
Getting Sugar Coating Right! The Role of the Golgi Trafficking Machinery in Glycosylation



Department of Physiology and Cell Biology, University of Arkansas for Medical Sciences, 4301 West Markham Street, Little Rock, AR 72205, USA
*
Authors to whom correspondence should be addressed.
Cells 2021, 10(12), 3275; https://doi.org/10.3390/cells10123275
Submission received: 15 October 2021
/
Revised: 17 November 2021
/
Accepted: 19 November 2021
/
Published: 23 November 2021
(This article belongs to the Special Issue Glycosylation and Deglycosylation in Animal Development)
Abstract
:The Golgi is the central organelle of the secretory pathway and it houses the majority of the glycosylation machinery, which includes glycosylation enzymes and sugar transporters. Correct compartmentalization of the glycosylation machinery is achieved by retrograde vesicular trafficking as the secretory cargo moves forward by cisternal maturation. The vesicular trafficking machinery which includes vesicular coats, small GTPases, tethers and SNAREs, play a major role in coordinating the Golgi trafficking thereby achieving Golgi homeostasis. Glycosylation is a template-independent process, so its fidelity heavily relies on appropriate localization of the glycosylation machinery and Golgi homeostasis. Mutations in the glycosylation enzymes, sugar transporters, Golgi ion channels and several vesicle tethering factors cause congenital disorders of glycosylation (CDG) which encompass a group of multisystem disorders with varying severities. Here, we focus on the Golgi vesicle tethering and fusion machinery, namely, multisubunit tethering complexes and SNAREs and their role in Golgi trafficking and glycosylation. This review is a comprehensive summary of all the identified CDG causing mutations of the Golgi trafficking machinery in humans.
1. Introduction
Eukaryotic cells have a remarkable feature of intracellular compartments or organelles enclosed by lipid membranes. Compartmental identity is determined by a number of factors, including the membrane lipid and luminal composition as well as by the transmembrane and peripheral proteins associated with the organelle [1,2]. The secretory pathway transports cargo not only to the extracellular space for secretion but also between organelles. The endoplasmic reticulum (ER), the Golgi apparatus (GA) and the endolysosomal system are key organelles of this pathway. Proteins of the secretory pathway collectively referred to as cargo are synthesized in the ER by ribosomes. Upon completion of synthesis, the Golgi receives incoming cargo from the ER. Within the lumen of the Golgi, proteins undergo posttranslational modifications and are finally sorted into appropriate carriers targeted to their destination.
The GA is the central hub of the secretory pathway. It is composed of at least four morphologically distinct cisternae named cis, medial, trans, and the trans-Golgi network (TGN), each having its own identity. The GA functions as a station that processes and packages cargo into appropriate membrane carriers. The cargo arrives at the GA continuously and bidirectionally. Biosynthetic cargo is transported in an anterograde manner from the ER to the Golgi and then to the post Golgi compartments, while retrograde cargo is brought to the Golgi by the endosomal system. The currently accepted cisternal maturation model of Golgi transport describes that the anterograde cargo stays in the Golgi cisternae and is carried forward by the gradual maturation of the cis-Golgi to the trans-Golgi while Golgi resident proteins are cycled back to newly formed cisternae [3,4,5,6]. This way, the identity of each compartment is maintained even as the cisternal maturation goes on.
Lipids and proteins transported by the secretory pathway undergo modifications such as glycosylation within the Golgi. The different known types of glycosylation are classified according to the amino acid residue to which the carbohydrate is attached. In humans, N-glycosylation and O-glycosylation are the two main types. N-glycans are where glycans are attached to Asn in the consensus peptide sequence Asn-X-Ser/Thr, while O-glycans are where glycans are attached to hydroxyl groups of serine or threonine. N-glycosylation is a well-defined process that begins in the ER while O-glycosylation is more varied and mostly begins in the Golgi [7,8,9,10]. Protein glycosylation includes the addition of N-linked glycans, O-linked glycans, phosphorylated glycans, glycosaminoglycans and glycosylphosphatidylinositol (GPI) anchors to peptide backbones as well as C-mannosylation of tryptophan residues [11]. The Golgi houses the majority of the glycosylation machinery within its cisternae. The glycosyltransferases, glycosidases together with their accessory machinery, modify and process glycans as they traverse the Golgi. At the TGN, fully processed glycoconjugates are packaged into carrier membranes and transported to appropriate destinations.
The glycosylation of macromolecules is involved in nearly every biological process and its defects give rise to various pathologies [11,12]. Glycosylation defects affect multiple organ systems with varying severities, and they fall under the umbrella of congenital disorders of glycosylation (CDG). CDGs are autosomal recessive inherited diseases due to mutations in genes involved in glycosylation [13,14]. CDGs are further subdivided into type I (CDG-I) and type II (CDG-II) [15,16]. CDG-I is due to defects in the transfer of preassembled dolichol-glycan in the ER (or cytosol). Defects in glycan processing in the Golgi give rise to CDG-II. Formerly, a CDG diagnosis was made only based on the isoelectric focusing of serum transferrin which only detected N-glycosylation defects. Nowadays, glycan mass spectrometry and/or ApoCIII IEF are often used to complement transferrin IEF testing [17].
2. Glycosylation Enzyme Compartmentalization
Apart from the glycosylation enzymes, the Golgi glycosylation machinery also includes ion channels and sugar transporters that maintain intraluminal pH and bring in cofactors and charged monosaccharides required for glycosylation. The key quality control for glycosylation is the proper cisternal compartmentalization of the glycosylation machinery [9,18,19,20]. Glycan processing/addition occurs in the sequence in which the enzymes encounter their substrates. As the cis-Golgi matures, eventually becoming the TGN and carrying cargo forward, the correct compartmentalization of the glycosylation machinery must be maintained within the framework of cisternal maturation. At the TGN, the cargo is sorted into appropriate anterograde carriers targeted to various destinations.
All Golgi glycosylation enzymes are transmembrane (TM) proteins and the majority of them are type II TM proteins. They possess a catalytic C-terminal domain in the lumen, a luminal stem region, a transmembrane domain (TMD) and a short N-terminal cytosolic tail. Evidence suggests that the cytoplasmic tail and TMD of Golgi enzymes, cytosolic adaptor and coat proteins, and membrane lipid composition play a role in the sorting and correct cisternal localization of the glycosylation machinery. The current models for sorting and retention at the Golgi have been described by Banfield et al. in [21,22] and more recently by Welch et al. in [23]. Here we summarize the retention and recycling mechanisms that have been described.
“Kin recognition” was the earliest proposed model for enzyme retention in the Golgi [24]. Kin oligomerization was first identified in the medial Golgi enzymes N-Acetylglucosaminyltransferase-1 (GlcNAcT1/MGAT1) and Mannosidase-2 (ManII/MAN2A1) [25]. These enzymes interact with each other via their TMDs and stem regions. Because enzyme oligomers would be too big to enter transport vesicles exiting the Golgi, they are retained in the Golgi. The enzyme α-2,6-sialyltransferase-1 (ST6Gal1) is believed to possess trans Golgi localization signals in its cytoplasmic tail, TMD and stem region because mutations in these regions lead to the secretion of the enzyme [26,27]. ST6Gal1 not only forms homodimers but also heteroligomerizes with β-1,4-Galactosyltransferase-1 (B4GALT1) [28]. All the currently known oligomers of the known Golgi enzymes in various glycosylation pathways have been described by de Graffenried et al. in [29].
Transmembrane domain-dependent sorting relies on compatibility between the TMD and membrane thickness [30]. Membrane thickness is believed to gradually increase from the ER to the PM due to different lipid compositions. Phospholipids are enriched in ER and Golgi membranes, while the enrichment of sphingolipids and sterols gradually increases in the TGN and post-Golgi compartments with the highest abundance in the PM. An extensive and detailed comparison of the TM proteins found that ER and Golgi residents have shorter TMDs compared to post-Golgi residents and bulkier residues were distributed closer to the exoplasmic side of the bilayer TMDs [31]. Simply stated, shorter TMDs reside in thinner membranes. Thus, Golgi enzymes are kept from entering the post-Golgi compartments by the mere incompatibility of their short TMDs in thicker membranes.
Golgi enzymes can cycle between the Golgi cisternae [32,33] as well as between the Golgi and ER [34], and the majority of recycling occurs in COPI-coated vesicles. Yeast α-1,6-mannosyltransferase (Mnn9p) is one such example [35,36]. This cis-Golgi enzyme is retrieved in COPI vesicles at the TGN [35]. A few other glycosylation enzyme complexes such as the Man-Pol-I (Mann9p and Van1p) and Man-Pol-II (Mnn9p, Hoc1p, Anp1, Mnn10p, and Mnn11p), in yeast, cycle through the ER in COPI vesicles. Selective sorting into retrograde COPI-coated carriers either occurs via direct interaction between the cytosolic tails or via an adaptor. Sorting signals—KKXX or RXR on ER residents [37] or φ(K/R)XLX(K/R) on some cis-Golgi enzymes—directly interacting with COPI subunits have been identified on the cytoplasmic tails of a subset of enzymes [38,39,40]. Vps74 is a COPI interacting protein that was identified as a protein essential for the proper localization of a subset of glycosylation enzymes in yeast [41]. Vps74 recognizes the ([F/L][L/I/V]XX[R/K]) peptide motif on the cytoplasmic tails of a subset of glycosyltransferases [42]. GOLPH3 and GOLPH3L are human homologs of Vps74. They have an affinity for PI4P enriched in the trans-Golgi and recognize membrane-proximal polybasic residues on the cytoplasmic tails of a subset of Golgi enzymes, including ST6Gal1 and N-acetylgalactosaminyltransferase-2 (GALNT2) [33,43]. About half the number of enzymes involved in glycosphingolipid synthesis have an LXX[R/K] consensus motif recognized by GOLPH3 on their cytosolic tails [44].
These mechanisms of enzyme compartmentalization within the Golgi are not mutually exclusive.
3. Membrane Trafficking Machinery
The membrane trafficking machinery moves the cargo in membrane-enclosed carriers to various destinations within the cell or releases secretory cargo at the plasma membrane. Every compartment in the secretory pathway is equipped with its own set of molecular players for transport. This machinery can be broadly categorized based on its function as budding, tethering and fusion machinery (Figure 1). The events of budding and fusion are controlled by a set of GTPases that modulate the activities of the trafficking machinery. Coat proteins together with cargo adaptors bind to the cargo, recruit them into specialized domains and bud these membrane domains off in the form of small vesicles or tubules [45,46]. COPI, COPII and Clathrin are the three known coat proteins. COPI is involved in the intra-Golgi and Golgi to ER retrograde trafficking, COPII is involved in the ER to Golgi anterograde trafficking and Clathrin is involved in TGN-plasma membrane bidirectional transport. Arf1 and Sar1 are small GTPases that are involved in vesicle formation. Membrane localized active Arf1-GTP recruits COPI and clathrin components while Sar1-GTP recruits the COPII components [47]. Membrane curvature is triggered by dimerization of Arf1-GTP [48]. Fission is thought to be driven by the interaction between the GTPases’ amphipathic helices and membrane lipids [48]. The membrane-enclosed cargo carrier is then tethered to its specific target membrane. Alignment of the fusion machinery on the two opposing membranes results in membrane fusion and the delivery of cargo to its destination. The function and intricate interactions between all the factors at play in cellular transport are yet to be fully understood. For this review, we elaborate on the tethering and fusion machinery at the Golgi and the disorders that arise due to genetic mutations in these trafficking factors.
3.1. Tethers (MTCs and Coiled-Coil Tethers)
Based on structure and function, tethers fall into two broad categories, namely coiled-coil tethers and multisubunit tethering complexes (MTCs).
3.1.1. Coiled-Coil Tethers
Coiled-coil tethers are structurally similar 100–200 nm long homodimers with large globular heads. Their length extends into the cytosol enabling the long-range capture of incoming cargo carriers and brings them close to their target membrane. At the Golgi, they may also function as stacking proteins holding the cisternae together (for a more in-depth review see [49] (Figure 2). The majority of the coiled-coil tethers that reside at the Golgi are called Golgins (see Table 1 for a complete list) (Figure 2). The cis-Golgins (USO1 (p115), GOLGA2 (GM130), TRIP11 (GMAP210), GOLGA3 (Golgin160), and GO45 (Golgin45)), and trans-Golgins (GOLGA1 (Golgin97), GOLGA4 (Goglin245), GCC1 (GCC88), GCC2 (GCC185), and TMF1 (TMF)), are soluble peripheral membrane proteins. GOLGB1 (Giantin), GOLGA5 (Golgin-84) and CUX1 (CASP) have a transmembrane domain at their carboxyl termini and localize at the rims of the Golgi stack. GORASP1 (GRASP65) and its homologue GORASP2 (GRASP55) are also Golgi matrix proteins but are not classified as Golgins due to their structural differences.
GOLGA2 (GM130 or Golgin95): This protein plays an important role in the development of vertebrates and invertebrates. Targeted or global depletion of GM130 in Drosophila, zebrafish and mice have shown developmental effects in the nervous and skeletal system [50,51,52]. Recently, GOLGA2 KO in mice has also been implicated in liver and lung fibrosis due to increased autophagy [53]. Two cases of GOLGA2 mutations have been identified in the human population. Both cases have neuromuscular disorders, including developmental delay, muscular dystrophy, seizures and microcephaly [51,54]. In a 2019 study, a lower level of GM130 expression was shown to cause aberrant O-glycosylation and was linked to Berger’s disease, also known as IgA neuropathy [55]. Though GOLGA2 CDG causing mutations in humans have not been reported, studies from cell lines do point to the essential role of GM130 in maintaining the Golgi structure and proper glycosylation [56,57].
TRIP11 (GMAP210): Early studies on GMAP210 point to its function in Golgi cisternal organization and positioning by anchoring microtubules at the cis-Golgi [58,59]. Its role in maintaining the Golgi stacks was also confirmed in HeLa cells wherein TRIP11 KD caused fragmentation and vesiculation of the Golgi [60]. It is required for proper anterograde and retrograde trafficking at the ER–Golgi interface [61]. It was also shown that GMAP210 can directly capture intra-Golgi vesicles [62]. Mutations in the thyroid hormone receptor interactor 11 (TRIP11) gene encoding the Golgi-associated microtubule-binding protein 210 (GMAP210) in the human population give rise to a lethal neonatal skeletal dysplasia termed as achondrogenesis (ACG1A) type IA or the much milder nonlethal odontochondrodysplasia (ODCD) depending on whether the mutation leads to a complete or partial loss of function [63,64,65,66,67]. In ACG1A and ODCD, Golgi disruption impairs the posttranslational processing and secretion of several extracellular matrix (ECM) components [64]. A mouse model bearing a nonsense mutation in TPIP11 with a complete loss of GMAP-210 resulted in ACG1A with fetuses showing an absence of mineralization. This affected chondrocyte differentiation, made them apoptotic, and reduced ECM production. Detailed analysis of cellular defects in the chondrocytes and osteoblasts revealed ER stress, vesicle aggregation and abnormal Golgi morphology. Studies on the glycosylation status in TRIP11 mutant mice fibroblasts using lectin staining confirmed incomplete glycan processing in the Golgi. Proteoglycan synthesis also differed in the mutant chondrocytes. The same study also investigated a 27-week-old human fetus with lethal ACG1A and found a similar phenotype as they did in their mouse model [63]. In hypomorphic mutations in TRIP11 causing ODCD, the milder counterpart of ACG1A, patient fibroblasts showed some degree of Golgi-localized GMAP210, which was completely absent in the ACG1A patient’s and the Golgi morphology was mostly normal. Patient fibroblasts also secreted reduced levels of ECM proteins. Both ACG1A and ODCD causing TRIP11 mutations have similar negative effects on glycosylation and proteoglycan modification of the ECM component decorin [64].
GOLGA3 (Golgin 160): Like GMAP210, Golgin160 is also localized to the cis-Golgi and functions in Golgi positioning and structure. It recruits the microtubule motor dynein to the Golgi [59,68]. Similar to the GMAP210 KD effects in HeLa cells, siRNA-induced KD of Golgin160 in the same cell line resulted in Golgi fragmentation and the dispersal of Golgi mini stacks [59]. A missense mutation in GOLGA3 has been linked to autism [69].
