
Mitochondrial Dysfunction in Cancer Cachexia: Impact on Muscle Health and Regeneration


1
Department of Clinical and Biological Sciences, University of Torino, 10125 Torino, Italy
2
Interuniversity Institute of Myology, 61029 Urbino, Italy
3
Department of Anatomy, Cell Biology and Physiology, Indiana University, Indianapolis, IN 46202, USA
*
Author to whom correspondence should be addressed.
Cells 2021, 10(11), 3150; https://doi.org/10.3390/cells10113150
Submission received: 18 August 2021
/
Revised: 5 November 2021
/
Accepted: 9 November 2021
/
Published: 12 November 2021
(This article belongs to the Section Intracellular and Plasma Membranes)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Cancer cachexia is a frequently neglected debilitating syndrome that, beyond representing a primary cause of death and cancer therapy failure, negatively impacts on patients’ quality of life. Given the complexity of its multisystemic pathogenesis, affecting several organs beyond the skeletal muscle, defining an effective therapeutic approach has failed so far. Revamped attention of the scientific community working on cancer cachexia has focused on mitochondrial alterations occurring in the skeletal muscle as potential triggers of the complex metabolic derangements, eventually leading to hypercatabolism and tissue wasting. Mitochondrial dysfunction may be simplistically viewed as a cause of energy failure, thus inducing protein catabolism as a compensatory mechanism; however, other peculiar cachexia features may depend on mitochondria. On the one side, chemotherapy also impacts on muscle mitochondrial function while, on the other side, muscle-impaired regeneration may result from insufficient energy production from damaged mitochondria. Boosting mitochondrial function could thus improve the energetic status and chemotherapy tolerance, and relieve the myogenic process in cancer cachexia. In the present work, a focused review of the available literature on mitochondrial dysfunction in cancer cachexia is presented along with preliminary data dissecting the potential role of stimulating mitochondrial biogenesis via PGC-1α overexpression in distinct aspects of cancer-induced muscle wasting.
Keywords:
cancer cachexia; muscle wasting; mitochondria; regeneration; myogenesis; PGC-1α; metabolism1. Pathogenesis of Cancer Cachexia
Cancer is one of the most relevant causes of death all over the world and its incidence is estimated to markedly increase globally in the next two decades (http://who.int/gho/database/en (accessed on 17 August 2021). Patients affected by cancer very frequently develop a complex multifactorial syndrome defined as ‘cachexia’. This is an urgent unmet medical need. Indeed, about 90% of cancer patients are at risk of developing this syndrome, with a mortality rate that can reach up to 80% [1]. Cachexia markedly complicates the management of cancer patients, reducing quality of life, tolerance to treatments, and survival rates, the more so when anticancer treatments are adopted [2]. Consistently, health care costs are higher in cachectic than in non-cachectic patients [3].
Loss of body weight and skeletal muscle mass, associated or not with reduced adipose tissue, altered metabolism, and low-grade chronic inflammation are hallmarks of cachexia [4]. Muscle wasting mainly results from an altered regulation of protein synthesis and degradation rates, the latter generally prevailing over the former. Although protein hypercatabolism is one of the main features of cachexia, specifically interfering with the different intracellular proteolytic systems is only marginally effective in protecting against cancer-induced loss of muscle mass and function [5,6].
In the last years, a number of studies focused on the possibility that altered energy metabolism could be the trigger of muscle wasting. Indeed, metabolomic analyses have shown that energy and protein metabolism are markedly perturbed in the skeletal muscle and in the liver of tumor-bearing animals [7,8]. Consistently, hypermetabolism associated with altered energy balance has been detected in about 50% of newly diagnosed cancer patients [9]. Particularly relevant to the impaired energy metabolism is the occurrence of mitochondrial dysfunction, resulting from the combined alteration of organelle biogenesis, dynamics, and disposal. Indeed, a considerably high number of studies reported perturbations of the mitochondrial system in the skeletal muscle of tumor hosts [10]. As an example, both the oxidative capacity and the production of ATP are impaired in the muscle of animals bearing cachexia-inducing tumors [11], a situation that is further exacerbated by chemotherapy [12]. Such mitochondrial alterations likely contribute to muscle protein hypercatabolism [13]. Indeed, in experimental cancer cachexia, the former precede the latter [14]. Consistent with data obtained on experimental models, a study reported that about 50% of hypermetabolic cancer patients do not report any body weight loss and impairment of performance status [9].
On the whole, the observations above suggest that mitochondria can become a potentially useful therapeutic target to tackle cachexia. Along this line, exercise training is one of the most explored strategies. While being quite a simple option, the benefits of exercise on mitochondrial health can be achieved only when training is systematic, which can be unachievable in cancer patients due to exercise intolerance as a consequence of co-morbidities and/or fatigue. In the last decade, drugs mimicking exercise have been developed and investigated [15]. On the one side, this pharmacological approach overcomes patient exercise intolerance, yet on the other it does not completely replace the beneficial effects of training. In addition to exercise mimetics, several drugs able to improve energy metabolism by indirectly impinging on mitochondria have been investigated for their protective action in experimental cancer cachexia [10]. In this regard, quite recent data provide evidence that the exposure of tumor-bearing mice to the Szeto-Schiller peptide (SS-31), a drug able to directly target mitochondria, results in improved body weight loss, restoration of muscle succinate dehydrogenase (SDH) activity and ATP levels, and preservation of mitochondrial mass (see the next chapter for further details) [8].
Beyond directly targeting mitochondrial function, other exercise-mimicking strategies have been tested in preclinical cancer cachexia models, potentially impacting on mitochondrial health. As previously described, muscle wasting results from excessive protein degradation, although autophagy degradation may be halted by saturation or exhaustion of the system. Consistently, pharmacological stimulation of autophagy using rapamycin or AICAR (a known PGC-1α inducer) counteracts C26-induced muscle loss and atrogene induction [16]. Additionally, antioxidants have been extensively tested for their potential anti-cachexia activity, acting on the one hand by reducing the oxidative stress (thus improving mitochondrial function) and slowing down inflammation, on the other hand. Among the mostly studied antioxidants, resveratrol [17] and quercetin [18] have produced more positive results. The other way round, the use of antioxidants in cancer patients is not considered free from potential side effects, also in terms of tumor growth promotion [19], limiting their use in multimodal anti-cachexia clinical trials.
In addition to the altered energy metabolism, and likely interconnected with the latter, delayed muscle regeneration has been shown to take place in both tumor-bearing animals and cancer patients [20,21,22,23], with the transcription factor NF-κB [18] and enhanced activation of ERK stress kinase playing a role [17]. Similarly, overexpression in myogenic precursors of Twist1, a transcription factor associated with the malignant progression of several tumors, or of the zinc transporter ZIP14, is associated with impaired differentiation and muscle atrophy in preclinical models of cancer cachexia [24,25]. While it is widely accepted that mitochondria play a crucial role in maintaining muscle metabolic fitness, it is still unclear if muscle metabolic homeostasis and regenerative capacity can be reciprocally regulated. A profound metabolic reprogramming, including the increase of mitochondrial mass, occurs in myogenic precursor cells after their activation [26,27], while during regeneration, mitochondrial function is progressively restored to normal levels [28]. Taken together, these observations suggest that energy metabolism, mitochondrial function in particular, is relevant to correctly achieve muscle regeneration. Consistently, the differentiation potential of human myogenic precursors can be enhanced by overexpression of peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α), a co-transcription factor working as a master regulator of mitochondrial biogenesis [29]. Along this line, our preliminary observations show that myogenic precursors isolated from transgenic mice with muscle-specific overexpression of PGC-1α (MCK-PGC-1α mice) and implanted into the muscle of wild-type (WT) animals fuse with existing myofibers and promote an oxidative phenotype shift more efficiently than cells obtained from non-transgenic animals (unpublished data).
2. Mitochondrial Alterations and Mitochondria-Targeted Anti-Cachexia Interventions
The unbalance between protein degradation and protein synthesis is one of the major culprits of skeletal muscle loss during cancer [30]. Different lines of evidence demonstrated that altered energy metabolism may potentially trigger or synergize with impaired protein turnover to affect muscle mass and function. Indeed, alterations of metabolites associated with glycolysis, TCA cycle, and fatty acid metabolism were detected in the skeletal muscle of tumor-bearing animals and even worsened in the presence of chemotherapy [31]. The metabolome unbalance found in the skeletal muscle was also corroborated by the alteration of the mitochondrial homeostasis in different preclinical models of cancer cachexia [31,32,33]. In this regard, protein levels of PGC-1α are found to be both decreased or unchanged in the skeletal muscle of animals carrying different types of cachectic tumors, and under different types of chemotherapeutic regimens [11,14,31,32,33,34,35]. Although a reduction of PGC-1α is not always detected in cachectic animals, mitochondrial alterations found in cancer cachexia cover a broad spectrum, from the organelle biogenesis and correct morphology to the disposal systems, ultimately affecting their quantity and function. In this regard, mitochondria are able to respond to muscle cues, remodeling their morphology through two different processes collectively defined as mitochondrial dynamics, namely mitochondrial fusion and fission [36]. In the muscle of tumor-bearing mice, the levels of mitochondrial dynamics indicators, such as Mfn2, Opa1, Fis1, and Drp1, are downregulated [11,31,32,37], suggesting an impairment of mitochondrial adaptation to muscle homeostatic changes. Together with altered mitochondrial biogenesis and dynamics, the disposal of mitochondria (mitophagy) is also affected in cachectic skeletal muscle, as suggested by the increase of bulk autophagy flux and of the mitophagy marker BNIP3 in the skeletal muscle of C26-bearing mice. In the same study, the lipidated form of LC3 II was found to be upregulated in mitochondrial-enriched fractions of cachectic muscles [13], suggesting an increase of molecular tagging of mitochondria towards degradation. In addition, another two independent reports demonstrated an increase in mitophagy-related proteins, such as PINK1 and Parkin, further suggesting an activation of mitochondrial degradation in the skeletal muscle of C26-bearing mice [11,34]. The alteration of mitochondrial biogenesis and disposal are in accordance with the common finding of reduced mitochondrial mass, commonly measured by quantifying protein or transcript levels of mitochondrial proteins, such as cytochrome c (cyt c), SDHA, COX IV, TFAM, and TOMM20, as well as the content of mitochondrial DNA [11,31,32,38]. The disrupted mitochondrial homeostasis found in cachectic animals results in reduced mitochondrial function combined with alterations in the levels of energy-related metabolites [8,11,12]. Indeed, different studies show an impairment of mitochondrial respiration ex vivo or in isolated mitochondria from cachectic muscles [8,13,14]. Altered mitochondrial function also leads to increased ROS production and oxidative stress, further promoting mitochondrial damage [10]. In this regard, the mitochondria-specific phospholipid cardiolipin is particularly affected by oxidative stress [39]. Cardiolipin is an essential constituent of the mitochondrial membrane and is crucial for proper mitochondrial function [40]. Our recent results demonstrated that cardiolipin and its precursor phosphatidylglycerol were reduced in C26-bearing mice, suggesting an intrinsic alteration of the mitochondrial membrane contributing to reduced mitochondrial function [8]. Another recent study confirmed these results by showing alteration of the muscle phospholipid profile, including cardiolipins, in the skeletal muscle of urothelial carcinoma-bearing mice [41]. Importantly, mitochondrial alterations have been validated in the skeletal muscle of patients affected by cancer cachexia. As compared to weight-stable cancer patients, cachectic patients showed increased muscle expression of Fis1 together with altered mitochondrial morphology [42]. Collectively, these studies implicate mitochondrial dysfunction as a key contributor to muscle wasting.
