
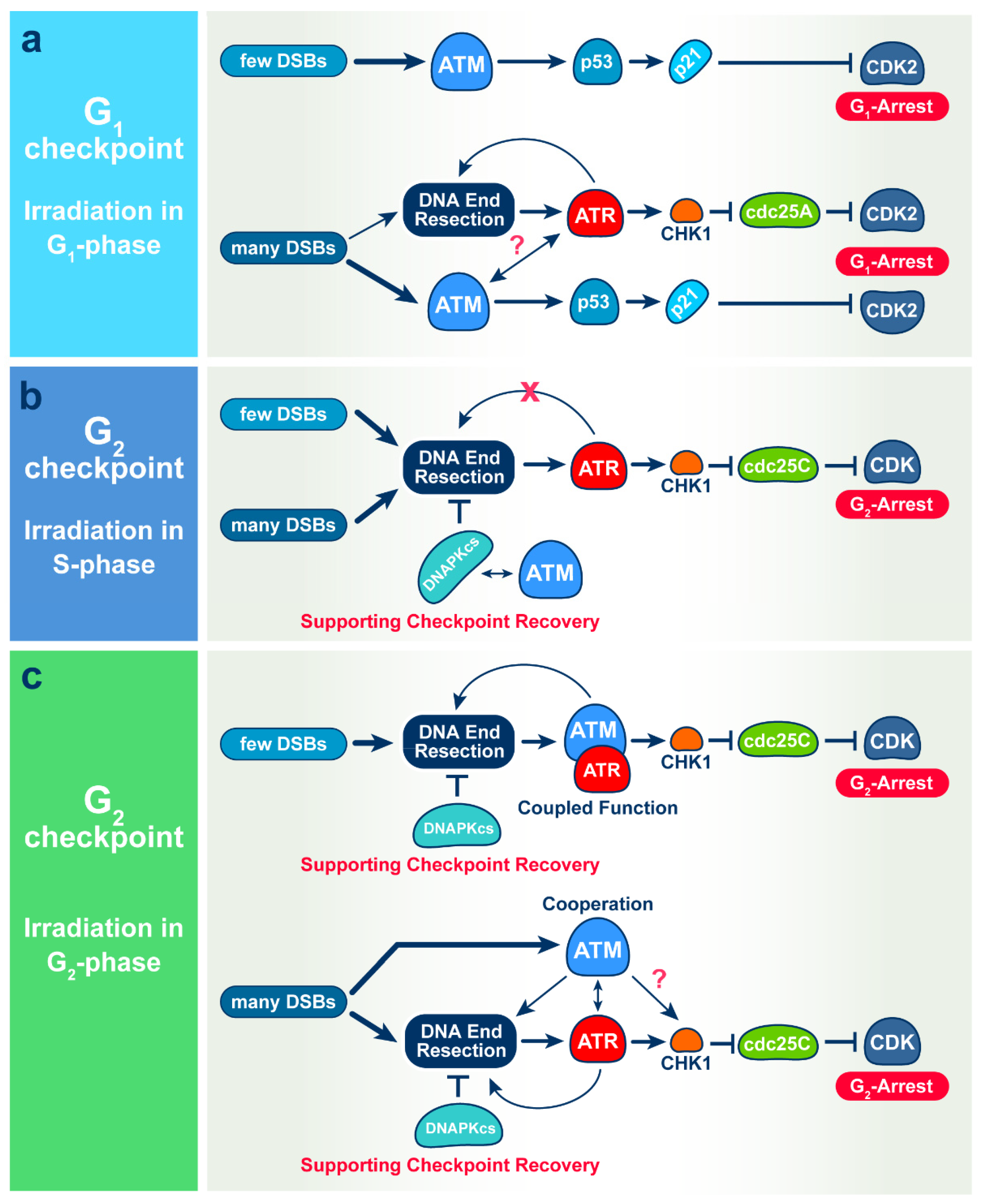
Shift in G1-Checkpoint from ATM-Alone to a Cooperative ATM Plus ATR Regulation with Increasing Dose of Radiation

 ,
, 
Abstract
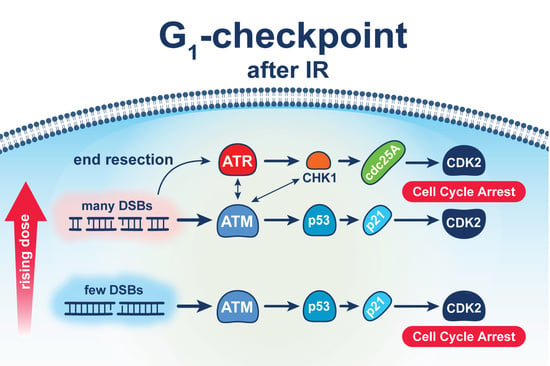
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Irradiation
2.2. Pyronin and Ki67 Staining
2.3. G1-Checkpoint Determination
2.4. Treatment of Cells with Kinase Inhibitors
2.5. Analysis of DNA End Resection at DSBs by Flow Cytometry
2.6. Indirect Immunofluorescence (IF) and Image Analysis
2.7. Western Blotting
2.8. Statistical Analyses
3. Results
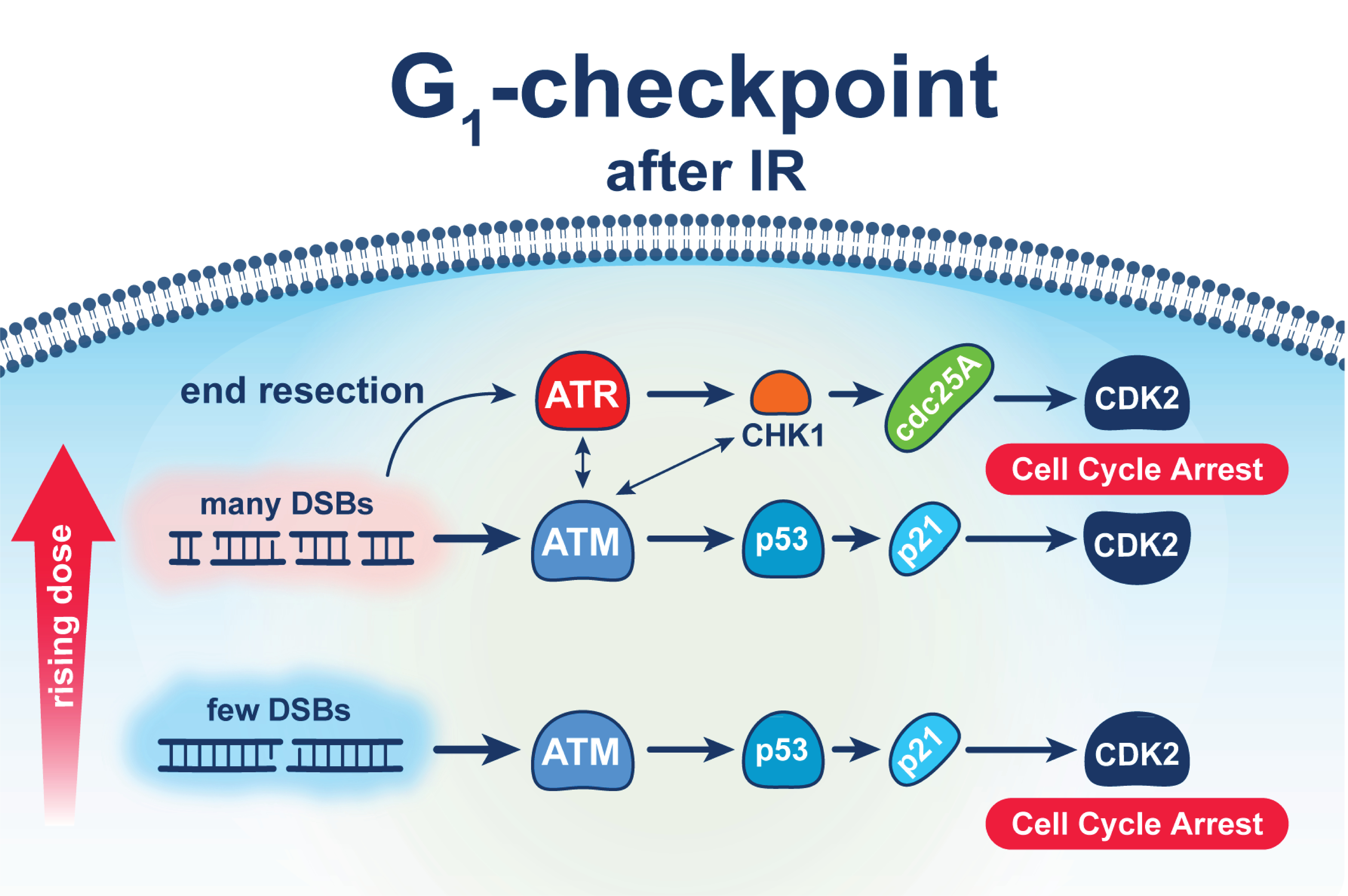
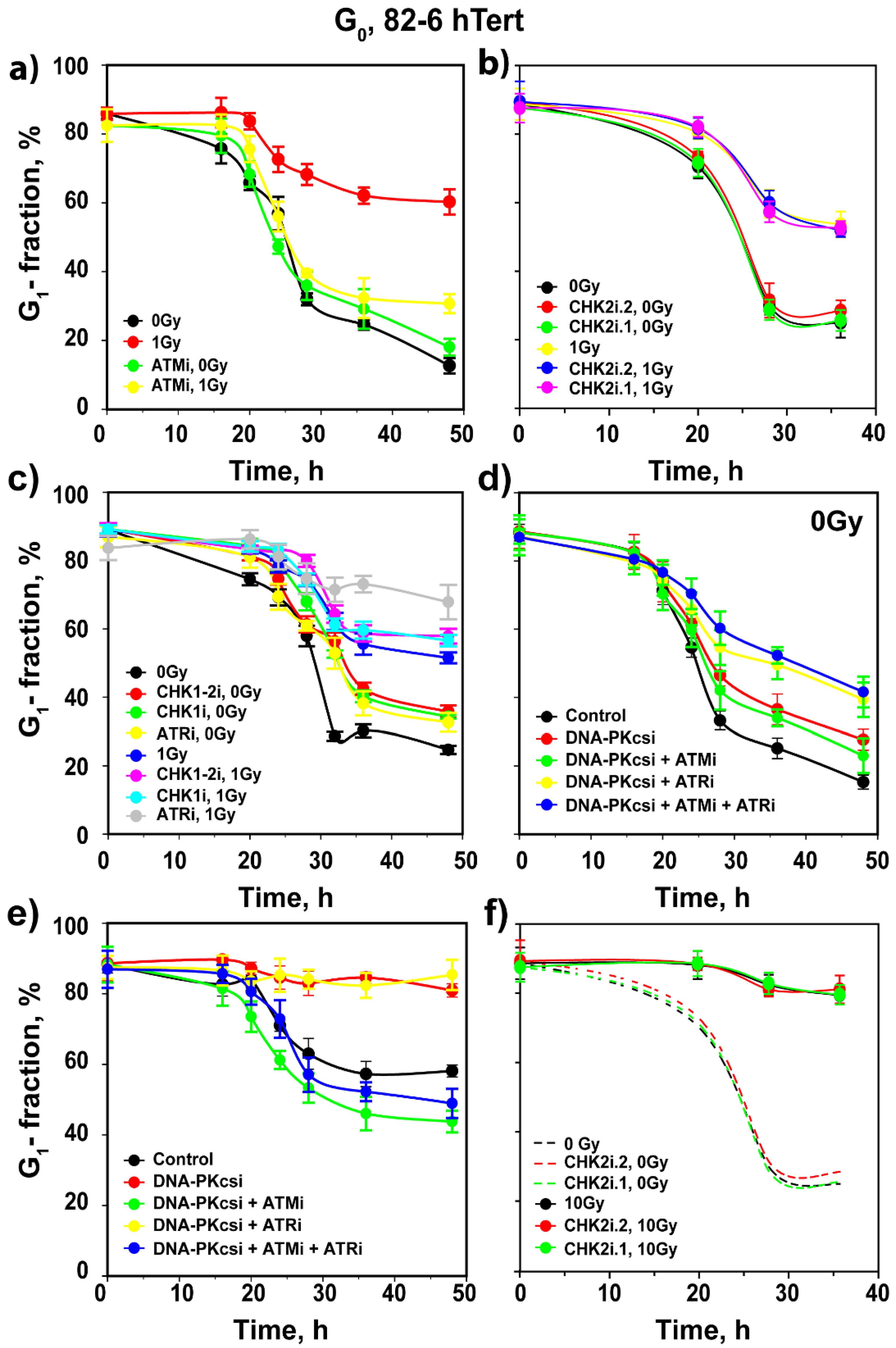
3.1. ATM Is the Main Regulator of G1 Checkpoint in Cells Irradiated in G1 Phase
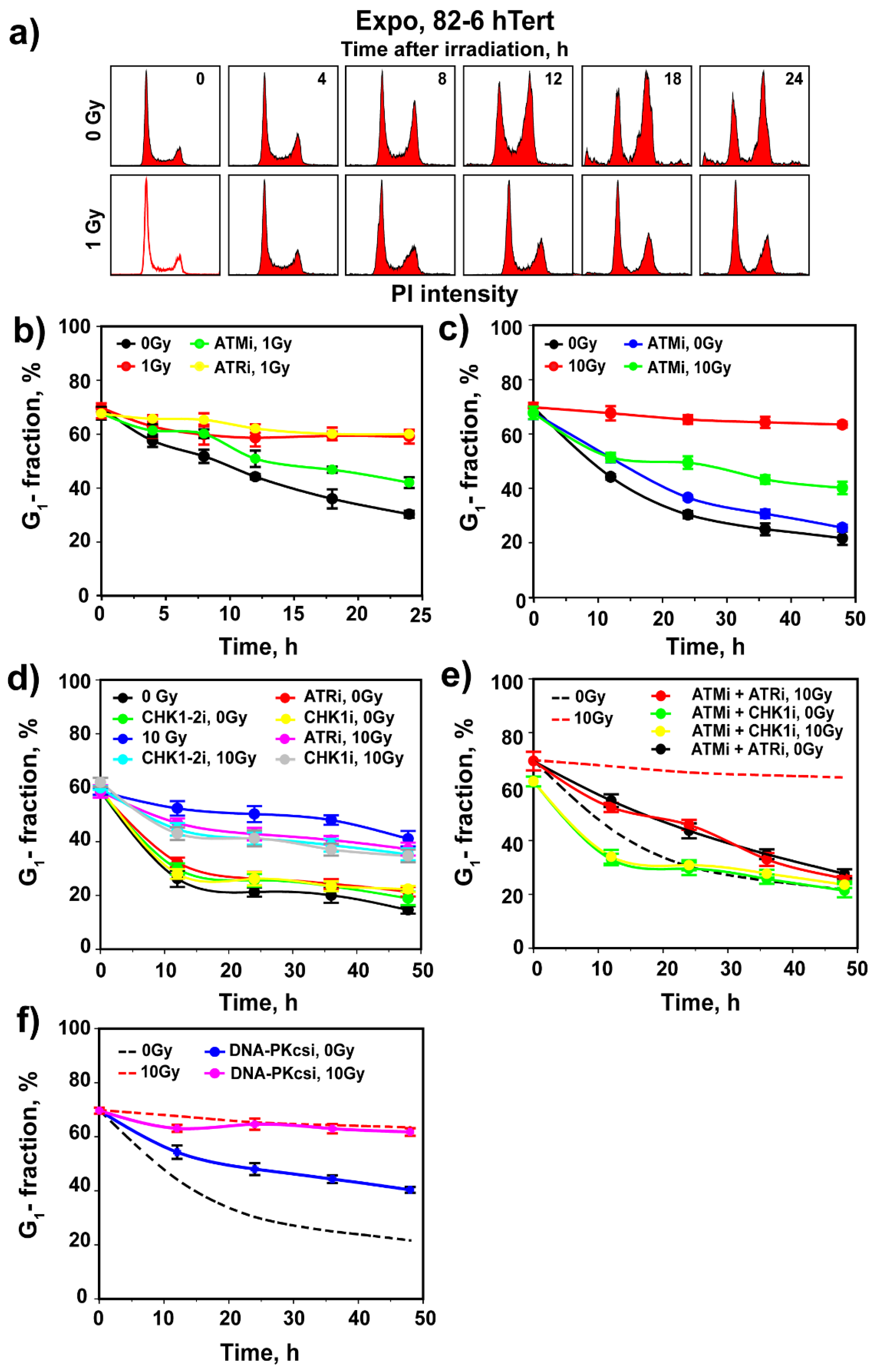
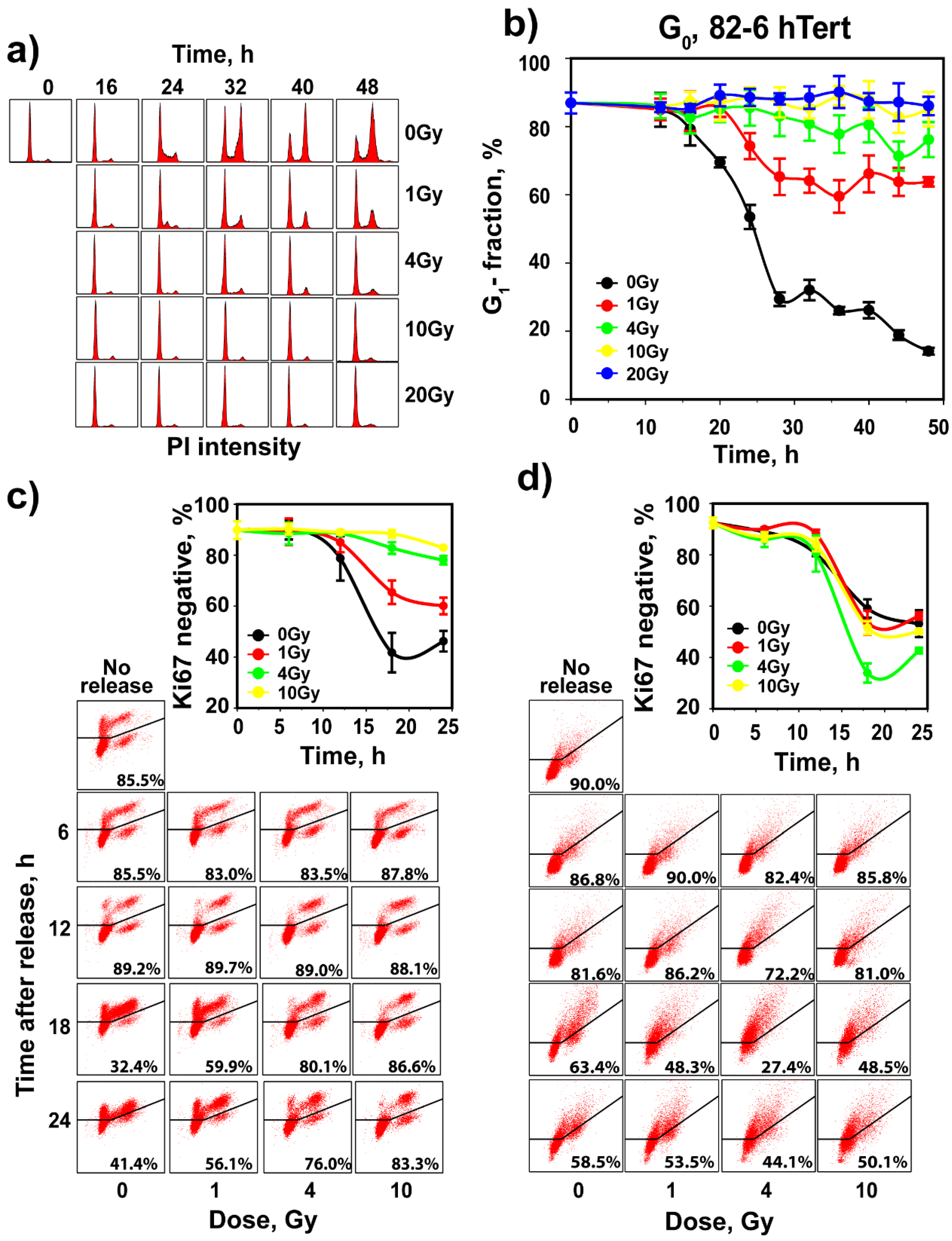
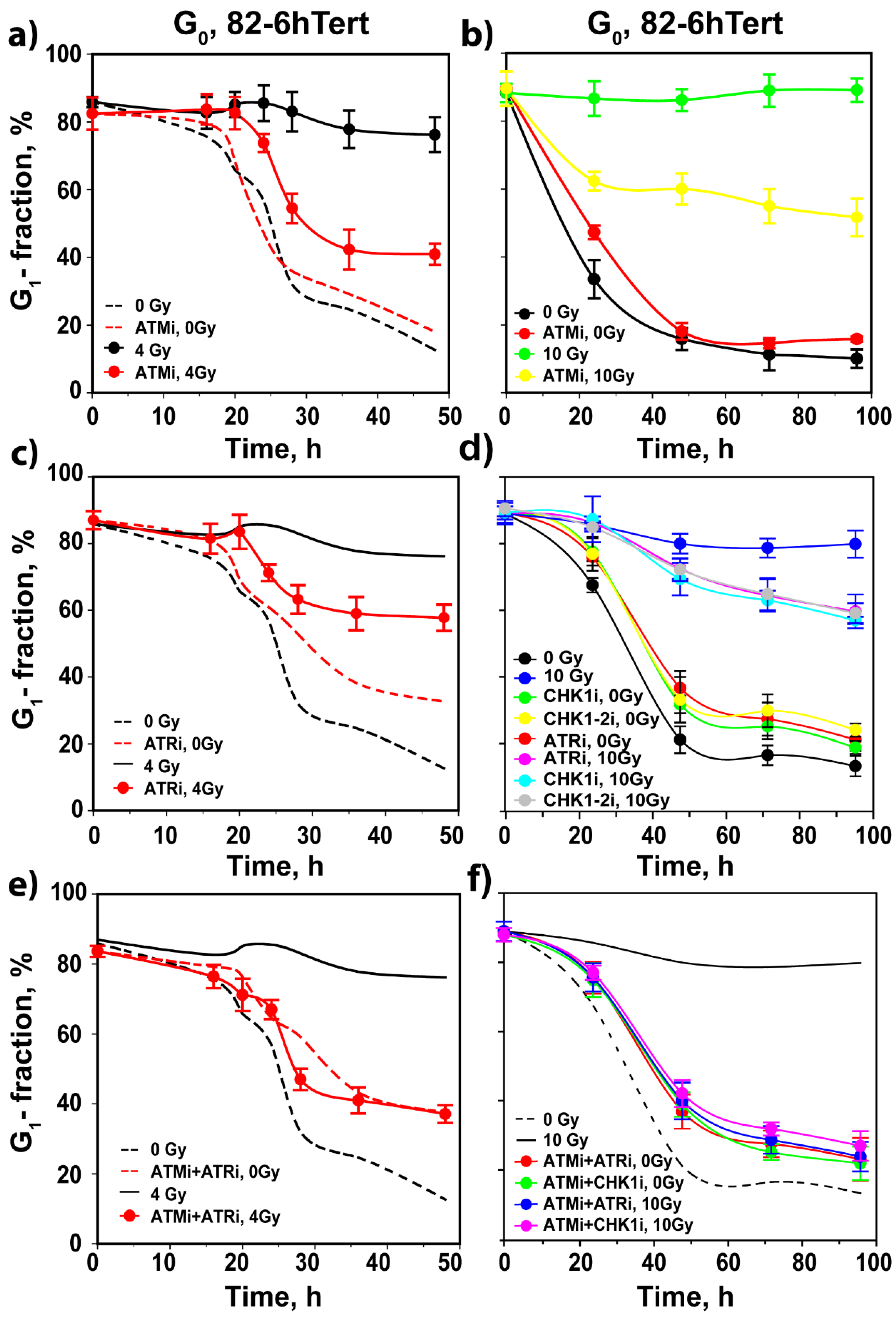
3.2. Regulation of G1 Checkpoint in G0-Irradiated Cells
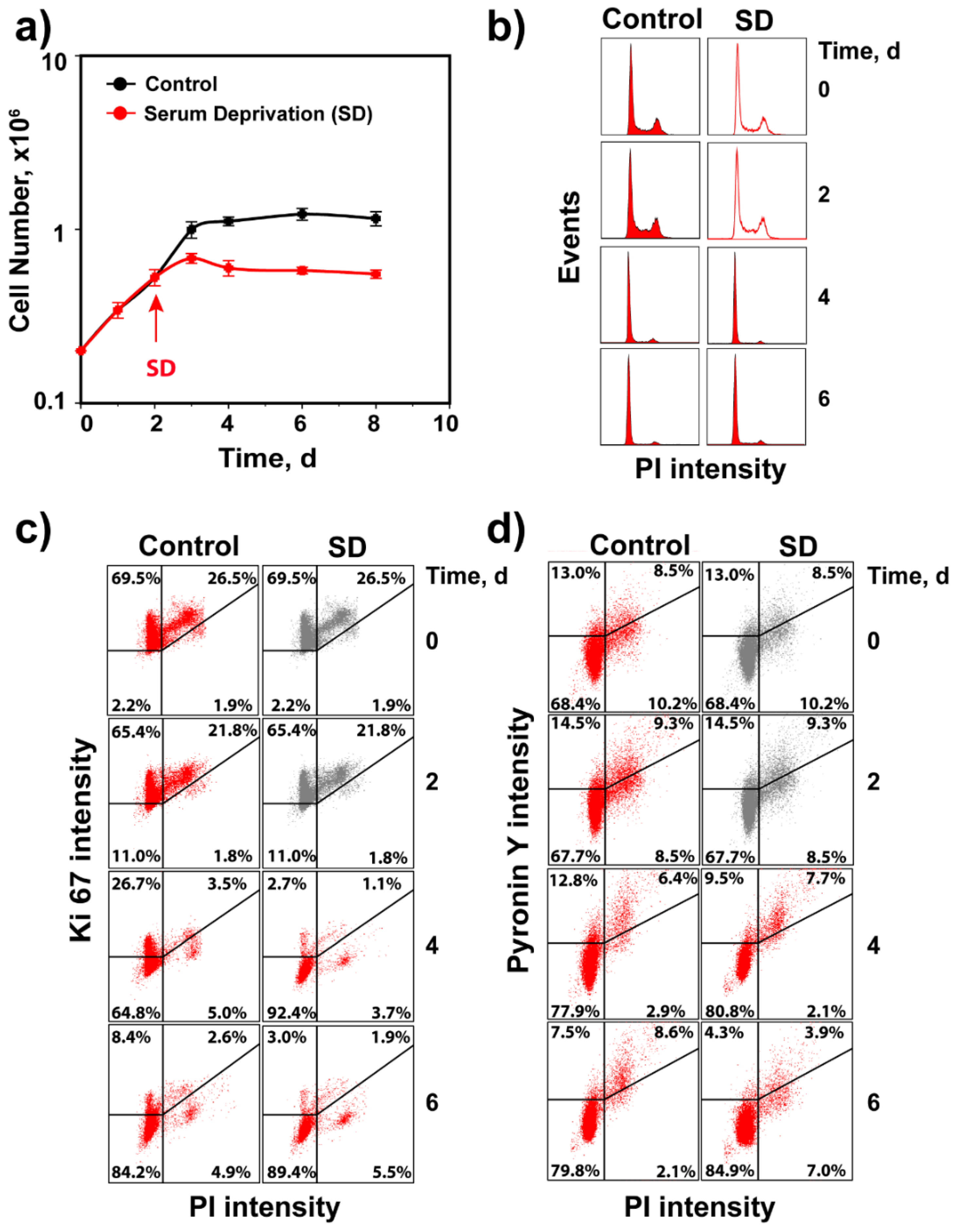
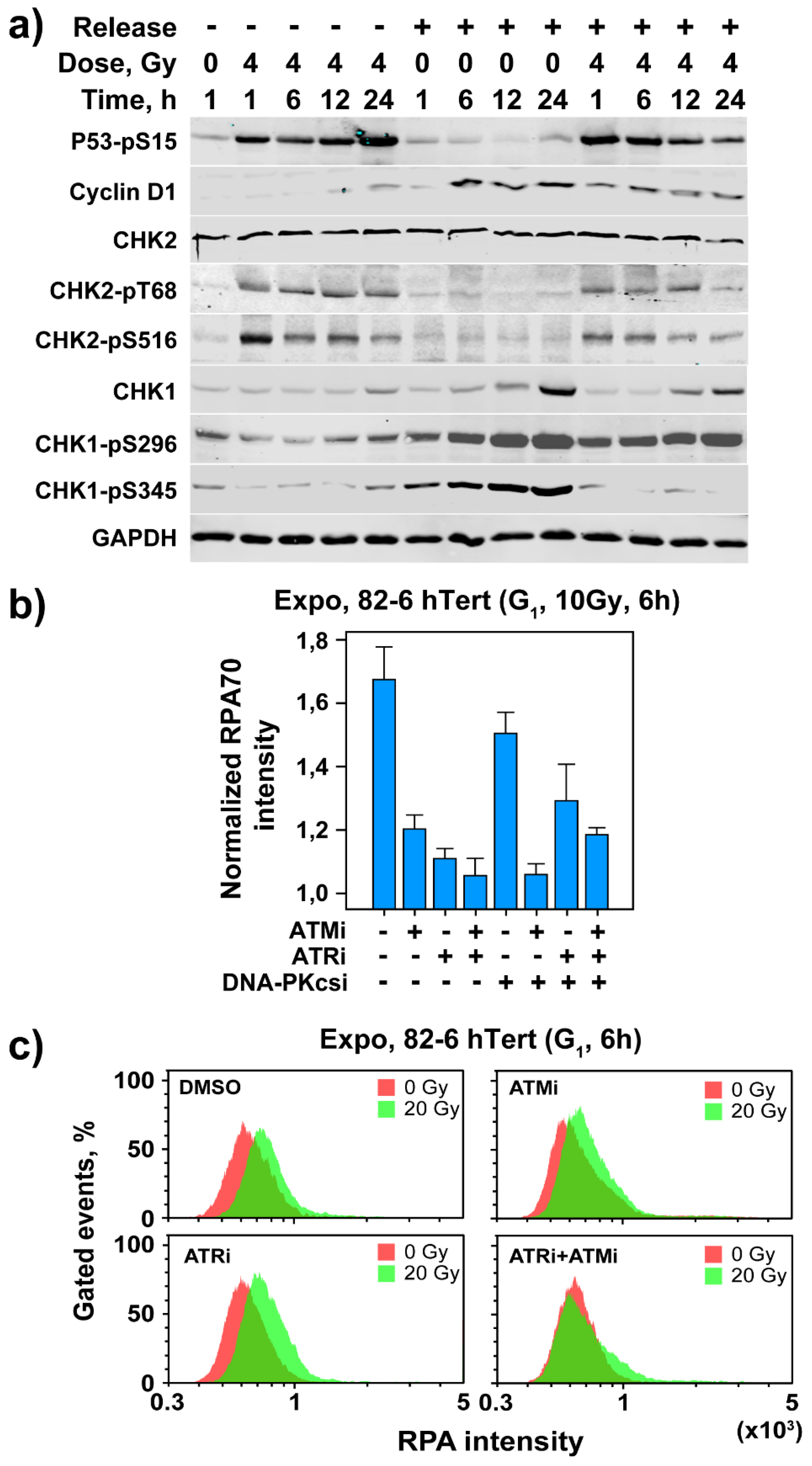
3.3. Activation of Cell Cycle and Checkpoint Proteins in Growth-Stimulated G0/G1 Cells
3.4. Resection in G1-Phase Activates ATR at High IR Doses
4. Discussion
4.1. Variable Functional Integration of ATM, ATR and DNA-PKcs in Checkpoint Regulation
4.2. Contributions of ATM, ATR and DNA-PKcs to the Checkpoint and DSB Processing throughout the Cell Cycle in Normal and Cancer Cell Lines
4.3. Putting Our Observations in the Context of the G1-Checkpoint Background
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferguson, D.O.; Alt, F.W. DNA Double Strand Break Repair and Chromosomal Translocation: Lessons from Animal Models. Oncogene 2001, 20, 5572–5579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliakis, G.; Mladenov, E.; Mladenova, V. Necessities in the Processing of DNA Double Strand Breaks and Their Effects on Genomic Instability and Cancer. Cancers 2019, 11, 1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA Double-Strand-Break Repair in Higher Eukaryotes and Its Role in Genomic Instability and Cancer: Cell Cycle and Proliferation-Dependent Regulation. Semin. Cancer Biol. 2016, 37–38, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA Double-Strand Break Repair as Determinant of Cellular Radiosensitivity to Killing and Target in Radiation Therapy. Front. Oncol. 2013, 3, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.P.; Bartek, J. The DNA-Damage Response in Human Biology and Disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.B.; Elledge, S.J. The DNA Damage Response: Putting Checkpoints in Perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Elledge, S.J. The DNA Damage Response: Ten Years After. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef]
