
Pharmacological Inhibition of Wee1 Kinase Selectively Modulates the Voltage-Gated Na+ Channel 1.2 Macromolecular Complex

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Plasmid Constructs
2.3. Cell Culture
2.4. Split-Luciferase Complementation Assay (LCA)
2.5. Whole-Cell Voltage-Clamp Recordings in Heterologous Cells
3. Results
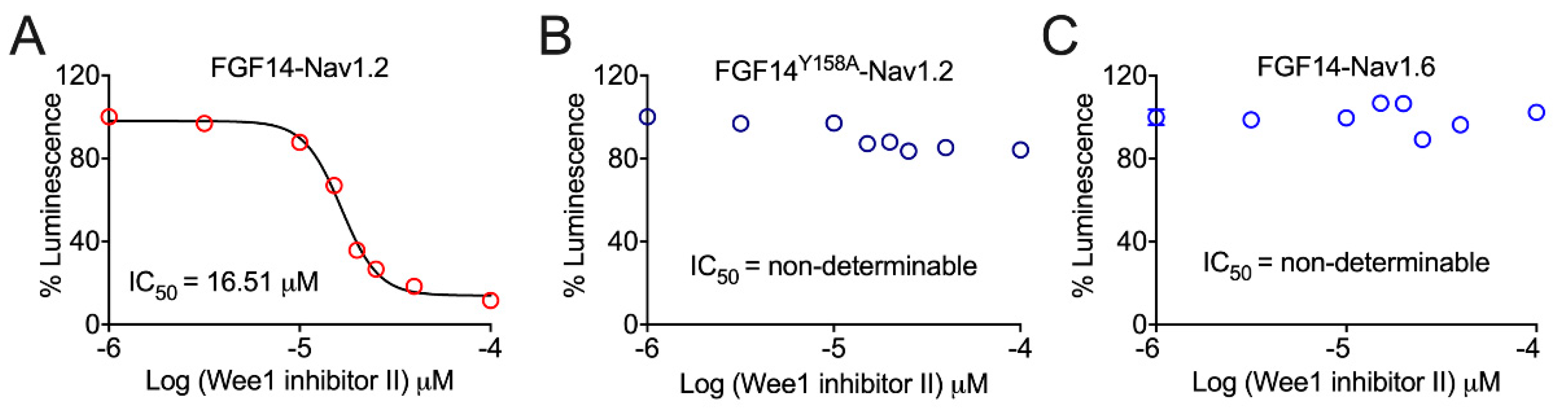
3.1. Pharmacological Inhibition of Wee1 Kinase Modulates FGF14:Nav1.2, but Not FGF14:Nav1.6, Complex Assembly in a FGF14Y158-Dependent Manner
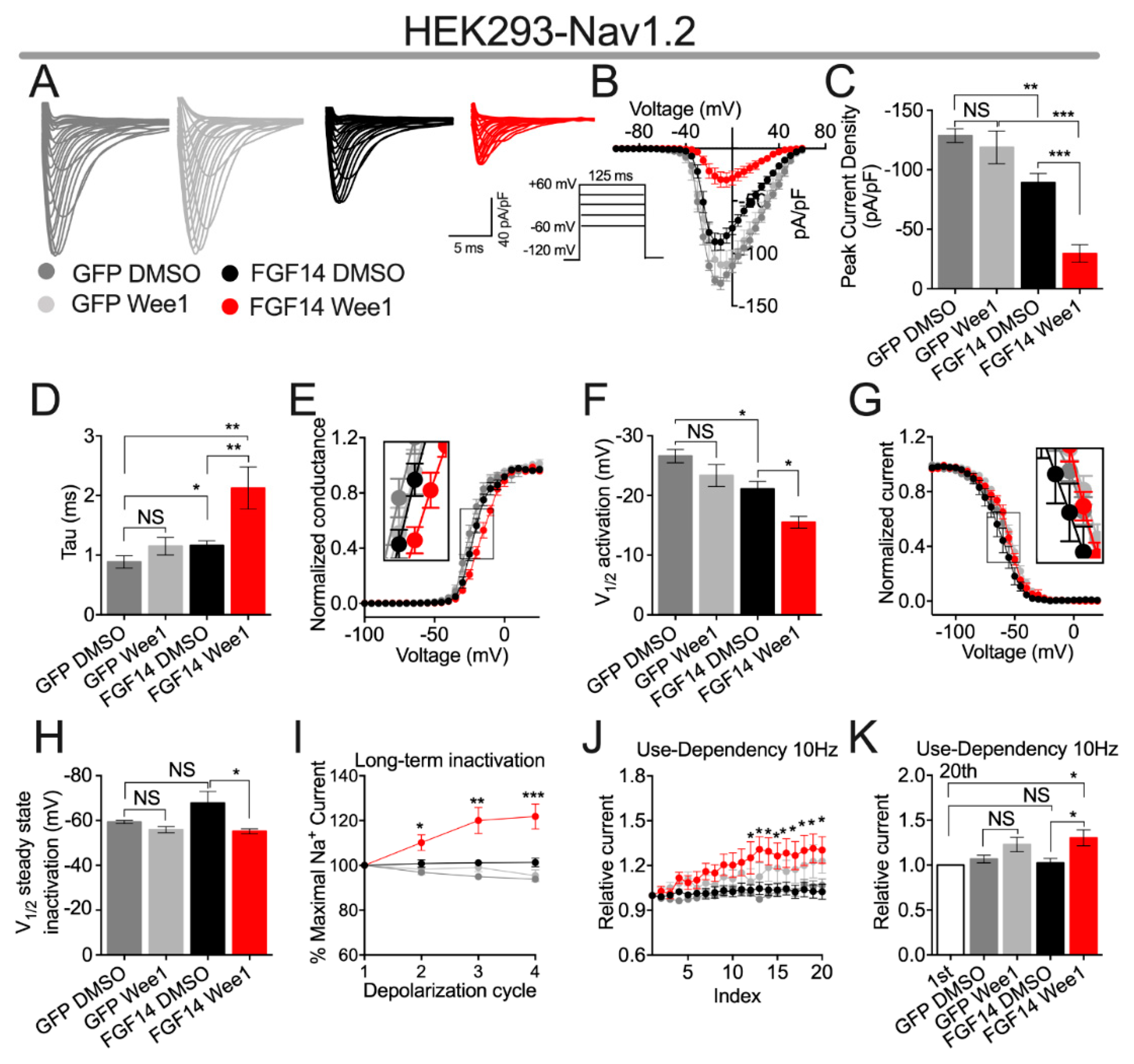
3.2. Pharmacological Inhibition of Wee1 Kinase Modulates FGF14-Mediated Regulation of Nav1.2 Channels
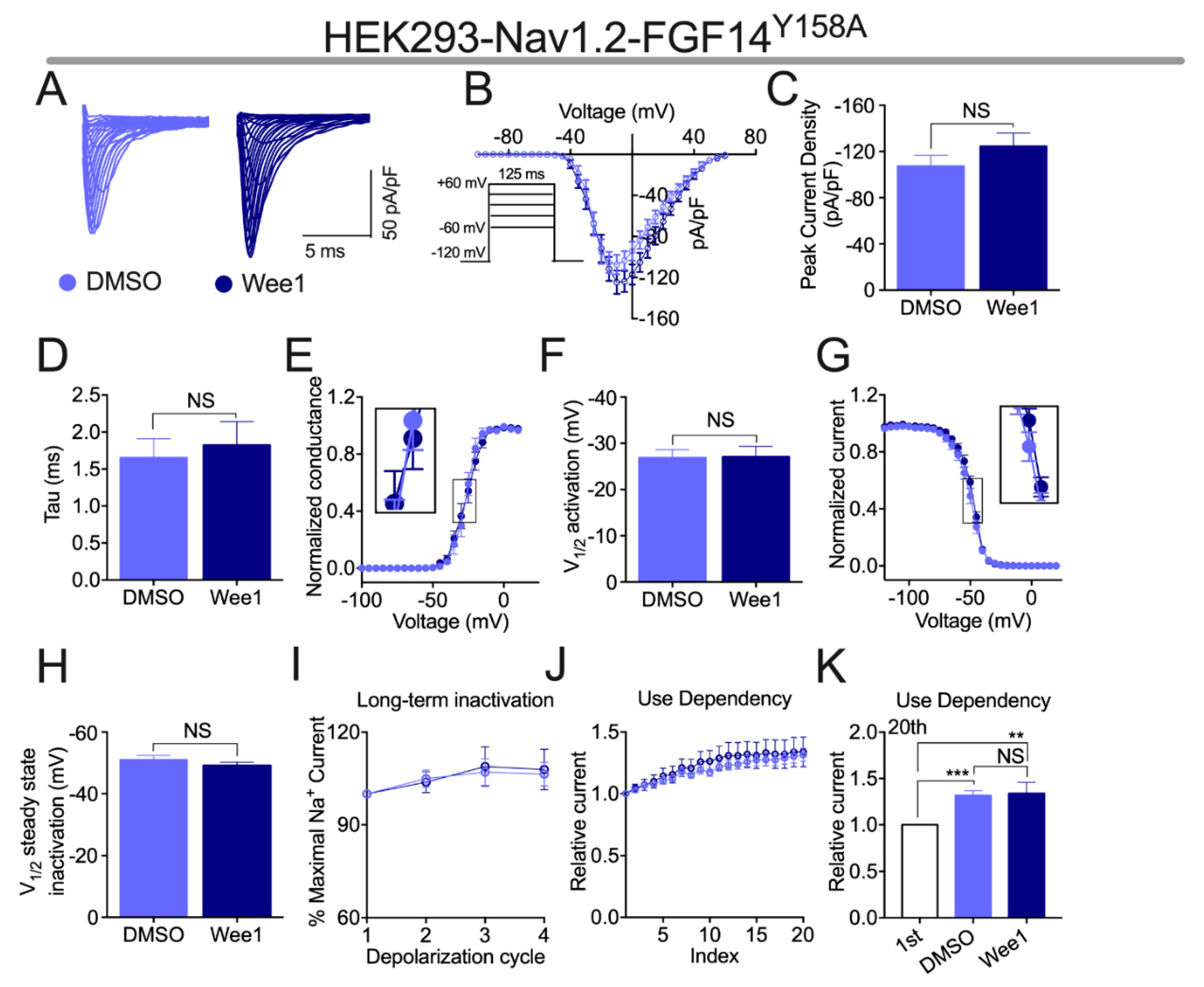
3.3. Effects of Wee1 Inhibitor II on FGF14-Mediated Regulation of Nav1.2 Channels Is Depedent upon the Presence of FGF14Y158
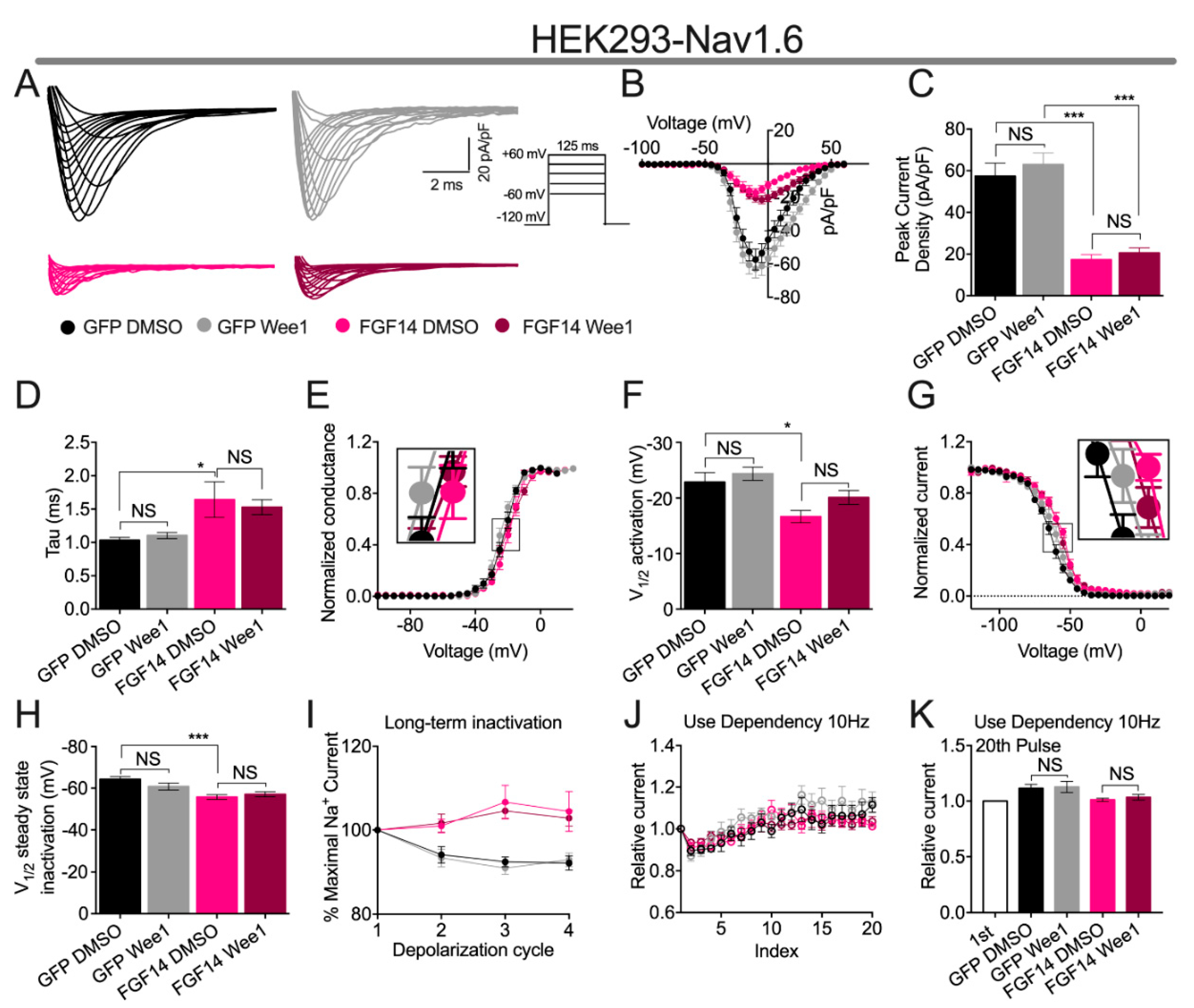
3.4. Wee1 Inhibitor II Does Not Affect FGF14-Mediated Regulation of the Nav1.6 Channel
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Catterall, W.A. Forty Years of Sodium Channels: Structure, Function, Pharmacology, and Epilepsy. Neurochem. Res. 2017, 42, 2495–2504. [Google Scholar] [CrossRef]
- Catterall, W.A. From Ionic Currents to Molecular Mechanisms: The Structure and Function of Voltage-Gated Sodium Channels. Neuron 2000, 26, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A.; Swanson, T.M. Structural Basis for Pharmacology of Voltage-Gated Sodium and Calcium Channels. Mol. Pharmacol. 2015, 88, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Tseng, T.-T.; McMahon, A.M.; Johnson, V.T.; Mangubat, E.Z.; Zahm, R.J.; Pacold, M.E.; Jakobsson, E. Sodium Channel Auxiliary Subunits. J. Mol. Microbiol. Biotechnol. 2007, 12, 249–262. [Google Scholar] [CrossRef]
- Pitt, G.S.; Lee, S.-Y. Current View on Regulation of Voltage-Gated Sodium Channels by Calcium and Auxiliary Proteins. Protein Sci. 2016, 25, 1573–1584. [Google Scholar] [CrossRef] [Green Version]