As briefly introduced above, exercise is a pleiotropic and non-pharmacologic approach that, among many different targets, modulates mitochondria homeostasis. Countless evidence exists on how both aerobic and resistance exercise, or the combination of both, are effective in counteracting muscle wasting in cachectic animals [11,16,33,34,43,44,45]. The effect on muscle mass and function by exercise may rely in part on the increase of mitochondrial quantity and quality. In this regard, increased PGC-1α levels were observed in exercised C26-bearing mice treated or not with chemotherapy [11,34,45]. In Apc+/Min and C26-bearing mice, exercise induces an increase of mitochondrial mass, suggested by increased levels of cyt c, SDHA, and COX IV [11,34,43,45]. Exercise impacts on mitochondrial homeostasis in chemotherapy-treated cachectic mice also by increasing the gene expression of the mitochondrial fusion protein Mfn2 and by suppressing mitophagy-related genes [11]. Exercise-dependent improvement of the whole mitochondrial machinery results in improved mitochondrial performance, indicated by increased SDH activity and ATP content in the skeletal muscle of cachectic mice [11,34]. The beneficial effects of exercise training on tumor-induced mitochondrial derangements provided an overview of different targets that may be modulated to improve mitochondrial homeostasis and possibly muscle wasting in cancer cachexia.
Despite the evidence supporting the relevance of cancer-induced disruption of skeletal muscle mitochondrial homeostasis, few studies have aimed to directly target mitochondrial alterations in cancer cachexia, so far. The above-mentioned mitochondrial-targeting peptide Szeto-Schiller-31 (SS-31) proved effective in reversing mitochondrial dysfunction in several diseases [46,47,48,49,50,51]. The efficacy of this compound relies on its ability to bind cardiolipin, protecting this key phospholipid from oxidation-induced damage, thus promoting mitochondrial function [39]. In C26-bearing mice treated with chemotherapy, SS-31 had protective effects against body and muscle wasting, along with improved mitochondrial activity [8], associated with decreased mitophagy. The metabolomic profile of SS-31-treated cachectic mice also revealed an increase of ATP levels, implying a partial restoration of energy metabolism in the skeletal muscle [8]. Interestingly, systemic administration of SS-31, beyond targeting the skeletal muscle, positively influenced both the liver and circulating metabolome by rescuing the levels of specific metabolites that are decreased in cachectic animals, including glucose and glycogen [8]. In addition, the depletion of liver glutathione observed in chemotherapy-treated C26-bearing mice was reversed by SS-31, suggesting an increase in the antioxidant capacity [8]. The improvement of the liver metabolome by SS-31 directs the attention to the relevance of mitochondria to other organs besides the skeletal muscle in correcting the systemic metabolic derangements induced in cancer cachexia. Other mitochondria-targeted compounds have been investigated for the treatment of cancer-induced cachexia. Among them, trimetazidine (TMZ) targets β-oxidation by inhibiting the 3-ketoacyl-CoA thiolase activity in the mitochondrial matrix [52]. TMZ shifts glucose metabolism towards mitochondrial respiration, reducing the lactate levels and increasing oxygen consumption [53]. In cachectic mice, TMZ prompted a metabolic shift towards oxidative muscle fibers, increasing SDH activity, and protein levels of PGC-1α and other mitochondrial-related proteins. Such improvements result in increased muscle mass and strength as compared to untreated tumor-bearing animals [38].
The complex metabolic impact of exercise or exercise mimetics renders the identification of the key genes controlling mitochondrial and metabolic dysfunction in cancer cachexia difficult. Additionally, only a few studies have implemented genetic approaches to target mitochondria or metabolism-related genes to counteract cachexia. A recent study investigated the role of the pyruvate dehydrogenase kinase 4 (PDK4), a mitochondrial enzyme critical for regulating cellular energy metabolism [54]. PDK4 inhibits the activity of the pyruvate dehydrogenase complex, responsible for the conversion of pyruvate into acetyl-coenzyme A, thus regulating the influx of the three-carbon molecule into the TCA cycle [54]. PDK4 has been found to be upregulated in the skeletal muscle of cachectic mice and this alteration was associated with reduced PDH and SDH activity [55]. In addition, the pharmacologic activation of PDK4 in healthy animals phenocopied the muscle wasting features as well as the alteration of mitochondrial-related proteins observed in cancer-induced cachexia [55]. Interestingly, in vitro deletion of PDK4 protected cultured myotubes from tumor-conditioned medium-induced atrophy, suggesting PDK4 as a target to improve the mitochondrial homeostasis and the energetic status of cachectic mice [55]. An overview of the currently investigated mitochondria targets is presented in Figure 1.
3. Role of Muscle PGC-1α in Cancer Cachexia
Starting from the observation that some of the exercise-based interventions and exercise mimetic effects in tumor-bearing animals are based on the ability to stimulate the expression and the activity of the master regulator of mitochondrial biogenesis PGC-1α, several studies have focused on the impact of this transcriptional coactivator on muscle phenotype, both in health and in wasting conditions, including cancer cachexia. Almost 20 years ago, Spiegelman’s group described for the first time PGC-1α preferential expression in oxidative fibers and characterized a transgenic mouse overexpressing PGC-1α specifically in the skeletal muscle (MCK promoter), showing a conversion towards slow-twitch muscle fibers [56]. On the contrary, myocyte-specific (Myogenin/MEF2 Cre) PGC-1α and β deletion impairs mitochondrial electron transport chain activity without impairing mitochondrial mass and physiological muscle function [57]. Despite being dispensable, PGC-1α mRNA rapidly decreases during atrophy induced experimentally by denervation, cancer cachexia in AH-130-bearing rats, streptozotocin-induced diabetes, and subtotal nephrectomy renal failure [58]. In the above-mentioned transgenic mice overexpressing PGC-1α, muscle mass loss and the induction of atrogenes induced by denervation and fasting were prevented via FoxO3 blockade [58], suggesting a mechanistic explanation for the anti-cachexia effect of exercise training. On the same line, PGC-1α overexpression was shown to prevent sarcopenia associated with aging or induced by hindlimb unloading [59,60].
Whether PGC-1α is also able to counteract cancer-induced muscle wasting is, however, still debated. Our previous observation showed that PGC-1α overexpression was either effective or not in preventing cancer-induced muscle wasting in young mice upon LLC or C26 tumor growth, respectively [11,33]. The protection observed in mice bearing the LLC tumor is in discordance with the data obtained by others [61] adopting apparently the same experimental conditions, where the lack of muscle wasting prevention may be potentially explained by the increased tumor growth observed in transgenic mice. Finally, an alternative splicing PGC-1α isoform (PGC-1α4) induced by resistance exercise was discovered [62] and muscle-specific overexpression determined muscle mass and strength increase, along with resistance to LLC-induced muscle wasting. The inconclusive observations may also depend on the fact that most preclinical proof of concept data come from studies using young animals, mainly for economic reasons, and not receiving chemotherapy, both conditions that limit the translational value of such models [63]. We considered such limitations and produced preliminary observations on the role of PGC-1α in an optimized preclinical model of cancer cachexia adopting both middle (14 months) and aged (22 months) mice bearing the LLC tumor and receiving doxorubicin (see Appendix A for details). Despite not protecting from body weight loss, PGC-1α overexpression counteracted muscle mass and strength loss in 14-month-old mice, while only improving muscle strength in 22-month-old mice. Despite the transgene being active only in the skeletal muscle, several parameters show a differential condition as compared to wild-type mice, including heart, white adipose tissue, and hematocrit, suggesting that boosting skeletal muscle mitochondrial biogenesis impacts on the whole body condition, deserving further investigation.
Although preliminary, our observations prompt new lines of investigation in the cancer cachexia field. The preservation of heart mass observed in transgenic mice is consistent with data obtained in experimental hearth failure [64,65], suggesting that PGC-1α overexpression could become a target to maintain exercise tolerance in cancer patients or at least normal daily physical activity, an essential point in the assessment of quality of life. Beyond heart function, PGC-1α could increase exercise tolerance by improving anemia. To date, no easy mechanistic explanation can be given to support this effect, although angiogenesis has been reported as one of the systemic effects of PGC-1α [66]. Increased hematocrit might be a consequence of enhanced muscle mitochondrial respiration and angiogenesis, in order to supply an adequate amount of oxygen to peripheral tissues. Overall, targeting PGC-1α in order to mimic exercise might be a promising strategy to contain the functional decline induced by both tumor growth and chemotherapy in cachectic patients.
4. Impact of Mitochondrial Dysfunction in Impaired Myogenesis during Cancer Cachexia
Muscle metabolism is mainly considered as the coordinated set of biochemical reactions occurring in adult muscle fibers, marginally considering the role of myogenic cell turnover and the impact of their metabolism on the overall muscle function and metabolism. Starting from the principle that the myogenic program is a highly energy-demanding process, it is possible that, beyond the tumor- and host-derived circulating factors that impair myogenesis [67], the alterations of energy metabolism present in cancer cachexia also impair muscle regeneration. The scenario is even more complex when considering that the muscle is not only composed of adult fibers and muscle-specific stem (satellite) cells, since other non-satellite muscle progenitor and accessory cells present in the muscle microenvironment have been causally associated with cachexia onset [68]. Both intrinsic (satellite cell) and extrinsic niche/muscle fiber dysfunctions may coordinately alter the myogenic program. The existence of an intrinsic satellite cell regeneration impairment has been recently questioned, since satellite cells derived from C26-bearing mice are increased as compared to healthy mice and retain their myogenic potential, growing and differentiating normally [22,69,70], suggesting that extrinsic factors, metabolism first, may determine the impaired myogenesis. The other way round, in all the above-mentioned papers, other accessory muscle cells were shown to be modulated in the muscle of C26-bearing mice, including innate immune cells, mesoangioblasts, and other interstitial cells, showing on the one side the canonical myogenic commitment (Pax7 expression, [22]) while, on the other, an adipocyte-like differentiation when cultured in vitro, suggestive of an aberrant metabolic and differentiative program [70]. The current hypothesis is that the alterations of the muscle microenvironment, mainly reflected by increased proinflammatory signals, do not allow satellite cell differentiation. However, inflammation might not be the only trigger, although very few studies have investigated how metabolic determinants in the stem cell niche impact on regeneration. The metabolic profile, the metabolism-associated gene signature, and the metabolic requirements of satellite cells in different myogenic states in young and old mice have recently been reported [71]. From this study it emerged that in aging, when the regeneration capacity is impaired, satellite cells have low oxidative phosphorylation and preferentially perform cytoplasmic glycolysis. Moreover, perturbing the ability to oxidize fatty acids in healthy mice, a process requiring efficient peroxisomes and mitochondria, impairs muscle regeneration [71]. The results are in accordance with previous ones showing the high metabolic demand of adult activated satellite cells, supported by higher mitochondrial activity and autophagy [26]. Mitochondrial function and energy availability in the satellite cell niche can thus represent limiting factors impairing the myogenic process during cachexia. Along this line, a recent report highlighted the importance of glutamine supplied locally from macrophages to support myogenesis after injury or rescuing the ischemia or age-related myogenic alterations [72].