USO1 (p115): p115 belongs to the golgin family of tethers. It is involved in transport between ER and Golgi. At the cis-Golgi it interacts with the golgins, GM130 and Giantin as well as the SNAREs, BET1 and Sec22b [70,71,72]. The golgin, p115, was discovered as a protein that stimulated the proper glycosylation of the cargo protein VSVG in the in vitro intra-Golgi trafficking setup [73]. It was later shown to play an important role in the formation of pre-Golgi ERGIC intermediates [74]. Early studies suggested that p115 is recruited by Rab1 to COPII vesicles budding from the ER where it interacts with COPII vesicle-associated SNAREs [75]. It interacts with the STX5 SNARE partners GOSR1/BET1L/YKT6 and GOSR2/BET1/Sec22b [76] and p115 is essential for cisternal stacking and cisternal regrowth, facilitating the initial tethering of Golgi cisternae [77]. Moreover, it has a vital role in the biogenesis and maintenance of the Golgi as it interacts with the COG complex and participates in vesicular trafficking at the Golgi [78]. Although the depletion of p115 in tissue culture cells leads to glycosylation defects [79], CDG-causing mutations have not been reported so far.
GOLGB1 (Giantin): Giantin is the largest and well-characterized cis-golgin. It is anchored at the cis-Golgi by its transmembrane domain and either has a short or no luminal domain at its C-terminus [80,81]. Regulation of the Golgi architecture is one of its key functions. Apart from p115, Giantin interacts with Rab1, Rab6, and GM130. Loss of Giantin can accelerate the anterograde transport instead of blocking it [76,81,82,83,84]. While Giantin may be dispensable in maintaining Golgi structure, its depletion inhibits lateral tethering between the cisternae [83,84]. GOLGB1 loss-of-function zebrafish [82] and mouse [85] models revealed that Giantin has specific functions in protein glycosylation and tissue morphogenesis. The expression of Golgi-resident glycosyltransferases like GALNT3 is altered due to a loss of Giantin [82].
GOLGA5 (Golgin-84): Human golgin-84 is well conserved in evolution with orthologs in plants, but not fungi. Golgin-84 directly interacts with another Golgi tether, CASP, [86] and the COG7 subunit of the COG complex [87], and its siRNA-driven depletion in HeLa cells causes glycosylation defects [88]. Surprisingly, GOLGA5 is dispensable for mouse embryonic development and postnatal survival [89], indicating the redundancy in Golgi coiled-coil tethers. It was also demonstrated that Golgin-84 can directly capture intra-Golgi vesicles carrying glycosylation enzymes via its N-terminus [62].
GOLGA1 (Golgin-97): This protein was identified as an autoantigen in patients with Sjögren’s syndrome [90]. Golgin-97 is localized at the TGN where it tethers endosome-derived vesicles [91]. It associates with the centrosome [92], plays a role in the retrograde trafficking from endosomes to TGN [93] and its silencing leads to Golgi fragmentation [94]. This protein has garnered particular interest in the field of virology due to its requirement in poxvirus replication [95].
GOLGA4 (Golgin-245): Like Golgin-97, Golgin-245 too was identified as an autoantigen in patients with Sjögren’s syndrome. It is involved in the retrograde trafficking from endosomes to TGN and contributes to Golgi positioning around the centrosome. In a HeLa cell line, siRNA-mediated depletion of Golgin-245 resulted in TGN dispersal and fragmentation of the Golgi ribbon into mini stacks [96]. Displacement of Golgin-245 from the TGN or siRNA silencing disrupts the anterograde transport of cell surface destined cargo from the TGN [97,98,99]. In a more recent study, this protein has also been shown to function in phagophore formation during autophagy [100]. It was also demonstrated that Golgin-245 could directly capture endosomal-derived vesicles via its N-terminus [62].
GCC1 (GCC88): This GRIP-domain containing TGN localized Golgin is involved in endosome to Golgi retrograde transport [101] and maintains the tubular organization of the TGN [102,103]. GCC88 KD in HeLa cells led to mislocalization of the Qc SNARE syntaxin 6 which prevented the recycling of TGN38 and CI-M6PR from endosomes to the TGN [101]. Due to impaired CI-M6PR recycling, lysosomal activity was perturbed and the lysosomal enzyme Cathepsin D’s processing was impaired [103].
GCC2 (GCC185): This is another GRIP domain-containing protein targeted to the TGN and has the dual functions of tethering vesicle and cytoskeletal organization by providing a microtubule attachment site [104]. It binds the TGN Qa SNARE STX16 [105]. Rab9 localizes GCC185 at the endosomes while Rab6 and Arl1 are suggested to localize it at the TGN [106,107,108], but Rab6 KD does not mislocalize GCC185 from the Golgi [109]. It functions in the CI-M6PRs recycling from endosomes to TGN. KD in HeLa cells fragments the Golgi [106,110]. Despite a fragmented Golgi in GCC185 KD cells, anterograde trafficking is unperturbed, but a subset of the retrograde cargo, Shiga toxin, for example, is blocked in endosomes [110].
TMF/ARA160: Tata element modulatory factor (TMF/ARA160) is a multifunctional Golgi tether that also functions as a transcriptional factor. Smg1, the yeast homologue, is recruited to the TGN by Ypt6-GTP and its deletion in yeast does not produce any trafficking defect [111,112]. TMF KD causes a dispersal of the Golgi cisternae [112], blocks endosome to TGN retrograde trafficking and mislocalizes GalNAc-T2 [113]. It directs the proteasomal degradation of Stat3, a cell growth regulator and p65 [114,115]. Interestingly TMF null male and female mice develop normally, but males are sterile with defects in the testis and epididymis. These mice produce acrosome-deficient-morphologically-defective immotile sperms, and have low testosterone levels [116,117]. It interacts with the microtubules and plays a critical role in Golgi orientation during spermatogenesis [118]. More recently, it was also found to be responsible for the transport of vesicles containing GLUT4, a receptor that drives glucose uptake in an insulin-dependent manner [119].
GRASP65 and GRASP55: The mammalian Golgi reassembly stacking proteins of 65 kDa (GRASP65/GORASP1) and 55 kDa (GRASP55/GORASP2) are two homologous Golgi transport proteins localized to the cis- and medial-trans cisternae, respectively [120,121]. GRASPs are required for the in vitro stacking of Golgi cisternae as well as Golgi ribbon formation by linking individual stacks [122,123,124]. Interestingly, a recent study reported that auxin-mediated acute depletion of GRASP55 and/or GRASP65 does not affect Golgi stack formation but causes the loss of lateral connectivity [125]. GRASP65 interacts with GM130 and GRASP55 forms a complex with Golgin-45 to regulate Golgi morphology and tethering [122,126]. Recently, a CRISPR/Cas9 mediated GRASP double knockout was reported to disperse the Golgi stack into single cisternae and tubulovesicular structures, increase protein trafficking and impair proteins and lipid glycosylation in HeLa and HEK293T cell lines [121,127]. Both the single and double GRASP knockout cell lines had reduced the cell surface binding of wheat germ agglutinin (WGA) that binds sialic acid and N-acetylglucosamine, and that of Maackia amurensis lectin (MAA), which binds α(2,3) sialic acid. In addition, LAMP1 and LAMP2 glycoproteins’ increased mobility indicated that they were not fully glycosylated [121]. Glycan mass spectrometry analysis confirmed that GRASP55 and GRASP65 depletion affects N-linked glycans in HeLa cells [127]. Furthermore, the altered cell surface binding of Shiga and Cholera toxins suggests that a GRASP knockout impairs the production and/or trafficking of glycolipids as well [121]. Besides protein and lipid glycosylation defects, GRASP depletion also causes sorting defects resulting in the production of immature Cathepsin D [127].
3.1.2. Multisubunit Tethering Complexes
Conserved Oligomeric Golgi Complex (COG)
The conserved oligomeric Golgi (COG) complex is the major CATCHR (complexes associated with tethering containing helical rods) vesicle tethering, hetero-octameric complex in the Golgi vicinity, with subunits COG1-COG8 [128,129,130,131]. It is divided into two sub-complexes named Lobe A (COGs 1–4) and Lobe B (COGs 5–8) with an interaction between COG1 and COG8 [128,132,133,134]. The COG complex appears to interact with all types of trafficking facilitators throughout the Golgi, such as SNAREs, SNARE-interacting proteins, Rabs, coiled-coil tethers, and vesicular coats [134]. The array of protein interactions suggests that COG, generally, orchestrates sequential processes triggered by activated Rab proteins beginning with vesicle attachment and concluding with the assembly of SNARE complexes [135]. Different COG subunits assigned as different “interaction hubs” such as COG2 and COG3 act as “tether hubs,” COG2, COG3, and COG4 act as “coat and motor hubs,” COG4, COG5, and COG6 act as “Rab hubs,” and COG4, COG6, COG7, and COG8 act as “SNARE hubs.” The COG complex has two potential conformations which were uncovered by electron microscopy (EM) in fixed and unfixed conditions. When unfixed, the COG complex has an extended and seemingly flexible structure (approximately 50–75 nm long) with multiple elongated, curved arms with globular or ‘hook like’ ends, whereas when fixed, the COG complex has a more globular appearance (∼37 nm in length) with rod-like connections between the two main lobes [128,129,130,135,136]. The exact conformation of the membrane-bound COG complex is unknown; however, live-cell super-resolution microscopy studies indicate that COG lobe A is preferentially Golgi bound, while lobe B is localized on vesicles [137].
COG mainly acts as a vesicular tether in retrograde intra-Golgi trafficking and recycles Golgi resident proteins (such as glycosylation enzymes) to maintain proper glycosylation of secretory proteins [138,139,140,141]. Recent studies on a complete set of COG subunit KOs in HEK293T cells, using a CRISPR-Cas9 approach, provided further insights into the COG’s function at the Golgi [142]. All COG deficient cells had glycosylation defects implicating the role of both lobes of the COG complex in maintaining glycosylation fidelity. Furthermore, each COG subunit-deficient cell line had defects in Golgi morphology, retrograde trafficking, sorting, and secretion, and accumulated enlarged endolysosomal structures (EELSs) [143,144]; however, the severity of these defects varied among subunits [142].
Almost one-third of the patients with congenital defects in the Golgi glycosylation have mutations in one of the subunits of the COG complex and this type of CDG is termed COG-CDG [10,145] (Table 1). Over a hundred individuals with 31 different mutations in different COG subunits (except COG3) have been identified to date, presenting heterogeneous clinical features and cellular phenotypes. COG-CDGs are rare and incurable diseases that belong to a group of autosomal recessive disorders and multi-systemic disorders with several distinguishable symptoms that include global developmental defects, including dysmorphic features, microcephaly, and failure to thrive. These deficits are often accompanied by liver and neurological impairment [13]. Lobe A (COG) mutations are proposed to be more deleterious than lobe B mutations [146], but it is still unclear because of the small number of known mutations and the lack of robust data on these patients. It has been reported that COG1 and COG4 patients present milder problems in comparison to COG7 and COG8 patients [147]. Among all the COG-CDGs, COG7 mutation is the most severe and COG6-CDG is nearly severe as 50% of the patients died within the first two years of life [148]. Neurological symptoms are most common in COG5 and COG8-CDG patients, while growth retardation is predominant in COG1 and COG7-CDG patients [145,149]; however, known COG mutations do not cause embryonic lethality suggesting that there is some degree of tolerance to COG malfunctions during embryonic development [131]. Clinically, prominent incomplete galactosylation and sialylation has been found in patients of COG-CDGs for variants of both the lobe A and B subunits [14]. Recent comprehensive reviews from ours and other groups have summarized the cellular and clinical manifestations of all the reported COG mutations up to 2020 [13,14,131].
Golgi-Associated Retrograde Protein Complex (GARP)
The GARP complex is another Golgi-localized multisubunit vesicle tethering complex of the CATCHR family [150,151,152]. Its subunits, VPS52, VPS53, and VPS54, were first identified in a Saccharomyces cerevisiae genetic screen which was designed to find new vacuolar protein sorting (VPS) genes essential in Carboxypeptidase Y (CPY) sorting [153]. These novel trimeric protein complexes were initially named as VPS fifty-three (VFT) [111]. Later, a fourth subunit, VPS51, was discovered in another genetic screen, a mutation of which has a similar defect in CPY sorting [154,155]. A co-immunoprecipitation experiment demonstrated that VPS51 belongs to the same complex as VFT [154]. Hence, this complex is composed of four different subunits named VPS51, VPS52, VPS53, and VPS54, and each of these four subunits forms an obligatory 1:1:1:1 complex [156,157]. All of these GARP subunits require the coiled-coil motif for assembly into the complex [154,155,158].
The GARP complex is evolutionary conserved, with homologues discovered in worms, flies, mammals, and plants [150,159,160,161]. GARP is localized at the TGN. In human cells, the small GTPases ARL5 and ARFRP1 localized at the TGN are responsible for the recruitment of GARP [162]. The GARP complex shares VPS51, VPS52, and VPS53 with another tethering complex-endosome associated recycling protein (EARP) whereas VPS50 is a unique subunit of EARP and VPS54 is a unique subunit of GARP [163]. The VPS54 subunit of the GARP complex has its N-terminal domain crucial in assembly and stability of the GARP complex, whereas the C-terminal domain mediates the localization to an early endocytic compartment [164].
GARP functions in the recycling receptors for lysosomal hydrolase precursors, transmembrane proteins, and SNAREs to the Golgi [157,165,166]. Depletion of the GARP subunit blocked the retrograde trafficking of the lysosomal hydrolase receptor Mannose-6-phosphate receptor, TGN protein TGN46, and the B subunit of the Shiga toxin. As a consequence of the blockade in retrograde trafficking, there is missorting of the precursor of the acid hydrolase Cathepsin D [166]. GARP is also essential for the maintenance of cholesterol and sphingolipid homeostasis, disruption of which can lead to neurodegenerative diseases known as “progressive cerebello–cerebral atrophy type 2” and “Hereditary spastic paraparesis” [167,168,169,170]. Auxin-induced acute depletion of yeast GARP subunits resulted in the missorting of plasma membrane proteins of two different functional groups, amino-phospholipid flippases and cell wall biosynthesis proteins [171].
Exome sequencing of a 6-year-old Caucasian female identified compound heterozygous mutations (c.1468C > T and c.2232delC) in the gene encoding the VPS51 resulting in a neurodevelopmental disorder [172]. The VPS52 subunit of GARP has shown to interact with Parkinson’s disease kinase LRRK2 (Leucine-rich repeat kinase 2) and contributes to intracellular trafficking by stabilizing the GARP–SNAREs complex formation [173]. Depletion of GARP exacerbates the neurodegeneration in C. elegans [173] (Table 1).