- Hustedt, N.; Durocher, D. The Control of DNA Repair by the Cell Cycle. Nat. Cell Biol. 2017, 19, 1–9. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN Complex for ATM Activation by DNA Damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [Green Version]
- Girard, P.-M.; Riballo, E.; Begg, A.C.; Waugh, A.; Jeggo, P.A. Nbs1 Promotes ATM Dependent Phosphorylation Events Including those Required for G2/S arrest. Oncogene 2002, 21, 4191–4199. [Google Scholar] [CrossRef] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiloh, Y. ATM: Expanding Roles as a Chief Guardian of Genome Stability. Exp. Cell Res. 2014, 329, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Menolfi, D.; Zha, S. ATM, ATR and DNA-PKcs Kinases-the Lessons from the Mouse Models: Inhibition not Equal Deletion. Cell Biosci. 2020, 10, 8. [Google Scholar] [CrossRef]
- Caron, P.; Choudjaye, J.; Clouaire, T.; Bugler, B.; Daburon, V.; Aguirrebengoa, M.; Mangeat, T.; Iacovoni, J.S.; Alvarez-Quilon, A.; Cortes-Ledesma, F.; et al. Non-Redundant Functions of ATM and DNA-PKcs in Response to DNA Double-Strand Breaks. Cell Rep. 2015, 13, 1598–1609. [Google Scholar] [CrossRef] [Green Version]
- Iliakis, G.; Wang, Y.; Guan, J.; Wang, H. DNA Damage Checkpoint Control in Cells Exposed to Ionizing Radiation. Oncogene 2003, 22, 5834–5847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mladenov, E.; Fan, X.; Paul-Konietzko, K.; Soni, A.; Iliakis, G. DNA-PKcs and ATM Epistatically Suppress DNA End Resection and Hyperactivation of ATR-Dependent G2-Checkpoint in S-Phase Irradiated Cells. Sci. Rep. 2019, 9, 14597. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Fan, X.; Dueva, R.; Soni, A.; Iliakis, G. Radiation-Dose-Dependent Functional Synergisms between ATM, ATR and DNA-PKcs in Checkpoint Control and Resection in G2-Phase. Sci. Rep. 2019, 9, 8255. [Google Scholar] [CrossRef] [Green Version]
- Mladenova, V.; Mladenov, E.; Scholz, M.; Stuschke, M.; Iliakis, G. Strong Shift to ATR-Dependent Regulation of the G2-Checkpoint after Exposure to High-LET Radiation. Life 2021, 11, 560. [Google Scholar] [CrossRef]