- Lou, J.-Y.; Laezza, F.; Gerber, B.R.; Xiao, M.; Yamada, K.A.; Hartmann, H.; Craig, A.M.; Nerbonne, J.M.; Ornitz, D.M. Fibroblast Growth Factor 14 Is an Intracellular Modulator of Voltage-Gated Sodium Channels. J. Physiol. 2005, 569, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Laezza, F.; Lampert, A.; Kozel, M.A.; Gerber, B.R.; Rush, A.M.; Nerbonne, J.M.; Waxman, S.G.; Dib-Hajj, S.D.; Ornitz, D.M. FGF14 N-Terminal Splice Variants Differentially Modulate Nav1.2 and Nav1.6-Encoded Sodium Channels. Mol. Cell. Neurosci. 2009, 42, 90–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, R.; Dover, K.; Laezza, F.; Shtraizent, N.; Huang, X.; Tchetchik, D.; Eliseenkova, A.V.; Xu, C.-F.; Neubert, T.A.; Ornitz, D.M.; et al. Crystal Structure of a Fibroblast Growth Factor Homologous Factor (FHF) Defines a Conserved Surface on FHFs for Binding and Modulation of Voltage-Gated Sodium Channels. J. Biol. Chem. 2009, 284, 17883–17896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Effraim, P.R.; Huang, J.; Lampert, A.; Stamboulian, S.; Zhao, P.; Black, J.A.; Dib-Hajj, S.D.; Waxman, S.G. Fibroblast Growth Factor Homologous Factor 2 (FGF-13) Associates with Nav1.7 in DRG Neurons and Alters Its Current Properties in an Isoform-Dependent Manner. Neurobiol. Pain 2019, 6, 100029. [Google Scholar] [CrossRef]
- Wittmack, E.K.; Rush, A.M.; Craner, M.J.; Goldfarb, M.; Waxman, S.G.; Dib-Hajj, S.D. Fibroblast Growth Factor Homologous Factor 2B: Association with Nav1.6 and Selective Colocalization at Nodes of Ranvier of Dorsal Root Axons. J. Neurosci. 2004, 24, 6765–6775. [Google Scholar] [CrossRef] [Green Version]
- White, H.V.; Brown, S.T.; Bozza, T.C.; Raman, I.M. Effects of FGF14 and NaVβ4 Deletion on Transient and Resurgent Na Current in Cerebellar Purkinje Neurons. J. Gen. Physiol. 2019, 151, 1300–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Pablo, J.L.; Wang, C.; Pitt, G.S. FGF14 Modulates Resurgent Sodium Current in Mouse Cerebellar Purkinje Neurons. eLife 2014, 3, e04193. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.-C.; Nenov, M.N.; Shavkunov, A.; Panova, N.; Zhan, M.; Laezza, F. Identifying a Kinase Network Regulating FGF14:Nav1.6 Complex Assembly Using Split-Luciferase Complementation. PLoS ONE 2015, 10, e0117246. [Google Scholar] [CrossRef] [Green Version]