In mice, a null mutation in the VPS54 subunit of the GARP complex causes embryonic lethality [174,175]. The “wobbler mouse” phenotype is caused by a spontaneous missense mutation L967Q near the C terminus of VPS54. The “wobbler mutations” characteristics, such as cellular transport abnormalities, progressive motor neuron degeneration, and neuroinflammation, are quite similar to human Amyotrophic lateral sclerosis, although there are no known GARP genetic abnormalities associated with human ALS patients. The “wobbler mutation” causes the VPS54 protein, as well as the whole GARP complex, to become unstable and impairs retrograde vesicular trafficking. This affects spermiogenesis and also leads to spinal muscular atrophy [176,177,178,179,180]
GARP is structurally and functionally similar to other multisubunit tethering complexes such as COG, Dsl1, and Exocyst [181]. Although the COG dysfunctions are associated with CDG, the connection between GARP function and Golgi glycosylation was discovered only recently. A case study of a 6-year-old patient carrying a mutation in VPS51 showed abnormal glycosylation testing, and her phenotype had substantial similarity with that seen in CDGs. Additionally, a significant reduction in the level of N- and O-glycosylated protein TGN46 were found in the patient’s fibroblasts. Moreover, immunostaining of LAMP2, a lysosomal protein in VPS51 mutated patient fibroblasts, demonstrated enlarged lysosomes, and immunostaining of the Mannose-6 phosphate receptor showed dispersed cytoplasmic staining in the patient fibroblasts, which is in contrast to bright juxtanuclear staining in control fibroblasts [172]. Consistently, deletion of the GARP complex subunits in tissue culture cells revealed defects in N- and O-glycosylation. These defects in the GARP complex alter the total level of the cis-Golgi protein GPP130, medial-trans-Golgi TMEM165, and the TGN-resident TGN46 Golgi proteins. In addition, lysosomal glycoprotein LAMP2, showed hypermobility on the SDS-PAGE of GARP deficient cells, indicating protein misglycosylation [143,182]. Misglycosylation of these Golgi glycoproteins can occur as a consequence of the reduction in Golgi enzymes that are responsible for N- and O- glycosylation [9]. Indeed, the protein level of the N-glycosylation Golgi enzymes α-1,3-mannosyl-glycoprotein 2-β-N-acetylglucosaminyltransferase (MGAT1), B4GALT1, ST6GAL1 and the O-glycosylation enzyme GALNT2 was reduced in GARP subunit depleted cells. Furthermore, a retention using selective hooks (RUSH) assay showed that B4GALT1 is not retained at the Golgi complex in GARP-KO cells but missorted to the endolysosomal system. The Golgi-resident enzyme ST6GAL1 was also not found to be retained in the Golgi in GARP impaired cells. This study also demonstrated that this defect in Golgi enzymes only occurs in GARP-deficient cells and not in EARP-deficient cells. The malfunction, mislocalization, and reduced stabilization of Golgi enzymes associated with a glycosylation defect were found to be rescued upon expression of the missing GARP subunit. Taken together, GARP plays a crucial role in normal Golgi glycosylation by mediating the maintenance of the Golgi glycosylation machinery [182]. While neurodevelopmental disorders are common in GARP mutated patients, it is crucial to investigate and characterize the role of the GARP complex in different cell types. The mechanisms of GARP function need to be further uncovered to elucidate how GARP is involved in the misglycosylation of proteins.
3.1.3. SNAREs
SNAREs (SNAP Receptors) complete the final step of docking and fusion of the carrier membrane at its target (Figure 3). There are a total of 36 known SNAREs in humans that are present throughout the secretory pathway and confer a degree of compartment identity. Upon vesicle docking at its target membrane, usually, a total of three or four SNAREs (bearing 4 SNARE motifs) on opposing membranes align, intertwine and form a hetero-oligomeric, parallel α helical complex. The SNARE motif–a distinctive feature of all SNAREs–is a coiled structure composed of about 60–70 amino acid residues. At the center of the SNARE motif lays a glutamate or arginine residue, leading to the categorization of SNAREs as Q and R SNAREs [183]. Q SNAREs are further subcategorized into Qa, Qb and Qc based on protein profiling [184]. Typically, each Qa-, Qb-, Qc- and R-SNARE provides one SNARE motif to the SNARE complex. As the name suggests, Qbc SNAREs possess a Qb and Qc SNARE motif. These SNAREs contribute two SNARE motifs to the four-helix SNARE complex. SNARE motifs align from the N to C terminus and form an extremely stable parallel α helical complex [185]. Aligned SNAREs on opposing membranes are termed trans-SNARE complexes. The energy released upon the formation of the trans-SNARE complex drives membrane fusion. After the initiation of membrane fusion, the SNARE complex then resides on the same membrane and is termed a cis-SNARE complex. This transition of the SNARE complex from trans to cis is thought to play an important role in the creation and enlargement of the fusion pore [186].
SNARE complexes have to be disassembled to participate in the next round of fusion. This is an enzymatic and energy-intensive process that uses the energy of ATP hydrolysis. The enzyme NEM sensitive factor (NSF), named after it, was found to be inactivated by N-ethyl maleimide (NEM), is an AAA + ATPase. NSF is recruited to SNARE complexes by the Soluble NSF Attachment Protein (SNAP). SNAP has three isoforms- α-, β- and γ- SNAP which are ubiquitously expressed in all tissues except β-SNAP which is brain-specific. SNAP is believed to bind to the trans-SNARE complex. From there on, the NSF binds SNAP. The free energy released upon SNARE dissociation is thought to contribute to driving membrane fusion.
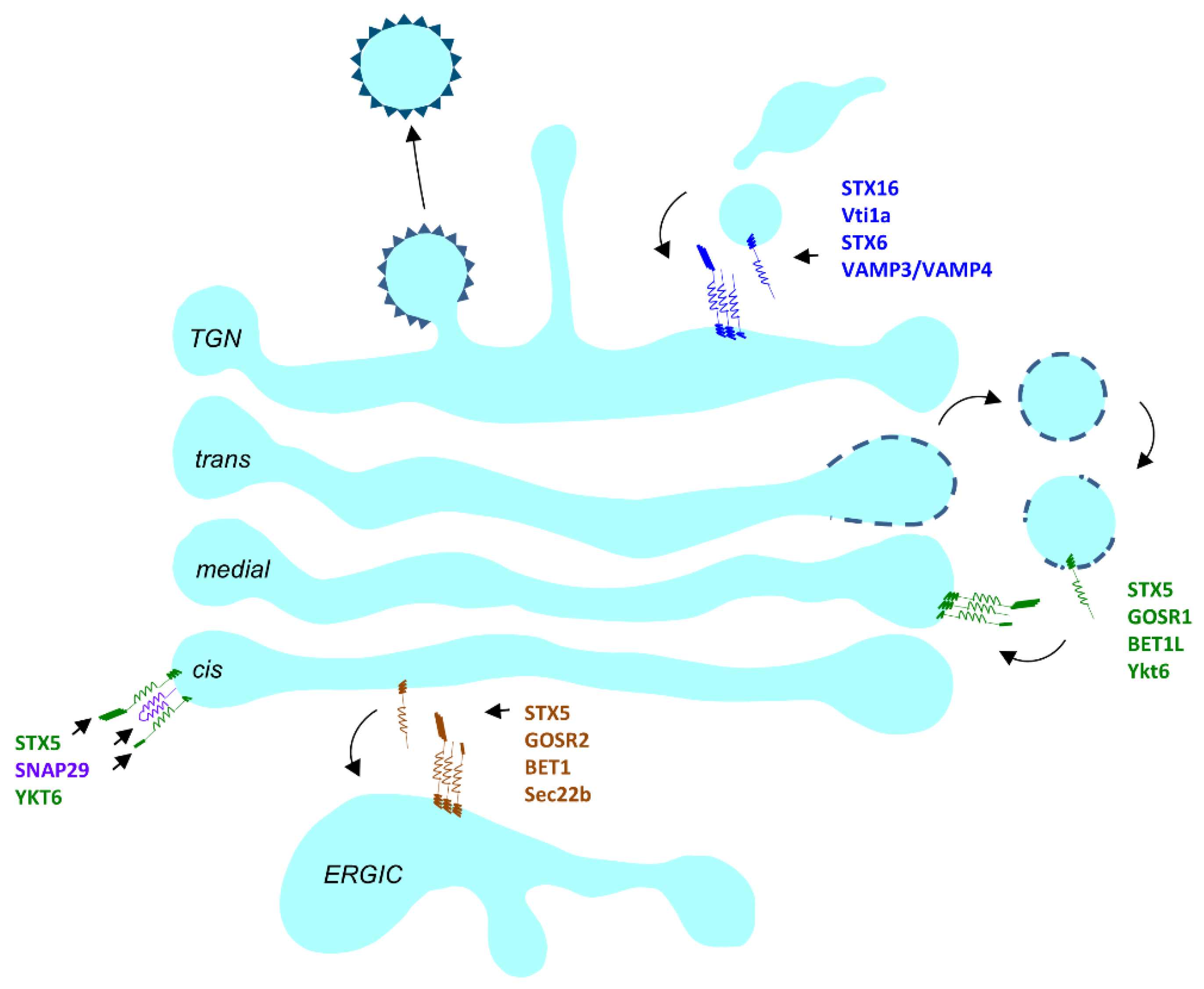
There are three major SNARE complexes operating in the Golgi (Figure 3). The STX5/GOSR2/Bet1/Sec22b complex mostly operates at the ERGIC compartment in mammalian cells [187]. The STX5/GOSR1/Bet1L/Ykt6 complex operates in retrograde trafficking at the Golgi [188] and the STX16/STX6/Vti1a/Vamp4 complex operates between the endosomal compartment and the trans side of the Golgi [189]. An additional Golgi-localized SNARE complex could be formed by STX5, SNAP29 and YKT6 [190,191].
In the STX5/GOSR2/ Bet1/Sec22b complex, GOSR2 mutations have been identified in the human population. GOSR2 (GS27/membrin) allelic variants in the human population have been associated with cardiovascular diseases and myocardial infarction [192,193,194]. A loss of function mutation (c.430G > T, p.Gly144Trp) in GOSR2 was reported to cause progressive myoclonus epilepsy with early ataxia [195], and heterozygous c.430G > T and c.82C > T is associated with muscular dystrophy [196]. The effects of GOSR2 mutations on the muscular system point to its essential role in development (Table 1).
Recently a CDG causing mutation, c.163 A > G, was identified in the Qa SNARE STX5 [197]. STX5 has two isoforms, long (STX5L) and short (STX5S), encoded by different start codons. The long isoform has an ER-retrieval sequence, while STX5S is Golgi localized. The c. 163 A > G (p.M55V) is a missense mutation at the start codon for the STX5S, leading to complete loss of STX5S protein. This mutation led to spontaneous abortion or death shortly after birth due to liver failure. Clinical presentations of this CDG include skeletal deformities, dysmorphia hypotonia, hepatomegaly, and elevated cholesterol. IEF of serum transferrin and ApoCIII together indicated N- and O- glycosylation defects which were confirmed by lectin binding analysis on patients’ fibroblasts.
Despite the essential role of SNAREs in Golgi trafficking, no other CDG has been reported so far. Embryonic lethality could result in mutations going undetected; however, due to redundancy and some degree of promiscuity among SNAREs, studies using KD or KO approaches in cell lines and other model organisms have yielded only mild phenotypes. Disruption of the Qb and Qc STX5 partners GOSR1 (GS28) and BET1L (GS15), only produce mild phenotypes. Overexpression of a BET1L lacking transmembrane domain caused Golgi disruption and mislocalization of the glycosylation enzyme MANN2 [188]. YKT6, the R SNARE in the Golgi STX5 SNARE complex, lacks a transmembrane domain and is integrated into the Golgi membrane via a lipid anchor, and its deletion is lethal in yeast [198]. Ykt6 has an essential role in Golgi organization which is disrupted when Ykt6 cannot localize to the Golgi [199].
SNAP29 is a Qbc SNARE that is reported to be membrane localized and has a cytosolic pool as well [200]. The Golgi is one of the membranes that SNAP29 is present on [190]. Among various other SNARE partners [201], it also partners promiscuously with the Golgi SNAREs STX5, STX6 [190]) and YKT6 [191], as well as the COG complex subunit, COG6 [202]. It has roles in synaptic transmission, endocytosis, cell cycle regulation and autophagy [203]. In humans, mutations in SNAP29 give rise to CEDNIK (cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma) syndrome or the neuromuscular disorder PMLD (Pelizaeus–Merzbacher-like disorder) [204,205]. Zebrafish carrying CEDNIK syndrome-causing mutations have multi-systemic developmental defects [206] and the mutant male mice carrying SNAP29 mutations, in addition to phenocopying the human mutant phenotypes, are also sterile [207]. SNAP29 deletion in Drosophila disrupts the Golgi organization [208] and controlled RNAi mediated silencing leads to lethality at high temperatures [209], but in C. elegans, it is the endosomal system that is disrupted and not the Golgi, upon SNAP29 KD [210]. Glycosylation defects in SNAP29 mutants have not yet been investigated.
At the TGN, KD of any of the STX16 SNARE complex subunits alters endosome to TGN transport. In humans, a 4.4-kb microdeletion in the STX16 locus leads to autosomal dominant pseudohypoparathyroidism Type Ib [211]. KD of any one of the four STX16 SNARE partners at the trans-Golgi leads to Golgi fragmentation [212] yet STX16 mice are viable [213]). Both Vti1a KO and Vti1b KO mice are viable, with the latter presenting with progressive neurodegeneration [214].
4. Conclusions
One of the most important tasks of the GA is to carry out proper processing of its cargos. Cargos traversing the Golgi are involved in diverse physiological roles. These cargos function as PM receptors, antibodies, ligands, signaling molecules including neurotransmitters and hormones, or as matrix proteins that function in organism structure. Hence, it is not surprising that defects in intra-Golgi trafficking result in multisystem disorders. Glycosylation disorders arising from improper glycan processing within the Golgi are categorized as CDG-II. Essentially, loss of function deletions of any component of the Golgi glycosylation machinery has the potential to produce a CDG, yet CDG-II is a rare disease. The clinical presentations and cellular phenotypes of CDGs are heterogeneous in nature which makes it difficult to compare the contributions of different tethers as well as different SNAREs.
Why do defects in Golgi tethering factors and SNAREs have an effect on glycosylation? The most likely explanation is that many of these factors are involved in capturing/tethering and fusion of intra-Golgi vesicles that recycle the Golgi glycosylation machinery. Golgi enzymes can be packaged in COPI vesicles in vitro [215], but how many types and varieties of intra-Golgi vesicles exist in different cells in vivo is unknown. Mutations in the tethering/fusion components may reduce or alter specific vesicle recognition, which will lead to the loss of recycling vesicles and a corresponding reduction in one or several components of Golgi glycosylation machinery. Another possibility is that the disruption of the Golgi structure can lead to the disruption of Golgi ion and lipid homeostasis, altering enzyme activity and retention. While coiled-coil tethers are mostly involved in maintaining Golgi structure, the Golgi MTCs, COG and GARP have been found to be primarily involved in delivering proper glycosylation enzymes by maintaining the trafficking in Golgi. The COG-CDGs have been well documented [131], and recently, our lab has demonstrated GARP’s requirement in the recycling of glycosylation enzymes [182]; however, we are yet to understand why COG and GARP mutations affect some organ systems more severely than others. A fundamental question that we are posed with is why do mutations affecting the Golgi trafficking machinery have various manifestations at the cellular, tissue, and organismal level? One would imagine that any dysfunctions of the Golgi trafficking machinery would affect Golgi homeostasis and thus glycosylation because of how intimately they are tied to each other, yet this is not the case. Certain Golgi proteins are more dispensable than others and the severity of their loss varies. Redundancy among the Golgi proteins could be one possible explanation.
An in-depth understanding of the contribution of the Golgi trafficking machinery will greatly benefit the development of therapeutic approaches for patients with life-threatening mutations. In the past, patient fibroblasts have served as a good cellular model for studying the disease phenotype, but this approach has many limitations. The main caveat with fibroblasts-based studies is a potential heterogeneity resulting from a diverse genetic background of the patients. In addition, fibroblast cell physiology may not reveal the specific defects manifested in nervous, ocular, bone, and other tissues severely affected in COG patients. To study COG mutations more elaborately, cell-based, vertebrate (zebrafish) and C. elegans models have been developed for COG4 mutations [216]. Another cell-based model for COG4-CDG disease has been developed recently using human RPE1 and HEK293T cell lines [217]. This strategy utilizes cells with the same genetic background to directly compare the effects of different human COG4 mutations on Golgi glycosylation and trafficking. Future studies could be directed to create the cell-based model for other COG mutations revealing the cause of specific CDG. This may help to generate more data regarding the trafficking and glycosylation abnormalities in human mutated cell lines, which will lead to the development of new treatment protocols for COG-CDGs.
In this review, we have summarized human diseases as well as the effects of experimental manipulations with the Golgi trafficking machinery and tried to shed light on the contribution of vesicular tethers and SNAREs in Golgi glycosylation (Table 1). While recent studies have made significant progress in delineating the trafficking machinery, it is difficult to outline the exact mechanism by which Golgi trafficking factors control glycosylation in specific cells and tissues. Newer approaches using inducible silencing of target proteins will certainly provide a better resolution into the intricate spatial and temporal coordination among the trafficking players to achieve proper glycosylation. Furthermore, similar approaches and other precise gene editing manipulations in the early embryos of various model organisms will provide invaluable insights into the role of the Golgi trafficking machinery in development.

{kind=link}
{kind=link}
{kind=link}
Table 1.
