
STK11/LKB1 Modulation of the Immune Response in Lung Cancer: From Biology to Therapeutic Impact
by
 , , , and
, , , and
Elvire Pons-Tostivint
1,2,*,† ,
,
Alexandre Lugat
2,†,
Jean-François Fontenau
2,
Marc Guillaume Denis
3 and
and
Jaafar Bennouna
2,4 1
Medical Oncology Department, Nantes University Hospital, 44000 Nantes, France
2
Center for Research in Cancerology and Immunology Nantes-Angers (CRCINA), University of Nantes, INSERM UMR 1232, 44000 Nantes, France
3
Department of Biochemistry, Nantes University Hospital, 44000 Nantes, France
4
Medical Oncology Department, Hopital Foch, 75073 Suresnes, France
*
Author to whom correspondence should be addressed.
†
E.P.-T. and A.L. equally contributed to the work.
Cells 2021, 10(11), 3129; https://doi.org/10.3390/cells10113129
Submission received: 27 September 2021
/
Revised: 28 October 2021
/
Accepted: 9 November 2021
/
Published: 11 November 2021
(This article belongs to the Special Issue Molecular and Cellular Mechanisms of Lung Cancers)
Abstract
:The STK11/LKB1 gene codes for liver kinase B1 (STK11/LKB1), a highly conserved serine/threonine kinase involved in many energy-related cellular processes. The canonical tumor-suppressive role for STK11/LKB1 involves the activation of AMPK-related kinases, a master regulator of cell survival during stress conditions. In pre-clinical models, inactivation of STK11/LKB1 leads to the progression of lung cancer with the acquisition of metastatic properties. Moreover, preclinical and clinical data have shown that inactivation of STK11/LKB1 is associated with an inert tumor immune microenvironment, with a reduced density of infiltrating cytotoxic CD8+ T lymphocytes, a lower expression of PD-(L)1, and a neutrophil-enriched tumor microenvironment. In this review, we first describe the biological function of STK11/LKB1 and the role of its inactivation in cancer cells. We report descriptive epidemiology, co-occurring genomic alterations, and prognostic impact for lung cancer patients. Finally, we discuss recent data based on pre-clinical models and lung cancer cohorts analyzing the results of STK11/LKB1 alterations on the immune system and response or resistance to immune checkpoint inhibitors.
1. Introduction
Germinal heterozygous mutations in the serine/threonine kinase 11 (STK11) gene were first identified as the causal mutation of the Peutz-Jeghers Syndrome, an autosomal dominant condition characterized by multiple hamartomatous polyps in the gastrointestinal tract and an increased cancer risk [1]. The STK11 gene codes for liver kinase B1 (LKB1), a highly conserved serine/threonine kinase that acts as a sensor of cellular energy, giving it a special role in cellular metabolism, especially in cancer cells [2]. Somatic STK11 alterations have been described in many different tumor types, and they represent the second most altered tumor suppressor gene after TP53 in non-small cell lung cancer (NSCLC).
Targeted therapies have significantly improved survival in non-squamous NSCLC patients expressing specific molecular alterations such as epidermal growth factor receptor (EGFR)-sensitizing mutations, ALK and ROS-1 rearrangements [3]. Outside of this selective context of oncogenic addiction, chemotherapy associated with immune checkpoint inhibitors (ICI) or ICI alone remains the first-line treatment option [3]. Unfortunately, most patients with advanced NSCLC relapse after treatment with PD-(L)1 (programmed death (ligand) 1) axis inhibitors. The landscape of primary and secondary resistance to PD-1 blockade is still largely unknown, while it is currently well known that molecular alterations of tumors are involved in the shaping of their immune microenvironment.
STK11/LKB1 alterations may have a major impact on the immune microenvironment of lung tumors, and evidence suggests its potential role in the resistance to ICI. This review aims to provide an overview of STK11/LKB1 biological functions and its implications in carcinogenesis. We present pre-clinical and clinical data evaluating the relevance of STK11/LKB1 alterations in NSCLC and its correlation with the immune system and cellular processes. Finally, we discuss available data reporting the therapeutic impact of STK11/LKB1 alterations on ICI efficacy in NSCLC patients.
2. STK11/LKB1: An Overview of Biological Functions
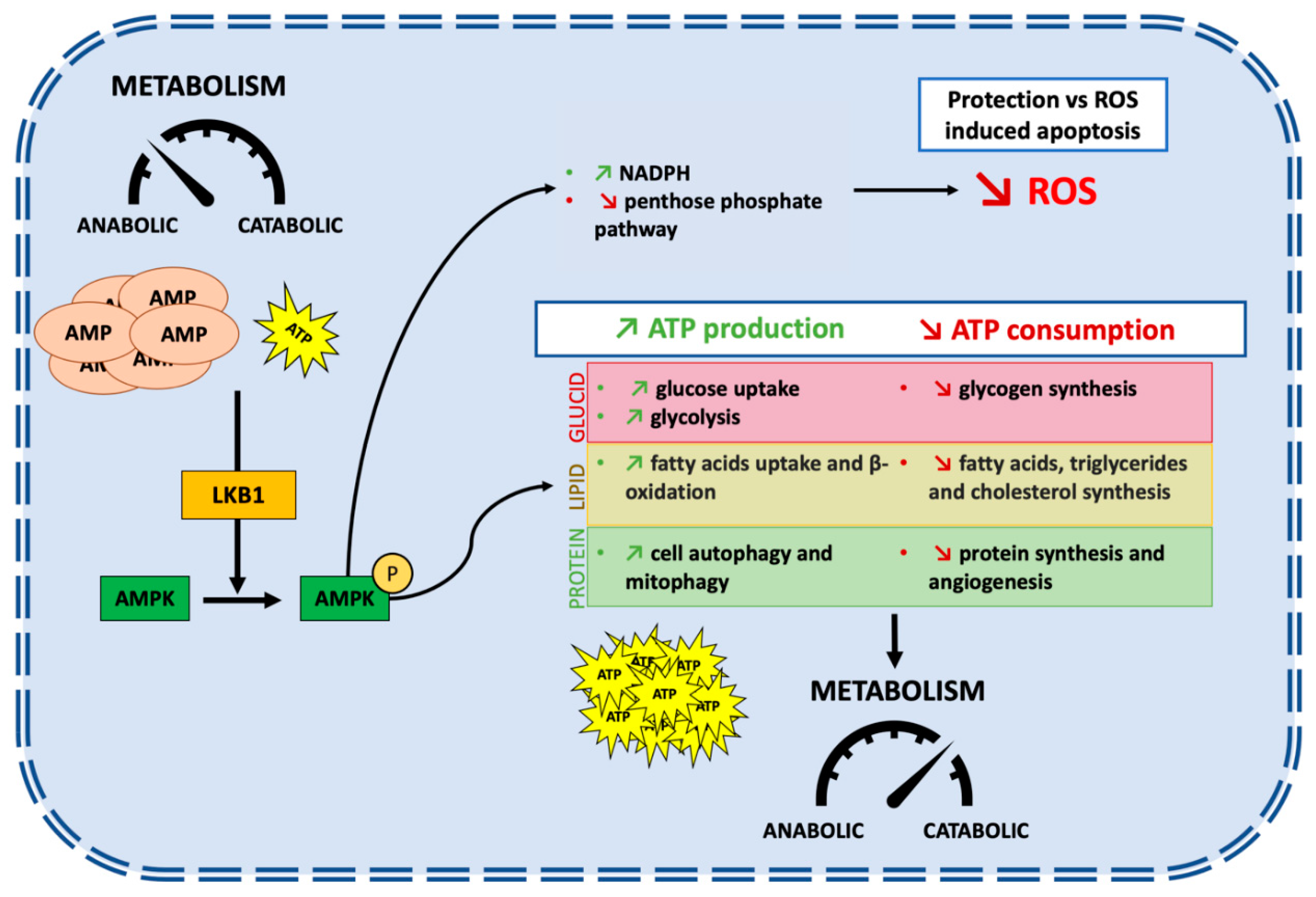
The STK11 gene is located at the telomeric region of the short arm of chromosome 19 (19p13.3) [4,5,6]. Nine exons code for a sequence of 443-amino acids which form the LKB1 protein [7]. STK11/LKB1 is an essential master upstream kinase that activates AMP-activated protein kinase (AMPK) in case of energy deprivation (Figure 1). AMPK is a central metabolic checkpoint in the cell that regulates glucose and lipid metabolism in response to nutrients and energy variations, as well as other cellular functions such as autophagy and polarity. Thus, under nutrient deprivation or hypoxia, an AMP accumulation occurs, in conjunction with ATP depletion, leading to the direct activation of AMPK by STK11/LKB1 [8]. Activation of AMPK in conjunction with other regulators allows a switch from an anabolic to a catabolic metabolism, promoting cell survival in energy stress conditions [9]. It triggers physiological processes that regenerate ATP, as well as activating 12 other kinases of the AMPK subfamily [10]. AMPK is involved in multiple metabolic ways to increase ATP cellular levels, both by promoting its production and reducing its consumption. Among others, AMPK promotes lipid catabolism by increasing fatty acid uptake and β-oxidation, leading to ATP and NADPH production [11,12,13,14]. LKB1/AMPK is also involved in the glucose catabolism by an increase in glucose uptake and the promotion of the glycolysis process [15,16].
Protein synthesis is energetically costly, requiring large amounts of ATP. It needs to be stopped to preserve cellular ATP. The large serine/threonine protein kinase mTOR (mechanistic target of rapamycin) is a central regulator of nutrient, energy, and growth factor signaling. It also regulates the activity of the translational machinery, deregulated in most solid tumors. Importantly, STK11/LKB1-AMPK activation inhibits the mTOR pathway leading to the inhibition of protein synthesis, thus limiting ATP intake [2]. Inhibition of mTOR also negatively regulates hypoxia-inducible factor 1 α (HIF-1α), a major regulator of genes involved in cellular metabolism and adaptation to hypoxic conditions by promoting angiogenesis [17]. Moreover, STK11/LKB1 via AMPK promotes autophagy and mitophagy, a mechanism that is initiated under nutrient starvation which increases intracellular metabolic reserves [18,19]. Autophagy is one of the key mechanisms that promotes cell survival in cancer under stress conditions [20].