Muscle disuse, due to both fatigue and bedrest, frequently occurring in cancer patients, was shown to stimulate satellite cell activation and a regenerative response [73]. Unfortunately, muscle disuse in the presence of cancer exacerbates muscle wasting, potentially via aberrant induction of myogenesis [74]. The satellite cell niche is strictly dependent on the type, amount, and organization of the collagen present in the extracellular matrix (ECM). During aging, the altered ECM drives a fibrogenic conversion of satellite cells at the expense of their ability to complete the myogenic program [75]. Such an effect is of particular relevance when considering the association between old age, cancer diagnosis, and cachexia. ECM remodeling occurring either during effective regeneration or cachexia is due to a complex interaction involving innate and adaptive immune cells, fibroblasts, fibroadipogenic precursors (FAPs), and vessel associated pericytes [76], making it clear that focusing only on proinflammatory mediators generates a reductionist view. The contribution of each cell type and signals released to the alterations of the complex orchestrated myogenic machinery is far from being fully elucidated, and will likely allow in the future the identification of new prospective therapeutic targets. The scenario is further complicated by recent evidence that the muscle of cachectic animals and cancer patients shows signs of impaired innervation [77,78,79]. Muscle denervation, either resulting from motor neuron or neuromuscular junction (NMJ) alteration, could thus represent the primary cause or at least contribute to both myofiber atrophy and regeneration in cancer cachexia. The other way round, sarcolemmal damage was reported in cancer cachexia [22] and recently associated with loss of the dystrophin glycoprotein complex interacting protein myocilin [80], providing another potential trigger for satellite cell activation.
In order to restore the myogenic process, distinct approaches have been proposed in the last years (Figure 2). MEK-ERK MAPK pathway inhibition, likely interfering with the proinflammatory cytokine signaling cascade, was the first approach that proved effective in preventing Pax7 accumulation and partially countering cachexia [21], also incidentally confirmed in cancer patients [81]. Potentially acting through a similar mechanism of downstream inflammatory mediators, NF-kB inhibition prevented Pax7-dependent inhibition of myoblast fusion [22]. Interfering with the cytokine milieu could thus impact on distinct aspects of cancer and cachexia, including impaired regeneration, as recently shown by interleukin (IL)-4 administration that counteracted cachexia and prevented the accumulation of satellite cells in C26-bearing mice, directly inducing myocyte differentiation [70]. Beyond cytokines, circulating extracellular vesicles and their microRNA cargo have also been implicated in impaired myogenesis [82] through toll-like 7 receptor (TLR7) signaling. Consistently, a TLR7/8/9 antagonist prevented Pax7 overexpression and cachexia onset [83]. Whether all these approaches rescue the myogenic program through a direct transcriptional regulation of the myogenic genes or other mechanisms is still unknown and the possibility that part of the action relies on cell metabolism is intriguing and will likely be clarified in future studies. On this line, the fact that physical exercise rescues muscle cell differentiation in tumor-bearing animals [84] suggests that metabolism modulation may provide an effective tool targeting cachexia from a new perspective. Another point in favor of such hypothesis is the presence in cachexia of myosteatosis and increased circulating free fatty acids that may produce lipotoxicity, leading to impaired muscle performance and regeneration [85]. For a long time, it has been demonstrated that satellite cell abundance is influenced by fiber metabolism, with the oxidative having more and the glycolytic less satellite cells [86]. The same authors showed that promoting a glycolytic to oxidative fiber shift leads to an increase in the satellite cell number. Such a mechanism also proved effective during cancer cachexia. Indeed, the above described TMZ anti-cachexia action, by promoting muscle oxidative metabolism via PGC1α overexpression, enhanced myogenesis in C26-bearing mice [87].
5. PGC-1α Overexpression Rescues Mitochondrial Content in Regenerating Muscles of Tumor-Bearing Mice
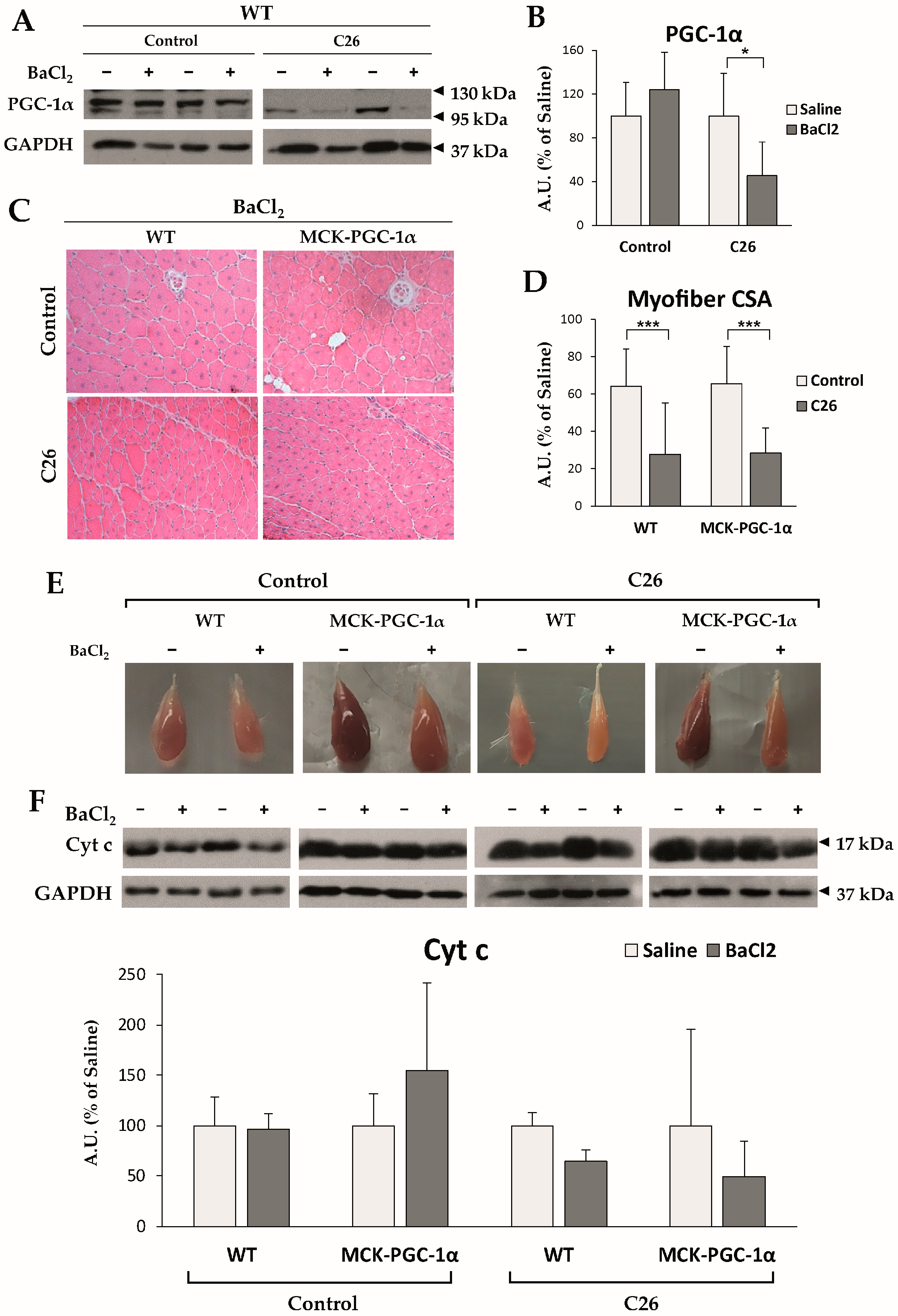
Although several lines of evidence suggest a connection between altered metabolism and impaired regeneration, it is still unclear whether transcription factors and coactivators involved in the control of muscle metabolism play a role in controlling muscle repair. As mentioned above, key mitochondrial players, such as PGC-1α, are found to be both unaffected or modulated among different preclinical cancer cachexia models and cachectic cancer patients. In details, chemotherapy-naïve animals bearing either the LLC or the C26 tumor present with no changes in PGC-1α levels [33], yet our observations presented here reveal that PGC-1α expression is significantly decreased in the skeletal muscle of C26-bearing mice only when a muscle injury has been inflicted (i.m. injection of BaCl2; Figure 3A,B, see Appendix A for methodological details). This new result suggests that PGC-1α repression might be partially accountable for defective muscle regeneration in cancer cachexia, with the muscle progenitor being the main cell type requiring PGC-1α expression to maintain skeletal muscle cellular turnover.
To further investigate this point, we tested whether forced overexpression of PGC-1α was sufficient to improve deficient regeneration in cancer cachexia by injuring the skeletal muscle of MCK-PGC-1α mice bearing the C26 tumor. The muscle architecture 14 days post-injury consisted of centrally nucleated myofibers (Figure 3C) with similar mean myofiber cross-sectional (CSA) area in both WT (64.1 ± 10.6% of uninjured muscle fibers) and MCK-PGC-1α (65.5 ± 27.5%) animals (Figure 3D), implying that PGC-1α overexpression does not accelerate muscle regeneration in healthy conditions. Consistent with previous reports [63], injured muscles of C26-bearing WT mice presented with delayed myofiber CSA recovery (32.3 ± 7.2%), which was not improved by PGC-1α overexpression (33.0 ± 13.6%; Figure 3D).
Although forced expression of PGC-1α was not sufficient to rescue myofiber maturation in cachectic animals, visual inspection revealed loss of pigmentation in injured muscles that was further aggravated in tumor bearers, suggestive of reduced mitochondrial content (Figure 3E). Macroscopic observations were supported by a decreased protein expression of cyt c in cachectic WT animals, indicating a decrease in mitochondrial mass (Figure 3F). On the contrary, injured muscles of cachectic MCK-PGC-1α mice presented with a marked brown coloring and spared levels of cyt c protein (Figure 3E,F), suggesting that PGC-1α overexpression preserves mitochondrial mass. On the whole, the present results indicate that PGC-1α overexpression is not sufficient to rescue muscle regeneration delay, although ameliorating the mitochondrial status, opening new possibilities to therapeutically modulate the metabolism in association with myogenesis-specific enhancers to impact on muscle regeneration in cancer cachexia.