- Bartek, J.; Lukas, J. Mammalian G1- and S-Phase Checkpoints in Response to DNA Damage. Curr. Opin. Cell Biol. 2001, 13, 738–747. [Google Scholar] [CrossRef]
- Cappell, S.D.; Chung, M.; Jaimovich, A.; Spencer, S.L.; Meyer, T. Irreversible APC(Cdh1) Inactivation Underlies the Point of No Return for Cell-Cycle Entry. Cell 2016, 166, 167–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappell, S.D.; Mark, K.G.; Garbett, D.; Pack, L.R.; Rape, M.; Meyer, T. EMI1 Switches from Being a Substrate to an Inhibitor of APC/CCDH1 to Start the Cell Cycle. Nature 2018, 558, 313–317. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, P.H. Quiescence: Early Evolutionary Origins and Universality Do Not Imply Uniformity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 3498–3507. [Google Scholar] [CrossRef] [Green Version]
- Pack, L.R.; Daigh, L.H.; Meyer, T. Putting the Brakes on the Cell Cycle: Mechanisms of Cellular Growth Arrest. Curr. Opin. Cell Biol. 2019, 60, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Planas-Silva, M.D.; Weinberg, R.A. The Restriction Point and Control of Cell Proliferation. Curr. Opin. Cell Biol. 1997, 9, 768–772. [Google Scholar] [CrossRef]
- Hume, S.; Dianov, G.L.; Ramadan, K. A Unified Model for the G1/S Cell Cycle Transition. Nucl. Acids Res. 2020, 48, 12483–12501. [Google Scholar] [CrossRef]
- Mladenov, E.; Staudt, C.; Soni, A.; Murmann-Konda, T.; Siemann-Loekes, M.; Iliakis, G. Strong Suppression of Gene Conversion with Increasing DNA Double-Strand Break Load Delimited by 53BP1 and RAD52. Nucl. Acids Res. 2020, 48, 1905–1924. [Google Scholar] [CrossRef] [Green Version]
- Kenny, M.K.; Schlegel, U.; Furneaux, H.; Hurwitz, J. The Role of Human Single-Stranded DNA Binding Protein and Its Individual Subunits in Simian Virus 40 DNA Replication. J. Biol. Chem. 1990, 265, 7693–7700. [Google Scholar] [CrossRef]
- Blasina, A.; Hallin, J.; Chen, E.; Arango, M.E.; Kraynov, E.; Register, J.; Grant, S.; Ninkovic, S.; Chen, P.; Nichols, T.; et al. Breaching the DNA Damage Checkpoint via PF-00477736, a Novel Small-Molecule Inhibitor of Checkpoint Kinase 1. Mol. Cancer Ther. 2008, 7, 2394–2404. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.A.; Parsels, L.A.; Zhao, L.; Parsels, J.D.; Davis, M.A.; Hassan, M.C.; Arumugarajah, S.; Hylander-Gans, L.; Morosini, D.; Simeone, D.M.; et al. Mechanism of Radiosensitization by the Chk1/2 Inhibitor AZD7762 Involves Abrogation of the G2 Checkpoint and Inhibition of Homologous Recombinational DNA Repair. Cancer Res. 2010, 70, 4972–4981. [Google Scholar] [CrossRef] [Green Version]