Besides its role in the cellular metabolism modification secondary to AMPK activation, STK11/LKB1 has a protective function against active stress. Reactive oxygen species (ROS) are highly reactive molecules produced by aerobic oxygen metabolism. Their accumulation triggers irreversible damages to DNA, especially under glucose starvation or hypoxia [21]. On the one hand, the accumulation of ROS may promote pro-oncogenic mutations and facilitate tumorigenesis; on the other hand, ROS may render cancer cells more vulnerable to cell death. High cellular levels of ROS activate the STK11/LKB1-AMPK pathway. It increases the production of NADPH, a key antioxidant, thus decreasing the level of ROS in order to prevent ROS-induced apoptosis [22,23]. Accordingly, STK11/LKB1 loss enhances levels of intracellular ROS and could lead to an increased sensibility to ROS-induced chemotherapies [24].
To sum up, STK11/LKB1 is an upstream kinase of the AMPK pathway, a major regulator of cell metabolism under stress conditions (Figure 1). STK11/LKB1 reschedules cell metabolism by restraining the activity of anabolic enzymes and by promoting the production of instantly available energy with ATP, thus slowing down cell growth and ATP-consuming processes. It also protects the genome from ROS-induced oxidation by regulating antioxidant gene products, and finally promotes cell survival under stress conditions.
3. STK11/LKB1: A Tumor Suppressor
3.1. Molecular Mechanisms of STK11/LKB1 Inactivation in Cancers
STK11/LKB1 loss of function has been found in several cancer types, mainly through somatic alterations in the STK11 gene such as non-sense mutation, loss of heterozygosity, insertions, intragenic deletions, or chromosomal deletions [25,26,27,28,29,30,31,32,33]. A recent study of 4446 patients with solid tumors found that the rate of STK11 alterations was 1.35% (n = 60) [34]. Forty-five percent of STK11-altered tumors (27/60) were found in NSCLC, 8% in breast, and 7% in head and neck cancer patients. While most mutations affect the kinase domain resulting in loss of kinase activity, others affect production, stability, or localization of the protein [35]. Meanwhile, other non-mutational mechanisms result in an alteration of STK11/LKB1 expression [36]. STK11/LKB1 expression can be negatively regulated by hyper-methylation of the STK11 promoter region as demonstrated in clear cell renal carcinoma [37], colorectal cancer [38], or melanoma [39]. Protein translation can also be reduced at the post-translational level by microRNAs, as shown in cervical [40] and head and neck cancer [41]. These non-mutational mechanisms should be considered in tumor characterization because such tumors could have growth and aggressiveness similar to STK11-mutant tumors [36]. Thus, while most clinical studies have used sequencing to characterize STK11 status, mutation analysis of the STK11 gene may not be sufficient to identify patients with impaired oncosuppressive STK11/LKB1 activity [36].
3.2. STK11/LKB1 Alterations Promote Lung Cancer Cell Survival and Invasion
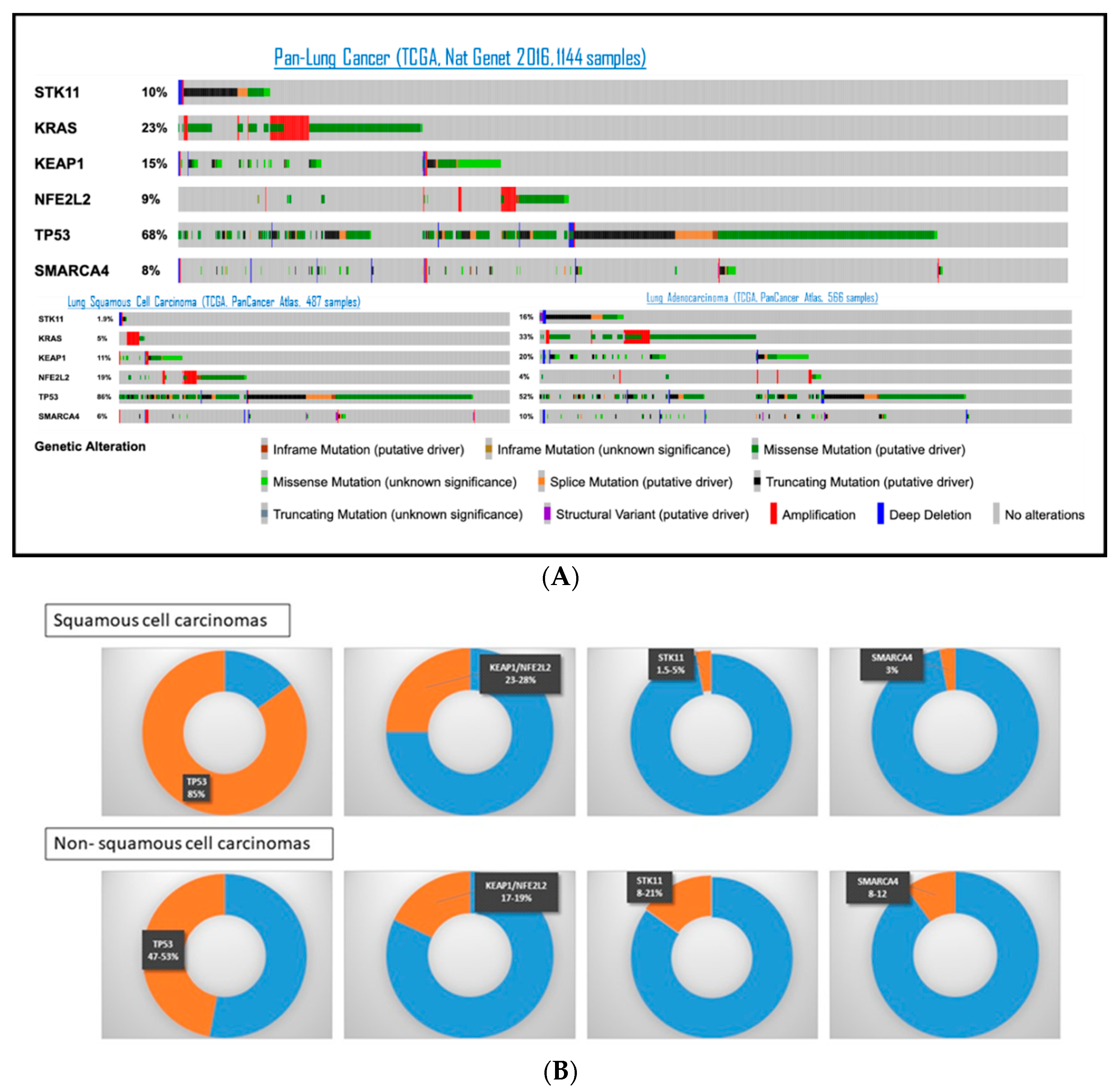
The most frequently altered tumor suppressor genes in NSCLC are resumed in Figure 2, based on TCGA (The Cancer Genome Atlas) and data from the literature. TP53 is the most frequently mutated in both lung adenocarcinoma (LUAD) and squamous-cell carcinomas (SqCC), in 47% and 85% respectively, followed by KEAP1 or NFE2L2, mutated in 17–19% of LUAD and 23–28% of SqCC [42,43]. The prevalence of STK11 alteration in LUAD is around 8–21% [42,44,45,46,47,48]. In SqCC, SKT11 alteration was only found in 1.5% to 5% of patients [48,49]. The use of different detection methods between studies (sequencing or immunohistochemistry) could in part explain differences in reported prevalence. Of note, SMARCA4, another gene on chromosome 19p, is also found altered at a frequency of 8–12% of NSCLC [50].
Whereas STK11/LKB1 loss alone was not sufficient to trigger oncogenesis, STK11 inactivation in a mutant KRAS-driven model of mouse lung cancer strongly stimulated growth and metastasis [51]. Latency until tumor growth and metastasis development were shortened compared to the STK11-wild-type (WT) counterpart. STK11-deficient tumors had increased expression of genes involved in angiogenesis and cell migration, such as NEDD9 (neural precursor cell expressed developmentally down-regulated 9) [51]. In vitro studies showed that STK11 inactivation increased cell motility, invasiveness, and favored epithelial–mesenchymal transition in lung cancer cells, thus enhancing metastatic potential [52,53]. Moreover, it was shown that STK11/LKB1 inactivation promotes cancer cell growth and survival via the upregulation of HIF-1α [54]. As explained earlier, inhibition of mTOR by STK11/LKB1 negatively regulates HIF-1α. Indeed, studies have shown that STK11/LKB1 inactivation in lung cancer cells led to an upregulated mTOR signaling providing growth advantages [55] associated with mitochondrial dysfunction by autophagy impairment [56]. A mouse model of Peutz-Jeghers syndrome (PJS) with STK11/LKB1 inactivation showed a dramatic HIF-1α increase via the mTOR pathway [56]. Lastly, mechanisms by which STK11/LKB1 constrains metastatic ability have been recently studied using LUAD primary tumors (55). Interestingly, STK11/LKB1 was characterized as a master regulator of chromatin accessibility, through the activation of the transcription factor SOX17.
In addition to its role as an AMPK regulator, STK11/LKB1 plays a role in glutamine metabolism. Glutamine is a major source of energy in cancer cells and acts as a nitrogen donor in pyrimidine and purine synthesis via the carbamoyl phosphate synthetase 1 (CPS1), a urea cycle enzyme [57,58]. It was demonstrated that STK11/LKB1 downregulates CPS1 transcription via AMPK-mediated effects and that KRAS/STK11 mutant lung cancer cells upregulate expression of CPS1, allowing cell division and tumor development [59].
Moreover, glutamine metabolism also plays a crucial role in redox homeostasis via glutathione, as glutathione is implicated in protecting cells from oxidative damage and maintaining redox homeostasis [60]. The enzyme glutamate cysteine ligase condenses glutamate and cysteine in ɣ-glutamylcysteine, then glutathione is produced via glutathione synthetase [61]. KEAP1 (Kelch-like ECH associated protein 1) is a gene encoding a ubiquitin ligase, a negative regulator of the proteasomal degradation of NRF2 (also known as NFE2L2). NRF2 is a transcription factor with a pivotal role in the cellular defense against oxidative stress, by regulating glutathione synthetase [62]. Thus, KEAP1 promotes transcription of genes encoding detoxifying enzymes and antioxidative stress proteins [43]. As previously mentioned, STK11/LBK1 loss implies a ROS accumulation.