6. Conclusions
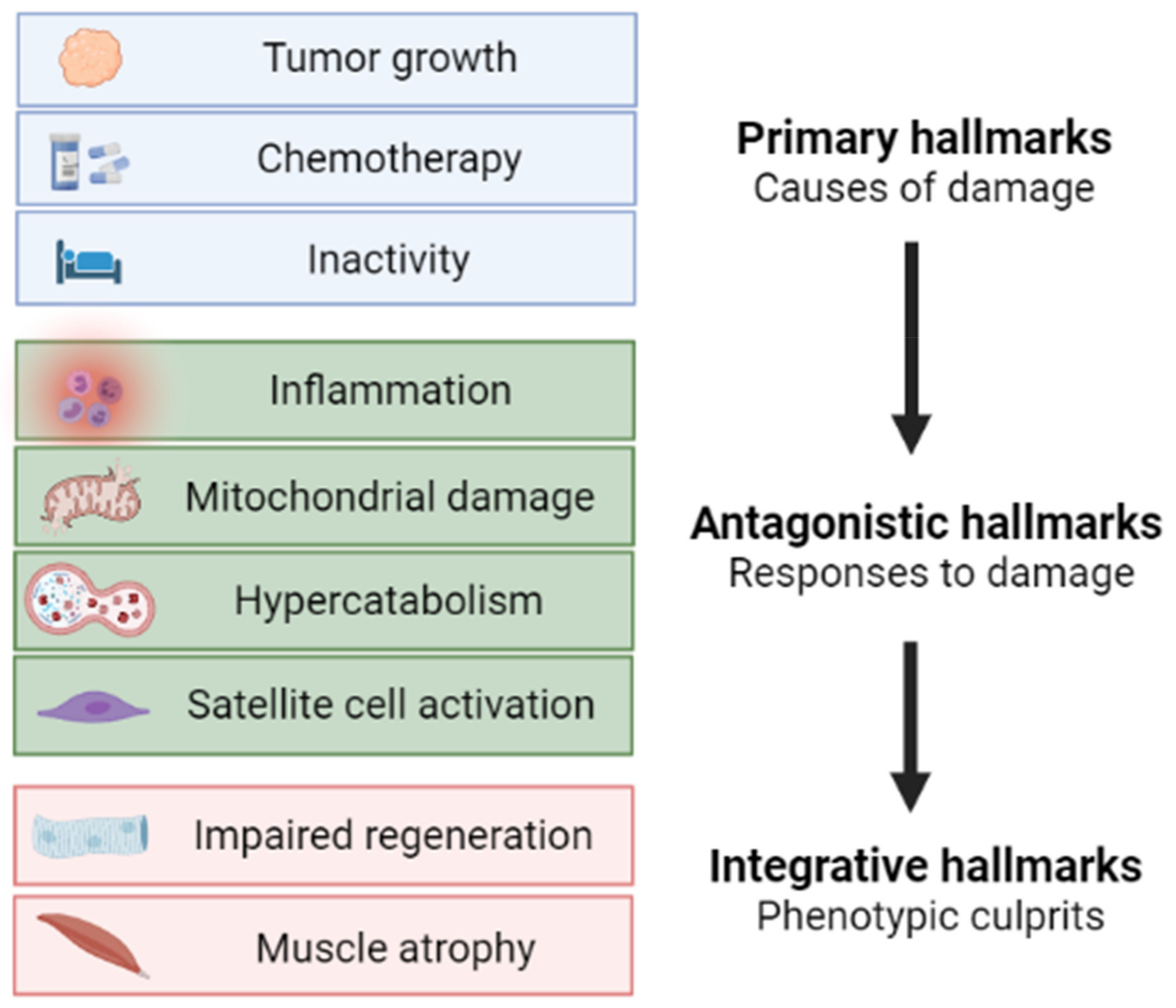
Based on the vast amount of data from both preclinical models and cancer patients, muscle wasting in cancer cachexia is now considered as a complex metabolic syndrome, with specific hallmarks, only in part shared with other wasting disorders. The emerging role of mitochondrial and metabolic alterations along with the metabolism-targeted approaches, including the genetic PGC-1α overexpression described herein, will likely lead to the optimization of new therapeutic options for cancer cachexia. Similarly to the hallmarks of aging [88], we here propose a scheme (Figure 4) describing muscle atrophy during cancer cachexia as a multi-phasic process characterized by initiating causes (primary hallmarks focused on cancer cachexia etiology), followed by the adaptive responses activated in both muscle fibers and the stem cell compartment (antagonistic hallmarks), eventually leading to the cachectic phenotype (integrative hallmarks).
In the future, new tools allowing single-cell analyses, including mass spectrometry imaging and single-cell metabolomics, will clarify how the distinct muscle components, from adult muscle fibers to myogenic precursors and accessory cells, are affected by both tumor growth and chemotherapy, potentially allowing the identification of new prospective therapeutic targets for cachexia directly impacting on cell metabolism.
Author Contributions
Conceptualization, P.C. and F.P. (Fabio Penna); investigation, M.B., F.P. (Fabrizio Pin), F.P. (Fabio Penna) and R.B.; writing original draft, M.B., F.P. (Fabrizio Pin), R.B., F.P. (Fabio Penna) and P.C.; review and editing, M.B., P.C. and F.P. (Fabio Penna); formal analysis, F.P. (Fabio Penna); funding acquisition, F.P. (Fabio Penna); project administration, F.P. (Fabio Penna); resources, F.P. (Fabio Penna). All authors have read and agreed to the published version of the manuscript.
Funding
The research leading to these results received funding from Fondazione AIRC under IG 2018—ID. 21963 project (Principal Investigator Fabio Penna). This study was supported by University of Torino (ex-60% funds) to Fabio Pennaand Paola Costelli.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Bioethical Committee of the University of Torino (Torino, Italy) and the Italian Ministry of Health (Aut. Nr. 579/2018-PR).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors have no relevant conflict of interest to disclose.
Appendix A
Appendix A.1. PGC-1α Overexpression Improves Cachexia in Aged Tumor-Bearing Mice Receiving Chemotherapy
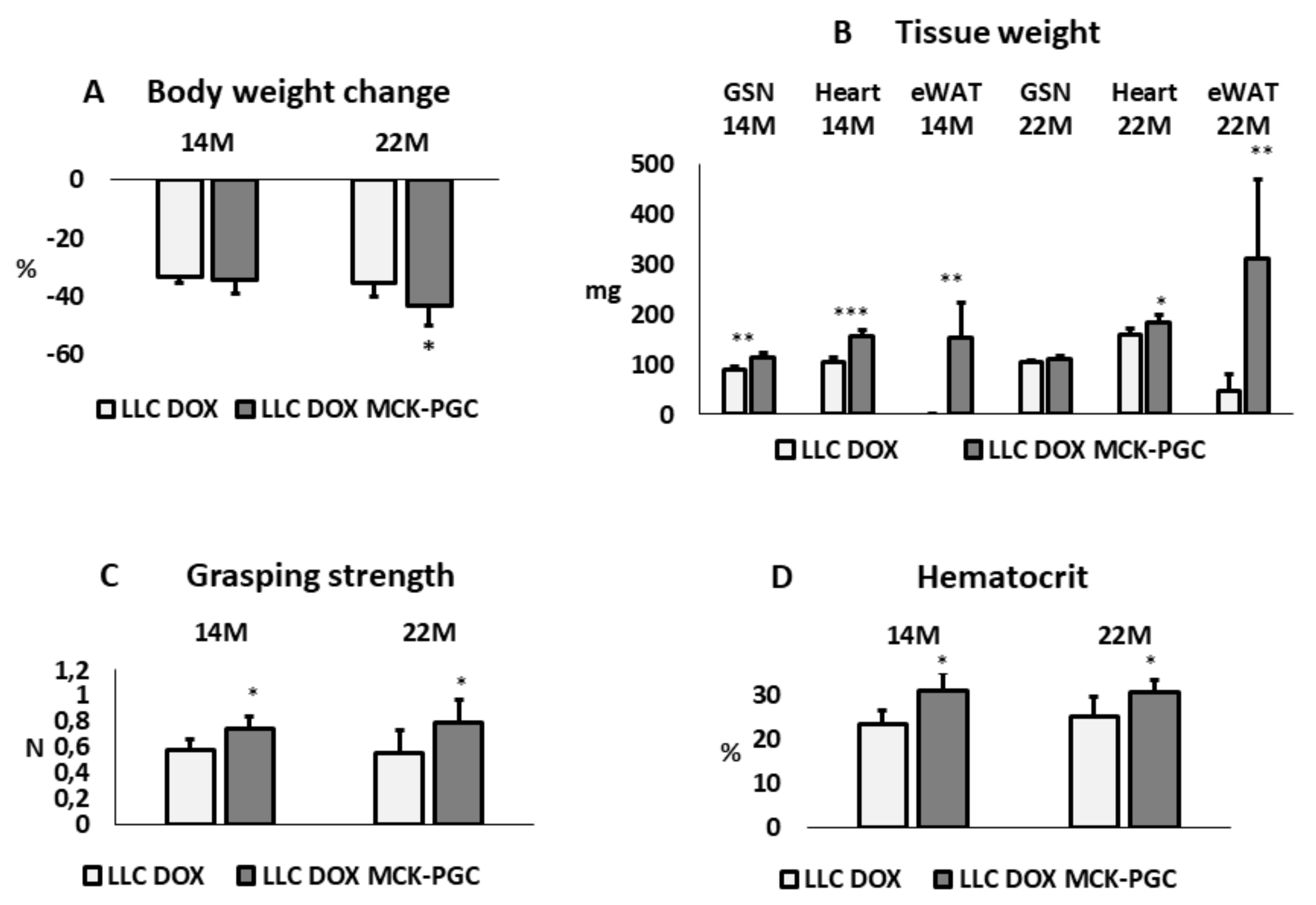
The impact of skeletal muscle-specific PGC-1α overexpression (MCK-PGC-1α mice) was assessed in 14- and 22-month-old LLC tumor-bearing mice receiving doxorubicin (DOX, 10 mg/kg BW, on days 10 and 19 after tumor implantation). It is noteworthy that before tumor injection, body weight was 30 to 40% higher in MCK-PGC-1α mice than in WT littermates, in both age cohorts. This observation is in contrast with data reported in the literature that do not show any difference in terms of body weight among WT and MCK-PGC-1α mice [59]. According to the extent of body weight loss (excluding the tumor mass), all the animals in the present study can be defined as overtly cachectic (Figure A1). Considering the results from a clinical point of view, muscle-specific PGC-1α overexpression does not prevent cancer cachexia, as body weight loss is assumed as the primary endpoint. Despite a similar percentage of body weight loss, it is fundamental to distinguish the impact on fat and lean mass in the onset of cachexia. The results show that in 14-month-old mice, both gastrocnemius (GSN) muscle and heart mass were protected in PGC-1α-overexpressing mice with respect to WT mice bearing the LLC tumor, whereas only heart mass was improved in 22-month-old mice (Figure A1B). Epididymal white adipose tissue almost disappeared in WT mice, while it was still abundant in transgenic animals in both age groups, possibly reflecting the different initial body fat content as reflected by the different initial body weight (Figure A1B). In PGC-1α-overexpressing mice, mitochondrial alterations in the skeletal muscle may be attenuated due to increased mitochondrial turnover, similar to the observations obtained in denervated mice, where PGC-1α promotes mitophagy [89], thus potentially reducing the accumulation of mitochondrial damage and accelerating DOX clearance.
Figure A1.
(A) Body weight change during tumor growth and chemotherapy experimental period of 14- and 22-month-old WT (n = 5; 7) and MCK-PGC-1α mice (n = 5; 5) bearing the LLC tumor. Data are reported as % ± SD of the initial body weight (IBW); (B) Gastrocnemius (GSN), heart, and epididymal white adipose tissue (eWAT) weight; data are expressed in mg ± SD; (C) Voluntary grasping strength; data (means ± SD) expressed in Newton. (D) Hematocrit. Data (means ± SD) expressed as percentages of total blood. For all panels, the significance of the differences was assessed by Student’s t-test for independent samples: * p < 0.05, ** p < 0.01, *** p < 0.001 vs. LLC DOX WT.
Figure A1.
(A) Body weight change during tumor growth and chemotherapy experimental period of 14- and 22-month-old WT (n = 5; 7) and MCK-PGC-1α mice (n = 5; 5) bearing the LLC tumor. Data are reported as % ± SD of the initial body weight (IBW); (B) Gastrocnemius (GSN), heart, and epididymal white adipose tissue (eWAT) weight; data are expressed in mg ± SD; (C) Voluntary grasping strength; data (means ± SD) expressed in Newton. (D) Hematocrit. Data (means ± SD) expressed as percentages of total blood. For all panels, the significance of the differences was assessed by Student’s t-test for independent samples: * p < 0.05, ** p < 0.01, *** p < 0.001 vs. LLC DOX WT.