- Wadsworth, P.A.; Singh, A.K.; Nguyen, N.; Dvorak, N.M.; Tapia, C.M.; Russell, W.K.; Stephan, C.; Laezza, F. JAK2 Regulates Nav1.6 Channel Function via FGF14(Y158) Phosphorylation. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2020, 1867, 118786. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, P.A.; Folorunso, O.; Nguyen, N.; Singh, A.K.; D’Amico, D.; Powell, R.T.; Brunell, D.; Allen, J.; Stephan, C.; Laezza, F. High-Throughput Screening against Protein:Protein Interaction Interfaces Reveals Anti-Cancer Therapeutics as Potent Modulators of the Voltage-Gated Na(+) Channel Complex. Sci. Rep. 2019, 9, 16890. [Google Scholar] [CrossRef] [PubMed]
- Scala, F.; Nenov, M.N.; Crofton, E.J.; Singh, A.K.; Folorunso, O.; Zhang, Y.; Chesson, B.C.; Wildburger, N.C.; James, T.F.; Alshammari, M.A.; et al. Environmental Enrichment and Social Isolation Mediate Neuroplasticity of Medium Spiny Neurons through the GSK3 Pathway. Cell Rep. 2018, 23, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Di Re, J.; Hsu, W.-C.J.; Kayasandik, C.B.; Fularczyk, N.; James, T.F.; Nenov, M.N.; Negi, P.; Marosi, M.; Scala, F.; Prasad, S.; et al. Inhibition of AKT Signaling Alters ΒIV Spectrin Distribution at the AIS and Increases Neuronal Excitability. Front. Mol. Neurosci. 2021, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Matheson, C.J.; Backos, D.S.; Reigan, P. Targeting WEE1 Kinase in Cancer. Trends Pharmacol. Sci. 2016, 37, 872–881. [Google Scholar] [CrossRef]
- Watanabe, N.; Arai, H.; Nishihara, Y.; Taniguchi, M.; Watanabe, N.; Hunter, T.; Osada, H. M-Phase Kinases Induce Phospho-Dependent Ubiquitination of Somatic Wee1 by SCFbeta-TrCP. Proc. Natl. Acad. Sci. USA 2004, 101, 4419–4424. [Google Scholar] [CrossRef] [Green Version]
- Li, V.S.W.; Ng, S.S.; Boersema, P.J.; Low, T.Y.; Karthaus, W.R.; Gerlach, J.P.; Mohammed, S.; Heck, A.J.R.; Maurice, M.M.; Mahmoudi, T.; et al. Wnt Signaling through Inhibition of β-Catenin Degradation in an Intact Axin1 Complex. Cell 2012, 149, 1245–1256. [Google Scholar] [CrossRef] [Green Version]
- Penas, C.; Mishra, J.K.; Wood, S.D.; Schürer, S.C.; Roush, W.R.; Ayad, N.G. GSK3 Inhibitors Stabilize Wee1 and Reduce Cerebellar Granule Cell Progenitor Proliferation. Cell Cycle 2015, 14, 417–424. [Google Scholar] [CrossRef] [Green Version]
