solute carrier family 2 member 4 gene; T2D, type 2 diabetes mellitus; TLR, toll-like receptors; TRAF2, TNF receptor-associated factor 2; XBP1, X-box-binding protein 1. Names and symbols of proteins are in accordance with the UNIPROT database (https://www.uniprot.org/, accessed on 2 November 2021). Parts of the figure were drawn from Servier Medical Art (https://smart.servier.com/, accessed on 2 November 2021).

Figure 2.
Advanced glycation end products-induced atherogenesis and CVD development: participation of inflammatory and endoplasmic reticulum stress. Glycated LDL reaches the arterial intima inducing circulating monocyte chemotaxis. Differentiated macrophages take up modified LDL by the scavenger receptors which induces intracellular cholesterol accumulation. In addition, advanced glycated albumin (AGE-albumin), modified in blood circulation or in the arterial intima is taken up by AGER. This induces the generation of ROS by NOX4 and mitochondrial respiratory chain activation. Moreover, AGEs induce inflammation and ER stress, with increased expression of BIP, ENPL, ATF6, EIF2AK3 and DDIT3 that relates to ABCA1 degradation by the ubiquitin-proteasomal and lysosomal systems. Ultimately, a dramatic decrease in ABCA1 protein content in macrophage foam cells is observed, leading to the reduction in the APOA1 and pre-beta HDL-mediated cholesterol efflux. Although AGEs do not alter ABCA1 mRNA, they reduce the gene expression of ABCG1, by an LXR-dependent mechanism, then compromising the HDL-mediated cholesterol efflux. The accumulation of cholesterol and oxysterols perpetuates the generation of ROS, inflammatory cytokines and AGEs, which culminates in a necrotic core formation, with apoptosis, pyroptosis, plaque instability and rupture. The deleterious effects of AGEs on macrophage homeostasis damage the cholesterol flux to the liver by the reverse cholesterol transport contributing to the independent relation of AGEs with CVD. Symbols are: ABCA1, phospholipid-transporting ATPase ABCA1; ABCG1, ATP binding cassette transporter G-1 AGE, advanced end product; AGER, advanced glycation end product-specific receptor, alias RAGE; APOA1, apolipoprotein A-1; ATF6, cyclic AMP-dependent transcription factor ATF-6; BIP, endoplasmic reticulum chaperone BIP, alias GRP78; CVD, cardiovascular disease; DDIT3, DNA damage-inducible transcript 3 protein, alias CHOP10/GADD153; EIF2AK3, eukaryotic translation initiation factor 2-alpha kinase 3, alias PERK; ENPL, endoplasmin, alias GRP94; HDL, high-density lipoprotein; LDL, low-density lipoprotein; LXR, liver X receptor; NOX4, NADPH oxidase 4; ROS, reactive oxygen species. Names and symbols of proteins are in accordance with the UNIPROT database (https://www.uniprot.org/, accessed on 2 November 2021). Parts of the figure were drawn from Servier Medical Art (https://smart.servier.com/, accessed on 2 November 2021).
Figure 2.
Advanced glycation end products-induced atherogenesis and CVD development: participation of inflammatory and endoplasmic reticulum stress. Glycated LDL reaches the arterial intima inducing circulating monocyte chemotaxis. Differentiated macrophages take up modified LDL by the scavenger receptors which induces intracellular cholesterol accumulation. In addition, advanced glycated albumin (AGE-albumin), modified in blood circulation or in the arterial intima is taken up by AGER. This induces the generation of ROS by NOX4 and mitochondrial respiratory chain activation. Moreover, AGEs induce inflammation and ER stress, with increased expression of BIP, ENPL, ATF6, EIF2AK3 and DDIT3 that relates to ABCA1 degradation by the ubiquitin-proteasomal and lysosomal systems. Ultimately, a dramatic decrease in ABCA1 protein content in macrophage foam cells is observed, leading to the reduction in the APOA1 and pre-beta HDL-mediated cholesterol efflux. Although AGEs do not alter ABCA1 mRNA, they reduce the gene expression of ABCG1, by an LXR-dependent mechanism, then compromising the HDL-mediated cholesterol efflux. The accumulation of cholesterol and oxysterols perpetuates the generation of ROS, inflammatory cytokines and AGEs, which culminates in a necrotic core formation, with apoptosis, pyroptosis, plaque instability and rupture. The deleterious effects of AGEs on macrophage homeostasis damage the cholesterol flux to the liver by the reverse cholesterol transport contributing to the independent relation of AGEs with CVD. Symbols are: ABCA1, phospholipid-transporting ATPase ABCA1; ABCG1, ATP binding cassette transporter G-1 AGE, advanced end product; AGER, advanced glycation end product-specific receptor, alias RAGE; APOA1, apolipoprotein A-1; ATF6, cyclic AMP-dependent transcription factor ATF-6; BIP, endoplasmic reticulum chaperone BIP, alias GRP78; CVD, cardiovascular disease; DDIT3, DNA damage-inducible transcript 3 protein, alias CHOP10/GADD153; EIF2AK3, eukaryotic translation initiation factor 2-alpha kinase 3, alias PERK; ENPL, endoplasmin, alias GRP94; HDL, high-density lipoprotein; LDL, low-density lipoprotein; LXR, liver X receptor; NOX4, NADPH oxidase 4; ROS, reactive oxygen species. Names and symbols of proteins are in accordance with the UNIPROT database (https://www.uniprot.org/, accessed on 2 November 2021). Parts of the figure were drawn from Servier Medical Art (https://smart.servier.com/, accessed on 2 November 2021).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Passarelli, M.; Machado, U.F.F. AGEs-Induced and Endoplasmic Reticulum Stress/Inflammation-Mediated Regulation of GLUT4 Expression and Atherogenesis in Diabetes Mellitus. Cells 2022, 11, 104. https://doi.org/10.3390/cells11010104
AMA Style
Passarelli M, Machado UFF. AGEs-Induced and Endoplasmic Reticulum Stress/Inflammation-Mediated Regulation of GLUT4 Expression and Atherogenesis in Diabetes Mellitus. Cells. 2022; 11(1):104. https://doi.org/10.3390/cells11010104
Chicago/Turabian StylePassarelli, Marisa, and Ubiratan Fabres Fabres Machado. 2022. "AGEs-Induced and Endoplasmic Reticulum Stress/Inflammation-Mediated Regulation of GLUT4 Expression and Atherogenesis in Diabetes Mellitus" Cells 11, no. 1: 104. https://doi.org/10.3390/cells11010104
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.