It has been shown that co-alterations of STK11/LKB1 and KEAP1 were significantly associated with lung tumors [49,63,64]. One explanation is the proximity of the corresponding genomic loci on the short arm of chromosome 19 (at 19p13.2 for KEAP1 and 19p13.3 for STK11/LKB1). In KRAS-mutated lung cancer, there is a synergic effect of STK11/LKB1 and KEAP1 loss: STK11/LKB1 loss results in an increased level of ROS and high redox stress with an inability to maintain ATP levels. This high redox and energetic stress induces a positive selection for KEAP1 loss. KEAP1 loss led to an upregulation of the NRF2 pathway with an increase in glutamate-cysteine ligase transcription catalyzing the production of glutathione. This activation of the NRF2 pathway allows STK11/LKB1-deficient cells to detoxify and promote survival in an STK11/LKB1-independent way [65].
In conclusion, while STK11/LKB1 favored cell survival under stress conditions, it has been shown that STK11/LKB1 loss enhanced cancer cell proliferation under energy deprivation. STK11/LKB1 deficiency promotes cancer cell growth, motility, and invasion. It also promotes metastasis, through activation of pro-metastatic genes, and regulation of chromatin accessibility.
4. STK11/LKB1 Alterations and Co-Occurring Mutations: Prognostic Impact in Lung Cancer Patients
4.1. STK11/LKB1 Co-Occurring Genomic Alterations in Lung Cancer
KRAS and TP53 alterations are present in about half of STK11-altered cancers [34,46,48]. KRAS mutations are an early mutagenic event in LUAD evolution, as demonstrated by multi-region sequencing that studied KRAS mutations in both minimally invasive adenocarcinoma and paired invasive carcinoma [66]. Moreover, 90% of samples with KRAS mutations were found to be clonal populations [63]. KRAS is the most common proto-oncogene mutated in NSCLC, found in 20–30% of LUAD patients and 5–7% of SqCC patients from Western countries [47,67,68]. Most of KRAS mutations occur at codons 12 (86% of patients) and 13 (7% of patients) [47,68]. The most common nucleotide change in metastatic LUAD is from guanine to thymidine (34 G > T or G12C), involving around 40–45% of patients, followed by G12V (15–20% of patients), G12D (16–18% of patients) and G12A (7–10% of patients). Other mutations are observed in less than 5% of patients (G13C, G13D, G12S, G12F, G12R, etc.) [47,67,68,69,70]. It was recently highlighted that KRAS-mutant tumors are a very heterogeneous disease, including different tumor subtypes with variable biological backgrounds, different prognoses, and clinical responses to immunotherapy. One hypothesis is that it is due to a greater molecular diversity of tumors defined by mutation subtypes and the presence of co-mutations. Less than 5% of KRAS-mutant NSCLC patients have another oncogenic-driven co-mutation, such as BRAF, EGFR, PIK3CA, or MET amplification. No concurrent ALK or ROS1 rearrangements have been described [47]. Rare EGFR mutations have been described with the STK11 aberration (1.5% STK11 mutations occurred in patients with EGFR mutations in the pooled analysis from OAK and POPLAR [46,49]). The frequency of STK11 mutation in each mutational status in NSCLC is reported in Table 1.
TP53, STK11, and KEAP1 were the three most commonly co-occurring mutations in KRAS-mutant tumors. Of note, the co-mutation patterns of STK11 and KEAP1 were similar between KRAS G12C mutant vs. KRAS-others [63,67]. Nearly half of KRAS-mutant patients had TP53 mutations, followed by STK11 (18% to 28%) and KEAP1 (24%) [42,46,47,64,67,69,71]. As previously described, the co-mutation frequency of STK11 and KEAP1 significantly co-occurred [49,63,64]. Furthermore, STK11 and KEAP1 alterations frequently co-occur in 30% to 60% of SMARCA4-mutant NSCLCs [49,72].
The impact of these co-occurring mutations KRAS/STK11/TP53 has been explored. Patients with NSCLC expressing KRAS and STK11 co-mutations were younger than patients with KRAS mutated NSCLC (median age 61 vs. 67 years, p = 0.08) [47]. No differences regarding gender, race, smoking history, or performance status were observed. Adenocarcinoma histology and tobacco exposure were documented to be positively associated with STK11 mutations [73]. Importantly, the distribution of KRAS-mutant alleles (G12V, G12C, or G12D) were not related to STK11 or TP53 status [64,67]. This suggests that the KRAS-specific mutation is not the primary driver of the subsequent mutations. Moreover, clonality analysis from the TCGA dataset found that genetic events such as STK11, TP53, and KEAP1 were clonal [64]. Co-occurrence of STK11 and TP53 mutations in KRAS-mutant tumors was rare in a cohort of untreated, resected LUAD [45]. In a cohort of chemorefractory patients, triple mutant KRAS/TP53/STK11 tumors were more common [64].
4.2. Prognostic Impact of STK11/LKB1 Alterations in Lung Cancer before the Era of Immunotherapy
The impact of STK11/LKB1 alterations was evaluated in localized, resected NSCLC patients. A retrospective series of 352 surgical NSCLC found that patients harboring mutations in TP53, STK11 or SMARCA4 had a worse overall survival (OS) after adjustment for confounding factors, with a hazard ratio (HR) for all causes of mortality respectively at 1.47 (95%CI 1.02–2.13), 1.66 (95%CI 1.05–2.61) and 2.1 (95%CI 1.22–3.61) [42]. Amongst patients with KRAS-mutant tumors, a significantly worse outcome was seen for the KRAS/STK11 co-mutant group in comparison to the WT group. These results were validated in a larger cohort from MSK-IMPACT [42]. Conversely, another analysis from the TCGA cohort in the setting of predominantly early-stage surgically resected tumors found that STK11 alterations were not associated with a decreased OS [64].
Several studies also evaluated the impact of STK11/LKB1 alterations in advanced NSCLC, before the advent of ICI. In a real-life cohort of advanced NSCLC patients treated with first-line chemotherapy, median OS was shorter in patients with STK11-mutant (n = 288) vs. STK11-WT tumors (n = 1849) (11.2 vs. 17.8 months; HR 1.4 (95%CI 1.2–1.6), p < 0.0001) [48]. In a smaller cohort of metastatic LUAD patients (17/92 (18%) had STK11 mutation), KRAS/STK11 co-mutations were associated with worse OS in univariate analyses (HR 2.66; p = 0.035) [47]. The STK11 mutation was found as a negative prognostic factor for OS in patients with either KRAS mutation (0.9 years vs. not reached) or KRAS-WT (1.46 vs. 2.03 years) [47]. Another retrospective analysis included advanced NSCLC patients who received platinum-based chemotherapy (25/302 (8%) had STK11 mutation) [46]. No differences in clinical characteristics according to STK11 status were reported. There was a trend toward shorter survival for the STK11-mutant vs. WT groups, with an OS of 10.4 months (95%CI 6.1–15.7) vs. 17.3 months (95%CI 14.0–21.1) (HR 1.53; 95%CI 0.94–2.49; p = 0.085), but it was not statistically significant.
Controversies exist regarding the independent prognostic impact of co-occurring mutations in STK11 with KRAS, especially after adjustment for concurrent KEAP1 and TP53 mutations and other clinical variables. As previously mentioned, it was demonstrated that KRAS/STK11 tumors have higher rates of KEAP1 mutational inactivation [64,69]. A large study has evaluated the impact of co-occurring mutations in 330 patients with advanced KRAS-mutant NSCLC [69]. The KRAS/STK11 co-mutation was associated with a shorter OS (12 vs. 21 months, HR 1.7; 95%CI 1.1–2.4; p = 0.002), but this was not significant after adjustment on major other prognostic factors. In multivariate analysis, only the concurrent mutation in KEAP1 or NFE2L2 was independently associated with shorter OS (HR 1.96; 95%CI 1.33–2.92; p < 0.001). This study suggested that it was the concurrent KEAP1 or NFE2L2 mutation which was associated with a worse prognostic, but not the SKT11 mutation. There was no impact of a concurrent mutation in TP53 (HR 0.9; 95%CI 0.6–1.2; p = 0.5) [69]. In contrast to STK11, KEAP1 mutations were infrequently observed among KRAS/TP53 co-mutant.