- Magin, S.; Meher, P.K.; Iliakis, G. Nucleoside Analogs Radiosensitize G0 Cells by Activating DNA End Resection and Alternative End-Joining. Radiat. Res. 2021, 195, 412–426. [Google Scholar] [CrossRef]
- Windhofer, F.; Wu, W.; Wang, M.; Singh, S.K.; Saha, J.; Rosidi, B.; Iliakis, G. Marked Dependence on Growth State of Backup Pathways of NHEJ. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G. Radiation Induced Potentially Lethal Damage: DNA Lesions Susceptible to Fixation. Int. J. Radiat. Biol. 1988, 53, 541–584. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G. Backup Pathways of NHEJ in Cells of Higher Eukaryotes: Cell Cycle Dependence. Radiother. Oncol. 2009, 92, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Wu, W.; Zhang, L.; Klammer, H.; Wang, M.; Iliakis, G. Widespread Dependence of Backup NHEJ on Growth State: Ramifications for the Use of DNA-PK Inhibitors. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 540–548. [Google Scholar] [CrossRef]
- Singh, S.K.; Bednar, T.; Zhang, L.; Wu, W.; Mladenov, E.; Iliakis, G. Inhibition of B-NHEJ in Plateau-Phase Cells Is Not a Direct Consequence of Suppressed Growth Factor Signaling. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, e237–e243. [Google Scholar] [CrossRef] [PubMed]
- Dueva, R.; Iliakis, G. Alternative Pathways of Non-Homologous End Joining (NHEJ) in Genomic Instability and Cancer. Transl. Cancer Res. 2013, 2, 163–177. [Google Scholar]
- Gerdes, J.; Schwab, U.; Lemke, H.; Stein, H. Production of a Mouse Monoclonal Antibody Reactive with a Human Nuclear Antigen Associated with Cell Proliferation. Int. J. Cancer 1983, 31, 13–20. [Google Scholar] [CrossRef]
- Miller, I.; Min, M.; Yang, C.; Tian, C.; Gookin, S.; Carter, D.; Spencer, S.L. Ki67 Is a Graded Rather than a Binary Marker of Proliferation versus Quiescence. Cell Rep. 2018, 24, 1105–1112.e1105. [Google Scholar] [CrossRef] [Green Version]