- Cen, L.; Carlson, B.L.; Schroeder, M.A.; Ostrem, J.L.; Kitange, G.J.; Mladek, A.C.; Fink, S.R.; Decker, P.A.; Wu, W.; Kim, J.-S.; et al. P16-Cdk4-Rb Axis Controls Sensitivity to a Cyclin-Dependent Kinase Inhibitor PD0332991 in Glioblastoma Xenograft Cells. Neuro-Oncology 2012, 14, 870–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shavkunov, A.; Panova, N.; Prasai, A.; Veselenak, R.; Bourne, N.; Stoilova-McPhie, S.; Laezza, F. Bioluminescence Methodology for the Detection of Protein-Protein Interactions within the Voltage-Gated Sodium Channel Macromolecular Complex. Assay Drug Dev. Technol. 2012, 10, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Shavkunov, A.S.; Wildburger, N.C.; Nenov, M.N.; James, T.F.; Buzhdygan, T.P.; Panova-Elektronova, N.I.; Green, T.A.; Veselenak, R.L.; Bourne, N.; Laezza, F. The Fibroblast Growth Factor 14·voltage-Gated Sodium Channel Complex Is a New Target of Glycogen Synthase Kinase 3 (GSK3). J. Biol. Chem. 2013, 288, 19370–19385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.R.; Singh, A.K.; Laezza, F. Identification of Amino Acid Residues in Fibroblast Growth Factor 14 (FGF14) Required for Structure-Function Interactions with Voltage-Gated Sodium Channel Nav1.6. J. Biol. Chem. 2016, 291, 11268–11284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.R.; Liu, Z.; Nenov, M.N.; Folorunso, O.; Singh, A.; Scala, F.; Chen, H.; James, T.F.; Alshammari, M.; Panova-Elektronova, N.I.; et al. Functional Modulation of Voltage-Gated Sodium Channels by a FGF14-Based Peptidomimetic. ACS Chem. Neurosci. 2018, 9, 976–987. [Google Scholar] [CrossRef]
- Singh, A.K.; Wadsworth, P.A.; Tapia, C.M.; Aceto, G.; Ali, S.R.; Chen, H.; D’Ascenzo, M.; Zhou, J.; Laezza, F. Mapping of the FGF14:Nav1.6 Complex Interface Reveals FLPK as a Functionally Active Peptide Modulating Excitability. Physiol. Rep. 2020, 8, e14505. [Google Scholar] [CrossRef]
- Singh, A.K.; Dvorak, N.M.; Tapia, C.M.; Mosebarger, A.; Ali, S.R.; Bullock, Z.; Chen, H.; Zhou, J.; Laezza, F. Differential Modulation of the Voltage-Gated Na+ Channel 1.6 by Peptides Derived From Fibroblast Growth Factor 14. Front. Mol. Biosci. 2021, 8, 860. [Google Scholar] [CrossRef] [PubMed]