5. Immune Impact of STK11/LKB1 Alterations in Lung Cancer Patients
5.1. Tumor-Extrinsic Impact of STK11/LKB1 Alteration: Interaction with Immune System
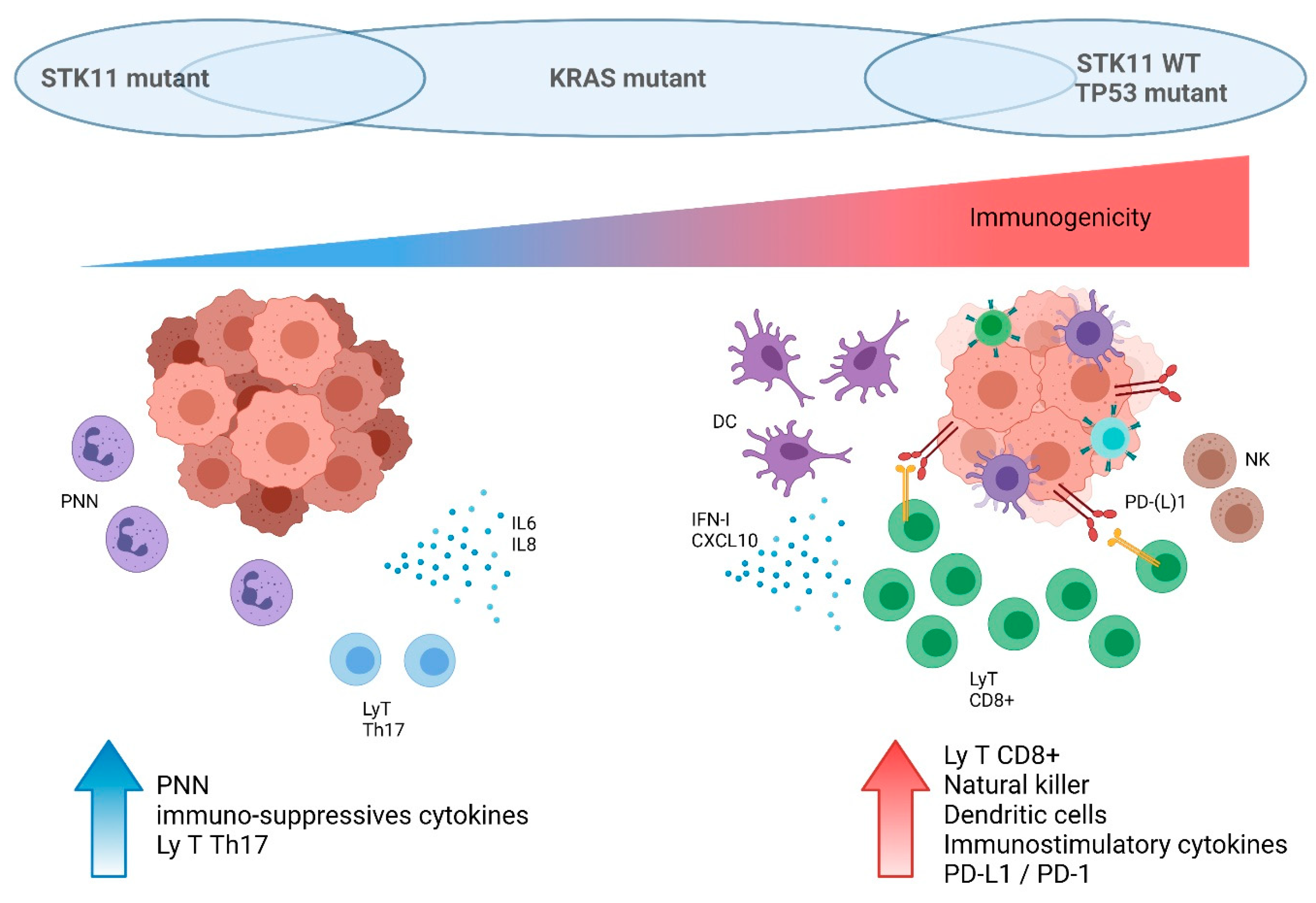
Recently, there is increasing evidence that loss of STK11/LKB1 may be involved in the modulation of the tumor immune microenvironment. To understand the underlying mechanisms of the immune surveillance mediated by STK11/LKB1 alterations, in vivo tumor models were generated with STK11 gene deletion by CRISPR/Cas9 editing. Genetic ablation of the STK11 locus in cell lines implanted in syngenic mice resulted in lower numbers of CD3+ CD8+ and CD3+ CD8+ /PD1+ T lymphocytes compared to their STK11-proficient counterpart, with no impact on CD45+ and CD3+ CD4 cell infiltrates [71]. Genetic ablation of STK11 in a KRAS-driven murine model of NSCLC demonstrated an accumulation of neutrophils with T-cell suppressive capacities associated with a decreased number of tumor-infiltrating lymphocytes and a reduced expression of PD-L1 in tumors [74]. In an in vitro model, STK11 extinction did not substantially modify the growth of KRAS-mutant colorectal tumors [75]. Conversely, in an immunocompetent mouse model, tumor growth increased, related to a loss of immune control [75]. Kitajima et al. showed that STK11 loss inactivates the stimulator of interferon genes (STING) expression via DNMT1 and EZH2 epigenetic activity. STING mediates cytosolic DNA-induced signaling events that trigger secretions of type I interferons and diverse chemokines. Without STING, tumor cells cannot detect mitochondrial or nuclear DNA in the cytoplasm leading to a lack of PD-L1 expression and a downregulation of chemokines such as CXCL10 that promote T cell recruitment [76,77]. Studies on cohorts of patients with NSCLC confirmed that STK11-mutant tumors could be classified as immunologically ignored, with less immune cell infiltration, as described in the following paragraph [44,45,71].
5.2. STK11/LKB1 Alterations Negatively Impact Immune Surveillance in NSCLC Patients
Studies on several cohorts of NSCLC patients retrospectively confirmed preclinical data regarding the impact of STK11/LKB1 status on the immune microenvironment. In-depth immune profiling was performed on 221 untreated resected LUAD [78]. STK11-mutant tumors were characterized by higher neutrophil density, lower CD8+ T cells and dendritic cell density, and lower PD-L1 expression. Conversely, TP53-mutant tumors were characterized by a higher density of CD8+ T cells, indicating a strong adaptive immune response. Moreover, in the TP53-mutant subgroup, co-occurring STK11 mutations were significantly associated with a reduced expression of PD-L1 and a lower CD8+ cell density. Importantly, KRAS mutations were not implicated in the modulation of the tumor immune microenvironment in this study [78]. Another analysis on resected LUAD found that STK11 mutations either individually or with KRAS mutations were strongly associated with lower NF-κB signature activity, while no such association was seen with TP53 mutations [45]. Skoulidis et al. focused their analyses on KRAS-mutant LUAD and demonstrated that different subsets exhibit distinct patterns of immune system engagement [64]. Tumors with KRAS/TP53 co-mutations had a higher immune system activation, with more T-cell infiltration (such as CD3+, CD8+, and CD45RO+ lymphocytes) and a higher expression of cell-intrinsic co-inhibitory signals such as PD-(L)1. Thus, the presence of TP53 mutations designated tumors with the strongest adaptive immune response and could potentially allow the identification of patients susceptible to being more sensitive to ICI. Comparatively, KRAS/STK11 mutated tumors had a lack of immune system engagement. This cold micro-environment could explain a lack of benefit from ICI, as discussed in the following paragraph. KRAS-only mutated tumors had a mixed immune microenvironment [64].
Biological markers such as tumor mutation burden (TMB) or PD-L1 expression were demonstrated to be associated with favorable outcomes to PD-1/PD-L1 blockade across diverse tumors [79]. NSCLC patients have among the highest TMB of all malignancies, after melanoma [80]. KRAS mutations are associated with an increased PD-L1 expression in NSCLC and are also associated with a high TMB in NSCLC [81,82]. Co-mutant KRAS/TP53 had a higher overall mutation load compared to co-mutant KRAS/STK11 regardless of exposure to tobacco [64]. Moreover, it was recently shown that patients with STK11 or KEAP1 alteration had significantly higher blood TMB compared with WT [49,73]. However, STK11 mutations were associated with a lack of PD-L1 expression in tumor cells, despite the presence of intermediate or high TMB, irrespective of KRAS status [71]. While tumor samples with STK11 mutations were enriched for negative PD-L1 staining [49,73], tumor samples with high PD-L1 expression over 50% were less frequently mutated for STK11 or KEAP1 [67,83].
In conclusion, STK11-mutant tumors were associated with T-cell exclusion, low PD-(L)1 levels, and higher density of immune suppressive cells. Thus, these tumors were mostly classified as immunologically ignored, or “cold” tumors, as resumed in Figure 3. The impact of this poor immune surveillance was then studied in NSCLC patients treated with ICI.
5.3. Impact of SKT11/LKB1 Alterations on ICI Efficacy in NSCLC Patients
ICI-based therapies are the new standard of care in NSCLC patients without oncogenic addiction, in monotherapy, or in combination with chemotherapy [3]. ICI demonstrated around a 30% reduction in the risk of death compared with chemotherapy for advanced NSCLC [84]. However, around half of the pre-treated patients did not have any benefit from ICI and developed early progression [85,86]. The impact of mutations such as KRAS and co-occurring mutations on disease behavior and clinical outcomes is an important matter for debate. Therefore, whether STK11 genomic alterations may predict a lack of clinical benefit from ICI was further investigated. In vivo studies showed that a syngenic mice model implanted with STK11 knockout cells was resistant to PD-1 blockade, contrary to their STK11-proficient counterpart [71,75]. Whereas cell lines with STK11-deficiency result in a higher TMB, co-occurring KRAS/STK11 mutations in a mouse model decreased the efficacy of anti-PD-1 therapy compared to the KRAS/TP53 co-mutant model [87]. This resistance to ICI was confirmed in different tumor cell types (colorectal CT26 KRAS-mutant or EMT6 mouse mammary KRAS-WT) [75].
In patients, several retrospective studies have evaluated the prognostic or predictive impact of STK11/LKB1 status on ICI efficacy (Table 2).
A cohort from the MSK-IMPACT included 240 NSCLC patients treated with anti-PD1 therapy alone or in combination with anti-CTLA4 and evaluated the impact of gene alterations identified by NGS on ICI efficacy [88]. Like other cohorts, SKT11 was the most enriched altered gene in PD-L1 negative tumors, but this association was not statistically significant. STK11 alterations were associated with a lack of benefit from ICI, mostly in pre-treated patients. Another study found that within a small cohort of 66 previously treated patients with a PD-L1 ≥ 1% receiving ICI, STK11-mutant tumors had significantly lower response rates vs. STK11-WT (0% vs. 34.5%, p = 0.026) regardless of KRAS status [71]. STK11 mutations were also associated with a shorter PFS (HR 4.76, 95%CI 2.0–11.1, p = 0.00012) and OS (HR 14.3, 95%CI 3.4–50.0, p < 0.0001) with ICI treatment [71].
However, studies of other cohorts found controversial results on the prognostic and/or predictive role of STK11. A study conducted among 86 KRAS-mutant NSCLC patients who received ICI concluded that neither the presence of TP53 nor SKT11 was associated with a difference in OS from the start of ICI [69]. Importantly, patients with a co-occurring mutation in KEAP1 or NFE2L2 were found to have a shorter OS in both univariate and multivariate analysis (HR 3.54, 95%CI 1.55–8.11, p = 0.003). Several larger cohorts compared ICI and also chemotherapy efficacy according to STK11 status. A real-world study including 2276 previously untreated patients demonstrated that STK11/KEAP1 mutations are more prognostic than predictive biomarkers of resistance to ICI [73]. Indeed, these mutations conferred a poor prognosis regardless of treatment class (ICI, chemotherapy, targeted therapies, or antiangiogenic therapies). Another large real-life cohort has evaluated the impact of STK11 status on first-line treatment efficacy, including 270 patients receiving first-line ICI-based therapy, and 770 patients ICI treated in the latter line [48]. There was a trend for a shorter OS in first-line therapy for patients with the STK11-mutant (n = 40) compared with STK11-WT tumors (n = 230) (11.2 vs. 17.7 months; HR 1.4 (95%CI 0.9–2.3), but it was more pronounced in the second-line ICI group for STK11-mutant (n = 211) vs. STK11-WT patients (n = 559) (6.3 vs. 12.0 months; HR 1.6 (95%CI 1.3–2.0). Finally, a pooled analysis from OAK and POPLAR studies showed that among patients receiving atezolizumab in second or third-line settings, those with STK11/KEAP1 tumor mutation had worse median OS compared with WT patients (7.3 vs. 15.6 months (p = 0.004)) [49]. Survival analysis among NSCLC patients harboring STK11 alterations demonstrated that atezolizumab did not improve OS compared to docetaxel, suggesting resistance to ICI treatment among STK11-mutant. However, importantly, this was also observed among patients receiving docetaxel. Thus, in these studies, STK11 mutations did not show different treatment-specific effects between atezolizumab and docetaxel.