- Melling, N.; Kowitz, C.M.; Simon, R.; Bokemeyer, C.; Terracciano, L.; Sauter, G.; Izbicki, J.R.; Marx, A.H. High Ki67 Expression Is an Independent Good Prognostic Marker in Colorectal Cancer. J. Clin. Pathol. 2016, 69, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Eddaoudi, A.; Canning, S.L.; Kato, I. Flow Cytometric Detection of G0 in Live Cells by Hoechst 33342 and Pyronin Y Staining. Methods Mol. Biol. 2018, 1686, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, H.M. Flow Cytometric Estimation of DNA and RNA Content in Intact Cells Stained with Hoechst 33342 and Pyronin Y. Cytometry 1981, 2, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agami, R.; Bernards, R. Distinct Initiation and Maintenance Mechanisms Cooperate to Induce G1 Cell Cycle Arrest in Response to DNA Damage. Cell 2000, 102, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Dueva, R.; Iliakis, G. Replication protein A: A Multifunctional Protein with Roles in DNA Replication, Repair and Beyond. NAR Cancer 2020, 2, zcaa022. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Mladenov, E.; Mortoga, S.; Iliakis, G. SCF(SKP2) Regulates APC/C(CDH1)-Mediated Degradation of CTIP to Adjust DNA-End Resection in G2-Phase. Cell Death Dis. 2020, 11, 548. [Google Scholar] [CrossRef] [PubMed]
- Murmann-Konda, T.; Soni, A.; Stuschke, M.; Iliakis, G. Analysis of Chromatid-Break-Repair Detects a Homologous Recombination to Non-Homologous End-Joining Switch with Increasing Load of DNA Double-Strand Breaks. Mutat. Res./Genet. Toxicol. Environ. Mutagenes. 2021, 867, 503372. [Google Scholar] [CrossRef] [PubMed]
- Soni, A.; Murmann-Konda, T.; Siemann-Loekes, M.; Pantelias, G.E.; Iliakis, G. Chromosome Breaks Generated by Low Doses of Ionizing Radiation in G2-Phase Are Processed Exclusively by Gene Conversion. DNA Repair (Amst.) 2020, 89, 102828. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, R.A.; Myler, L.R.; Soniat, M.M.; Makharashvili, N.; Lee, L.; Lees-Miller, S.P.; Finkelstein, I.J.; Paull, T.T. DNA-Dependent Protein Kinase Promotes DNA End Processing by MRN and CtIP. Sci. Adv. 2020, 6, eaay0922. [Google Scholar] [CrossRef] [Green Version]
- Sak, A.; Groneberg, M.; Stuschke, M. DNA-Dependent Protein Kinase: Effect on DSB Repair, G2/M Checkpoint and Mode of Cell Death in NSCLC Cell Lines. Int. J. Radiat. Biol. 2019, 95, 1205–1219. [Google Scholar] [CrossRef]
- Wang, H.; Wang, H.; Powell, S.N.; Iliakis, G.; Wang, Y. ATR Affecting Cell Radiosensitivity Is Dependent on Homologous Recombination Repair But Independent of Nonhomologous End Joining. Cancer Res. 2004, 64, 7139–7143. [Google Scholar] [CrossRef] [Green Version]