- James, T.F.; Nenov, M.N.; Wildburger, N.C.; Lichti, C.F.; Luisi, J.; Vergara, F.; Panova-Electronova, N.I.; Nilsson, C.L.; Rudra, J.S.; Green, T.A.; et al. The Nav1.2 Channel Is Regulated by GSK3. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2015, 1850, 832–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Wadsworth, P.A.; Dvorak, N.M.; Singh, A.K.; Chen, H.; Liu, Z.; Zhou, R.; Holthauzen, L.M.F.; Zhou, J.; Laezza, F. Design, Synthesis, and Pharmacological Evaluation of Analogues Derived from the PLEV Tetrapeptide as Protein–Protein Interaction Modulators of Voltage-Gated Sodium Channel 1.6. J. Med. Chem. 2020, 63, 11522–11547. [Google Scholar] [CrossRef]
- Dvorak, N.M.; Wadsworth, P.A.; Wang, P.; Chen, H.; Zhou, J.; Laezza, F. Bidirectional Modulation of the Voltage-Gated Sodium (Nav1.6) Channel by Rationally Designed Peptidomimetics. Molecules 2020, 25, 3365. [Google Scholar] [CrossRef]
- Dong, X.-W.; Priestley, T. Electrophysiological Analysis of Tetrodotoxin-Resistant Sodium Channel Pharmacology. Curr. Protoc. Pharmacol. 2004, 23, 11.8.1–11.8.33. [Google Scholar] [CrossRef]
- Tapia, C.M.; Folorunso, O.; Singh, A.K.; McDonough, K.; Laezza, F. Effects of Deltamethrin Acute Exposure on Nav1.6 Channels and Medium Spiny Neurons of the Nucleus Accumbens. Toxicology 2020, 440, 152488. [Google Scholar] [CrossRef]
- Palmer, B.D.; Thompson, A.M.; Booth, R.J.; Dobrusin, E.M.; Kraker, A.J.; Lee, H.H.; Lunney, E.A.; Mitchell, L.H.; Ortwine, D.F.; Smaill, J.B.; et al. 4-Phenylpyrrolo[3,4-c]Carbazole-1,3(2H,6H)-Dione Inhibitors of the Checkpoint Kinase Wee1. Structure−Activity Relationships for Chromophore Modification and Phenyl Ring Substitution. J. Med. Chem. 2006, 49, 4896–4911. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, M.; Schoorlemmer, J.; Williams, A.; Diwakar, S.; Wang, Q.; Huang, X.; Giza, J.; Tchetchik, D.; Kelley, K.; Vega, A.; et al. Fibroblast Growth Factor Homologous Factors Control Neuronal Excitability through Modulation of Voltage-Gated Sodium Channels. Neuron 2007, 55, 449–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Wang, C.; Hoch, E.G.; Pitt, G.S. Identification of Novel Interaction Sites That Determine Specificity between Fibroblast Growth Factor Homologous Factors and Voltage-Gated Sodium Channels. J. Biol. Chem. 2011, 286, 24253–24263. [Google Scholar] [CrossRef] [Green Version]