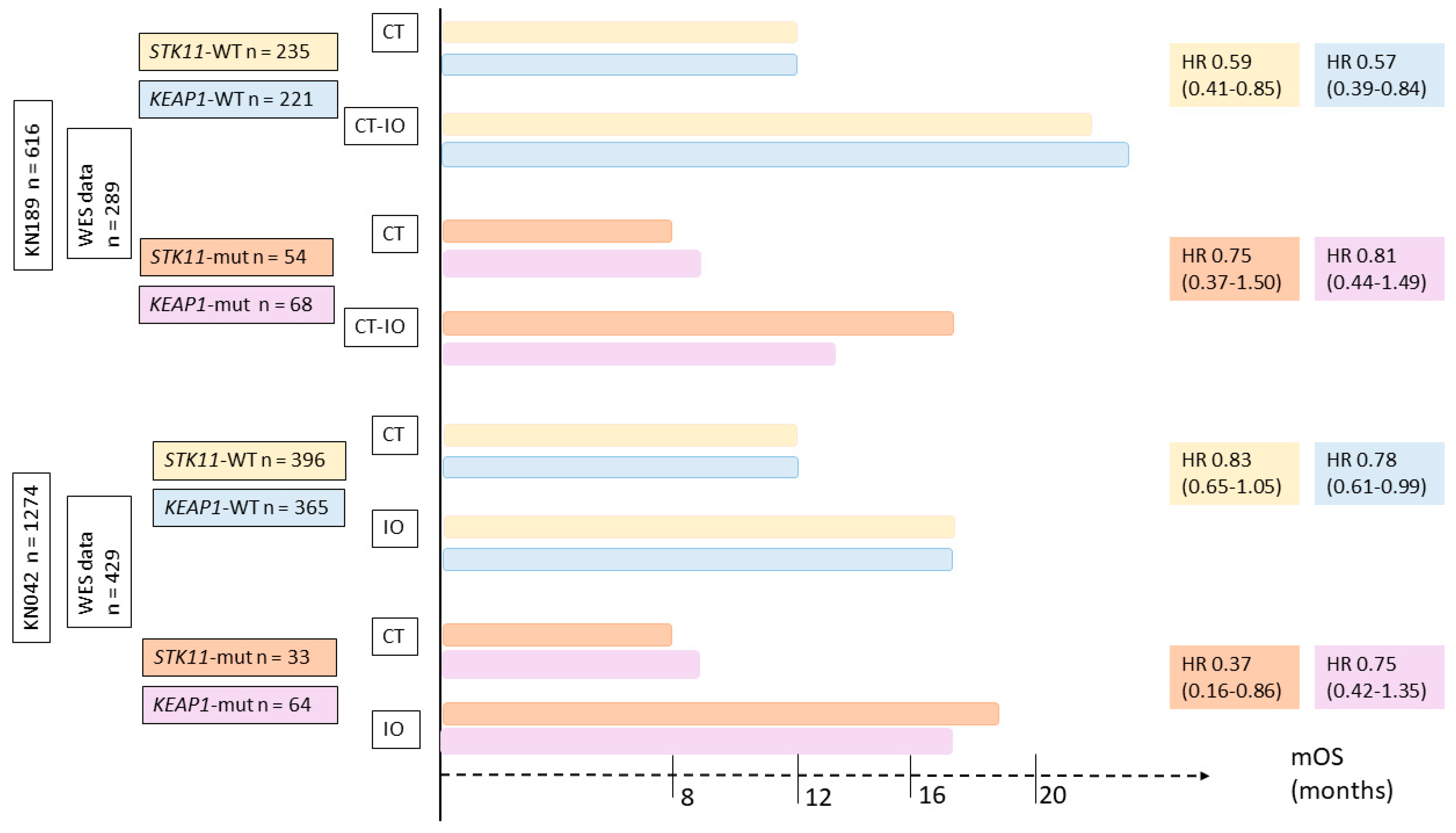
Recently, the impact of STK11 or KEAP1 status at diagnosis on immune resistance during first-line therapy has been investigated. Exploratory analyses from two phase 3 trials were presented during AACR 2020 [89,90]. The first study, KEYNOTE-189, evaluated chemotherapy +/− pembrolizumab or placebo as first-line therapy for non-squamous NSCLC. The other study was KEYNOTE-042, which evaluated pembrolizumab monotherapy vs. chemotherapy as first-line for PD-L1-positive NSCLC. Whole-exome sequencing data from tumor and normal DNA was available for 47% and 34% of patients, respectively.
For both studies, monotherapy with ICI or in combination with chemotherapy was associated with better outcomes than chemotherapy alone, regardless of STK11 or KEAP1 status. Results on OS by treatment and mutation type are summarized in Figure 4.
In conclusion, STK11 status was not identified as a predictive biomarker of resistance to ICI, but more as a negative prognostic factor regardless of treatment type. These exploratory analyses strongly support the use of ICI (alone or in combination) in first-line settings for NSCLC patients, regardless of STK11, or KEAP1, status. These results highlighted the fact that each individual mutation is insufficient to predict ICI response or resistance.
6. Conclusions
STK11/LKB1 acts as a major regulator of energy sensing and cellular metabolism. It activates the canonical AMPK pathway, a well-described central metabolic sensor that regulates lipid and glucose metabolisms. Intracellular signaling downstream of STK11/LKB1 also regulates the mTOR pathway to modulate protein synthesis and cellular proliferation. Moreover, STK11/LKB1 has been reported to have tumor suppressor activity. Inactivating somatic mutations of STK11 have been observed in different types of solid tumors, especially in NSCLC, where it modulates cancer cell differentiation, invasion, and metastasis. STK11 mutations in tumor cells also have tumor-extrinsic functions. Indeed, STK11-mutant tumors were associated with low PD-(L)1 levels, cytotoxic T-cell exclusion, and a higher density of the immuno-suppressive cell population. The biological function of STK11/LKB1 and the impact of its loss under stress conditions are resumed in Figure 5.
Despite sharing common clinical-pathological factors, NSCLC patients have distinct phenotypes linked to a unique tumor microenvironment that regulates tumor behavior and response to therapies. Thus, alterations in the STK11 gene in NSCLC have recently emerged as an important regulator of immune response and a potential resistance biomarker to ICI. However, the effect of STK11/LKB1 inactivation on clinical response to PD-(L)1 is a subject of much debate. While some authors suggest that STK11/LKB1 inactivation could favor primary resistance to ICI, several questions have emerged. Are STK11/LKB1 alterations really a predictive biomarker, or only a prognostic one? Are they confounding factors due to co-occurring mutations, such as KEAP1? The clinical and biological significance of associated genetic events is unclear. Will physicians have to adapt their therapeutic choices in the future based on STK11/LKB1 tumoral status?
So far, guidelines do not recommend to systematically characterize STK11/LKB1 status as it has not been demonstrated to impact therapeutic strategy. It is important to highlight the fact that exploratory analyses support the use of ICI (alone or in combination) in first-line settings for all NSCLC patients without oncogenic addiction, regardless of STK11 or KEAP1 status when available. However, the impact of tumor suppressor genes such as STK11, KEAP1, and TP53 co-mutations may help refine response prediction algorithms in both PD-L1 positive and negative tumors. Whether it could impact the decision to add chemotherapy to ICI in high-PDL1 patients is still unknown.
Author Contributions
Conceptualization, E.P.-T., A.L., J.B.; methodology, E.P.-T. and A.L.; validation, E.P.-T., A.L., J.-F.F., M.G.D., J.B.; formal analysis, E.P.-T. and A.L.; writing—original draft preparation, E.P.-T. and A.L.; writing—review and editing, E.P.-T., A.L., M.G.D., J.-F.F., J.B.; supervision, J.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Beggs, A.D.; Latchford, A.R.; Vasen, H.F.A.; Moslein, G.; Alonso, A.; Aretz, S.; Bertario, L.; Blanco, I.; Bulow, S.; Burn, J.; et al. Peutz-Jeghers syndrome: A systematic review and recommendations for management. Gut 2010, 59, 975–986. [Google Scholar] [CrossRef] [Green Version]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumor suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, A.; Tomlinson, I.; Markie, D.; Järvinen, H.; Sistonen, P.; Björkqvist, A.M.; Knuutila, S.; Salovaara, R.; Bodmer, W.; Shibata, D.; et al. Localization of a susceptibility locus for Peutz-Jeghers syndrome to 19p using comparative genomic hybridization and targeted linkage analysis. Nat. Genet. 1997, 15, 87–90. [Google Scholar] [CrossRef]
- Hemminki, A.; Markie, D.; Tomlinson, I.; Avizienyte, E.; Roth, S.; Loukola, A.; Bignell, G.; Warren, W.; Aminoff, M.; Höglund, P.; et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998, 391, 184–187. [Google Scholar] [CrossRef]
- Jenne, D.E.; Reimann, H.; Nezu, J.; Friedel, W.; Loff, S.; Jeschke, R.; Müller, O.; Back, W.; Zimmer, M. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat. Genet. 1998, 18, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Launonen, V. Mutations in the human LKB1/STK11 gene. Hum. Mutat. 2005, 26, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.-S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.-L.; Wu, Y.-Q.; Li, T.Y.; Liang, Y.; Lu, Z.; et al. The Lysosomal v-ATPase-Ragulator Complex Is a Common Activator for AMPK and mTORC1, Acting as a Switch between Catabolism and Anabolism. Cell Metab. 2014, 20, 526–540. [Google Scholar] [CrossRef] [Green Version]
- Lizcano, J.M.; Göransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Mäkelä, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843. [Google Scholar] [CrossRef] [Green Version]
- Munday, M.R. Regulation of mammalian acetyl-CoA carboxylase. Biochem. Soc. Trans. 2002, 30, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Ferré, P.; Foufelle, F. SREBP-1c transcription factor and lipid homeostasis: Clinical perspective. Horm. Res. 2007, 68, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, S.; Zhai, A.; Zhang, B.; Tian, G. AMPK-Mediated Regulation of Lipid Metabolism by Phosphorylation. Biol. Pharm. Bull. 2018, 41, 985–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsin, A.-S.; Bouzin, C.; Bertrand, L.; Hue, L. The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP-activated protein kinase and inducible 6-phosphofructo-2-kinase. J. Biol. Chem. 2002, 277, 30778–30783. [Google Scholar] [CrossRef] [Green Version]
- Wojtaszewski, J.F.P.; Nielsen, J.N.; Jørgensen, S.B.; Frøsig, C.; Birk, J.B.; Richter, E.A. Transgenic models--a scientific tool to understand exercise-induced metabolism: The regulatory role of AMPK (5′-AMP-activated protein kinase) in glucose transport and glycogen synthase activity in skeletal muscle. Biochem. Soc. Trans. 2003, 31, 1290–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Ji, J.; Yan, X.-H. Cross-Talk between AMPK and mTOR in Regulating Energy Balance. Crit. Rev. Food Sci. Nutr. 2012, 52, 373–381. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Onorati, A.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in Biology and Medicine. React. Oxyg. Species (Apex, NC) 2016, 1, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whang, Y.M.; Park, S.I.; Trenary, I.A.; Egnatchik, R.A.; Fessel, J.P.; Kaufman, J.M.; Carbone, D.P.; Young, J.D. LKB1 deficiency enhances sensitivity to energetic stress induced by erlotinib treatment in non-small-cell lung cancer (NSCLC) cells. Oncogene 2016, 35, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.-M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [Green Version]
- Bonanno, L.; Zulato, E.; Pavan, A.; Attili, I.; Pasello, G.; Conte, P.; Indraccolo, S. LKB1 and Tumor Metabolism: The Interplay of Immune and Angiogenic Microenvironment in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Cespedes, M.; Parrella, P.; Esteller, M.; Nomoto, S.; Trink, B.; Engles, J.M.; Westra, W.H.; Herman, J.G.; Sidransky, D. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002, 62, 3659–3662. [Google Scholar] [PubMed]