Impaired physical function resulting from muscle wasting is one of the factors that mainly affects patients’ quality of life [90] and functional assays can be easily performed in experimental settings, increasing the translational value. In this study, the grasping strength test revealed a significant improvement in muscle strength in MCK-PGC-1α versus WT mice in both age cohorts (Figure A1C), consistent with an improvement in muscle mass. Another frequently occurring symptom in both experimental and human cancer cachexia is anemia [91]. Consistently, WT mice bearing the LLC tumor and treated with DOX revealed a dramatic reduction in the hematocrit in both age cohorts (Figure A1D), similarly to young LLC-bearing mice, as previously reported [92]. By contrast, hematocrit was significantly higher in MCK-PGC-1α than in WT animals, irrespective of mouse age, suggesting that muscle health can control erythrocyte turnover during cachexia.
Appendix A.2. Animals and Experimental Design
Animals were maintained on a regular dark-light cycle of 12:12 h with a controlled temperature (18–23 °C) and free access to food and water during the whole experimental period.
- –
- –
- For animal experiment 2 (Figure 3, Balb/c-MCK-PGC-1α mice were generated by backcrossing with WT Balb/c mice (Charles River, Calco, Italy). The colony was maintained by breeding mice as hemizygotes for 8 generations. Both WT and MCK-PGC-1α male mice of 3 months of age were used in this study.
Either 5 × 105 Lewis-Lung (LLC) or Colon26 (C26) carcinoma cells were injected subcutaneously into tumor-bearing groups. Both C26 and LLC cells were grown in vitro in DMEM supplemented with 10% FBS, 100 U/mL penicillin, 100 µg/mL streptomycin, 100 µg/mL sodium pyruvate, 2 mM L-glutamine, at 37 °C in a humidified atmosphere of 5% CO2 in air. The day of tumor implantation, C26 cells were detached with trypsin, resuspended in sterile saline, and implanted in the back of the animals. Two doses of doxorubicin (DOX, 10 mg/kg of body weight) were administered intraperitoneally 10 and 19 days after tumor implantation. Injury experiments were performed by intramuscular (i.m.) injection of 30 µL of 1.2% BaCl2 in the TA muscle. Contralateral TA muscles were injected with filtered 0.9% NaCl solution and used as controls (saline). During all subcutaneous and i.m. injections, the animals were anesthetized with 2% isoflurane in O2.
Body weight and food intake was periodically monitored, and grasping force was assessed using a Panlab-Harvard Apparatus device (Panlab, Barcelona, Spain) before euthanasia. Animals were sacrificed under anesthesia either 28 (LLC) or 14 days (C26) after tumor implantation. After intracardiac blood collection, euthanasia was applied by means of cervical dislocation. Skeletal muscles were excised, weighted, frozen in liquid nitrogen, and stored at −80 °C for further analyses.
Appendix A.3. Western Blotting
Total protein homogenates from tibialis anterior muscles were obtained by mechanical tissue disruption in RIPA buffer containing protease inhibitors. Homogenates were sonicated for 10 s at low intensity, centrifuged at 15,000× g for 5 min at 4 °C, and the supernatant was collected. Total protein concentration was quantified with Bradford reagent (Bio-Rad). Equal amounts of protein (10–30 μg) were heat-denatured in sample-loading buffer, resolved by SDS-PAGE, and transferred to nitrocellulose membranes (Bio-Rad). The filters were blocked with Tris-buffered saline (TBS) containing 0.05% Tween and 5% non-fat dry milk and then incubated overnight with antibodies directed against specific proteins: anti-cyt c (556433, BD Biosciences, Franklin Lakes, NJ, USA), anti-PGC-1α (AB3242, Merck-Sigma, Darmstadt, Germany), and anti-GAPDH (G8795, Merck-Sigma, Darmstadt, Germany). Peroxidase-conjugated IgGs (Bio-Rad) were used as secondary antibodies. Quantification of the bands was performed by densitometric analysis using a specific software (TotalLab, NonLinear Dynamics, Newcastle upon Tyne, UK).
Appendix A.4. H&E Staining
TA muscles were frozen in melting isopentane cooled in liquid nitrogen and stored at −80 °C. Transverse sections of 10 µm from the midbelly region were cut on a cryostat, left at room temperature for 10 min, and stored at −80 °C for later staining. Hematoxylin/eosin staining (H&E) was performed following standard procedures. Slides were then dehydrated using ethanol scale and xylene, and mounted using Eukitt Quick-hardening mounting medium (03959, Merck-Sigma, Darmstadt, Germany). Images were captured by a Leica DM750 optical microscope (Leica Camera AG, Wetzlar, Germany).
References
- Von Haehling, S.; Anker, M.; Anker, S. Prevalence and clinical impact of cachexia in chronic illness in Europe, USA, and Japan: Facts and numbers update 2016. J. Cachexia Sarcopenia Muscle 2016, 7, 507–509. [Google Scholar] [CrossRef] [PubMed]
- Muscaritoli, M.; Lucia, S.; Farcomeni, A.; Lorusso, V.; Saracino, V.; Barone, C.; Plastino, F.; Gori, S.; Magarotto, R.; Carteni, G.; et al. Prevalence of malnutrition in patients at first medical oncology visit: The PreMiO study. Oncotarget 2017, 8, 79884–79896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, S.T.; Van Doren, B.A.; Roy, D.; Noone, J.M.; Zacherle, E.; Blanchette, C.M. Cachexia among US cancer patients. J. Med. Econ. 2016, 19, 874–880. [Google Scholar] [CrossRef] [PubMed]
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef]
- Penna, F.; Bonetto, A.; Aversa, Z.; Minero, V.G.; Rossi Fanelli, F.; Costelli, P.; Muscaritoli, M. Effect of the specific proteasome inhibitor bortezomib on cancer-related muscle wasting. J. Cachexia Sarcopenia Muscle 2016, 7, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Pin, F.; Minero, V.G.; Penna, F.; Muscaritoli, M.; De Tullio, R.; Baccino, F.M.; Costelli, P. Interference with Ca2+-Dependent Proteolysis Does Not Alter the Course of Muscle Wasting in Experimental Cancer Cachexia. Front. Physiol. 2017, 8, 213. [Google Scholar] [CrossRef]
- Pötgens, S.A.; Thibaut, M.M.; Joudiou, N.; Sboarina, M.; Neyrinck, A.M.; Cani, P.D.; Claus, S.P.; Delzenne, N.M.; Bindels, L.B. Multi-compartment metabolomics and metagenomics reveal major hepatic and intestinal disturbances in cancer cachectic mice. J. Cachexia Sarcopenia Muscle 2021, 12, 456–475. [Google Scholar] [CrossRef]
- Ballarò, R.; Lopalco, P.; Audrito, V.; Beltrà, M.; Pin, F.; Angelini, R.; Costelli, P.; Corcelli, A.; Bonetto, A.; Szeto, H.H.; et al. Targeting Mitochondria by SS-31 Ameliorates the Whole Body Energy Status in Cancer- and Chemotherapy-Induced Cachexia. Cancers 2021, 13, 850. [Google Scholar] [CrossRef]
- Jouinot, A.; Ulmann, G.; Vazeille, C.; Durand, J.P.; Boudou-Rouquette, P.; Arrondeau, J.; Tlemsani, C.; Fournel, L.; Alifano, M.; Wislez, M.; et al. Hypermetabolism is an independent prognostic factor of survival in metastatic non-small cell lung cancer patients. Clin. Nutr. 2020, 39, 1893–1899. [Google Scholar] [CrossRef]
- Penna, F.; Ballarò, R.; Costelli, P. The Redox Balance: A Target for Interventions against Muscle Wasting in Cancer Cachexia? Antioxid. Redox Signal. 2020, 33, 542–558. [Google Scholar] [CrossRef]
- Ballarò, R.; Beltrà, M.; De Lucia, S.; Pin, F.; Ranjbar, K.; Hulmi, J.J.; Costelli, P.; Penna, F. Moderate exercise in mice improves cancer plus chemotherapy-induced muscle wasting and mitochondrial alterations. FASEB J. 2019, 33, 5482–5494. [Google Scholar] [CrossRef]
- Pin, F.; Barreto, R.; Couch, M.E.; Bonetto, A.; O’Connell, T.M.; Oconnell, T.; Barreto, R.; Couch, M.E.; Bonetto, A.; O’Connell, T.M. Cachexia induced by cancer and chemotherapy yield distinct perturbations to energy metabolism. J. Cachexia Sarcopenia Muscle 2019, 10, 140–154. [Google Scholar] [CrossRef] [Green Version]
- Penna, F.; Ballarò, R.; Martinez-Cristobal, P.; Sala, D.; Sebastian, D.; Busquets, S.; Muscaritoli, M.; Argilés, J.M.; Costelli, P.; Zorzano, A. Autophagy Exacerbates Muscle Wasting in Cancer Cachexia and Impairs Mitochondrial Function. J. Mol. Biol. 2019, 431, 2674–2686. [Google Scholar] [CrossRef]
- Brown, J.L.; Rosa-Caldwell, M.E.; Lee, D.E.; Blackwell, T.A.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Hardee, J.P.; Carson, J.A.; Wiggs, M.P.; et al. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J. Cachexia Sarcopenia Muscle 2017, 8, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Pin, F.; Ballarò, R.; Baccino, F.M.; Costelli, P. Novel investigational drugs mimicking exercise for the treatment of cachexia. Expert Opin. Investig. Drugs 2016, 25, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Pigna, E.; Berardi, E.; Aulino, P.; Rizzuto, E.; Zampieri, S.; Carraro, U.; Kern, H.; Merigliano, S.; Gruppo, M.; Mericskay, M.; et al. Aerobic Exercise and Pharmacological Treatments Counteract Cachexia by Modulating Autophagy in Colon Cancer. Sci. Rep. 2016, 6, 26991. [Google Scholar] [CrossRef] [PubMed]
- Shadfar, S.; Couch, M.E.; McKinney, K.A.; Weinstein, L.J.; Yin, X.; Rodriguez, J.E.; Guttridge, D.C.; Willis, M.; Rodríguez, J.E.; Guttridge, D.C.; et al. Oral resveratrol therapy inhibits cancer-induced skeletal muscle and cardiac atrophy in vivo. Nutr. Cancer 2011, 63, 749–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vel, K.T.; Enos, R.T.; Narsale, A.A.; Puppa, M.J.; Davis, J.M.; Murphy, E.A.; Carson, J.A.; Velázquez, K.T. Quercetin Supplementation Attenuates the Progression of Cancer Cachexia in ApcMin/+ Mice. J. Nutr. 2014, 144, 868–875. [Google Scholar] [CrossRef] [Green Version]