- Soni, A.; Mladenov, E.; Iliakis, G. Proficiency in Homologous Recombination Repair Is Prerequisite for Activation of G2-Checkpoint at Low Radiation Doses. DNA Repair 2021, 101, 103076. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.P.; Uematsu, N.; Kobayashi, J.; Lerenthal, Y.; Krempler, A.; Yajima, H.; Lobrich, M.; Shiloh, Y.; Chen, D.J. Ataxia Telangiectasia Mutated (ATM) Is Essential for DNA-PKcs Phosphorylations at the Thr-2609 Cluster upon DNA Double Strand Break. J. Biol. Chem. 2007, 282, 6582–6587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Lee, J.H.; Jiang, W.; Crowe, J.L.; Zha, S.; Paull, T.T. Regulation of the DNA Damage Response by DNA-PKcs Inhibitory Phosphorylation of ATM. Mol. Cell 2017, 65, 91–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finzel, A.; Grybowski, A.; Strasen, J.; Cristiano, E.; Loewer, A. Hyperactivation of ATM upon DNA-PKcs Inhibition Modulates p53 Dynamics and Cell Fate in Response to DNA Damage. Mol. Biol. Cell 2016, 27, 2360–2367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Woods, R.G.; Beamish, H.; Ye, R.; Lees-Miller, S.P.; Lavin, M.F.; Bedford, J.S. Deficiency in the Catalytic Subunit of DNA-Dependent Protein Kinase Causes Down-Regulation of ATM. Cancer Res. 2005, 65, 1670–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arlander, S.J.; Greene, B.T.; Innes, C.L.; Paules, R.S. DNA Protein Kinase-Dependent G2 Checkpoint Revealed Following Knockdown of Ataxia-Telangiectasia Mutated in Human Mammary Epithelial Cells. Cancer Res. 2008, 68, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schipler, A.; Mladenova, V.; Soni, A.; Nikolov, V.; Saha, J.; Mladenov, E.; Iliakis, G. Chromosome Thripsis by DNA Double Strand Break Clusters Causes Enhanced Cell Lethality, Chromosomal Translocations and 53BP1-Recruitment. Nucl. Acids Res. 2016, 44, 7673–7690. [Google Scholar] [CrossRef] [Green Version]
- Biehs, R.; Steinlage, M.; Barton, O.; Juhasz, S.; Kunzel, J.; Spies, J.; Shibata, A.; Jeggo, P.A.; Lobrich, M. DNA Double-Strand Break Resection Occurs during Non-Homologous End Joining in G1 but Is Distinct from Resection during Homologous Recombination. Mol. Cell 2017, 65, 671–684.e675. [Google Scholar] [CrossRef] [Green Version]