- Hezel, A.F.; Gurumurthy, S.; Granot, Z.; Swisa, A.; Chu, G.C.; Bailey, G.; Dor, Y.; Bardeesy, N.; Depinho, R.A. Pancreatic LKB1 deletion leads to acinar polarity defects and cystic neoplasms. Mol. Cell. Biol. 2008, 28, 2414–2425. [Google Scholar] [CrossRef] [Green Version]
- Wingo, S.N.; Gallardo, T.D.; Akbay, E.A.; Liang, M.-C.; Contreras, C.M.; Boren, T.; Shimamura, T.; Miller, D.S.; Sharpless, N.E.; Bardeesy, N.; et al. Somatic LKB1 mutations promote cervical cancer progression. PLoS ONE 2009, 4, e5137. [Google Scholar] [CrossRef]
- Gill, R.K.; Yang, S.-H.; Meerzaman, D.; Mechanic, L.E.; Bowman, E.D.; Jeon, H.-S.; Roy Chowdhuri, S.; Shakoori, A.; Dracheva, T.; Hong, K.-M.; et al. Frequent homozygous deletion of the LKB1/STK11 gene in non-small cell lung cancer. Oncogene 2011, 30, 3784–3791. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.M.; Choi, J.E.; Na, Y.K.; Lee, E.J.; Lee, W.K.; Choi, Y.Y.; Yoon, G.S.; Jeon, H.-S.; Kim, D.S.; Park, J.Y. Genetic and epigenetic alterations of the LKB1 gene and their associations with mutations in TP53 and EGFR pathway genes in Korean non-small cell lung cancers. Lung Cancer 2013, 81, 194–199. [Google Scholar] [CrossRef]
- Tanwar, P.S.; Mohapatra, G.; Chiang, S.; Engler, D.A.; Zhang, L.; Kaneko-Tarui, T.; Ohguchi, Y.; Birrer, M.J.; Teixeira, J.M. Loss of LKB1 and PTEN tumor suppressor genes in the ovarian surface epithelium induces papillary serous ovarian cancer. Carcinogenesis 2014, 35, 546–553. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, J.; Li, P.; Mao, X.; Li, W.; Yang, J.; Liu, P. Loss of LKB1 disrupts breast epithelial cell polarity and promotes breast cancer metastasis and invasion. J. Exp. Clin. Cancer Res. 2014, 33, 70. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-Y.; Jiang, S.-H.; Liu, D.-J.; Yang, X.-M.; Huo, Y.-M.; Li, J.; Hua, R.; Zhang, Z.-G.; Sun, Y.-W. Decreased LKB1 predicts poor prognosis in Pancreatic Ductal Adenocarcinoma. Sci. Rep. 2015, 5, 10575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Yin, L.; Song, G.; Han, X.; Yin, Z.; Luo, D. LKB1 loss cooperating with BRAF V600E promotes melanoma cell invasion and migration by up-regulation MMP-2 via PI3K/Akt/mTOR pathway. Oncotarget 2017, 8, 113847–113857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, N.; Goodman, A.M.; Barkauskas, D.A.; Kurzrock, R. STK11 alterations in the pan-cancer setting: Prognostic and therapeutic implications. Eur. J. Cancer 2021, 148, 215–229. [Google Scholar] [CrossRef]
- Granado-Martínez, P.; Garcia-Ortega, S.; González-Sánchez, E.; McGrail, K.; Selgas, R.; Grueso, J.; Gil, R.; Naldaiz-Gastesi, N.; Rhodes, A.C.; Hernandez-Losa, J.; et al. STK11 (LKB1) missense somatic mutant isoforms promote tumor growth, motility and inflammation. Commun. Biol. 2020, 3, 366. [Google Scholar] [CrossRef]
- Borzi, C.; Galli, G.; Ganzinelli, M.; Signorelli, D.; Vernieri, C.; Garassino, M.C.; Sozzi, G.; Moro, M. Beyond LKB1 Mutations in Non-Small Cell Lung Cancer: Defining LKB1less Phenotype to Optimize Patient Selection and Treatment. Pharmaceuticals 2020, 13, 385. [Google Scholar] [CrossRef]
- Zheng, F.; Yuan, X.; Chen, E.; Ye, Y.; Li, X.; Dai, Y. Methylation of STK11 promoter is a risk factor for tumor stage and survival in clear cell renal cell carcinoma. Oncol. Lett. 2017, 14, 3065–3070. [Google Scholar] [CrossRef] [Green Version]
- Trojan, J.; Brieger, A.; Raedle, J.; Esteller, M.; Zeuzem, S. 5′-CpG island methylation of the LKB1/STK11 promoter and allelic loss at chromosome 19p13.3 in sporadic colorectal cancer. Gut 2000, 47, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, X.; Song, G.; Luo, D. Prognostic significance of LKB1 promoter methylation in cutaneous malignant melanoma. Oncol. Lett. 2017, 14, 2075–2080. [Google Scholar] [CrossRef] [Green Version]
- Lao, G.; Liu, P.; Wu, Q.; Zhang, W.; Liu, Y.; Yang, L.; Ma, C. Mir-155 promotes cervical cancer cell proliferation through suppression of its target gene LKB1. Tumour Biol. 2014, 35, 11933–11938. [Google Scholar] [CrossRef]
- Figueroa-González, G.; Carrillo-Hernández, J.F.; Perez-Rodriguez, I.; Cantú de León, D.; Campos-Parra, A.D.; Martínez-Gutiérrez, A.D.; Coronel-Hernández, J.; García-Castillo, V.; López-Camarillo, C.; Peralta-Zaragoza, O.; et al. Negative Regulation of Serine Threonine Kinase 11 (STK11) through miR-100 in Head and Neck Cancer. Genes 2020, 11, 1058. [Google Scholar] [CrossRef] [PubMed]
- La Fleur, L.; Falk-Sörqvist, E.; Smeds, P.; Berglund, A.; Sundström, M.; Mattsson, J.S.; Brandén, E.; Koyi, H.; Isaksson, J.; Brunnström, H.; et al. Mutation patterns in a population-based non-small cell lung cancer cohort and prognostic impact of concomitant mutations in KRAS and TP53 or STK11. Lung Cancer 2019, 130, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, R.; Scheffler, M.; Merkelbach-Bruse, S.; Ihle, M.A.; Kron, A.; Rauer, M.; Ueckeroth, F.; König, K.; Michels, S.; Fischer, R.; et al. Clinical and Pathological Characteristics of KEAP1- and NFE2L2-Mutated Non-Small Cell Lung Carcinoma (NSCLC). Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 3087–3096. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Ding, B.; Xu, J.; Mao, K.; Zhang, P.; Xue, Q. Relevance of STK11 Mutations Regarding Immune Cell Infiltration, Drug Sensitivity, and Cellular Processes in Lung Adenocarcinoma. Front. Oncol. 2020, 10, 580027. [Google Scholar] [CrossRef]
- Schabath, M.B.; Welsh, E.A.; Fulp, W.J.; Chen, L.; Teer, J.K.; Thompson, Z.J.; Engel, B.E.; Xie, M.; Berglund, A.E.; Creelan, B.C.; et al. Differential association of STK11 and TP53 with KRAS mutation-associated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene 2016, 35, 3209–3216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facchinetti, F.; Bluthgen, M.V.; Tergemina-Clain, G.; Faivre, L.; Pignon, J.-P.; Planchard, D.; Remon, J.; Soria, J.-C.; Lacroix, L.; Besse, B. LKB1/STK11 mutations in non-small cell lung cancer patients: Descriptive analysis and prognostic value. Lung Cancer Amst. Neth. 2017, 112, 62–68. [Google Scholar] [CrossRef]
- El Osta, B.; Behera, M.; Kim, S.; Berry, L.D.; Sica, G.; Pillai, R.N.; Owonikoko, T.K.; Kris, M.G.; Johnson, B.E.; Kwiatkowski, D.J.; et al. Characteristics and Outcomes of Patients With Metastatic KRAS-Mutant Lung Adenocarcinomas: The Lung Cancer Mutation Consortium Experience. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2019, 14, 876–889. [Google Scholar] [CrossRef]
- Shire, N.J.; Klein, A.B.; Golozar, A.; Collins, J.M.; Fraeman, K.H.; Nordstrom, B.L.; McEwen, R.; Hembrough, T.; Rizvi, N.A. STK11 (LKB1) mutations in metastatic NSCLC: Prognostic value in the real world. PLoS ONE 2020, 15, e0238358. [Google Scholar] [CrossRef]
- Shang, X.; Li, Z.; Sun, J.; Zhao, C.; Lin, J.; Wang, H. Survival analysis for non-squamous NSCLC patients harbored STK11 or KEAP1 mutation receiving atezolizumab. Lung Cancer 2021, 154, 105–112. [Google Scholar] [CrossRef]
- Armon, S.; Hofman, P.; Ilié, M. Perspectives and Issues in the Assessment of SMARCA4 Deficiency in the Management of Lung Cancer Patients. Cells 2021, 10, 1920. [Google Scholar] [CrossRef]
- Ji, H.; Ramsey, M.R.; Hayes, D.N.; Fan, C.; McNamara, K.; Kozlowski, P.; Torrice, C.; Wu, M.C.; Shimamura, T.; Perera, S.A.; et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 2007, 448, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.C.; Kohno, T.; Iwakawa, R.; Moriguchi, T.; Kiyono, T.; Morishita, K.; Sanchez-Cespedes, M.; Akiyama, T.; Yokota, J. Involvement of LKB1 in epithelial-mesenchymal transition (EMT) of human lung cancer cells. Lung Cancer 2010, 70, 136–145. [Google Scholar] [CrossRef]
- Yao, Y.-H.; Cui, Y.; Qiu, X.-N.; Zhang, L.-Z.; Zhang, W.; Li, H.; Yu, J.-M. Attenuated LKB1-SIK1 signaling promotes epithelial-mesenchymal transition and radioresistance of non-small cell lung cancer cells. Chin. J. Cancer 2016, 35, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giatromanolaki, A.; Koukourakis, M.I.; Sivridis, E.; Turley, H.; Talks, K.; Pezzella, F.; Gatter, K.C.; Harris, A.L. Relation of hypoxia inducible factor 1 alpha and 2 alpha in operable non-small cell lung cancer to angiogenic/molecular profile of tumours and survival. Br. J. Cancer 2001, 85, 881–890. [Google Scholar] [CrossRef] [Green Version]