Golgi tethering factors and SNAREs.
| Gene | Alternative Names | Biological Role | Glycosylation Defects If Depleted | Known Human Mutations | References |
|---|---|---|---|---|---|
| GOLGA1 | golgin-97 | Endosome to TGN retrograde transport | No | - | [90,94] |
| GOLGA2 | GM130 | Maintenance of Golgi structure and ER to Golgi traffic | Yes (‘O’ glycosylation defects) | c.2251 C > T (p.Gln751Ter) c.629G > A (p.Arg210His) c.1266_1269del (p.Glu423Argfs*6) | [52,55,218] |
| GOLGA3 | golgin-160 | Maintain Golgi integrity and trafficking of plasma membrane protein | No | - | [219,220] |
| GOLGA4 | golgin-245 | Regulatory transport from TGN to plasma membrane | No | - | [100,221] |
| GOLGA5 | Golgin-84 | Tethering of intra-Golgi vesicles | Yes | - | [87,222] |
| GOLGA7 | GCP16 | - | No | - | [223] |
| GOLGB1 | Giantin, GCP364 | Maintaining Golgi structure and intra-Golgi retrograde trafficking | Yes (both ‘N’ and ‘O’ glycosylation defects) | (Golgb1ivs9+1G > A) | [81,85,224,225] |
| GCC1 | GCC88 | Efficient retrograde transport of cargo from the early endosomes to the TGN | [62,102] | ||
| GCC2 | GCC185 | Endosome-to-Golgi transport and maintenance of Golgi structure | [110,226] | ||
| TRIP11 | GMAP210 | Cisternal organization and anterograde, retrograde trafficking at ER-Golgi interface | Yes (‘N’ glycosylation defects) | c.5003T→A (p.L1668X) | [63] |
| TMF1 | ARA160 | Golgi organization | [118] | ||
| CUX1 | CASP | Forms a complex with Golgin 84 and tethers intra-Golgi recycling vesicles | [86,227] | ||
| USO1 | p115 | ER to Golgi trafficking | Yes | [228] | |
| GO45 | Golgin-45, BLZF1 | Golgi structure maintenance and secretion | - | [126] | |
| GORASP1 | GRASP65 | Maintaining the Golgi structure and protein trafficking, glycosylation | Yes | [229,230] | |
| GORASP2 | GRASP55 | Maintaining the Golgi structure and protein trafficking, glycosylation | Yes | [123] | |
| COG1 | KIAA1381, LDLB | Intra-Golgi retrograde trafficking | Yes (‘N’ Glycosylation defect) | 2659–2660insC (p.P888fsX900) c.1070 + 5G > A | [145,231,232] |
| COG2 | LDLC | Intra-Golgi retrograde trafficking | Yes (‘N’ and ‘O’-glycosylation defect) | c.701dup (p.Y234*), c.1900 T > G (p.W634G) | [231,233] |
| COG3 | hSec34 | Intra-Golgi retrograde trafficking | Yes | [234] | |
| COG4 | Intra-Golgi retrograde trafficking | Yes (‘N’ and ‘O’-glycosylation defect) | c.2185C > T (p.R729W) 16q22 deletion, c.697G > T (p.E233X) [de novo], c.2318 T > G (p.L773R), c.1546G > A or c.1546G > C (p.G516R) | [128,147,216,235] | |
| COG5 | GOLTC1, GTC90 | Intra-Golgi retrograde trafficking | Yes (‘N’-glycosylation defect) | c.1669-15 T > C, c.556_560delAGTAAinsCT (p.S186_K187delinsL), c.1856 T > C (p.I619T) c.95T > G (p.M32R), c.2518G > T (p.E840X), c.189delG (p.C64Vfs*6), c.2338_2340dupATT (p.I780dup), c.1780G > T (p.V594F), c.1209delG (p.M403IfsX3), c.2324C > T (p.P775L), c.330delT (p.V111Lfs*22), c.1290C > A (p.Y430X), c.2077A > C (p.T693P), c.2324C > T (p.P775L), c.1508dup (p.G505Wfs*3) | [236,237,238,239,240,241] |
| COG6 | KIAA1134 | Intra-Golgi retrograde trafficking | Yes (‘N’-glycosylation defect) | c. 1646G > T (p.G549V), c.1167-24A > G (p.G390FfsX6), c.511C > T (p.R171*), c.1746 + 2 T > G, c.1238_1239insA (p.F414Lfs*4), c.1646G > T (p.G549V), c.785A > G (p.Y262C), c.511C > T (p.R171*), c.1746 + 2 T > G | [148,242,243,244,245,246] |
| COG7 | UNQ3082/PRO10013 | Intra-Golgi retrograde trafficking | Yes (‘N’ and ‘O’ glycosylation defects) | IVS1 + 4 A → C aka c.169 + 4A > C, c.170-7A > G (p.56–57insAT) | [247,248,249,250] |
| COG8 | Intra-Golgi retrograde trafficking | Yes (‘N’ and ‘O’ glycosylation defects) | c.1611C > G (p.Y537X), IVS3 + 1G > A, TT 1687–1688, c.171dupG (p.L58Afs*29), c.1656dupC (p.A553Rfs*15), c.1583-1G > A | [251,252,253,254] | |
| VPS51 | ANG2, C11orf2, C11orf3, FFR | Retrograde transport from endosomes to TGN | Yes (‘N’ and ‘O’ glycosylation defects) | c.1468C > T, c.2232delC (c.1419_1421del; (p (Phe474del))) | [172,255] |
| VPS52 | SACM2L | Retrograde transport from endosomes to TGN | - | ||
| VPS53 | Hit1 | Retrograde transport from endosomes to TGN | Yes (‘N’ and ‘O’ glycosylation defects) | c.2084A > G c.1556+5G > A | [167] |
| VPS54 | CGP1, LUV1, RKI1, TCS3 | Retrograde transport from endosomes to TGN | Yes (‘N’ and ‘O’ glycosylation defects) | ||
| STX5 | Intra-Golgi retrograde transport | Yes (‘N’ and ‘O’ glycosylation defects) | c.163A > G (p.(Met55Val)) | [197] | |
| GOSR1 | GS28 | Intra-Golgi retrograde transport | - | - | |
| GOSR2 | GS27/membrin | ER-Golgi transport | - | c.430G > T (p.Gly144Trp) | [196,256,257] |
| YKT6 | Golgi organization and autophagy | ||||
| SNAP29 | Golgi trafficking and autophagy | - | 22q11.2 deletion | [208,210] | |
| STX16 | Endosome to TGN transport and maintaining Golgi structure | - | 3kb deletion in STX16 gene | [211] |
Author Contributions
Conceptualization, Z.D. and V.L.; writing—original draft preparation, Z.D., F.T.S. and A.K.; writing—review and editing, Z.D., F.T.S., A.K. and V.L.; supervision, V.L.; project administration, Z.D. and V.L.; funding acquisition, V.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Institute of General Medical Sciences, grant number R01 GM083144.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Casares, D.; Escriba, P.V.; Rossello, C.A. Membrane Lipid Composition: Effect on Membrane and Organelle Structure, Function and Compartmentalization and Therapeutic Avenues. Int. J. Mol. Sci. 2019, 20, 2167. [Google Scholar] [CrossRef] [Green Version]
- Park, K.; Ju, S.; Kim, N.; Park, S.Y. The Golgi complex: A hub of the secretory pathway. BMB Rep. 2021, 54, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, R.; Sticco, L.; Luini, A. Regulation of cargo export and sorting at the trans-Golgi network. FEBS Lett. 2019, 593, 2306–2318. [Google Scholar] [CrossRef] [Green Version]
- Glick, B.S.; Luini, A. Models for Golgi traffic: A critical assessment. Cold Spring Harb. Perspect. Biol. 2011, 3, a005215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luini, A. A brief history of the cisternal progression-maturation model. Cell. Logist. 2011, 1, 6–11. [Google Scholar] [CrossRef] [Green Version]
- Fisher, P.; Thomas-Oates, J.; Wood, A.J.; Ungar, D. The N-Glycosylation Processing Potential of the Mammalian Golgi Apparatus. Front. Cell Dev. Biol. 2019, 7, 157. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Wang, H.; Gai, J.; Tian, X.; Zhang, X.; Lv, Y.; Jian, Y. Evolution of protein N-glycosylation process in Golgi apparatus which shapes diversity of protein N-glycan structures in plants, animals and fungi. Sci. Rep. 2017, 7, 40301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanley, P. Golgi glycosylation. Cold Spring Harb. Perspect. Biol. 2011, 3, a005199. [Google Scholar] [CrossRef] [PubMed]
- Reynders, E.; Foulquier, F.; Annaert, W.; Matthijs, G. How Golgi glycosylation meets and needs trafficking: The case of the COG complex. Glycobiology 2011, 21, 853–863. [Google Scholar] [CrossRef] [Green Version]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef] [Green Version]
- Ondruskova, N.; Cechova, A.; Hansikova, H.; Honzik, T.; Jaeken, J. Congenital disorders of glycosylation: Still “hot” in 2020. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129751. [Google Scholar] [CrossRef] [PubMed]
- Linders, P.T.A.; Peters, E.; Ter Beest, M.; Lefeber, D.J.; van den Bogaart, G. Sugary Logistics Gone Wrong: Membrane Trafficking and Congenital Disorders of Glycosylation. Int. J. Mol. Sci. 2020, 21, 4654. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J. Congenital disorders of glycosylation (CDG): It’s (nearly) all in it! J. Inherit. Metab. Dis. 2011, 34, 853–858. [Google Scholar] [CrossRef]
- Jaeken, J.; Hennet, T.; Matthijs, G.; Freeze, H.H. CDG nomenclature: Time for a change! Biochim. Biophys. Acta 2009, 1792, 825–826. [Google Scholar] [CrossRef] [Green Version]
- Lefeber, D.J.; Morava, E.; Jaeken, J. How to find and diagnose a CDG due to defective N-glycosylation. J. Inherit. Metab. Dis. 2011, 34, 849–852. [Google Scholar] [CrossRef] [Green Version]
- Berninsone, P.M.; Hirschberg, C.B. Nucleotide sugar transporters of the Golgi apparatus. Curr. Opin. Struct. Biol. 2000, 10, 542–547. [Google Scholar] [CrossRef]
- Maeda, Y.; Ide, T.; Koike, M.; Uchiyama, Y.; Kinoshita, T. GPHR is a novel anion channel critical for acidification and functions of the Golgi apparatus. Nat. Cell Biol. 2008, 10, 1135–1145. [Google Scholar] [CrossRef]
- Maeda, Y.; Kinoshita, T. The acidic environment of the Golgi is critical for glycosylation and transport. Methods Enzymol. 2010, 480, 495–510. [Google Scholar] [CrossRef]
- Banfield, D.K. Mechanisms of protein retention in the Golgi. Cold Spring Harb. Perspect. Biol. 2011, 3, a005264. [Google Scholar] [CrossRef]
- Tu, L.; Banfield, D.K. Localization of Golgi-resident glycosyltransferases. Cell. Mol. Life Sci. CMLS 2010, 67, 29–41. [Google Scholar] [CrossRef]
- Welch, L.G.; Munro, S. A tale of short tails, through thick and thin: Investigating the sorting mechanisms of Golgi enzymes. FEBS Lett. 2019, 593, 2452–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, T.; Slusarewicz, P.; Hoe, M.H.; Warren, G. Kin recognition. A model for the retention of Golgi enzymes. FEBS Lett. 1993, 330, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, T.; Hoe, M.H.; Slusarewicz, P.; Rabouille, C.; Watson, R.; Hunte, F.; Watzele, G.; Berger, E.G.; Warren, G. Kin recognition between medial Golgi enzymes in HeLa cells. EMBO J. 1994, 13, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Munro, S. Sequences within and adjacent to the transmembrane segment of alpha-2,6-sialyltransferase specify Golgi retention. EMBO J. 1991, 10, 3577–3588. [Google Scholar] [CrossRef] [PubMed]
- Colley, K.J.; Lee, E.U.; Paulson, J.C. The signal anchor and stem regions of the beta-galactoside alpha 2,6-sialyltransferase may each act to localize the enzyme to the Golgi apparatus. J. Biol. Chem. 1992, 267, 7784–7793. [Google Scholar] [CrossRef]
- Khoder-Agha, F.; Harrus, D.; Brysbaert, G.; Lensink, M.F.; Harduin-Lepers, A.; Glumoff, T.; Kellokumpu, S. Assembly of B4GALT1/ST6GAL1 heteromers in the Golgi membranes involves lateral interactions via highly charged surface domains. J. Biol. Chem. 2019, 294, 14383–14393. [Google Scholar] [CrossRef] [PubMed]
- de Graffenried, C.L.; Bertozzi, C.R. The roles of enzyme localisation and complex formation in glycan assembly within the Golgi apparatus. Curr. Opin. Cell Biol. 2004, 16, 356–363. [Google Scholar] [CrossRef]
- Munro, S. An investigation of the role of transmembrane domains in Golgi protein retention. EMBO J. 1995, 14, 4695–4704. [Google Scholar] [CrossRef]
- Sharpe, H.J.; Stevens, T.J.; Munro, S. A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell 2010, 142, 158–169. [Google Scholar] [CrossRef] [Green Version]
- Frappaolo, A.; Karimpour-Ghahnavieh, A.; Sechi, S.; Giansanti, M.G. The Close Relationship between the Golgi Trafficking Machinery and Protein Glycosylation. Cells 2020, 9, 2652. [Google Scholar] [CrossRef] [PubMed]
- Welch, L.G.; Peak-Chew, S.Y.; Begum, F.; Stevens, T.J.; Munro, S. GOLPH3 and GOLPH3L are broad-spectrum COPI adaptors for sorting into intra-Golgi transport vesicles. J. Cell Biol. 2021, 220. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P.; Satpute-Krishnan, P.; Seo, A.Y.; Burnette, D.T.; Patterson, G.H.; Lippincott-Schwartz, J. ER trapping reveals Golgi enzymes continually revisit the ER through a recycling pathway that controls Golgi organization. Proc. Natl. Acad. Sci. USA 2015, 112, E6752–E6761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, S.L.; Waters, M.G. Localization of a yeast early Golgi mannosyltransferase, Och1p, involves retrograde transport. J. Cell Biol. 1996, 132, 985–998. [Google Scholar] [CrossRef]
- Todorow, Z.; Spang, A.; Carmack, E.; Yates, J.; Schekman, R. Active recycling of yeast Golgi mannosyltransferase complexes through the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2000, 97, 13643–13648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munro, S.; Pelham, H.R. A C-terminal signal prevents secretion of luminal ER proteins. Cell 1987, 48, 899–907. [Google Scholar] [CrossRef]
- McMahon, H.T.; Mills, I.G. COP and clathrin-coated vesicle budding: Different pathways, common approaches. Curr. Opin. Cell Biol. 2004, 16, 379–391. [Google Scholar] [CrossRef]
- Michelsen, K.; Yuan, H.; Schwappach, B. Hide and run. Arginine-based endoplasmic-reticulum-sorting motifs in the assembly of heteromultimeric membrane proteins. EMBO Rep. 2005, 6, 717–722. [Google Scholar] [CrossRef]
- Liu, L.; Doray, B.; Kornfeld, S. Recycling of Golgi glycosyltransferases requires direct binding to coatomer. Proc. Natl. Acad. Sci. USA 2018, 115, 8984–8989. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, K.R.; Liu, J.; Li, S.; Setty, T.G.; Wood, C.S.; Burd, C.G.; Ferguson, K.M. Golgi localization of glycosyltransferases requires a Vps74p oligomer. Dev. Cell 2008, 14, 523–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, L.; Tai, W.C.; Chen, L.; Banfield, D.K. Signal-mediated dynamic retention of glycosyltransferases in the Golgi. Science 2008, 321, 404–407. [Google Scholar] [CrossRef]
- Eckert, E.S.; Reckmann, I.; Hellwig, A.; Rohling, S.; El-Battari, A.; Wieland, F.T.; Popoff, V. Golgi phosphoprotein 3 triggers signal-mediated incorporation of glycosyltransferases into coatomer-coated (COPI) vesicles. J. Biol. Chem. 2014, 289, 31319–31329. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, R.; Russo, D.; Kurokawa, K.; Sahu, P.; Lombardi, B.; Supino, D.; Zhukovsky, M.A.; Vocat, A.; Pothukuchi, P.; Kunnathully, V.; et al. Golgi maturation-dependent glycoenzyme recycling controls glycosphingolipid biosynthesis and cell growth via GOLPH3. EMBO J. 2021, 40, e107238. [Google Scholar] [CrossRef] [PubMed]