- Assi, M.; Rébillard, A. The Janus-Faced Role of Antioxidants in Cancer Cachexia: New Insights on the Established Concepts. Oxid. Med. Cell. Longev. 2016, 2016, 9579868. [Google Scholar] [CrossRef] [Green Version]
- Ramamoorthy, S.; Donohue, M.; Buck, M. Decreased Jun-D and myogenin expression in muscle wasting of human cachexia. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E392–E401. [Google Scholar] [CrossRef] [Green Version]
- Penna, F.; Costamagna, D.; Fanzani, A.; Bonelli, G.; Baccino, F.M.; Costelli, P. Muscle wasting and impaired Myogenesis in tumor bearing mice are prevented by ERK inhibition. PLoS ONE 2010, 5, e13604. [Google Scholar] [CrossRef]
- He, W.A.; Berardi, E.; Cardillo, V.M.; Acharyya, S.; Aulino, P.; Thomas-Ahner, J.; Wang, J.; Bloomston, M.; Muscarella, P.; Nau, P.; et al. NF-κB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Investig. 2013, 123, 4821–4835. [Google Scholar] [CrossRef] [Green Version]
- Arneson-Wissink, P.C.; Ducharme, A.M.; Doles, J.D. A novel transplantable model of lung cancer-associated tissue loss and disrupted muscle regeneration. Skelet. Muscle 2020, 10, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parajuli, P.; Kumar, S.; Loumaye, A.; Singh, P.; Eragamreddy, S.; Nguyen, T.L.; Ozkan, S.; Razzaque, M.S.; Prunier, C.; Thissen, J.P.; et al. Twist1 Activation in Muscle Progenitor Cells Causes Muscle Loss Akin to Cancer Cachexia. Dev. Cell 2018, 45, 712–725.e6. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Biswas, A.K.; Ma, W.; Kandpal, M.; Coker, C.; Grandgenett, P.M.; Hollingsworth, M.A.; Jain, R.; Tanji, K.; López-Pintado, S.; et al. Metastatic cancers promote cachexia through ZIP14 upregulation in skeletal muscle. Nat. Med. 2018, 24, 770–781. [Google Scholar] [CrossRef]
- Tang, A.; Rando, T. Induction of autophagy supports the bioenergetic demands of quiescent muscle stem cell activation. EMBO J. 2014, 33, 2782–2797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dell’Orso, S.; Juan, A.H.; Ko, K.D.; Naz, F.; Perovanovic, J.; Gutierrez-Cruz, G.; Feng, X.; Sartorelli, V. Single cell analysis of adult mouse skeletal muscle stem cells in homeostatic and regenerative conditions. Development 2019, 146, dev174177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duguez, S.; Féasson, L.; Denis, C.; Freyssenet, D. Mitochondrial biogenesis during skeletal muscle regeneration. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E802–E809. [Google Scholar] [CrossRef] [PubMed]
- Haralampieva, D.; Salemi, S.; Dinulovic, I.; Sulser, T.; Ametamey, S.M.; Handschin, C.; Eberli, D. Human muscle precursor cells overexpressing PGC-1α enhance early skeletal muscle tissue formation. Cell Transplant. 2017, 26, 1103–1114. [Google Scholar] [CrossRef] [Green Version]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H.H. Cancer-associated cachexia. Nat. Rev. Dis. Prim. 2018, 4, 17105. [Google Scholar] [CrossRef]
- Barreto, R.; Mandili, G.; Witzmann, F.A.; Novelli, F.; Zimmers, T.A.; Bonetto, A. Cancer and Chemotherapy Contribute to Muscle Loss by Activating Common Signaling Pathways. Front. Physiol. 2016, 7, 472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.P.; Baltgalvis, K.A.; Puppa, M.J.; Sato, S.; Baynes, J.W.; Carson, J.A. Muscle oxidative capacity during IL-6-dependent cancer cachexia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R201–R211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pin, F.; Busquets, S.; Toledo, M.; Camperi, A.; Lopez-Soriano, F.J.; Costelli, P.; Argilés, J.M.; Penna, F. Combination of exercise training and erythropoietin prevents cancer-induced muscle alterations. Oncotarget 2015, 6, 43202–43215. [Google Scholar] [CrossRef] [PubMed]
- Ballarò, R.; Penna, F.; Pin, F.; Gómez-Cabrera, M.C.; Viña, J.; Costelli, P. Moderate exercise improves experimental cancer cachexia by modulating the redox homeostasis. Cancers 2019, 11, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.P.; Puppa, M.J.; Sato, S.; Gao, S.; Price, R.L.; Baynes, J.W.; Kostek, M.C.; Matesic, L.E.; Carson, J.A. IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet. Muscle 2012, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2015, 78, 505–531. [Google Scholar] [CrossRef]
- Van der Ende, M.; Grefte, S.; Plas, R.; Meijerink, J.; Witkamp, R.F.; Keijer, J.; van Norren, K. Mitochondrial dynamics in cancer-induced cachexia. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 137–150. [Google Scholar] [CrossRef]
- Molinari, F.; Pin, F.; Gorini, S.; Chiandotto, S.; Pontecorvo, L.; Penna, F.; Rizzuto, E.; Pisu, S.; Musarò, A.; Costelli, P.; et al. The mitochondrial metabolic reprogramming agent trimetazidine as an “exercise mimetic” in cachectic C26-bearing mice. J. Cachexia Sarcopenia Muscle 2017, 8, 954–973. [Google Scholar] [CrossRef]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef] [Green Version]
- Dudek, J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Biol. 2017, 5, 90. [Google Scholar] [CrossRef] [Green Version]
- Montero-Bullon, J.F.; Melo, T.; Ferreira, R.; Padrão, A.I.; Oliveira, P.A.; Domingues, M.R.M.; Domingues, P. Exercise training counteracts urothelial carcinoma-induced alterations in skeletal muscle mitochondria phospholipidome in an animal model. Sci. Rep. 2019, 9, 13423. [Google Scholar] [CrossRef] [Green Version]
- De Castro, G.S.; Simoes, E.; Lima, J.D.C.C.; Ortiz-Silva, M.; Festuccia, W.T.; Tokeshi, F.; Alcântara, P.S.; Otoch, J.P.; Coletti, D.; Seelaender, M. Human cachexia induces changes in mitochondria, autophagy and apoptosis in the skeletal muscle. Cancers 2019, 11, 1264. [Google Scholar] [CrossRef] [Green Version]
- Puppa, M.J.; White, J.P.; Velázquez, K.T.; Baltgalvis, K.A.; Sato, S.; Baynes, J.W.; Carson, J.A. The effect of exercise on IL-6-induced cachexia in the ApcMin/+ mouse. J. Cachexia Sarcopenia Muscle 2012, 3, 117–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padilha, C.S.; Borges, F.H.; Costa Mendes da Silva, L.E.; Frajacomo, F.T.T.; Jordao, A.A.; Duarte, J.A.; Cecchini, R.; Guarnier, F.A.; Deminice, R. Resistance exercise attenuates skeletal muscle oxidative stress, systemic pro-inflammatory state, and cachexia in Walker-256 tumor-bearing rats. Appl. Physiol. Nutr. Metab. 2017, 42, 916–923. [Google Scholar] [CrossRef]
- Ranjbar, K.; Ballaro, R.; Bover, Q.; Pin, F.; Beltra, M.; Penna, F.; Costelli, P. Combined Exercise Training Positively Affects Muscle Wasting in Tumor-bearing Mice. Med. Sci. Sports Exerc. 2019, 51, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Eirin, A.; Ebrahimi, B.; Kwon, S.H.; Fiala, J.A.; Williams, B.J.; Woollard, J.R.; He, Q.; Gupta, R.C.; Sabbah, H.N.; Prakash, Y.S.; et al. Restoration of mitochondrial cardiolipin attenuates cardiac damage in swine renovascular hypertension. J. Am. Heart Assoc. 2016, 5, e003118. [Google Scholar] [CrossRef] [Green Version]
- Escribano-Lopez, I.; Diaz-Morales, N.; Iannantuoni, F.; Lopez-Domenech, S.; de Marañon, A.M.; Abad-Jimenez, Z.; Bañuls, C.; Rovira-Llopis, S.; Herance, J.R.; Rocha, M.; et al. The mitochondrial antioxidant SS-31 increases SIRT1 levels and ameliorates inflammation, oxidative stress and leukocyte-endothelium interactions in type 2 diabetes. Sci. Rep. 2018, 8, 15862. [Google Scholar] [CrossRef]
- Zhao, W.; Xu, Z.; Cao, J.; Fu, Q.; Wu, Y.; Zhang, X.; Long, Y.; Zhang, X.; Yang, Y.; Li, Y.; et al. Elamipretide (SS-31) improves mitochondrial dysfunction, synaptic and memory impairment induced by lipopolysaccharide in mice. J. Neuroinflammat. 2019, 16, 230. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.D.; Duan, J.; Samuelson, A.T.; Gaffrey, M.J.; Merrihew, G.E.; Egertson, J.D.; Wang, L.; Bammler, T.K.; Moore, R.J.; White, C.C.; et al. Improving mitochondrial function with SS-31 reverses age-related redox stress and improves exercise tolerance in aged mice. Free Radic. Biol. Med. 2019, 134, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Chiao, Y.A.; Zhang, H.; Sweetwyne, M.; Whitson, J.; Ting, Y.S.; Basisty, N.; Pino, L.K.; Quarles, E.; Nguyen, N.H.; Campbell, M.D.; et al. Late-life restoration of mitochondrial function reverses cardiac dysfunction in old mice. eLife 2020, 9, e55513. [Google Scholar] [CrossRef]
- Liu, Y.; Fu, H.; Wu, Y.; Nie, B.; Liu, F.; Wang, T.; Xiao, W.; Yang, S.; Kan, M.; Fan, L. Elamipretide (SS-31) Improves Functional Connectivity in Hippocampus and Other Related Regions Following Prolonged Neuroinflammation Induced by Lipopolysaccharide in Aged Rats. Front. Aging Neurosci. 2021, 13, 600484. [Google Scholar] [CrossRef]
- Lopaschuk, D.G.; Barr, R.; Panakkezhum, D.T.; Dyck, R.J. Beneficial Effects of Trimetazidine in Ex Vivo Working Ischemic Hearts Are Due to a Stimulation of Glucose Oxidation Secondary to Inhibition of Long-Chain 3-Ketoacyl Coenzyme A Thiolase. Circ. Res. 2003, 93, e33–e37. [Google Scholar] [CrossRef] [Green Version]
- Dyck, J.R.B. Malonyl Coenzyme A Decarboxylase Inhibition Protects the Ischemic Heart by Inhibiting Fatty Acid Oxidation and Stimulating Glucose Oxidation. Circ. Res. 2004, 94, e78–e84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Hulver, M.W.; McMillan, R.P.; Cline, M.A.; Gilbert, E.R. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr. Metab. 2014, 11, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pin, F.; Novinger, L.J.; Huot, J.R.; Harris, R.A.; Couch, M.E.; O’Connell, T.M.; Bonetto, A. PDK4 drives metabolic alterations and muscle atrophy in cancer cachexia. FASEB J. 2019, 33, 7778–7790. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.-Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef]