- Momcilovic, M.; Shackelford, D.B. Targeting LKB1 in cancer—Exposing and exploiting vulnerabilities. Br. J. Cancer 2015, 113, 574–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shackelford, D.B.; Abt, E.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 2013, 23, 143–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Le, A. Glutamine Metabolism in Cancer. Adv. Exp. Med. Biol. 2018, 1063, 13–32. [Google Scholar] [CrossRef] [PubMed]
- Charlier, D.; Nguyen Le Minh, P.; Roovers, M. Regulation of carbamoylphosphate synthesis in Escherichia coli: An amazing metabolite at the crossroad of arginine and pyrimidine biosynthesis. Amino Acids 2018, 50, 1647–1661. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Hu, Z.; Cai, L.; Li, K.; Choi, E.; Faubert, B.; Bezwada, D.; Rodriguez-Canales, J.; Villalobos, P.; Lin, Y.-F.; et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 2017, 546, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.C. Regulation of Glutathione Synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.D.; Caso, R.; Tan, K.S.; Mastrogiacomo, B.; Sanchez-Vega, F.; Liu, Y.; Connolly, J.G.; Murciano-Goroff, Y.R.; Bott, M.J.; Adusumilli, P.S.; et al. KRASG12C Mutation Is Associated with Increased Risk of Recurrence in Surgically Resected Lung Adenocarcinoma. Clin. Cancer Res. 2021, 27, 2604–2612. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.-H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef]
- Izumchenko, E.; Chang, X.; Brait, M.; Fertig, E.; Kagohara, L.T.; Bedi, A.; Marchionni, L.; Agrawal, N.; Ravi, R.; Jones, S.; et al. Targeted sequencing reveals clonal genetic changes in the progression of early lung neoplasms and paired circulating DNA. Nat. Commun. 2015, 6, 8258. [Google Scholar] [CrossRef] [PubMed]
- Arbour, K.C.; Rizvi, H.; Plodkowski, A.J.; Hellmann, M.D.; Knezevic, A.; Heller, G.; Yu, H.A.; Ladanyi, M.; Kris, M.G.; Arcila, M.E.; et al. Treatment Outcomes and Clinical Characteristics of Patients with KRAS-G12C-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 2209-2015. [Google Scholar] [CrossRef]
- Reck, M.; Carbone, D.P.; Garassino, M.; Barlesi, F. Targeting KRAS in non-small-cell lung cancer: Recent progress and new approaches. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, S0923-753402045-7. [Google Scholar] [CrossRef]
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.A.; Sima, C.S.; Shen, R.; Kass, S.; Gainor, J.; Shaw, A.; Hames, M.; Iams, W.; Aston, J.; Lovly, C.M.; et al. Prognostic impact of KRAS mutation subtypes in 677 patients with metastatic lung adenocarcinomas. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2015, 10, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessi, J.V.; Ricciuti, B.; Spurr, L.F.; Gupta, H.; Li, Y.Y.; Glass, C.; Nishino, M.; Cherniack, A.D.; Lindsay, J.; Sharma, B.; et al. SMARCA4 and Other SWItch/Sucrose NonFermentable Family Genomic Alterations in NSCLC: Clinicopathologic Characteristics and Outcomes to Immune Checkpoint Inhibition. J. Thorac. Oncol. 2021, 16, 1176–1187. [Google Scholar] [CrossRef]
- Papillon-Cavanagh, S.; Doshi, P.; Dobrin, R.; Szustakowski, J.; Walsh, A.M. STK11 and KEAP1 mutations as prognostic biomarkers in an observational real-world lung adenocarcinoma cohort. ESMO Open 2020, 5, e000706. [Google Scholar] [CrossRef] [Green Version]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pore, N.; Wu, S.; Standifer, N.; Jure-Kunkel, M.; de Los Reyes, M.; Shrestha, Y.; Halpin, R.; Rothstein, R.; Mulgrew, K.; Blackmore, S.; et al. Resistance to durvalumab and durvalumab plus tremelimumab is associated with functional STK11 mutations in non-small-cell lung cancer patients and is reversed by STAT3 knockdown. Cancer Discov. 2021, candisc.1543.2020. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; An, X.; Zhang, X.; Qiao, Y.; Zheng, T.; Li, X. STING: A master regulator in the cancer-immunity cycle. Mol. Cancer 2019, 18, 152. [Google Scholar] [CrossRef] [Green Version]
- Biton, J.; Mansuet-Lupo, A.; Pécuchet, N.; Alifano, M.; Ouakrim, H.; Arrondeau, J.; Boudou-Rouquette, P.; Goldwasser, F.; Leroy, K.; Goc, J.; et al. TP53, STK11, and EGFR Mutations Predict Tumor Immune Profile and the Response to Anti-PD-1 in Lung Adenocarcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 5710–5723. [Google Scholar] [CrossRef] [Green Version]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Fang, W.; Lin, Z.; Peng, P.; Wang, J.; Zhan, J.; Hong, S.; Huang, J.; Liu, L.; Sheng, J.; et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol. Immunother. 2017, 66, 1175–1187. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zheng, S.; Jin, R.; Wang, X.; Wang, F.; Zang, R.; Xu, H.; Lu, Z.; Huang, J.; Lei, Y.; et al. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 2020, 470, 95–105. [Google Scholar] [CrossRef]
- Schoenfeld, A.J.; Rizvi, H.; Bandlamudi, C.; Sauter, J.L.; Travis, W.D.; Rekhtman, N.; Plodkowski, A.J.; Perez-Johnston, R.; Sawan, P.; Beras, A.; et al. Clinical and molecular correlates of PD-L1 expression in patients with lung adenocarcinomas. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Man, J.; Lord, S.; Cooper, W.; Links, M.; Gebski, V.; Herbst, R.S.; Gralla, R.J.; Mok, T.; Yang, J.C.-H. Clinical and Molecular Characteristics Associated with Survival among Patients Treated with Checkpoint Inhibitors for Advanced Non-Small Cell Lung Carcinoma: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 210–216. [Google Scholar] [CrossRef]
- Dudnik, E.; Moskovitz, M.; Daher, S.; Shamai, S.; Hanovich, E.; Grubstein, A.; Shochat, T.; Wollner, M.; Bar, J.; Merimsky, O.; et al. Effectiveness and safety of nivolumab in advanced non-small cell lung cancer: The real-life data. Lung Cancer 2018, 126, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Morita, R.; Okishio, K.; Shimizu, J.; Saito, H.; Sakai, H.; Kim, Y.H.; Hataji, O.; Yomota, M.; Nishio, M.; Aoe, K.; et al. Real-world effectiveness and safety of nivolumab in patients with non-small cell lung cancer: A multicenter retrospective observational study in Japan. Lung Cancer 2020, 140, 8–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salehi-Rad, R.; Li, R.; Tran, L.M.; Lim, R.J.; Abascal, J.; Momcilovic, M.; Park, S.J.; Ong, S.L.; Shabihkhani, M.; Huang, Z.L.; et al. Novel Kras-mutant murine models of non-small cell lung cancer possessing co-occurring oncogenic mutations and increased tumor mutational burden. Cancer Immunol. Immunother. 2021, 70, 2389–2400. [Google Scholar] [CrossRef]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D.; Plodkowski, A.; Long, N.; Sauter, J.L.; Rekhtman, N.; et al. Molecular Determinants of Response to Anti–Programmed Cell Death (PD)-1 and Anti–Programmed Death-Ligand 1 (PD-L1) Blockade in Patients With Non–Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing. J. Clin. Oncol. 2018, 36, 633–641. [Google Scholar] [CrossRef]
- Gadgeel, S.M.; Rodriguez-Abreu, D.; Felip, E.; Esteban, E.; Speranza, G.; Reck, M.; Hui, R.; Boyer, M.; Garon, E.B.; Horinouchi, H.; et al. Abstract LB-397: Pembrolizumab plus pemetrexed and platinum vs. placebo plus pemetrexed and platinum as first-line therapy for metastatic nonsquamous NSCLC: Analysis of KEYNOTE-189 by STK11 and KEAP1 status. Cancer Res. 2020, 80, LB-397. [Google Scholar] [CrossRef]
- Cho, B.C.; Lopes, G.; Kowalski, D.M.; Kasahara, K.; Wu, Y.-L.; Castro, G.; Turna, H.Z.; Cristescu, R.; Aurora-Garg, D.; Loboda, A.; et al. Abstract CT084: Relationship between STK11 and KEAP1 mutational status and efficacy in KEYNOTE-042: Pembrolizumab monotherapy versus platinum-based chemotherapy as first-line therapy for PD-L1-positive advanced NSCLC. Cancer Res. 2020, 80, CT084. [Google Scholar] [CrossRef]
Figure 1.
STK11/LBK1 promotes cell survival under stress conditions by allowing a switch from an anabolic to a catabolic metabolism. Under energy deprivation, the accumulation of AMP and the decreased levels of ATP lead to activation of LKB1 which phosphorylate AMPK at Thr-172 in an ⍺ subunit resulting in its activation. AMPK is involved in multiple metabolic pathways to increase ATP production and to stop ATP consumption switching metabolism from an anabolic state to a catabolic state. This pathway promotes cell survival under stress conditions but impairs cell growth and proliferation.