- Kirchhausen, T. Three ways to make a vesicle. Nat. Rev. Mol. Cell Biol. 2000, 1, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Glick, B.S. The mechanisms of vesicle budding and fusion. Cell 2004, 116, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Cevher-Keskin, B. ARF1 and SAR1 GTPases in endomembrane trafficking in plants. Int. J. Mol. Sci. 2013, 14, 18181–18199. [Google Scholar] [CrossRef]
- Zhukovsky, M.A.; Filograna, A.; Luini, A.; Corda, D.; Valente, C. Protein Amphipathic Helix Insertion: A Mechanism to Induce Membrane Fission. Front. Cell Dev. Biol. 2019, 7, 291. [Google Scholar] [CrossRef] [PubMed]
- Witkos, T.M.; Lowe, M. The Golgin Family of Coiled-Coil Tethering Proteins. Front. Cell Dev. Biol. 2015, 3, 86. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Chang, J.; Wang, X.; Savelieff, M.G.; Zhao, Y.; Ke, S.; Ye, B. GM130 is required for compartmental organization of dendritic golgi outposts. Curr. Biol. CB 2014, 24, 1227–1233. [Google Scholar] [CrossRef] [Green Version]
- Shamseldin, H.E.; Bennett, A.H.; Alfadhel, M.; Gupta, V.; Alkuraya, F.S. GOLGA2, encoding a master regulator of golgi apparatus, is mutated in a patient with a neuromuscular disorder. Hum. Genet. 2016, 135, 245–251. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Mei, M.; Li, Q.; Roboti, P.; Pang, Q.; Ying, Z.; Gao, F.; Lowe, M.; Bao, S. Loss of the golgin GM130 causes Golgi disruption, Purkinje neuron loss, and ataxia in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 346–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Kim, S.; Kim, M.J.; Hong, Y.; Lee, A.Y.; Lee, H.; Tran, Q.; Kim, M.; Cho, H.; Park, J.; et al. GOLGA2 loss causes fibrosis with autophagy in the mouse lung and liver. Biochem. Biophys. Res. Commun. 2018, 495, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, U.; Mistri, M.; Shah, N.; Shah, P.S.; Gupta, V.A. Bi-allelic loss of function variants in GOLGA2 are associated with a complex neurological phenotype: Report of a second family. Clin. Genet. 2021, 100, 748–751. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ye, M.; Zhao, Q.; Xia, M.; Liu, D.; He, L.; Chen, G.; Peng, Y.; Liu, H. Loss of the Golgi Matrix Protein 130 Cause Aberrant IgA1 Glycosylation in IgA Nephropathy. Am. J. Nephrol. 2019, 49, 307–316. [Google Scholar] [CrossRef]
- Puthenveedu, M.A.; Bachert, C.; Puri, S.; Lanni, F.; Linstedt, A.D. GM130 and GRASP65-dependent lateral cisternal fusion allows uniform Golgi-enzyme distribution. Nat. Cell Biol. 2006, 8, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Petrosyan, A.; Ali, M.F.; Cheng, P.W. Glycosyltransferase-specific Golgi-targeting mechanisms. J. Biol. Chem. 2012, 287, 37621–37627. [Google Scholar] [CrossRef] [Green Version]
- Rios, R.M.; Sanchis, A.; Tassin, A.M.; Fedriani, C.; Bornens, M. GMAP-210 recruits gamma-tubulin complexes to cis-Golgi membranes and is required for Golgi ribbon formation. Cell 2004, 118, 323–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, S.; Puri, S.; Linstedt, A.D. A primary role for Golgi positioning in directed secretion, cell polarity, and wound healing. Mol. Biol. Cell 2009, 20, 1728–1736. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Roboti, P.; Mironov, A.A.; Lowe, M. Coupling of vesicle tethering and Rab binding is required for in vivo functionality of the golgin GMAP-210. Mol. Biol. Cell 2015, 26, 537–553. [Google Scholar] [CrossRef] [Green Version]
- Roboti, P.; Sato, K.; Lowe, M. The golgin GMAP-210 is required for efficient membrane trafficking in the early secretory pathway. J. Cell Sci. 2015, 128, 1595–1606. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.; Gillingham, A.K.; Munro, S. The golgin coiled-coil proteins capture different types of transport carriers via distinct N-terminal motifs. BMC Biol. 2017, 15, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smits, P.; Bolton, A.D.; Funari, V.; Hong, M.; Boyden, E.D.; Lu, L.; Manning, D.K.; Dwyer, N.D.; Moran, J.L.; Prysak, M.; et al. Lethal skeletal dysplasia in mice and humans lacking the golgin GMAP-210. N. Engl. J. Med. 2010, 362, 206–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehrle, A.; Witkos, T.M.; Unger, S.; Schneider, J.; Follit, J.A.; Hermann, J.; Welting, T.; Fano, V.; Hietala, M.; Vatanavicharn, N.; et al. Hypomorphic mutations of TRIP11 cause odontochondrodysplasia. JCI Insight 2019, 4, e124701. [Google Scholar] [CrossRef]
- Medina, C.T.N.; Sandoval, R.; Oliveira, G.; da Costa Silveira, K.; Cavalcanti, D.P.; Pogue, R. Pathogenic variants in the TRIP11 gene cause a skeletal dysplasia spectrum from odontochondrodysplasia to achondrogenesis 1A. Am. J. Med. Genet. Part A 2020, 182, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Del Pino, M.; Sanchez-Soler, M.J.; Parron-Pajares, M.; Aza-Carmona, M.; Heath, K.E.; Fano, V. Description of four patients with TRIP11 variants expand the clinical spectrum of odontochondroplasia (ODCD) and demonstrate the existence of common variants. Eur. J. Med. Genet. 2021, 64, 104198. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Hu, G.; Chen, M.; Liu, B.; Yan, K.; Zhou, C.; Yu, Y.; Dong, M. Novel deep intronic and frameshift mutations causing a TRIP11-related disorder. Am. J. Med. Genet. Part A 2021, 185, 2482–2487. [Google Scholar] [CrossRef]
- Yadav, S.; Puthenveedu, M.A.; Linstedt, A.D. Golgin160 recruits the dynein motor to position the Golgi apparatus. Dev. Cell 2012, 23, 153–165. [Google Scholar] [CrossRef] [Green Version]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef]
- Nelson, D.S.; Alvarez, C.; Gao, Y.S.; Garcia-Mata, R.; Fialkowski, E.; Sztul, E. The membrane transport factor TAP/p115 cycles between the Golgi and earlier secretory compartments and contains distinct domains required for its localization and function. J. Cell Biol. 1998, 143, 319–331. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, C.; Garcia-Mata, R.; Hauri, H.P.; Sztul, E. The p115-interactive proteins GM130 and giantin participate in endoplasmic reticulum-Golgi traffic. J. Biol. Chem. 2001, 276, 2693–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Grabski, R.; Sztul, E.; Hay, J.C. p115-SNARE interactions: A dynamic cycle of p115 binding monomeric SNARE motifs and releasing assembled bundles. Traffic 2015, 16, 148–171. [Google Scholar] [CrossRef] [Green Version]
- Sapperstein, S.K.; Walter, D.M.; Grosvenor, A.R.; Heuser, J.E.; Waters, M.G. p115 is a general vesicular transport factor related to the yeast endoplasmic reticulum to Golgi transport factor Uso1p. Proc. Natl. Acad. Sci. USA 1995, 92, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, C.; Fujita, H.; Hubbard, A.; Sztul, E. ER to Golgi transport: Requirement for p115 at a pre-Golgi VTC stage. J. Cell Biol. 1999, 147, 1205–1222. [Google Scholar] [CrossRef]
- Allan, B.B.; Moyer, B.D.; Balch, W.E. Rab1 recruitment of p115 into a cis-SNARE complex: Programming budding COPII vesicles for fusion. Science 2000, 289, 444–448. [Google Scholar] [CrossRef]
- Shorter, J.; Beard, M.B.; Seemann, J.; Dirac-Svejstrup, A.B.; Warren, G. Sequential tethering of Golgins and catalysis of SNAREpin assembly by the vesicle-tethering protein p115. J. Cell Biol. 2002, 157, 45–62. [Google Scholar] [CrossRef] [Green Version]
- Shorter, J.; Warren, G. A role for the vesicle tethering protein, p115, in the post-mitotic stacking of reassembling Golgi cisternae in a cell-free system. J. Cell Biol. 1999, 146, 57–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohda, M.; Misumi, Y.; Yoshimura, S.; Nakamura, N.; Fusano, T.; Ogata, S.; Sakisaka, S.; Ikehara, Y. The interaction of two tethering factors, p115 and COG complex, is required for Golgi integrity. Traffic 2007, 8, 270–284. [Google Scholar] [CrossRef]
- Sohda, M.; Misumi, Y.; Yoshimura, S.; Nakamura, N.; Fusano, T.; Sakisaka, S.; Ogata, S.; Fujimoto, J.; Kiyokawa, N.; Ikehara, Y. Depletion of vesicle-tethering factor p115 causes mini-stacked Golgi fragments with delayed protein transport. Biochem. Biophys. Res. Commun. 2005, 338, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.A.; Short, B. Golgins in the structure and dynamics of the Golgi apparatus. Curr. Opin. Cell Biol. 2003, 15, 405–413. [Google Scholar] [CrossRef]
- Koreishi, M.; Gniadek, T.J.; Yu, S.; Masuda, J.; Honjo, Y.; Satoh, A. The golgin tether giantin regulates the secretory pathway by controlling stack organization within Golgi apparatus. PLoS ONE 2013, 8, e59821. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, N.L.; Bergen, D.J.M.; Skinner, R.E.H.; Kague, E.; Martin-Silverstone, E.; Robson Brown, K.A.; Hammond, C.L.; Stephens, D.J. Giantin-knockout models reveal a feedback loop between Golgi function and glycosyltransferase expression. J. Cell Sci. 2017, 130, 4132–4143. [Google Scholar] [CrossRef] [Green Version]
- Lowe, M. The Physiological Functions of the Golgin Vesicle Tethering Proteins. Front. Cell Dev. Biol. 2019, 7, 94. [Google Scholar] [CrossRef]
- Stevenson, N.L.; Bergen, D.J.M.; Lu, Y.; Prada-Sanchez, M.E.; Kadler, K.E.; Hammond, C.L.; Stephens, D.J. Giantin is required for intracellular N-terminal processing of type I procollagen. J. Cell Biol. 2021, 220, e202005166. [Google Scholar] [CrossRef]
- Lan, Y.; Zhang, N.; Liu, H.; Xu, J.; Jiang, R. Golgb1 regulates protein glycosylation and is crucial for mammalian palate development. Development 2016, 143, 2344–2355. [Google Scholar] [CrossRef] [Green Version]
- Malsam, J.; Satoh, A.; Pelletier, L.; Warren, G. Golgin tethers define subpopulations of COPI vesicles. Science 2005, 307, 1095–1098. [Google Scholar] [CrossRef] [PubMed]
- Sohda, M.; Misumi, Y.; Yamamoto, A.; Nakamura, N.; Ogata, S.; Sakisaka, S.; Hirose, S.; Ikehara, Y.; Oda, K. Interaction of Golgin-84 with the COG complex mediates the intra-Golgi retrograde transport. Traffic 2010, 11, 1552–1566. [Google Scholar] [CrossRef]
- Munro, S. The golgin coiled-coil proteins of the Golgi apparatus. Cold Spring Harb. Perspect. Biol. 2011, 3, a005256. [Google Scholar] [CrossRef] [Green Version]
- McGee, L.J.; Jiang, A.L.; Lan, Y. Golga5 is dispensable for mouse embryonic development and postnatal survival. Genesis 2017, 55, e23039. [Google Scholar] [CrossRef]
- Griffith, K.J.; Chan, E.K.; Lung, C.C.; Hamel, J.C.; Guo, X.; Miyachi, K.; Fritzler, M.J. Molecular cloning of a novel 97-kd Golgi complex autoantigen associated with Sjogren’s syndrome. Arthritis Rheum. 1997, 40, 1693–1702. [Google Scholar] [CrossRef]
- Shin, J.J.H.; Gillingham, A.K.; Begum, F.; Chadwick, J.; Munro, S. Author Correction: TBC1D23 is a bridging factor for endosomal vesicle capture by golgins at the trans-Golgi. Nat. Cell Biol. 2018, 20, 222. [Google Scholar] [CrossRef] [Green Version]
- Takatsuki, A.; Nakamura, M.; Kono, Y. Possible implication of Golgi-nucleating function for the centrosome. Biochem. Biophys. Res. Commun. 2002, 291, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Jing, J.; Junutula, J.R.; Wu, C.; Burden, J.; Matern, H.; Peden, A.A.; Prekeris, R. FIP1/RCP binding to Golgin-97 regulates retrograde transport from recycling endosomes to the trans-Golgi network. Mol. Biol. Cell 2010, 21, 3041–3053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Tai, G.; Hong, W. Autoantigen Golgin-97, an effector of Arl1 GTPase, participates in traffic from the endosome to the trans-golgi network. Mol. Biol. Cell 2004, 15, 4426–4443. [Google Scholar] [CrossRef] [Green Version]
- Alzhanova, D.; Hruby, D.E. A host cell membrane protein, golgin-97, is essential for poxvirus morphogenesis. Virology 2007, 362, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, A.; Setty, S.R.; Poynton, C.; Whiteman, E.L.; Saint-Pol, A.; Burd, C.G.; Johannes, L.; Holzbaur, E.L.; Koval, M.; McCaffery, J.M.; et al. tGolgin-1 (p230, golgin-245) modulates Shiga-toxin transport to the Golgi and Golgi motility towards the microtubule-organizing centre. J. Cell Sci. 2005, 118, 2279–2293. [Google Scholar] [CrossRef] [Green Version]
- Kakinuma, T.; Ichikawa, H.; Tsukada, Y.; Nakamura, T.; Toh, B.H. Interaction between p230 and MACF1 is associated with transport of a glycosyl phosphatidyl inositol-anchored protein from the Golgi to the cell periphery. Exp. Cell Res. 2004, 298, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Lock, J.G.; Hammond, L.A.; Houghton, F.; Gleeson, P.A.; Stow, J.L. E-cadherin transport from the trans-Golgi network in tubulovesicular carriers is selectively regulated by golgin-97. Traffic 2005, 6, 1142–1156. [Google Scholar] [CrossRef] [PubMed]
- Lieu, Z.Z.; Lock, J.G.; Hammond, L.A.; La Gruta, N.L.; Stow, J.L.; Gleeson, P.A. A trans-Golgi network golgin is required for the regulated secretion of TNF in activated macrophages in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 3351–3356. [Google Scholar] [CrossRef] [Green Version]
- Sohda, M.; Misumi, Y.; Ogata, S.; Sakisaka, S.; Hirose, S.; Ikehara, Y.; Oda, K. Trans-Golgi protein p230/golgin-245 is involved in phagophore formation. Biochem. Biophys. Res. Commun. 2015, 456, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Lieu, Z.Z.; Derby, M.C.; Teasdale, R.D.; Hart, C.; Gunn, P.; Gleeson, P.A. The golgin GCC88 is required for efficient retrograde transport of cargo from the early endosomes to the trans-Golgi network. Mol. Biol. Cell 2007, 18, 4979–4991. [Google Scholar] [CrossRef] [Green Version]
- Luke, M.R.; Kjer-Nielsen, L.; Brown, D.L.; Stow, J.L.; Gleeson, P.A. GRIP domain-mediated targeting of two new coiled-coil proteins, GCC88 and GCC185, to subcompartments of the trans-Golgi network. J. Biol. Chem. 2003, 278, 4216–4226. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Yang, Z.; Teasdale, R.D. A role of GCC88 in the retrograde transport of CI-M6PR and the maintenance of lysosomal activity. Cell Biol. Int. 2019, 43, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Efimov, A.; Kharitonov, A.; Efimova, N.; Loncarek, J.; Miller, P.M.; Andreyeva, N.; Gleeson, P.; Galjart, N.; Maia, A.R.; McLeod, I.X.; et al. Asymmetric CLASP-dependent nucleation of noncentrosomal microtubules at the trans-Golgi network. Dev. Cell 2007, 12, 917–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganley, I.G.; Espinosa, E.; Pfeffer, S.R. A syntaxin 10-SNARE complex distinguishes two distinct transport routes from endosomes to the trans-Golgi in human cells. J. Cell Biol. 2008, 180, 159–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, J.V.; Burguete, A.S.; Sridevi, K.; Ganley, I.G.; Nottingham, R.M.; Pfeffer, S.R. A functional role for the GCC185 golgin in mannose 6-phosphate receptor recycling. Mol. Biol. Cell 2006, 17, 4353–4363. [Google Scholar] [CrossRef] [Green Version]