- Laezza, F.; Gerber, B.R.; Lou, J.-Y.; Kozel, M.A.; Hartman, H.; Craig, A.M.; Ornitz, D.M.; Nerbonne, J.M. The FGF14(F145S) Mutation Disrupts the Interaction of FGF14 with Voltage-Gated Na+ Channels and Impairs Neuronal Excitability. J. Neurosci. 2007, 27, 12033–12044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Re, J.; Wadsworth, P.A.; Laezza, F. Intracellular Fibroblast Growth Factor 14: Emerging Risk Factor for Brain Disorders. Front. Cell. Neurosci. 2017, 11, 103. [Google Scholar] [CrossRef]
- Wildburger, N.C.; Ali, S.R.; Hsu, W.-C.J.; Shavkunov, A.S.; Nenov, M.N.; Lichti, C.F.; LeDuc, R.D.; Mostovenko, E.; Panova-Elektronova, N.I.; Emmett, M.R.; et al. Quantitative Proteomics Reveals Protein-Protein Interactions with Fibroblast Growth Factor 12 as a Component of the Voltage-Gated Sodium Channel 1.2 (Nav1.2) Macromolecular Complex in Mammalian Brain. Mol. Cell. Proteom. 2015, 14, 1288–1300. [Google Scholar] [CrossRef]
- Dover, K.; Solinas, S.; D’Angelo, E.; Goldfarb, M. Long-Term Inactivation Particle for Voltage-Gated Sodium Channels. J. Physiol. 2010, 588, 3695–3711. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.J.; Dib-Hajj, S.D.; Waxman, S.G. Fibroblast Growth Factor Homologous Factor 1B Binds to the C Terminus of the Tetrodotoxin-Resistant Sodium Channel RNav1.9a (NaN). J. Biol. Chem. 2001, 276, 18925–18933. [Google Scholar] [CrossRef] [Green Version]
- Hennessey, J.A.; Marcou, C.A.; Wang, C.; Wei, E.Q.; Wang, C.; Tester, D.J.; Torchio, M.; Dagradi, F.; Crotti, L.; Schwartz, P.J.; et al. FGF12 Is a Candidate Brugada Syndrome Locus. Heart Rhythm 2013, 10, 1886–1894. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Dong, F.; Yang, Q.; Yang, P.-F.; Wu, R.; Wu, Q.-F.; Wu, D.; Li, C.-L.; Zhong, Y.-Q.; Lu, Y.-J.; et al. FGF13 Selectively Regulates Heat Nociception by Interacting with Na(v)1.7. Neuron 2017, 93, 806–821.e9. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yang, J.; Wang, H.; Shan, B.; Yin, C.; Yu, H.; Zhang, X.; Dong, Z.; Yu, Y.; Zhao, R.; et al. Fibroblast Growth Factor 13 Stabilizes Microtubules to Promote Na+ Channel Function in Nociceptive DRG Neurons and Modulates Inflammatory Pain. J. Adv. Res. 2021, 31, 97–111. [Google Scholar] [CrossRef]