Figure 1.
STK11/LBK1 promotes cell survival under stress conditions by allowing a switch from an anabolic to a catabolic metabolism. Under energy deprivation, the accumulation of AMP and the decreased levels of ATP lead to activation of LKB1 which phosphorylate AMPK at Thr-172 in an ⍺ subunit resulting in its activation. AMPK is involved in multiple metabolic pathways to increase ATP production and to stop ATP consumption switching metabolism from an anabolic state to a catabolic state. This pathway promotes cell survival under stress conditions but impairs cell growth and proliferation.

Figure 2.
Frequency of most common genomic alterations in tumor suppressor genes among NSCLC patients, based on TCGA (A) and the literature (B) Panel (A) is adapted from https://www.cbioportal.org/ (accessed on 1 October 2021).
Figure 2.
Frequency of most common genomic alterations in tumor suppressor genes among NSCLC patients, based on TCGA (A) and the literature (B) Panel (A) is adapted from https://www.cbioportal.org/ (accessed on 1 October 2021).

Figure 3.
Schematic impact of KRAS, STK11, and TP53 status on immune cell tumor microenvironment in lung cancer. This figure was created with BioRender.com (accessed on 1 October 2021).
Figure 3.
Schematic impact of KRAS, STK11, and TP53 status on immune cell tumor microenvironment in lung cancer. This figure was created with BioRender.com (accessed on 1 October 2021).

Figure 4.
Impact of STK11 or KEAP1 status on immune resistance during first-line therapy based on two phase III trials, evaluated chemotherapy versus immunotherapy (KEYNOTE 042) or versus chemo-immunotherapy (KEYNOTE 189). CT = chemotherapy, IO = immunotherapy.
Figure 4.
Impact of STK11 or KEAP1 status on immune resistance during first-line therapy based on two phase III trials, evaluated chemotherapy versus immunotherapy (KEYNOTE 042) or versus chemo-immunotherapy (KEYNOTE 189). CT = chemotherapy, IO = immunotherapy.

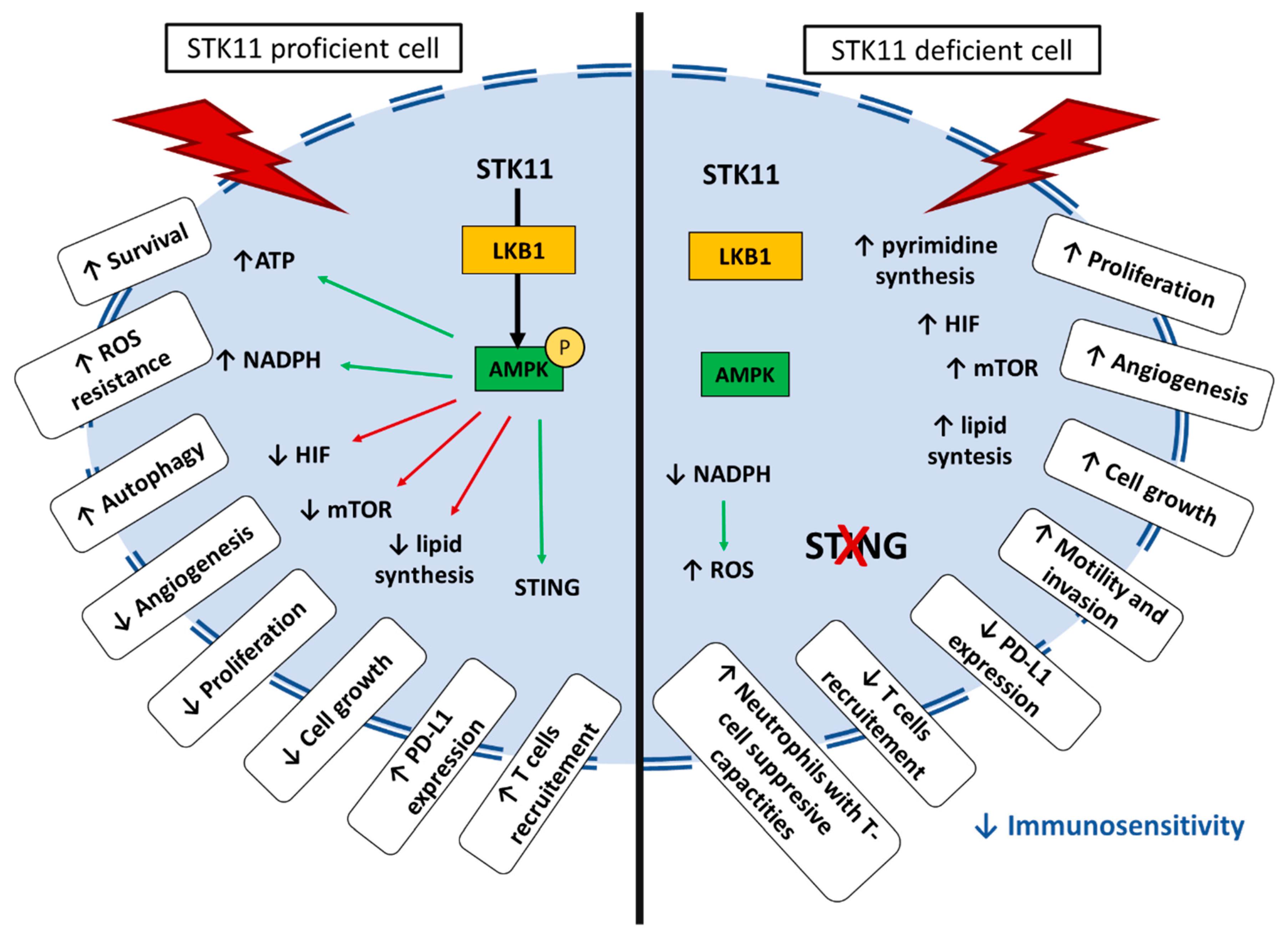
Figure 5.
Schematic representation of impact of STK11-proficient or deficient cell under stress condition.
Figure 5.
Schematic representation of impact of STK11-proficient or deficient cell under stress condition.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Frequency of STK11 mutation in each mutational status in NSCLC. TCGA. Nat Genet 2016. 1144 samples. HD: homozygous deletion) from https://www.cbioportal.org/ (accessed on 1 October 2021).
Table 1.
Frequency of STK11 mutation in each mutational status in NSCLC. TCGA. Nat Genet 2016. 1144 samples. HD: homozygous deletion) from https://www.cbioportal.org/ (accessed on 1 October 2021).
| Mutational Status | Total Number of Mutated Patients | Number of Patients with Co-Mutation Gene/STK11 | % Co-Mutation |
|---|---|---|---|
| KRAS | 259 | 63 | 24.32 |
| NTRK1 | 96 | 18 | 18.75 |
| NRAS | 30 | 4 | 13.33 |
| SMARCA4 | 94 | 11 | 11.70 |
| BRAF | 80 | 9 | 11.25 |
| NKX2-1 | 99 | 11 | 11.11 |
| ALK | 57 | 5 | 8.77 |
| ROS1 | 71 | 6 | 8.45 |
| CDKN2A (HD) | 342 | 25 | 7.29 |
| TP53 | 776 | 50 | 6.44 |
| RET | 48 | 3 | 6.25 |
| MAP2K1 | 19 | 1 | 5.26 |
| EGFR | 164 | 7 | 4.27 |
| ERBB2 | 50 | 2 | 4.00 |
| MET | 57 | 2 | 3.51 |
| PIK3CA | 276 | 9 | 3.26 |
Table 2.
Major retrospective studies evaluating prognostic or predictive impact of STK11 status on ICI efficacy.
Table 2.
Major retrospective studies evaluating prognostic or predictive impact of STK11 status on ICI efficacy.
| Publication | Population | Number of Patients | Impact of STK11 Mutation on OS | Conclusion |
|---|---|---|---|---|
| Rivizi J Clin Oncol 2018 | MSK-IMPACT Cohort
| n = 240 received ICI Comparison to a non-ICI group (n = 608) | STK11-mutant tumors were significantly enriched in the no durable benefit group (progressive disease or stable disease shorter than 6 moths), compared to the non-ICI group | Alterations in STK11 were associated with lack of benefit from ICI |
| Skoulidis Cancer Discov 2018 | MDACC cohort
| n = 66 All patients received ICI (no other groups) | STK11 mutations were associated with shorter OS
| Alterations in STK11 were associated with lack of benefit from ICI |
| Papillon-Cavanagh ESMO Open 2020 | Retrospective real-world cohort
| n= 2276 574 (25%) received first-line ICI-based therapies | STK11 mutations were associated with shorter OS
| STK11 confers a poor prognosis, regardless of treatment class with ICI or chemotherapy or targeted therapies |
| Shire Plos One 2020 | Retrospective real-word cohort
| n = 2407 patients 270 (11%) received first-line ICI-based therapies | Median OS was shorter for patients with STK11-mut compared with patients with STK11 WT:
| STK11 confers a poor prognosis, regardless of treatment class with ICI or chemotherapy |
| Shang Lung Cancer 2021 | Selected population from the POPLAR and OAK studies
| n = 598 304 received atezolizumab 294 received docetaxel | Median OS was shorter for patients with STK11-mut compared with patients with STK11 WT:
| STK11 confers a poor prognosis, regardless of treatment class with ICI (atezolizumab) or chemotherapy (docetaxel) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pons-Tostivint, E.; Lugat, A.; Fontenau, J.-F.; Denis, M.G.; Bennouna, J. STK11/LKB1 Modulation of the Immune Response in Lung Cancer: From Biology to Therapeutic Impact. Cells 2021, 10, 3129. https://doi.org/10.3390/cells10113129
AMA Style
Pons-Tostivint E, Lugat A, Fontenau J-F, Denis MG, Bennouna J. STK11/LKB1 Modulation of the Immune Response in Lung Cancer: From Biology to Therapeutic Impact. Cells. 2021; 10(11):3129. https://doi.org/10.3390/cells10113129
Chicago/Turabian StylePons-Tostivint, Elvire, Alexandre Lugat, Jean-François Fontenau, Marc Guillaume Denis, and Jaafar Bennouna. 2021. "STK11/LKB1 Modulation of the Immune Response in Lung Cancer: From Biology to Therapeutic Impact" Cells 10, no. 11: 3129. https://doi.org/10.3390/cells10113129
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.