- Rowe, G.C.; Patten, I.S.; Zsengeller, Z.K.; El-Khoury, R.; Okutsu, M.; Bampoh, S.; Koulisis, N.; Farrell, C.; Hirshman, M.F.; Yan, Z.; et al. Disconnecting Mitochondrial Content from Respiratory Chain Capacity in PGC-1-Deficient Skeletal Muscle. Cell Rep. 2013, 3, 1449–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandri, M.; Lin, J.; Handschin, C.; Yang, W.; Arany, Z.P.; Lecker, S.H.; Goldberg, A.L.; Spiegelman, B.M. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 16260–16265. [Google Scholar] [CrossRef] [Green Version]
- Wenz, T.; Rossi, S.G.; Rotundo, R.L.; Spiegelman, B.M.; Moraes, C.T. Increased muscle PGC-1α expression protects from sarcopenia and metabolic disease during aging. Proc. Natl. Acad. Sci. USA 2009, 106, 20405–20410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannavino, J.; Brocca, L.; Sandri, M.; Grassi, B.; Bottinelli, R.; Pellegrino, M.A. The role of alterations in mitochondrial dynamics and PGC-1α over-expression in fast muscle atrophy following hindlimb unloading. J. Physiol. 2015, 593, 1981–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Pickrell, A.M.; Zimmers, T.A.; Moraes, C.T. Increase in muscle mitochondrial biogenesis does not prevent muscle loss but increased tumor size in a mouse model of acute cancer-induced cachexia. PLoS ONE 2012, 7, e33426. [Google Scholar] [CrossRef]
- Ruas, J.L.; White, J.P.; Rao, R.R.; Kleiner, S.; Brannan, K.T.; Harrison, B.C.; Greene, N.P.; Wu, J.; Estall, J.L.; Irving, B.A.; et al. A PGC-1α isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 2012, 151, 1319–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penna, F.; Busquets, S.; Argilés, J.M.J.M. Experimental cancer cachexia: Evolving strategies for getting closer to the human scenario. Semin. Cell Dev. Biol. 2016, 54, 20–27. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, H.; Li, X.; Yang, T.; Jiang, Q. Effects of PPARα/PGC-1α on the myocardial energy metabolism during heart failure in the doxorubicin induced dilated cardiomyopathy in mice. Int. J. Clin. Exp. Med. 2014, 7, 2435–2442. [Google Scholar] [PubMed]
- Guo, J.; Guo, Q.; Fang, H.; Lei, L.; Zhang, T.; Zhao, J.; Peng, S. Cardioprotection against doxorubicin by metallothionein Is associated with preservation of mitochondrial biogenesis involving PGC-1α pathway. Eur. J. Pharmacol. 2014, 737, 117–124. [Google Scholar] [CrossRef]
- Correia, J.C.; Ferreira, D.M.S.; Ruas, J.L. Intercellular: Local and systemic actions of skeletal muscle PGC-1s. Trends Endocrinol. Metab. 2015, 26, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.M.; Kempen, L.J.A.P.; Hardy, R.S.; Langen, R.C.J. Inflammation and Skeletal Muscle Wasting During Cachexia. Front. Physiol. 2020, 11, 597675. [Google Scholar] [CrossRef]
- Talbert, E.E.; Guttridge, D.C. Impaired Regeneration: A Role for the Muscle Microenvironment in Cancer Cachexia. Semin. Cell Dev. Biol. 2015, 54, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Inaba, S.; Hinohara, A.; Tachibana, M.; Tsujikawa, K.; Fukada, S. Muscle regeneration is disrupted by cancer cachexia without loss of muscle stem cell potential. PLoS ONE 2018, 13, e0205467. [Google Scholar] [CrossRef] [Green Version]
- Costamagna, D.; Duelen, R.; Penna, F.; Neumann, D.; Costelli, P.; Sampaolesi, M. Interleukin-4 administration improves muscle function, adult myogenesis, and lifespan of colon carcinoma-bearing mice. J. Cachexia Sarcopenia Muscle 2020, 11, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Pala, F.; Di Girolamo, D.; Mella, S.; Yennek, S.; Chatre, L.; Ricchetti, M.; Tajbakhsh, S. Distinct metabolic states govern skeletal muscle stem cell fates during prenatal and postnatal myogenesis. J. Cell Sci. 2018, 131, jcs212977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, M.; Cappellesso, F.; Amorim, R.; Serneels, J.; Virga, F.; Eelen, G.; Carobbio, S.; Rincon, M.Y.; Maechler, P.; De Bock, K.; et al. Macrophage-derived glutamine boosts satellite cells and muscle regeneration. Nature 2020, 587, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Guitart, M.; Lloreta, J.; Mañas-Garcia, L.; Barreiro, E. Muscle regeneration potential and satellite cell activation profile during recovery following hindlimb immobilization in mice. J. Cell. Physiol. 2018, 233, 4360–4372. [Google Scholar] [CrossRef] [Green Version]
- Mañas-García, L.; Penedo-Vázquez, A.; López-Postigo, A.; Deschrevel, J.; Durán, X.; Barreiro, E. Prolonged immobilization exacerbates the loss of muscle mass and function induced by cancer-associated cachexia through enhanced proteolysis in mice. Int. J. Mol. Sci. 2020, 21, 8167. [Google Scholar] [CrossRef] [PubMed]
- Stearns-Reider, K.M.; D’Amore, A.; Beezhold, K.; Rothrauff, B.; Cavalli, L.; Wagner, W.R.; Vorp, D.A.; Tsamis, A.; Shinde, S.; Zhang, C.; et al. Aging of the skeletal muscle extracellular matrix drives a stem cell fibrogenic conversion. Aging Cell 2017, 16, 518–528. [Google Scholar] [CrossRef] [Green Version]
- VanderVeen, B.N.; Murphy, E.A.; Carson, J.A. The Impact of Immune Cells on the Skeletal Muscle Microenvironment During Cancer Cachexia. Front. Physiol. 2020, 11, 1037. [Google Scholar] [CrossRef]
- Daou, N.; Hassani, M.; Matos, E.; De Castro, G.S.; Costa, R.G.F.; Seelaender, M.; Moresi, V.; Rocchi, M.; Adamo, S.; Li, Z.; et al. Displaced myonuclei in cancer cachexia suggest altered innervation. Int. J. Mol. Sci. 2020, 21, 1092. [Google Scholar] [CrossRef] [Green Version]
- Sartori, R.; Hagg, A.; Zampieri, S.; Armani, A.; Winbanks, C.E.; Viana, L.R.; Haidar, M.; Watt, K.I.; Qian, H.; Pezzini, C.; et al. Perturbed BMP signaling and denervation promote muscle wasting in cancer cachexia. Sci. Transl. Med. 2021, 13, eaay9592. [Google Scholar] [CrossRef] [PubMed]
- Huot, J.R.; Pin, F.; Bonetto, A. Muscle weakness caused by cancer and chemotherapy is associated with loss of motor unit connectivity. Am. J. Cancer Res. 2021, 11, 2990–3001. [Google Scholar]
- Judge, S.M.; Deyhle, M.R.; Neyroud, D.; Nosacka, R.L.; D’Lugos, A.C.; Cameron, M.E.; Vohra, R.S.; Smuder, A.J.; Roberts, B.M.; Callaway, C.S.; et al. MEF2c-dependent downregulation of myocilin mediates cancer-induced muscle wasting and associates with cachexia in patients with cancer. Cancer Res. 2021, 80, 1861–1874. [Google Scholar] [CrossRef] [Green Version]
- Prado, C.M.M.; Bekaii-Saab, T.; Doyle, L.A.; Shrestha, S.; Ghosh, S.; Baracos, V.E.; Sawyer, M.B. Skeletal muscle anabolism is a side effect of therapy with the MEK inhibitor: Selumetinib in patients with cholangiocarcinoma. Br. J. Cancer 2012, 106, 1583–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.A.; Calore, F.; Londhe, P.; Canella, A.; Guttridge, D.C.; Croce, C.M. Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proc. Natl. Acad. Sci. USA 2014, 111, 4525–4529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calore, F.; Londhe, P.; Fadda, P.; Nigita, G.; Casadei, L.; Marceca, G.P.; Fassan, M.; Lovat, F.; Gasparini, P.; Rizzotto, L.; et al. The TLR7/8/9 antagonist IMO-8503 inhibits cancer-induced cachexia. Cancer Res. 2018, 78, 6680–6690. [Google Scholar] [CrossRef] [Green Version]
- Coletti, D.; Aulino, P.; Pigna, E.; Barteri, F.; Moresi, V.; Annibali, D.; Adamo, S.; Berardi, E. Spontaneous physical activity downregulates pax7 in cancer cachexia. Stem Cells Int. 2016, 2016, 6729268. [Google Scholar] [CrossRef] [Green Version]
- Tamilarasan, K.P.; Temmel, H.; Das, S.K.; Al Zoughbi, W.; Schauer, S.; Vesely, P.W.; Hoefler, G. Skeletal muscle damage and impaired regeneration due to LPL-mediated lipotoxicity. Cell Death Dis. 2012, 3, e354. [Google Scholar] [CrossRef] [Green Version]
- Putman, C.T.; Sultan, K.R.; Wassmer, T.; Bamford, J.A.; Skorjanc, D.; Pette, D. Fiber-type transitions and satellite cell activation in low-frequency-stimulated muscles of young and aging rats. J. Gerontol.—Ser. A Biol. Sci. Med. Sci. 2001, 56, B510–B519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatta, L.; Vitiello, L.; Gorini, S.; Chiandotto, S.; Costelli, P.; Giammarioli, A.M.; Malorni, W.; Rosano, G.; Ferraro, E. Modulating the metabolism by trimetazidine enhances myoblast differentiation and promotes myogenesis in cachectic tumorbearing c26 mice. Oncotarget 2017, 8, 113938–113956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vainshtein, A.; Desjardins, E.M.; Armani, A.; Sandri, M.; Hood, D.A. PGC-1α modulates denervation-induced mitophagy in skeletal muscle. Skelet. Muscle 2015, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Argilés, J.M.; López-Soriano, F.J.; Toledo, M.; Betancourt, A.; Serpe, R.; Busquets, S.; Van Hauwermeiren, F.; Armaka, M.; Karagianni, N.; Kranidioti, K.; et al. Definition of cancer cachexia: Effect of weight loss, reduced food intake, and systemic inflammation on functional status and. J. Cell Biol. 2009, 24, 7–26. [Google Scholar] [CrossRef]
- Knight, K.; Wade, S.; Balducci, L. Prevalence and outcomes of anemia in cancer: A systematic review of the literature. Am. J. Med. 2004, 116 (Suppl. 1), 11–26. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Busquets, S.; Toledo, M.; Pin, F.; Massa, D.; López-Soriano, F.J.; Costelli, P.; Argilés, J.M. Erythropoietin administration partially prevents adipose tissue loss in experimental cancer cachexia models. J. Lipid Res. 2013, 54, 3045–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
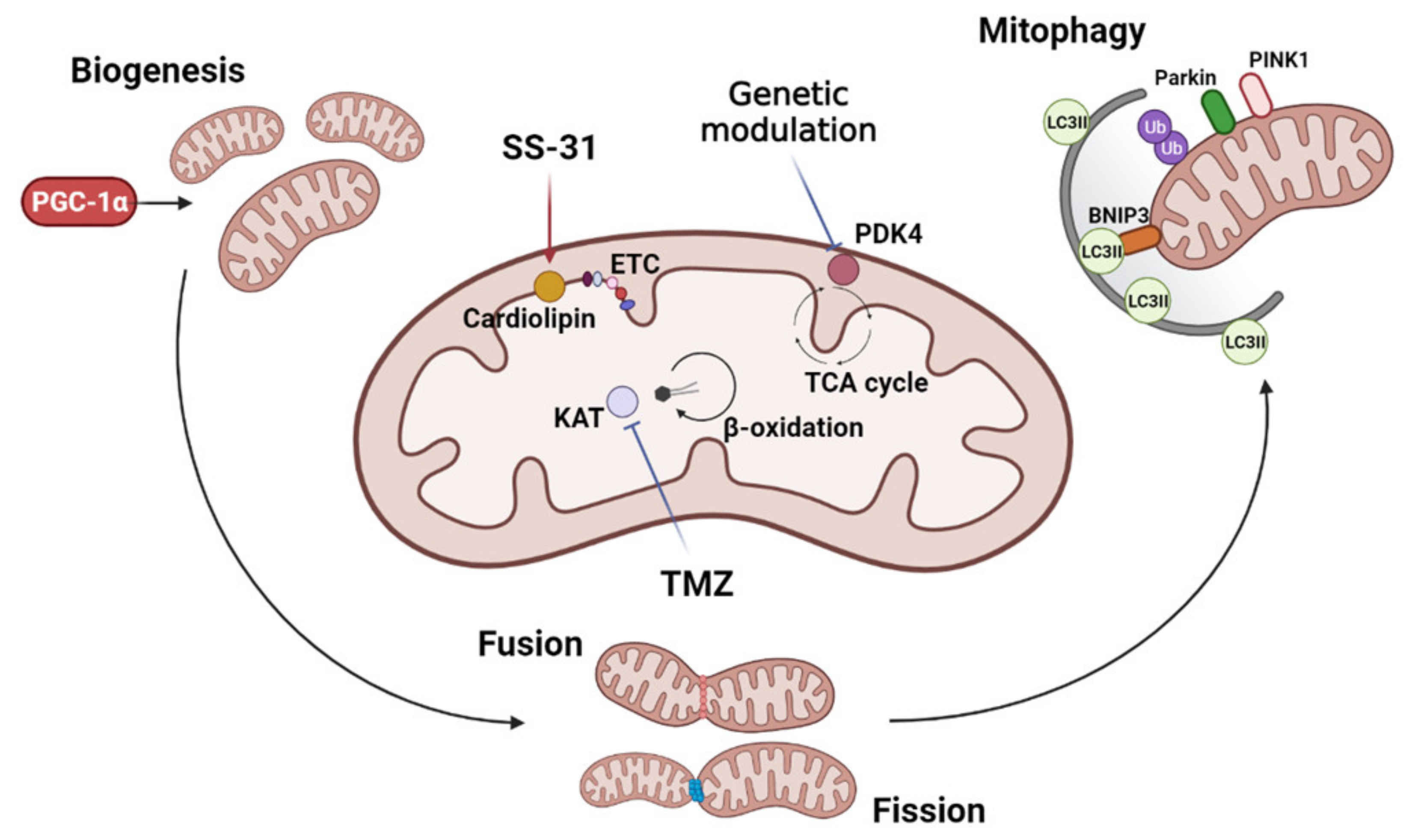
Figure 1.