- Burguete, A.S.; Fenn, T.D.; Brunger, A.T.; Pfeffer, S.R. Rab and Arl GTPase family members cooperate in the localization of the golgin GCC185. Cell 2008, 132, 286–298. [Google Scholar] [CrossRef] [Green Version]
- Hayes, G.L.; Brown, F.C.; Haas, A.K.; Nottingham, R.M.; Barr, F.A.; Pfeffer, S.R. Multiple Rab GTPase binding sites in GCC185 suggest a model for vesicle tethering at the trans-Golgi. Mol. Biol. Cell 2009, 20, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Houghton, F.J.; Chew, P.L.; Lodeho, S.; Goud, B.; Gleeson, P.A. The localization of the Golgin GCC185 is independent of Rab6A/A’ and Arl1. Cell 2009, 138, 787–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derby, M.C.; Lieu, Z.Z.; Brown, D.; Stow, J.L.; Goud, B.; Gleeson, P.A. The trans-Golgi network golgin, GCC185, is required for endosome-to-Golgi transport and maintenance of Golgi structure. Traffic 2007, 8, 758–773. [Google Scholar] [CrossRef] [PubMed]
- Siniossoglou, S.; Pelham, H.R. An effector of Ypt6p binds the SNARE Tlg1p and mediates selective fusion of vesicles with late Golgi membranes. EMBO J. 2001, 20, 5991–5998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridmann-Sirkis, Y.; Siniossoglou, S.; Pelham, H.R. TMF is a golgin that binds Rab6 and influences Golgi morphology. BMC Cell Biol. 2004, 5, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamane, J.; Kubo, A.; Nakayama, K.; Yuba-Kubo, A.; Katsuno, T.; Tsukita, S.; Tsukita, S. Functional involvement of TMF/ARA160 in Rab6-dependent retrograde membrane traffic. Exp. Cell Res. 2007, 313, 3472–3485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, E.; Tsruya, R.; Levitsky, P.; Pomp, O.; Taller, M.; Weisberg, S.; Parris, W.; Kulkarni, S.; Malovani, H.; Pawson, T.; et al. TMF/ARA160 is a BC-box-containing protein that mediates the degradation of Stat3. Oncogene 2004, 23, 8908–8919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrham, G.; Volpe, M.; Shpungin, S.; Nir, U. TMF/ARA160 downregulates proangiogenic genes and attenuates the progression of PC3 xenografts. Int. J. Cancer 2009, 125, 43–53. [Google Scholar] [CrossRef]
- Lerer-Goldshtein, T.; Bel, S.; Shpungin, S.; Pery, E.; Motro, B.; Goldstein, R.S.; Bar-Sheshet, S.I.; Breitbart, H.; Nir, U. TMF/ARA160: A key regulator of sperm development. Dev. Biol. 2010, 348, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Elkis, Y.; Bel, S.; Lerer-Goldstein, T.; Nyska, A.; Creasy, D.M.; Shpungin, S.; Nir, U. Testosterone deficiency accompanied by testicular and epididymal abnormalities in TMF(-/-) mice. Mol. Cell. Endocrinol. 2013, 365, 52–63. [Google Scholar] [CrossRef]
- Elkis, Y.; Bel, S.; Rahimi, R.; Lerer-Goldstein, T.; Levin-Zaidman, S.; Babushkin, T.; Shpungin, S.; Nir, U. TMF/ARA160 Governs the Dynamic Spatial Orientation of the Golgi Apparatus during Sperm Development. PLoS ONE 2015, 10, e0145277. [Google Scholar] [CrossRef]
- Rahimi, R.; Malek, I.; Lerrer-Goldshtein, T.; Elkis, Y.; Shoval, I.; Jacob, A.; Shpungin, S.; Nir, U. TMF1 is upregulated by insulin and is required for a sustained glucose homeostasis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21295. [Google Scholar] [CrossRef]
- Kummel, D.; Reinisch, K.M. Structure of Golgi transport proteins. Cold Spring Harb. Perspect. Biol. 2011, 3, a007609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekier, M.E., 2nd; Wang, L.; Li, J.; Huang, H.; Tang, D.; Zhang, X.; Wang, Y. Knockout of the Golgi stacking proteins GRASP55 and GRASP65 impairs Golgi structure and function. Mol. Biol. Cell 2017, 28, 2833–2842. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.A.; Nakamura, N.; Warren, G. Mapping the interaction between GRASP65 and GM130, components of a protein complex involved in the stacking of Golgi cisternae. EMBO J. 1998, 17, 3258–3268. [Google Scholar] [CrossRef]
- Shorter, J.; Watson, R.; Giannakou, M.E.; Clarke, M.; Warren, G.; Barr, F.A. GRASP55, a second mammalian GRASP protein involved in the stacking of Golgi cisternae in a cell-free system. EMBO J. 1999, 18, 4949–4960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabouille, C.; Linstedt, A.D. GRASP: A Multitasking Tether. Front. Cell Dev. Biol. 2016, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Seemann, J. Rapid degradation of GRASP55 and GRASP65 reveals their immediate impact on the Golgi structure. J. Cell Biol. 2021, 220, e202007052. [Google Scholar] [CrossRef] [PubMed]
- Short, B.; Preisinger, C.; Korner, R.; Kopajtich, R.; Byron, O.; Barr, F.A. A GRASP55-rab2 effector complex linking Golgi structure to membrane traffic. J. Cell Biol. 2001, 155, 877–883. [Google Scholar] [CrossRef]
- Xiang, Y.; Zhang, X.; Nix, D.B.; Katoh, T.; Aoki, K.; Tiemeyer, M.; Wang, Y. Regulation of protein glycosylation and sorting by the Golgi matrix proteins GRASP55/65. Nat. Commun. 2013, 4, 1659. [Google Scholar] [CrossRef] [Green Version]
- Ungar, D.; Oka, T.; Brittle, E.E.; Vasile, E.; Lupashin, V.V.; Chatterton, J.E.; Heuser, J.E.; Krieger, M.; Waters, M.G. Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J. Cell Biol. 2002, 157, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.A.; Yip, C.K.; Walz, T.; Hughson, F.M. Molecular organization of the COG vesicle tethering complex. Nat. Struct. Mol. Biol. 2010, 17, 1292–1297. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, J.B.; D’Souza, Z.; Lupashin, V.V. Maintaining order: COG complex controls Golgi trafficking, processing, and sorting. FEBS Lett. 2019, 593, 2466–2487. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, Z.; Taher, F.S.; Lupashin, V.V. Golgi inCOGnito: From vesicle tethering to human disease. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129694. [Google Scholar] [CrossRef] [PubMed]
- Whyte, J.R.; Munro, S. The Sec34/35 Golgi transport complex is related to the exocyst, defining a family of complexes involved in multiple steps of membrane traffic. Dev. Cell 2001, 1, 527–537. [Google Scholar] [CrossRef]
- Fotso, P.; Koryakina, Y.; Pavliv, O.; Tsiomenko, A.B.; Lupashin, V.V. Cog1p plays a central role in the organization of the yeast conserved oligomeric Golgi complex. J. Biol. Chem. 2005, 280, 27613–27623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willett, R.; Ungar, D.; Lupashin, V. The Golgi puppet master: COG complex at center stage of membrane trafficking interactions. Histochem. Cell Biol. 2013, 140, 271–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungar, D.; Oka, T.; Vasile, E.; Krieger, M.; Hughson, F.M. Subunit architecture of the conserved oligomeric Golgi complex. J. Biol. Chem. 2005, 280, 32729–32735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, J.Y.; Chou, H.T.; Ungar, D.; Yip, C.K.; Walz, T.; Hughson, F.M. Molecular architecture of the complete COG tethering complex. Nat. Struct. Mol. Biol. 2016, 23, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Willett, R.; Blackburn, J.B.; Climer, L.; Pokrovskaya, I.; Kudlyk, T.; Wang, W.; Lupashin, V. COG lobe B sub-complex engages v-SNARE GS15 and functions via regulated interaction with lobe A sub-complex. Sci. Rep. 2016, 6, 29139. [Google Scholar] [CrossRef] [Green Version]
- Shestakova, A.; Zolov, S.; Lupashin, V. COG complex-mediated recycling of Golgi glycosyltransferases is essential for normal protein glycosylation. Traffic 2006, 7, 191–204. [Google Scholar] [CrossRef]
- Steet, R.; Kornfeld, S. COG-7-deficient Human Fibroblasts Exhibit Altered Recycling of Golgi Proteins. Mol. Biol. Cell 2006, 17, 2312–2321. [Google Scholar] [CrossRef] [Green Version]
- Pokrovskaya, I.D.; Willett, R.; Smith, R.D.; Morelle, W.; Kudlyk, T.; Lupashin, V.V. Conserved oligomeric Golgi complex specifically regulates the maintenance of Golgi glycosylation machinery. Glycobiology 2011, 21, 1554–1569. [Google Scholar] [CrossRef] [Green Version]
- Willett, R.; Pokrovskaya, I.; Kudlyk, T.; Lupashin, V. Multipronged interaction of the COG complex with intracellular membranes. Cell. Logist. 2014, 4, e27888. [Google Scholar] [CrossRef] [Green Version]
- Bailey Blackburn, J.; Pokrovskaya, I.; Fisher, P.; Ungar, D.; Lupashin, V.V. COG Complex Complexities: Detailed Characterization of a Complete Set of HEK293T Cells Lacking Individual COG Subunits. Front. Cell Dev. Biol. 2016, 4, 23. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, J.B.; Kudlyk, T.; Pokrovskaya, I.; Lupashin, V.V. More than just sugars: Conserved oligomeric Golgi complex deficiency causes glycosylation-independent cellular defects. Traffic 2018, 19, 463–480. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, Z.; Blackburn, J.B.; Kudlyk, T.; Pokrovskaya, I.D.; Lupashin, V.V. Defects in COG-Mediated Golgi Trafficking Alter Endo-Lysosomal System in Human Cells. Front. Cell Dev. Biol. 2019, 7, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeevaert, R.; Foulquier, F.; Jaeken, J.; Matthijs, G. Deficiencies in subunits of the Conserved Oligomeric Golgi (COG) complex define a novel group of Congenital Disorders of Glycosylation. Mol. Genet. Metab. 2008, 93, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Haijes, H.A.; Jaeken, J.; Foulquier, F.; van Hasselt, P.M. Hypothesis: Lobe A (COG1-4)-CDG causes a more severe phenotype than lobe B (COG5-8)-CDG. J. Med. Genet. 2018, 55, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Reynders, E.; Foulquier, F.; Leao Teles, E.; Quelhas, D.; Morelle, W.; Rabouille, C.; Annaert, W.; Matthijs, G. Golgi function and dysfunction in the first COG4-deficient CDG type II patient. Hum. Mol. Genet. 2009, 18, 3244–3256. [Google Scholar] [CrossRef]
- Rymen, D.; Winter, J.; Van Hasselt, P.M.; Jaeken, J.; Kasapkara, C.; Gokcay, G.; Haijes, H.; Goyens, P.; Tokatli, A.; Thiel, C.; et al. Key features and clinical variability of COG6-CDG. Mol. Genet. Metab. 2015, 116, 163–170. [Google Scholar] [CrossRef]
- Peanne, R.; de Lonlay, P.; Foulquier, F.; Kornak, U.; Lefeber, D.J.; Morava, E.; Perez, B.; Seta, N.; Thiel, C.; Van Schaftingen, E.; et al. Congenital disorders of glycosylation (CDG): Quo vadis? Eur. J. Med. Genet. 2018, 61, 643–663. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Hierro, A. Transport according to GARP: Receiving retrograde cargo at the trans-Golgi network. Trends Cell Biol. 2011, 21, 159–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, H.T.; Dukovski, D.; Chambers, M.G.; Reinisch, K.M.; Walz, T. CATCHR, HOPS and CORVET tethering complexes share a similar architecture. Nat. Struct. Mol. Biol. 2016, 23, 761–763. [Google Scholar] [CrossRef] [Green Version]
- Santana-Molina, C.; Gutierrez, F.; Devos, D.P. Homology and Modular Evolution of CATCHR at the Origin of the Eukaryotic Endomembrane System. Genome Biol. Evol. 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Conibear, E.; Stevens, T.H. Vps52p, Vps53p, and Vps54p form a novel multisubunit complex required for protein sorting at the yeast late Golgi. Mol. Biol. Cell 2000, 11, 305–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siniossoglou, S.; Pelham, H.R. Vps51p links the VFT complex to the SNARE Tlg1p. J. Biol. Chem. 2002, 277, 48318–48324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conibear, E.; Cleck, J.N.; Stevens, T.H. Vps51p mediates the association of the GARP (Vps52/53/54) complex with the late Golgi t-SNARE Tlg1p. Mol. Biol. Cell 2003, 14, 1610–1623. [Google Scholar] [CrossRef] [Green Version]
- Brocker, C.; Engelbrecht-Vandre, S.; Ungermann, C. Multisubunit tethering complexes and their role in membrane fusion. Curr. Biol. CB 2010, 20, R943–R952. [Google Scholar] [CrossRef] [Green Version]
- Perez-Victoria, F.J.; Schindler, C.; Magadan, J.G.; Mardones, G.A.; Delevoye, C.; Romao, M.; Raposo, G.; Bonifacino, J.S. Ang2/fat-free is a conserved subunit of the Golgi-associated retrograde protein complex. Mol. Biol. Cell 2010, 21, 3386–3395. [Google Scholar] [CrossRef] [Green Version]
- Liewen, H.; Meinhold-Heerlein, I.; Oliveira, V.; Schwarzenbacher, R.; Luo, G.; Wadle, A.; Jung, M.; Pfreundschuh, M.; Stenner-Liewen, F. Characterization of the human GARP (Golgi associated retrograde protein) complex. Exp. Cell Res. 2005, 306, 24–34. [Google Scholar] [CrossRef]
- Walter, L.; Stark, S.; Helou, K.; Flugge, P.; Levan, G.; Gunther, E. Identification, characterization and cytogenetic mapping of a yeast Vps54 homolog in rat and mouse. Gene 2002, 285, 213–220. [Google Scholar] [CrossRef]
- Lobstein, E.; Guyon, A.; Ferault, M.; Twell, D.; Pelletier, G.; Bonhomme, S. The putative Arabidopsis homolog of yeast vps52p is required for pollen tube elongation, localizes to Golgi, and might be involved in vesicle trafficking. Plant Physiol. 2004, 135, 1480–1490. [Google Scholar] [CrossRef] [Green Version]
- Oka, T.; Krieger, M. Multi-component protein complexes and Golgi membrane trafficking. J. Biochem. 2005, 137, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Ishida, M.; Bonifacino, J.S. ARFRP1 functions upstream of ARL1 and ARL5 to coordinate recruitment of distinct tethering factors to the trans-Golgi network. J. Cell Biol. 2019, 218, 3681–3696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindler, C.; Chen, Y.; Pu, J.; Guo, X.; Bonifacino, J.S. EARP is a multisubunit tethering complex involved in endocytic recycling. Nat. Cell Biol. 2015, 17, 639–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quenneville, N.R.; Chao, T.Y.; McCaffery, J.M.; Conibear, E. Domains within the GARP subunit Vps54 confer separate functions in complex assembly and early endosome recognition. Mol. Biol. Cell 2006, 17, 1859–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonifacino, J.S.; Rojas, R. Retrograde transport from endosomes to the trans-Golgi network. Nat. Rev. Mol. Cell Biol. 2006, 7, 568–579. [Google Scholar] [CrossRef]