- Schipler, A.; Iliakis, G. DNA Double-Strand-Break Complexity Levels and Their Possible Contributions to the Probability for Error-Prone Processing and Repair Pathway Choice. Nucl. Acids Res. 2013, 41, 7589–7605. [Google Scholar] [CrossRef] [Green Version]
- Bartek, J.; Bartkova, J.; Lukas, J. The Retinoblastoma Protein Pathway and the Restriction Point. Curr. Opin. Cell Biol. 1996, 8, 805–814. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.P.; Levine, A.J. Surfing the p53 Network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, C. Pathways Governing G1/S Transition and Their Response to DNA Damage. FEBS Lett. 2001, 490, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Ryan, K.M.; Phillips, A.C.; Vousden, K.H. Regulation and Function of the p53 Tumor Suppressor Protein. Curr. Opin. Cell Biol. 2001, 13, 332–337. [Google Scholar] [CrossRef]
- Buschmann, T.; Fuchs, S.Y.; Lee, C.-G.; Pan, Z.-Q.; Ronai, Z. SUMO-1 Modification of Mdm2 Prevents Its Self-Ubiquitination and Increases Mdm2 Ability to Ubiquitinate p53. Cell 2000, 101, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Giannakakou, P.; Sackett, D.L.; Ward, Y.; Webster, K.R.; Blagosklonny, M.V.; Fojo, T. p53 Is Associated with Cellular Microtubules and Is Transported to the Nucleus by Dynein. Nat. Cell Biol. 2000, 2, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Hirao, A.; Kong, Y.Y.; Matsuoka, S.; Wakeham, A.; Ruland, J.; Yoshida, H.; Liu, D.; Elledge, S.J.; Mak, T.W. DNA Damage-Induced Activation of p53 by the Checkpoint Kinase Chk2. Science 2000, 287, 1824–1827. [Google Scholar] [CrossRef] [PubMed]
- Shieh, S.Y.; Ahn, J.; Tamai, K.; Taya, Y.; Prives, C. The Human Homologs of Checkpoint Kinases Chk1 and Cds1 (Chk2) Phosphorylate p53 at Multiple DNA Damage-Inducible Sites. Genes Dev. 2000, 14, 289–300. [Google Scholar] [CrossRef]
- Maya, R.; Balass, M.; Kim, S.T.; Shkedy, D.; Leal, J.F.; Shifman, O.; Moas, M.; Buschmann, T.; Ronai, Z.; Shiloh, Y.; et al. ATM-Dependent Phosphorylation of Mdm2 on Serine 395: Role in p53 Activation by DNA Damage. Genes Dev. 2001, 15, 1067–1077. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xiong, Y. A p53 Amino-Terminal Nuclear Export Signal Inhibited by DNA Damage-Induced Phosphorylation. Science 2001, 292, 1910–1915. [Google Scholar] [CrossRef] [Green Version]
- Shiloh, Y. ATM and ATR: Networking Cellular Responses to DNA Damage. Curr. Opin. Genet. Dev. 2001, 11, 71–77. [Google Scholar] [CrossRef]