Mitochondria-targeted anti-cachexia interventions. Interventions focused on mitochondria to improve cancer and chemotherapy-induced muscle wasting may include the modulation of mitochondrial biogenesis, dynamics (fusion and fission), and mitophagy. Different strategies have been adopted to directly target mitochondria: SS-31 protects cardiolipin, enhancing mitochondrial activity and energy production through increased electron transport chain (ETC) efficiency; trimetazidine (TMZ) inhibits the 3-ketoacyl-CoA thiolase (KAT) activity, shifting the mitochondrial metabolism towards oxidative respiration and increasing mitochondrial function; the genetic deletion of pyruvate dehydrogenase kinase 4 (PDK4) enhances the influx of pyruvate into the TCA cycle, likely improving mitochondrial homeostasis. Image created with BioRender.com (accessed on 17 August 2021).
Figure 1.
Mitochondria-targeted anti-cachexia interventions. Interventions focused on mitochondria to improve cancer and chemotherapy-induced muscle wasting may include the modulation of mitochondrial biogenesis, dynamics (fusion and fission), and mitophagy. Different strategies have been adopted to directly target mitochondria: SS-31 protects cardiolipin, enhancing mitochondrial activity and energy production through increased electron transport chain (ETC) efficiency; trimetazidine (TMZ) inhibits the 3-ketoacyl-CoA thiolase (KAT) activity, shifting the mitochondrial metabolism towards oxidative respiration and increasing mitochondrial function; the genetic deletion of pyruvate dehydrogenase kinase 4 (PDK4) enhances the influx of pyruvate into the TCA cycle, likely improving mitochondrial homeostasis. Image created with BioRender.com (accessed on 17 August 2021).

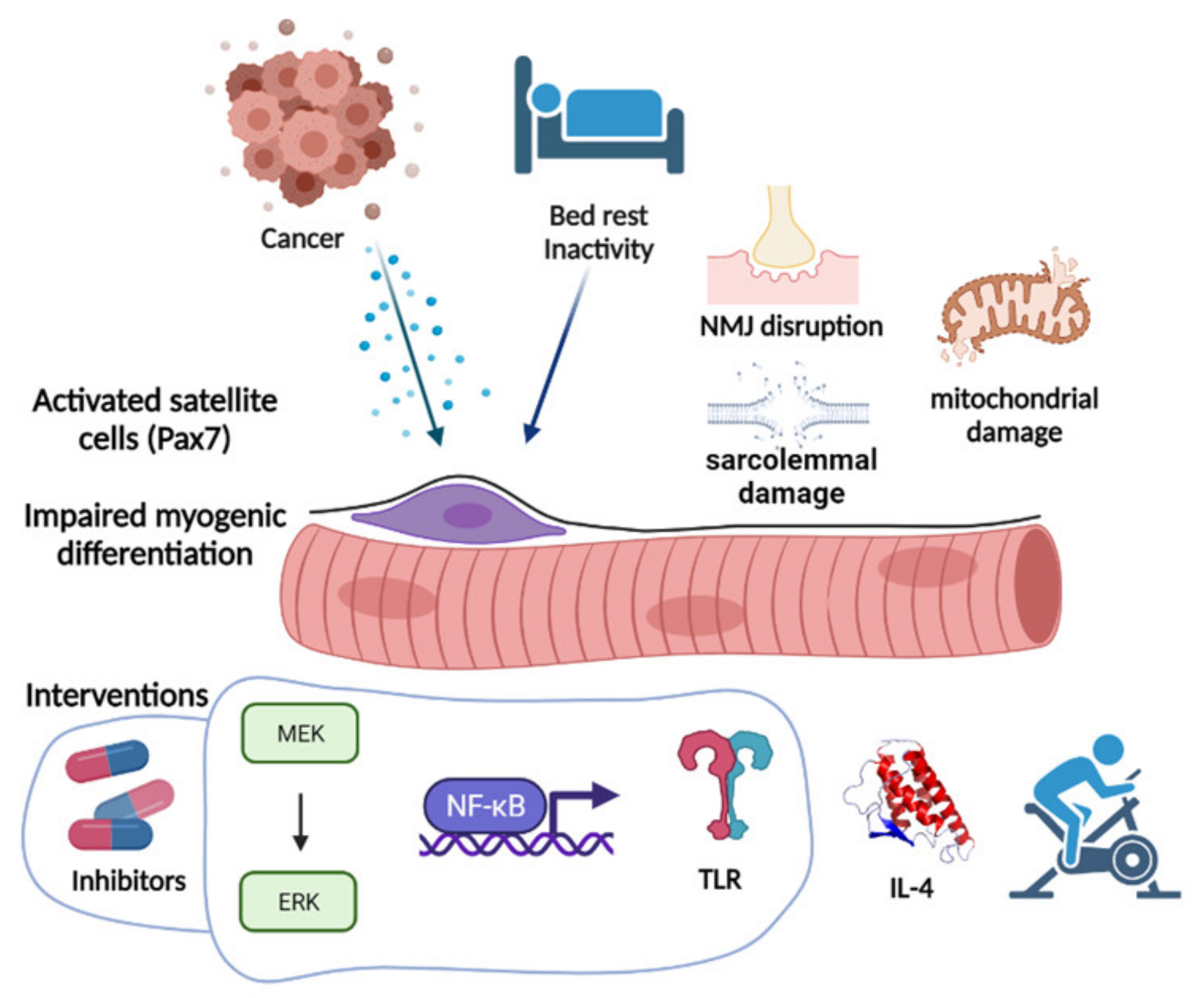
Figure 2.
Cancer and the consequent inactivity impair the myogenic process, either directly via cytokine release or through several mechanisms including neuromuscular junction (NMJ) disruption, and mitochondrial and sarcolemmal damage. Several interventions have been proposed, mainly focused on signaling pathway inhibition, cytokine modulation, and exercise or exercise-mimicking metabolism modulators. Image created with BioRender.com (accessed on 17 August 2021).
Figure 2.
Cancer and the consequent inactivity impair the myogenic process, either directly via cytokine release or through several mechanisms including neuromuscular junction (NMJ) disruption, and mitochondrial and sarcolemmal damage. Several interventions have been proposed, mainly focused on signaling pathway inhibition, cytokine modulation, and exercise or exercise-mimicking metabolism modulators. Image created with BioRender.com (accessed on 17 August 2021).

Figure 3.
(A,B) Representative Western blotting bands and densitometric analysis of PGC-1α protein expression, normalized by GAPDH. Data are reported as % ± SD of the uninjured control group; (C,D) Representative H&E panels and CSA quantification of injured muscles of either healthy or cachectic WT and MCK-PGC-1α mice. Data are expressed as % ± SD of the uninjured WT control group; (E) Representative pictures of tibialis anterior muscles according to its specific experimental condition. (F) Representative Western blotting bands and densitometric analysis of cyt c protein, normalized by GAPDH expression. Data are reported as % ± SD of uninjured muscle in each experimental condition. For all panels, the significance of the differences was assessed by Student’s t-test for independent samples: * p < 0.05, *** p < 0.001.
Figure 3.
(A,B) Representative Western blotting bands and densitometric analysis of PGC-1α protein expression, normalized by GAPDH. Data are reported as % ± SD of the uninjured control group; (C,D) Representative H&E panels and CSA quantification of injured muscles of either healthy or cachectic WT and MCK-PGC-1α mice. Data are expressed as % ± SD of the uninjured WT control group; (E) Representative pictures of tibialis anterior muscles according to its specific experimental condition. (F) Representative Western blotting bands and densitometric analysis of cyt c protein, normalized by GAPDH expression. Data are reported as % ± SD of uninjured muscle in each experimental condition. For all panels, the significance of the differences was assessed by Student’s t-test for independent samples: * p < 0.05, *** p < 0.001.

Figure 4.
Proposed schematic pathogenesis of muscle wasting in cancer cachexia according to primary, antagonistic, and integrative hallmarks. Image created with BioRender.com (accessed on 17 August 2021).
Figure 4.
Proposed schematic pathogenesis of muscle wasting in cancer cachexia according to primary, antagonistic, and integrative hallmarks. Image created with BioRender.com (accessed on 17 August 2021).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Beltrà, M.; Pin, F.; Ballarò, R.; Costelli, P.; Penna, F. Mitochondrial Dysfunction in Cancer Cachexia: Impact on Muscle Health and Regeneration. Cells 2021, 10, 3150. https://doi.org/10.3390/cells10113150
AMA Style
Beltrà M, Pin F, Ballarò R, Costelli P, Penna F. Mitochondrial Dysfunction in Cancer Cachexia: Impact on Muscle Health and Regeneration. Cells. 2021; 10(11):3150. https://doi.org/10.3390/cells10113150
Chicago/Turabian StyleBeltrà, Marc, Fabrizio Pin, Riccardo Ballarò, Paola Costelli, and Fabio Penna. 2021. "Mitochondrial Dysfunction in Cancer Cachexia: Impact on Muscle Health and Regeneration" Cells 10, no. 11: 3150. https://doi.org/10.3390/cells10113150
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.