- Perez-Victoria, F.J.; Mardones, G.A.; Bonifacino, J.S. Requirement of the human GARP complex for mannose 6-phosphate-receptor-dependent sorting of cathepsin D to lysosomes. Mol. Biol. Cell 2008, 19, 2350–2362. [Google Scholar] [CrossRef] [Green Version]
- Feinstein, M.; Flusser, H.; Lerman-Sagie, T.; Ben-Zeev, B.; Lev, D.; Agamy, O.; Cohen, I.; Kadir, R.; Sivan, S.; Leshinsky-Silver, E.; et al. VPS53 mutations cause progressive cerebello-cerebral atrophy type 2 (PCCA2). J. Med. Genet. 2014, 51, 303–308. [Google Scholar] [CrossRef] [Green Version]
- Frohlich, F.; Petit, C.; Kory, N.; Christiano, R.; Hannibal-Bach, H.K.; Graham, M.; Liu, X.; Ejsing, C.S.; Farese, R.V.; Walther, T.C. The GARP complex is required for cellular sphingolipid homeostasis. eLife 2015, 4, e08712. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhang, Y.Y.; Luo, J.; Wang, J.Q.; Zhou, Y.X.; Miao, H.H.; Shi, X.J.; Qu, Y.X.; Xu, J.; Li, B.L.; et al. The GARP Complex Is Involved in Intracellular Cholesterol Transport via Targeting NPC2 to Lysosomes. Cell Rep. 2017, 19, 2823–2835. [Google Scholar] [CrossRef] [Green Version]
- Petit, C.S.; Lee, J.J.; Boland, S.; Swarup, S.; Christiano, R.; Lai, Z.W.; Mejhert, N.; Elliott, S.D.; McFall, D.; Haque, S.; et al. Inhibition of sphingolipid synthesis improves outcomes and survival in GARP mutant wobbler mice, a model of motor neuron degeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 10565–10574. [Google Scholar] [CrossRef] [PubMed]
- Eising, S.; Thiele, L.; Frohlich, F. A systematic approach to identify recycling endocytic cargo depending on the GARP complex. eLife 2019, 8, e42837. [Google Scholar] [CrossRef] [PubMed]
- Gershlick, D.C.; Ishida, M.; Jones, J.R.; Bellomo, A.; Bonifacino, J.S.; Everman, D.B. A neurodevelopmental disorder caused by mutations in the VPS51 subunit of the GARP and EARP complexes. Hum. Mol. Genet. 2019, 28, 1548–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beilina, A.; Bonet-Ponce, L.; Kumaran, R.; Kordich, J.J.; Ishida, M.; Mamais, A.; Kaganovich, A.; Saez-Atienzar, S.; Gershlick, D.C.; Roosen, D.A.; et al. The Parkinson’s Disease Protein LRRK2 Interacts with the GARP Complex to Promote Retrograde Transport to the trans-Golgi Network. Cell Rep. 2020, 31, 107614. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, P.; Droce, A.; Moser, J.M.; Cuhlmann, S.; Padilla, C.O.; Heimann, P.; Bartsch, J.W.; Fuchtbauer, A.; Fuchtbauer, E.M.; Schmitt-John, T. Loss of vps54 function leads to vesicle traffic impairment, protein mis-sorting and embryonic lethality. Int. J. Mol. Sci. 2013, 14, 10908–10925. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-John, T.; Drepper, C.; Mussmann, A.; Hahn, P.; Kuhlmann, M.; Thiel, C.; Hafner, M.; Lengeling, A.; Heimann, P.; Jones, J.M.; et al. Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse. Nat. Genet. 2005, 37, 1213–1215. [Google Scholar] [CrossRef]
- Perez-Victoria, F.J.; Abascal-Palacios, G.; Tascon, I.; Kajava, A.; Magadan, J.G.; Pioro, E.P.; Bonifacino, J.S.; Hierro, A. Structural basis for the wobbler mouse neurodegenerative disorder caused by mutation in the Vps54 subunit of the GARP complex. Proc. Natl. Acad. Sci.USA 2010, 107, 12860–12865. [Google Scholar] [CrossRef] [Green Version]
- Paiardi, C.; Pasini, M.E.; Gioria, M.; Berruti, G. Failure of acrosome formation and globozoospermia in the wobbler mouse, a Vps54 spontaneous recessive mutant. Spermatogenesis 2011, 1, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Moser, J.M.; Bigini, P.; Schmitt-John, T. The wobbler mouse, an ALS animal model. Mol. Genet. Genom. MGG 2013, 288, 207–229. [Google Scholar] [CrossRef] [Green Version]
- Jockusch, H.; Holland, A.; Staunton, L.; Schmitt-John, T.; Heimann, P.; Dowling, P.; Ohlendieck, K. Pathoproteomics of testicular tissue deficient in the GARP component VPS54: The wobbler mouse model of globozoospermia. Proteomics 2014, 14, 839–852. [Google Scholar] [CrossRef]
- Schmitt-John, T. VPS54 and the wobbler mouse. Front. Neurosci. 2015, 9, 381. [Google Scholar] [CrossRef] [Green Version]
- Vasan, N.; Hutagalung, A.; Novick, P.; Reinisch, K.M. Structure of a C-terminal fragment of its Vps53 subunit suggests similarity of Golgi-associated retrograde protein (GARP) complex to a family of tethering complexes. Proc. Natl. Acad. Sci.USA 2010, 107, 14176–14181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khakurel, A.; Kudlyk, T.; Bonifacino, J.S.; Lupashin, V.V. The Golgi-associated retrograde protein (GARP) complex plays an essential role in the maintenance of the Golgi glycosylation machinery. Mol. Biol. Cell 2021, 32, 1594–1610. [Google Scholar] [CrossRef] [PubMed]
- Fasshauer, D.; Sutton, R.B.; Brunger, A.T.; Jahn, R. Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc. Natl. Acad. Sci. USA 1998, 95, 15781–15786. [Google Scholar] [CrossRef] [Green Version]
- Bock, J.B.; Matern, H.T.; Peden, A.A.; Scheller, R.H. A genomic perspective on membrane compartment organization. Nature 2001, 409, 839–841. [Google Scholar] [CrossRef] [Green Version]
- Lou, X.; Shin, Y.K. SNARE zippering. Biosci. Rep. 2016, 36, e00327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Pluhackova, K.; Bockmann, R.A. The Multifaceted Role of SNARE Proteins in Membrane Fusion. Front. Physiol. 2017, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Joglekar, A.P.; Williams, A.L.; Hay, J.C. Subunit structure of a mammalian ER/Golgi SNARE complex. J. Biol. Chem. 2000, 275, 39631–39639. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Martin, S.; James, D.E.; Hong, W. GS15 forms a SNARE complex with syntaxin 5, GS28, and Ykt6 and is implicated in traffic in the early cisternae of the Golgi apparatus. Mol. Biol. Cell 2002, 13, 3493–3507. [Google Scholar] [CrossRef] [Green Version]
- Mallard, F.; Tang, B.L.; Galli, T.; Tenza, D.; Saint-Pol, A.; Yue, X.; Antony, C.; Hong, W.; Goud, B.; Johannes, L. Early/recycling endosomes-to-TGN transport involves two SNARE complexes and a Rab6 isoform. J. Cell Biol. 2002, 156, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.H.; Xu, Y.; Zhang, T.; Griffiths, G.; Lowe, S.L.; Subramaniam, V.N.; Seow, K.T.; Hong, W. GS32, a novel Golgi SNARE of 32 kDa, interacts preferentially with syntaxin 6. Mol. Biol. Cell 1999, 10, 119–134. [Google Scholar] [CrossRef] [Green Version]
- Yong, C.Q.Y.; Tang, B.L. Another longin SNARE for autophagosome-lysosome fusion-how does Ykt6 work? Autophagy 2019, 15, 352–357. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.; Nakayama, T.; Sato, N.; Izumi, Y.; Soma, M.; Aoi, N.; Ma, Y. A haplotype of the GOSR2 gene is associated with essential hypertension in Japanese men. Clin. Biochem. 2013, 46, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Lahm, H.; Jia, M.; Dressen, M.; Wirth, F.; Puluca, N.; Gilsbach, R.; Keavney, B.D.; Cleuziou, J.; Beck, N.; Bondareva, O.; et al. Congenital heart disease risk loci identified by genome-wide association study in European patients. J. Clin. Investig. 2021, 131, e141837. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Guan, G.C.; Lv, Y.; Liu, Z.W.; Liu, F.Q.; Zhang, Y.; Zhu, S.M.; Zhang, R.H.; Zhao, N.; Shi, S.; et al. G-T haplotype established by rs3785889-rs16941382 in GOSR2 gene is associated with coronary artery disease in Chinese Han population. Oncotarget 2017, 8, 82165–82173. [Google Scholar] [CrossRef] [Green Version]
- Corbett, M.A.; Schwake, M.; Bahlo, M.; Dibbens, L.M.; Lin, M.; Gandolfo, L.C.; Vears, D.F.; O’Sullivan, J.D.; Robertson, T.; Bayly, M.A.; et al. A mutation in the Golgi Qb-SNARE gene GOSR2 causes progressive myoclonus epilepsy with early ataxia. Am. J. Hum. Genet. 2011, 88, 657–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henige, H.; Kaur, S.; Pappas, K. Compound heterozygous variants in GOSR2 associated with congenital muscular dystrophy: A case report. Eur. J. Med. Genet. 2021, 64, 104184. [Google Scholar] [CrossRef]
- Peter, T.A.; Linders, E.C.F.G.; Ashikov, A.; Vals, M.-A.; de Boer, R.; Revelo, N.H.; Arts, R.; Baerenfaenger, M.; Zijlstra, F.; Huijben, K.; et al. Congenital disorder of glycosylation caused by starting site-specific variant in syntaxin-5. Nat. Commun. 2021, 12, 6227. [Google Scholar] [CrossRef] [Green Version]
- McNew, J.A.; Sogaard, M.; Lampen, N.M.; Machida, S.; Ye, R.R.; Lacomis, L.; Tempst, P.; Rothman, J.E.; Sollner, T.H. Ykt6p, a prenylated SNARE essential for endoplasmic reticulum-Golgi transport. J. Biol. Chem. 1997, 272, 17776–17783. [Google Scholar] [CrossRef] [Green Version]
- Shirakawa, R.; Goto-Ito, S.; Goto, K.; Wakayama, S.; Kubo, H.; Sakata, N.; Trinh, D.A.; Yamagata, A.; Sato, Y.; Masumoto, H.; et al. A SNARE geranylgeranyltransferase essential for the organization of the Golgi apparatus. EMBO J. 2020, 39, e104120. [Google Scholar] [CrossRef] [PubMed]
- Steegmaier, M.; Yang, B.; Yoo, J.S.; Huang, B.; Shen, M.; Yu, S.; Luo, Y.; Scheller, R.H. Three novel proteins of the syntaxin/SNAP-25 family. J. Biol. Chem. 1998, 273, 34171–34179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohenstein, A.C.; Roche, P.A. SNAP-29 is a promiscuous syntaxin-binding SNARE. Biochem. Biophys. Res. Commun. 2001, 285, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Kudlyk, T.; Willett, R.; Pokrovskaya, I.D.; Lupashin, V. COG6 interacts with a subset of the Golgi SNAREs and is important for the Golgi complex integrity. Traffic 2013, 14, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Mastrodonato, V.; Morelli, E.; Vaccari, T. How to use a multipurpose SNARE: The emerging role of Snap29 in cellular health. Cell Stress 2018, 2, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Sprecher, E.; Ishida-Yamamoto, A.; Mizrahi-Koren, M.; Rapaport, D.; Goldsher, D.; Indelman, M.; Topaz, O.; Chefetz, I.; Keren, H.; O’Brien, T.J.; et al. A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma. Am. J. Hum. Genet. 2005, 77, 242–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llaci, L.; Ramsey, K.; Belnap, N.; Claasen, A.M.; Balak, C.D.; Szelinger, S.; Jepsen, W.M.; Siniard, A.L.; Richholt, R.; Izat, T.; et al. Compound heterozygous mutations in SNAP29 is associated with Pelizaeus-Merzbacher-like disorder (PMLD). Hum. Genet. 2019, 138, 1409–1417. [Google Scholar] [CrossRef]
- Mastrodonato, V.; Beznoussenko, G.; Mironov, A.; Ferrari, L.; Deflorian, G.; Vaccari, T. A genetic model of CEDNIK syndrome in zebrafish highlights the role of the SNARE protein Snap29 in neuromotor and epidermal development. Sci. Rep. 2019, 9, 1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keser, V.; Lachance, J.B.; Alam, S.S.; Lim, Y.; Scarlata, E.; Kaur, A.; Zhang, T.F.; Lv, S.; Lachapelle, P.; O’Flaherty, C.; et al. Snap29 mutant mice recapitulate neurological and ophthalmological abnormalities associated with 22q11 and CEDNIK syndrome. Commun. Biol. 2019, 2, 375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morelli, E.; Ginefra, P.; Mastrodonato, V.; Beznoussenko, G.V.; Rusten, T.E.; Bilder, D.; Stenmark, H.; Mironov, A.A.; Vaccari, T. Multiple functions of the SNARE protein Snap29 in autophagy, endocytic, and exocytic trafficking during epithelial formation in Drosophila. Autophagy 2014, 10, 2251–2268. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Mohtashami, M.; Stewart, B.; Boulianne, G.; Trimble, W.S. Drosophila SNAP-29 is an essential SNARE that binds multiple proteins involved in membrane traffic. PLoS ONE 2014, 9, e91471. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Bai, Z.; Zegarek, M.H.; Grant, B.D.; Lee, J. Essential roles of snap-29 in C. elegans. Dev. Biol. 2011, 355, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Turan, S.; Ignatius, J.; Moilanen, J.S.; Kuismin, O.; Stewart, H.; Mann, N.P.; Linglart, A.; Bastepe, M.; Juppner, H. De novo STX16 deletions: An infrequent cause of pseudohypoparathyroidism type Ib that should be excluded in sporadic cases. J. Clin. Endocrinol. Metab. 2012, 97, E2314–E2319. [Google Scholar] [CrossRef] [Green Version]
- Shitara, A.; Shibui, T.; Okayama, M.; Arakawa, T.; Mizoguchi, I.; Sakakura, Y.; Takuma, T. VAMP4 is required to maintain the ribbon structure of the Golgi apparatus. Mol. Cell. Biochem. 2013, 380, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Frohlich, L.F.; Bastepe, M.; Ozturk, D.; Abu-Zahra, H.; Juppner, H. Lack of Gnas epigenetic changes and pseudohypoparathyroidism type Ib in mice with targeted disruption of syntaxin-16. Endocrinology 2007, 148, 2925–2935. [Google Scholar] [CrossRef] [PubMed]
- Kunwar, A.J.; Rickmann, M.; Backofen, B.; Browski, S.M.; Rosenbusch, J.; Schoning, S.; Fleischmann, T.; Krieglstein, K.; Fischer von Mollard, G. Lack of the endosomal SNAREs vti1a and vti1b led to significant impairments in neuronal development. Proc. Natl. Acad. Sci. USA 2011, 108, 2575–2580. [Google Scholar] [CrossRef] [Green Version]
- Adolf, F.; Rhiel, M.; Hessling, B.; Gao, Q.; Hellwig, A.; Bethune, J.; Wieland, F.T. Proteomic Profiling of Mammalian COPII and COPI Vesicles. Cell Rep. 2019, 26, 250–265.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, C.R.; Xia, Z.J.; Clement, A.; Parry, D.A.; Davids, M.; Taylan, F.; Sharma, P.; Turgeon, C.T.; Blanco-Sanchez, B.; Ng, B.G.; et al. A Recurrent De Novo Heterozygous COG4 Substitution Leads to Saul-Wilson Syndrome, Disrupted Vesicular Trafficking, and Altered Proteoglycan Glycosylation. Am. J. Hum. Genet. 2018, 103, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Sumya, F.T.; Pokrovskaya, I.D.; Lupashin, V. Development and Initial Characterization of Cellular Models for COG Complex-Related CDG-II Diseases. Front. Genet. 2021, 12, 733048. [Google Scholar] [CrossRef]
- Nakamura, N.; Rabouille, C.; Watson, R.; Nilsson, T.; Hui, N.; Slusarewicz, P.; Kreis, T.E.; Warren, G. Characterization of a cis-Golgi matrix protein, GM130. J. Cell Biol. 1995, 131, 1715–1726. [Google Scholar] [CrossRef] [Green Version]