- Hsu, W.-C.J.; Scala, F.; Nenov, M.N.; Wildburger, N.C.; Elferink, H.; Singh, A.K.; Chesson, C.B.; Buzhdygan, T.; Sohail, M.; Shavkunov, A.S.; et al. CK2 Activity Is Required for the Interaction of FGF14 with Voltage-Gated Sodium Channels and Neuronal Excitability. FASEB J. 2016, 30, 2171–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spratt, P.W.E.; Alexander, R.P.D.; Ben-Shalom, R.; Sahagun, A.; Kyoung, H.; Keeshen, C.M.; Sanders, S.J.; Bender, K.J. Paradoxical Hyperexcitability from NaV1.2 Sodium Channel Loss in Neocortical Pyramidal Cells. Cell Rep. 2021, 36, 109483. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Tian, C.; Li, T.; Yang, M.; Hou, H.; Shu, Y. Distinct Contributions of Na(v)1.6 and Na(v)1.2 in Action Potential Initiation and Backpropagation. Nat. Neurosci. 2009, 12, 996–1002. [Google Scholar] [CrossRef]
- Osorio, N.; Cathala, L.; Meisler, M.H.; Crest, M.; Magistretti, J.; Delmas, P. Persistent Nav1.6 Current at Axon Initial Segments Tunes Spike Timing of Cerebellar Granule Cells. J. Physiol. 2010, 588, 651–670. [Google Scholar] [CrossRef]
- Royeck, M.; Horstmann, M.-T.; Remy, S.; Reitze, M.; Yaari, Y.; Beck, H. Role of Axonal NaV1.6 Sodium Channels in Action Potential Initiation of CA1 Pyramidal Neurons. J. Neurophysiol. 2008, 100, 2361–2380. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Peak Density | Activation | Kact | Steady-State Inactivation | Kinact | Tau (τ) |
|---|---|---|---|---|---|---|
| pA/pF | mV | mV | mV | mV | ms | |
| GFP DMSO | −128.7 ± 5.7 (11) | −26.61 ± 1.1 (11) | 4.22 ± 0.4 (11) | −59.32 ± 0.7 (8) | 5.17 ± 0.28 (8) | 0.88 ± 0.10 (11) |
| GFP Wee1 | −118.8 ± 13.7 (9) ns | −23.34 ± 1.8 (9) | 4.50 ± 0.3 (9) | −55.92 ± 1.4 (10) | 5.38 ± 0.29 (10) | 1.15 ± 0.14 (10) |
| FGF14 DMSO | −89.24 ± 7.7 (11) a | −21.09 ± 1.2 (11) d | 4.47 ± 0.5 (11) | −67.76 ± 5.2 (8) | 6.04 ± 0.81 (8) | 1.16 ± 0.08 (11) h |
| FGF14 Wee1 | −29.6 ± 7.29 (9) b,c | −15.51 ± 0.9 (8) e | 6.58 ± 0.8 (8) f | −55.14 ± 1.1 (7) g | 7.58 ± 0.61 (7) | 2.13 ± 0.35 (9) i |
| FGF14Y158A DMSO | −107.3 ± 9.52 (10) | −26.88 ± 1.7 (10) | 3.04 ± 0.3 (10) | −51 ± 1.4 (9) | 5.76 ± 0.69 (9) | 1.65 ± 0.25 (10) |
| FGF14Y158A Wee1 | −124.5 ± 11.4 (11) ns | −27.08 ± 2.3 (11) | 3.74 ± 0.4 (11) | −49.07 ± 1.2 (11) | 5.35 ± 0.50 (10) | 1.82 ± 0.32 (11) |
| Condition | LTI (% Maximal Na+ Current) | ||
|---|---|---|---|
| 2nd Pulse | 3rd Pulse | 4th Pulse | |
| GFP DMSO | 96.87 ± 0.7 (7) | 94.95 ± 0.7 (7) | 93.82 ± 0.5 (7) |
| GFP Wee1 | 98.23 ± 2.3 (10) | 99.1 ± 1.9 (10) | 95.47 ± 2.6 (10) |