- Dip, R.; Naegeli, H. More than just Strand Breaks: The Recognition of Structural DNA Discontinuities by DNA-Dependent Protein Kinase Catalytic Subunit. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jack, M.T.; Woo, R.A.; Motoyama, N.; Takai, H.; Lee, P.W.K. DNA-Dependent Protein Kinase and Checkpoint Kinase 2 Synergistically Activate a Latent Population of p53 upon DNA Damage. J. Biol. Chem. 2004, 279, 15269–15273. [Google Scholar] [CrossRef] [Green Version]
- Kachnic, L.A.; Wu, B.; Wunsch, H.; Mekeel, K.L.; DeFrank, J.S.; Tang, W.; Powell, S.N. The Ability of p53 to Activate Downstream Genes p21WAF1/cip1 and MDM2, and Cell Cycle Arrest Following DNA Damage Is Delayed and Attenuated in Scid Cells Deficient in the DNA-Dependent Protein Kinase. J. Biol. Chem. 1999, 274, 13111–13117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailand, N.; Falck, J.; Lukas, C.; Syljuasen, R.G.; Welcker, M.; Bartek, J.; Lukas, J. Rapid Destruction of Human Cdc25A in Response to DNA Damage [In Process Citation]. Science 2000, 288, 1425–1429. [Google Scholar] [CrossRef] [PubMed]
- Nghiem, P.; Park, P.K.; Kim, Y.; Vaziri, C.; Schreiber, S.L. ATR Inhibition Selectively Sensitizes G1 Checkpoint-Deficient Cells to Lethal Premature Chromatin Condensation. Proc. Natl. Acad. Sci. USA 2001, 98, 9092–9097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamper, A.M.; Rofougaran, R.; Watkins, S.C.; Greenberger, J.S.; Beumer, J.H.; Bakkenist, C.J. ATR Kinase Activation in G1 Phase Facilitates the Repair of Ionizing Radiation-Induced DNA Damage. Nucl. Acids Res. 2013, 41, 10334–10344. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, F.; Mladenov, E.; Dueva, R.; Stuschke, M.; Timmermann, B.; Iliakis, G. Shift in G1-Checkpoint from ATM-Alone to a Cooperative ATM Plus ATR Regulation with Increasing Dose of Radiation. Cells 2022, 11, 63. https://doi.org/10.3390/cells11010063
Li F, Mladenov E, Dueva R, Stuschke M, Timmermann B, Iliakis G. Shift in G1-Checkpoint from ATM-Alone to a Cooperative ATM Plus ATR Regulation with Increasing Dose of Radiation. Cells. 2022; 11(1):63. https://doi.org/10.3390/cells11010063
Chicago/Turabian StyleLi, Fanghua, Emil Mladenov, Rositsa Dueva, Martin Stuschke, Beate Timmermann, and George Iliakis. 2022. "Shift in G1-Checkpoint from ATM-Alone to a Cooperative ATM Plus ATR Regulation with Increasing Dose of Radiation" Cells 11, no. 1: 63. https://doi.org/10.3390/cells11010063