- Fritzler, M.J.; Hamel, J.C.; Ochs, R.L.; Chan, E.K. Molecular characterization of two human autoantigens: Unique cDNAs encoding 95- and 160-kD proteins of a putative family in the Golgi complex. J. Exp. Med. 1993, 178, 49–62. [Google Scholar] [CrossRef] [Green Version]
- Hicks, S.W.; Machamer, C.E. Isoform-specific interaction of golgin-160 with the Golgi-associated protein PIST. J. Biol. Chem. 2005, 280, 28944–28951. [Google Scholar] [CrossRef] [Green Version]
- Fritzler, M.J.; Lung, C.C.; Hamel, J.C.; Griffith, K.J.; Chan, E.K. Molecular characterization of Golgin-245, a novel Golgi complex protein containing a granin signature. J. Biol. Chem. 1995, 270, 31262–31268. [Google Scholar] [CrossRef] [Green Version]
- Bascom, R.A.; Srinivasan, S.; Nussbaum, R.L. Identification and characterization of golgin-84, a novel Golgi integral membrane protein with a cytoplasmic coiled-coil domain. J. Biol. Chem. 1999, 274, 2953–2962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, E.; Misumi, Y.; Sohda, M.; Fujiwara, T.; Yano, A.; Ikehara, Y. Identification and characterization of GCP16, a novel acylated Golgi protein that interacts with GCP170. J. Biol. Chem. 2003, 278, 51957–51967. [Google Scholar] [CrossRef] [Green Version]
- Linstedt, A.D.; Hauri, H.P. Giantin, a novel conserved Golgi membrane protein containing a cytoplasmic domain of at least 350 kDa. Mol. Biol. Cell 1993, 4, 679–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, A.; Hayashi-Nishino, M.; Shakuno, T.; Masuda, J.; Koreishi, M.; Murakami, R.; Nakamura, Y.; Nakamura, T.; Abe-Kanoh, N.; Honjo, Y.; et al. The Golgin Protein Giantin Regulates Interconnections Between Golgi Stacks. Front. Cell Dev. Biol. 2019, 7, 160. [Google Scholar] [CrossRef] [Green Version]
- Cheung, P.Y.; Limouse, C.; Mabuchi, H.; Pfeffer, S.R. Protein flexibility is required for vesicle tethering at the Golgi. eLife 2015, 4, e12790. [Google Scholar] [CrossRef] [PubMed]
- MacNeil, A.J.; Pohajdak, B. Getting a GRASP on CASP: Properties and role of the cytohesin-associated scaffolding protein in immunity. Immunol. Cell Biol. 2009, 87, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Waters, M.G.; Clary, D.O.; Rothman, J.E. A novel 115-kD peripheral membrane protein is required for intercisternal transport in the Golgi stack. J. Cell Biol. 1992, 118, 1015–1026. [Google Scholar] [CrossRef] [Green Version]
- Barr, F.A.; Puype, M.; Vandekerckhove, J.; Warren, G. GRASP65, a protein involved in the stacking of Golgi cisternae. Cell 1997, 91, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, Y. Nonredundant Roles of GRASP55 and GRASP65 in the Golgi Apparatus and Beyond. Trends Biochem. Sci. 2020, 45, 1065–1079. [Google Scholar] [CrossRef]
- Kingsley, D.M.; Kozarsky, K.F.; Segal, M.; Krieger, M. Three types of low density lipoprotein receptor-deficient mutant have pleiotropic defects in the synthesis of N-linked, O-linked, and lipid-linked carbohydrate chains. J. Cell Biol. 1986, 102, 1576–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foulquier, F.; Vasile, E.; Schollen, E.; Callewaert, N.; Raemaekers, T.; Quelhas, D.; Jaeken, J.; Mills, P.; Winchester, B.; Krieger, M.; et al. Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proc. Natl. Acad. Sci. USA 2006, 103, 3764–3769. [Google Scholar] [CrossRef] [Green Version]
- Kodera, H.; Ando, N.; Yuasa, I.; Wada, Y.; Tsurusaki, Y.; Nakashima, M.; Miyake, N.; Saitoh, S.; Matsumoto, N.; Saitsu, H. Mutations in COG2 encoding a subunit of the conserved oligomeric golgi complex cause a congenital disorder of glycosylation. Clin. Genet. 2015, 87, 455–460. [Google Scholar] [CrossRef]
- Suvorova, E.S.; Kurten, R.C.; Lupashin, V.V. Identification of a human orthologue of Sec34p as a component of the cis-Golgi vesicle tethering machinery. J. Biol. Chem. 2001, 276, 22810–22818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, B.G.; Sharma, V.; Sun, L.; Loh, E.; Hong, W.; Tay, S.K.; Freeze, H.H. Identification of the first COG-CDG patient of Indian origin. Mol. Genet. Metab. 2011, 102, 364–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paesold-Burda, P.; Maag, C.; Troxler, H.; Foulquier, F.; Kleinert, P.; Schnabel, S.; Baumgartner, M.; Hennet, T. Deficiency in COG5 causes a moderate form of congenital disorders of glycosylation. Hum. Mol. Genet. 2009, 18, 4350–4356. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.W.; Matthijs, G.; Sturiale, L.; Garozzo, D.; Wong, K.Y.; Wong, R.; Wong, V.; Jaeken, J. COG5-CDG with a Mild Neurohepatic Presentation. JIMD Rep. 2012, 3, 67–70. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.O.; Yun, M.; Jeong, J.H.; Choi, S.M.; Kim, S.K.; Yoon, W.; Park, C.; Hong, Y.; Woo, Y.J. A Mild Form of COG5 Defect Showing Early-Childhood-Onset Friedreich’s-Ataxia-Like Phenotypes with Isolated Cerebellar Atrophy. J. Korean Med. Sci. 2017, 32, 1885–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, S.; Gong, L.; Qiu, H.; Zhao, Y.; Zhang, Y.; Liu, C.; Jiang, H.; Mao, Y.; Kong, L.Y.; Liang, B.; et al. Novel compound heterozygous COG5 mutations in a Chinese male patient with severe clinical symptoms and type IIi congenital disorder of glycosylation: A case report. Exp. Ther. Med. 2019, 18, 2695–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Han, L.; Wang, X.Y.; Wang, J.H.; Li, X.M.; Jin, C.H.; Wang, L. Identification of Two Novel Mutations in COG5 Causing Congenital Disorder of Glycosylation. Front. Genet. 2020, 11, 168. [Google Scholar] [CrossRef] [Green Version]
- Cherot, E.; Keren, B.; Dubourg, C.; Carre, W.; Fradin, M.; Lavillaureix, A.; Afenjar, A.; Burglen, L.; Whalen, S.; Charles, P.; et al. Using medical exome sequencing to identify the causes of neurodevelopmental disorders: Experience of 2 clinical units and 216 patients. Clin. Genet. 2018, 93, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Lubbehusen, J.; Thiel, C.; Rind, N.; Ungar, D.; Prinsen, B.H.; de Koning, T.J.; van Hasselt, P.M.; Korner, C. Fatal outcome due to deficiency of subunit 6 of the conserved oligomeric Golgi complex leading to a new type of congenital disorders of glycosylation. Hum. Mol. Genet. 2010, 19, 3623–3633. [Google Scholar] [CrossRef]
- Ciftci, H.; Celepkolo, B.; Dilmec, F.; Koksal, M.; Yeni, E.; Yagmur, I.; Gumus, K. Genetic polymorphisms of hspa1b and hspa1l in infertile men. JPMA J. Pak. Med. Assoc. 2015, 65, 701–704. [Google Scholar] [PubMed]
- Huybrechts, S.; De Laet, C.; Bontems, P.; Rooze, S.; Souayah, H.; Sznajer, Y.; Sturiale, L.; Garozzo, D.; Matthijs, G.; Ferster, A.; et al. Deficiency of Subunit 6 of the Conserved Oligomeric Golgi Complex (COG6-CDG): Second Patient, Different Phenotype. JIMD Rep. 2012, 4, 103–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaheen, R.; Ansari, S.; Alshammari, M.J.; Alkhalidi, H.; Alrukban, H.; Eyaid, W.; Alkuraya, F.S. A novel syndrome of hypohidrosis and intellectual disability is linked to COG6 deficiency. J. Med. Genet. 2013, 50, 431–436. [Google Scholar] [CrossRef]
- Althonaian, N.; Alsultan, A.; Morava, E.; Alfadhel, M. Secondary Hemophagocytic Syndrome Associated with COG6 Gene Defect: Report and Review. JIMD Rep. 2018, 42, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Steet, R.A.; Bohorov, O.; Bakker, J.; Newell, J.; Krieger, M.; Spaapen, L.; Kornfeld, S.; Freeze, H.H. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat. Med. 2004, 10, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Spaapen, L.J.; Bakker, J.A.; van der Meer, S.B.; Sijstermans, H.J.; Steet, R.A.; Wevers, R.A.; Jaeken, J. Clinical and biochemical presentation of siblings with COG-7 deficiency, a lethal multiple O- and N-glycosylation disorder. J. Inherit. Metab. Dis. 2005, 28, 707–714. [Google Scholar] [CrossRef]
- Morava, E.; Zeevaert, R.; Korsch, E.; Huijben, K.; Wopereis, S.; Matthijs, G.; Keymolen, K.; Lefeber, D.J.; De Meirleir, L.; Wevers, R.A. A common mutation in the COG7 gene with a consistent phenotype including microcephaly, adducted thumbs, growth retardation, VSD and episodes of hyperthermia. Eur. J. Hum. Genet. EJHG 2007, 15, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Zeevaert, R.; Foulquier, F.; Cheillan, D.; Cloix, I.; Guffon, N.; Sturiale, L.; Garozzo, D.; Matthijs, G.; Jaeken, J. A new mutation in COG7 extends the spectrum of COG subunit deficiencies. Eur. J. Med. Genet. 2009, 52, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Foulquier, F.; Ungar, D.; Reynders, E.; Zeevaert, R.; Mills, P.; Garcia-Silva, M.T.; Briones, P.; Winchester, B.; Morelle, W.; Krieger, M.; et al. A new inborn error of glycosylation due to a Cog8 deficiency reveals a critical role for the Cog1-Cog8 interaction in COG complex formation. Hum. Mol. Genet. 2007, 16, 717–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranz, C.; Ng, B.G.; Sun, L.; Sharma, V.; Eklund, E.A.; Miura, Y.; Ungar, D.; Lupashin, V.; Winkel, R.D.; Cipollo, J.F.; et al. COG8 deficiency causes new congenital disorder of glycosylation type IIh. Hum. Mol. Genet. 2007, 16, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Cho, S.Y.; Jang, J.H.; Kim, J.; Kim, S.Z.; Lee, B.H.; Yoo, H.W.; Jin, D.K. Further delineation of COG8-CDG: A case with novel compound heterozygous mutations diagnosed by targeted exome sequencing. Clin. Chim. Acta Int. J. Clin. Chem. 2017, 471, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Arora, V.; Puri, R.D.; Bhai, P.; Sharma, N.; Bijarnia-Mahay, S.; Dimri, N.; Baijal, A.; Saxena, R.; Verma, I. The first case of antenatal presentation in COG8-congenital disorder of glycosylation with a novel splice site mutation and an extended phenotype. Am. J. Med. Genet. Part A 2019, 179, 480–485. [Google Scholar] [CrossRef]
- Uwineza, A.; Caberg, J.H.; Hitayezu, J.; Wenric, S.; Mutesa, L.; Vial, Y.; Drunat, S.; Passemard, S.; Verloes, A.; El Ghouzzi, V.; et al. VPS51 biallelic variants cause microcephaly with brain malformations: A confirmatory report. Eur. J. Med. Genet. 2019, 62, 103704. [Google Scholar] [CrossRef]
- Fusella, A.; Micaroni, M.; Di Giandomenico, D.; Mironov, A.A.; Beznoussenko, G.V. Segregation of the Qb-SNAREs GS27 and GS28 into Golgi vesicles regulates intra-Golgi transport. Traffic 2013, 14, 568–584. [Google Scholar] [CrossRef]
- Dibbens, L.M.; Rubboli, G. GOSR2: A progressive myoclonus epilepsy gene. Epileptic Disord. Int. Epilepsy J. Videotape 2016, 18, 111–114. [Google Scholar] [CrossRef]
Figure 1.
Schematic outline of the steps involved in Golgi enzyme recycling: 1. Membrane budding at the donor membrane is initiated by recruitment of coat proteins which directly or indirectly interact with cargo (glycosylation enzyme) 2. and 3. After budding, the coat protein partially falls off and the vesicle is captured by long coiled-coil tethers. 4. Among other interactions, MTCs interact with the SNAREs on the vesicle and target membrane, thereby facilitating SNARE alignment 5. and 6. Priming of the SNAREs and formation of the trans-SNARE complex leads to vesicle fusion at the target membrane and cargo delivery.
Figure 1.
Schematic outline of the steps involved in Golgi enzyme recycling: 1. Membrane budding at the donor membrane is initiated by recruitment of coat proteins which directly or indirectly interact with cargo (glycosylation enzyme) 2. and 3. After budding, the coat protein partially falls off and the vesicle is captured by long coiled-coil tethers. 4. Among other interactions, MTCs interact with the SNAREs on the vesicle and target membrane, thereby facilitating SNARE alignment 5. and 6. Priming of the SNAREs and formation of the trans-SNARE complex leads to vesicle fusion at the target membrane and cargo delivery.

Figure 2.
Schematic outline of localization of the coiled-coil tethers and the MTCs at the Golgi. Coiled-coil tethers are depicted in red. Golgi stacking proteins GRASP55 and GRASP65 are depicted in pink. The conserved oligomeric Golgi (COG) complex is the MTC that operates at the Golgi and is depicted in green, while the (Golgi associated retrograde protein) GARP complex operates at the TGN and is depicted in purple.
Figure 2.
Schematic outline of localization of the coiled-coil tethers and the MTCs at the Golgi. Coiled-coil tethers are depicted in red. Golgi stacking proteins GRASP55 and GRASP65 are depicted in pink. The conserved oligomeric Golgi (COG) complex is the MTC that operates at the Golgi and is depicted in green, while the (Golgi associated retrograde protein) GARP complex operates at the TGN and is depicted in purple.

Figure 3.
Schematic representation of the SNARE complexes participating at the Golgi. The STX5/GOSR1/BET1L/YKT6 (green) Golgi SNARE complex operates throughout the Golgi. STX5/SNAP29/YKT6 is another SNARE complex. STX5/GOSR2/BET1/Sec22b operates between the ER/ERGIC and Golgi. The STX16/Vti1a/STX6/VAMP3 or VAMP4 (purple) operates at the TGN.
Figure 3.
Schematic representation of the SNARE complexes participating at the Golgi. The STX5/GOSR1/BET1L/YKT6 (green) Golgi SNARE complex operates throughout the Golgi. STX5/SNAP29/YKT6 is another SNARE complex. STX5/GOSR2/BET1/Sec22b operates between the ER/ERGIC and Golgi. The STX16/Vti1a/STX6/VAMP3 or VAMP4 (purple) operates at the TGN.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
D’Souza, Z.; Sumya, F.T.; Khakurel, A.; Lupashin, V. Getting Sugar Coating Right! The Role of the Golgi Trafficking Machinery in Glycosylation. Cells 2021, 10, 3275. https://doi.org/10.3390/cells10123275
AMA Style
D’Souza Z, Sumya FT, Khakurel A, Lupashin V. Getting Sugar Coating Right! The Role of the Golgi Trafficking Machinery in Glycosylation. Cells. 2021; 10(12):3275. https://doi.org/10.3390/cells10123275
Chicago/Turabian StyleD’Souza, Zinia, Farhana Taher Sumya, Amrita Khakurel, and Vladimir Lupashin. 2021. "Getting Sugar Coating Right! The Role of the Golgi Trafficking Machinery in Glycosylation" Cells 10, no. 12: 3275. https://doi.org/10.3390/cells10123275
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.