| FGF14 DMSO | 100.8 ± 1.6 (14) | 101.2 ± 1.4 (14) | 101.3 ± 1.9 (14) |
| FGF14 Wee1 | 110.2 ± 3.5 (8) a | 120 ± 5.8 (8) b | 121.8 ± 5.5 (8) c |
| FGF14Y158A DMSO | 105 ± 2.8 (11) | 107.1 ± 4.3 (11) | 106.4 ± 3.8 (11) |
| FGF14Y158A Wee1 | 103.8 ± 3.4 (9) | 108.9 ± 6.4 (9) | 107.9 ± 6.5 (9) |
| Condition | Peak Density | Activation | Kact | Steady-State Inactivation | Kinact | Tau (τ) |
|---|---|---|---|---|---|---|
| pA/pF | mV | mV | mV | mV | ms | |
| GFP DMSO | −57.38 ± 6.3 (8) | −22.87 ± 1.69 (8) | 4.79 ± 0.46 (8) | −60.4 ± 1.67 (8) | 5.93 ± 0.48 (8) | 1.03 ± 0.04 (8) |
| GFP Wee1 | −62.99 ± 5.5 (8) | −24.38 ± 1.19 (8) | 4.29 ± 0.43 (8) | −60.84 ± 1.67 (8) | 6.15 ± 0.40 (8) | 1.11 ± 0.04 (8) |
| FGF14 DMSO | −17.31 ± 2.5 (10) a | −18.15 ± 1.03 (8) c | 5.05 ± 0.51 (8) | −55.77 ± 1.10 (8) d | 5.654 ± 0.57 (8) | 1.64 ± 0.26 (8) e |
| FGF14 Wee1 | −20.57 ± 2.5 (10) ns,b | −20.11 ± 1.25 (8) | 5.89 ± 0.57 (8) | −57.12 ± 1.19 (8) | 6.91 ± 0.80 (7) | 1.53 ± 0.11 (9) |
| Condition | LTI (% Maximal Na+ Current) | ||
|---|---|---|---|
| 2nd Pulse | 3rd Pulse | 4th Pulse | |
| GFP DMSO | 94.11 ± 1.9 (8) | 92.43 ± 1.3 (8) | 92.18 ± 1.6 (8) |
| GFP Wee1 | 93.35 ± 2.1 (9) | 91.02 ± 1.5 (9) | 92.94 ± 1.5 (9) |
| FGF14 DMSO | 101.0 ± 1.6 (8) a | 106.7 ± 4.0 (8) b | 104.5 ± 4.7 (8) c |
| FGF14 Wee1 | 101.7 ± 2.2 (8) | 104.6 ± 1.7 (8) | 102.9 ± 1.9 (8) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dvorak, N.M.; Tapia, C.M.; Baumgartner, T.J.; Singh, J.; Laezza, F.; Singh, A.K. Pharmacological Inhibition of Wee1 Kinase Selectively Modulates the Voltage-Gated Na+ Channel 1.2 Macromolecular Complex. Cells 2021, 10, 3103. https://doi.org/10.3390/cells10113103
Dvorak NM, Tapia CM, Baumgartner TJ, Singh J, Laezza F, Singh AK. Pharmacological Inhibition of Wee1 Kinase Selectively Modulates the Voltage-Gated Na+ Channel 1.2 Macromolecular Complex. Cells. 2021; 10(11):3103. https://doi.org/10.3390/cells10113103
Chicago/Turabian StyleDvorak, Nolan M., Cynthia M. Tapia, Timothy J. Baumgartner, Jully Singh, Fernanda Laezza, and Aditya K. Singh. 2021. "Pharmacological Inhibition of Wee1 Kinase Selectively Modulates the Voltage-Gated Na+ Channel 1.2 Macromolecular Complex" Cells 10, no. 11: 3103. https://doi.org/10.3390/cells10113103